

Abstract
Alzheimer’s disease (AD) is a progressive brain disorder characterized by memory loss and the accumulation of two insoluble protein aggregates, tau neurofibrillary tangles and beta-amyloid plaques. Widespread mitochondrial dysfunction also occurs and mitochondria from AD patients display changes in number, ultrastructure, and enzyme activities. Mitochondrial dysfunction in AD presumably links in some way to its other disease characteristics, either as a cause or consequence. This review characterizes AD-associated mitochondrial perturbations and considers their position in its pathologic hierarchy. It focuses on the crosstalk that occurs between mitochondria, nuclear gene expression, and cytosolic signaling pathways that serves to maintain cell homeostasis. To this point, recent evidence indicates mitochondria trigger retrograde responses that influence cell proteostasis in general and AD proteostasis specifically. Potentially pertinent retrograde responses include the mitochondrial unfolded protein response (mtUPR), integrated stress response (ISR), autophagy/mitophagy, and proteasome function. A fuller perspective of mitochondrial dysfunction in AD, and its relation to protein aggregation, could enhance our overall understanding of this disease.
Keywords: Alzheimer’s disease, aggregation, metabolism, mitochondria, proteostasis, mitophagy, mitochondrial DNA
1. Introduction
Alzheimer’s disease (AD) accounts for ~80% of all cases of dementia, making AD the most common form of dementia (Thies and Bleiler, 2011). Clinically, AD is defined by cognitive impairment pervasive enough to interfere with a person’s ability to work or complete daily activities. The main histologic characteristics include amyloid plaques that largely contain amyloid beta (Aβ) protein and neurofibrillary tangles (NFTs) that consist of hyperphosphorylated tau protein. There is often concomitant brain atrophy in the temporal lobe and medial parietal cortex, and decreased glucose utilization that at least early on predominates in posterior brain regions (McKhann et al., 2011). Current AD treatments confer a slight benefit but ultimately do not prevent progression (Grossberg et al., 2019).
Different AD variants are recognized. Most cases are genetically influenced but not considered genetically determined, at least not in a Mendelian sense (Swerdlow, 2007). Advancing age is particularly relevant in these cases, and prevalence and incidence increase in older populations. There are also Mendelian forms, which typically manifest in middle age within an autosomal dominant context. Autosomal dominant AD is caused by mutations in the amyloid precursor protein (APP) gene that carries within it the Aβ amino acid sequence, the presenilin 1 (PS1) gene which encodes a subunit of the gamma secretase that cleaves APP, and the presenilin 2 (PS2) gene that is homologous to PS1 (Goate et al., 1991; Kelleher and Shen, 2017; Murrell et al., 1991).
Synapse loss and brain atrophy correlate well with cognitive decline, and brain volume measurements can predict disease progression and differentiate AD subtypes (DeKosky and Scheff, 1990; Risacher et al., 2017). Brain glucose utilization, measured using (18F)-2-fluoro-deoxy-D-glucose positron emission tomography (FDG-PET), also correlates with cognitive decline (Hoffman et al., 2000; Nestor et al., 2003; Silverman et al., 2001). Evidence suggests brain hypometabolism may be useful in early AD detection and diagnosis, as well as differential dementia diagnosis (Mosconi et al., 2008a; Mosconi et al., 2008b). Brain hypometabolism certainly occurs early in AD and worsens as the disease progresses. It is possible that brain hypometabolism may occur in the absence of discernable Aβ or tau pathology (Minoshima et al., 1997; Mosconi et al., 2007; Reiman et al., 1996).
2. Metabolism-pertinent associations and alterations
Metabolism-related health parameters including diabetes, hypertension, and hypercholesterolemia associate with AD (Rosendorff et al., 2007; Schrijvers et al., 2010). Obesity during middle age increases AD risk three times, while overweight status increases AD risk two-fold (Whitmer et al., 2007).
Insulin receptor levels increase in the AD brain compared to age-matched controls (Frolich et al., 1998), but AD brains also display reduced glucose transporter levels, even after correcting for neuronal loss (Simpson et al., 1994). Brain derived neurotrophic factor (BDNF), an important regulator of neuronal development, survival, and metabolism is altered in the AD brain (Phillips et al., 1991).
Metabolomic studies provide more specific information regarding AD metabolic deficiencies (An et al., 2018). Studies utilizing nuclear magnetic resonance spectroscopy to assay metabolites consistently associate reduced N-acetylaspartate (NAA) with neurodegeneration (Barba et al., 2008). NAA’s biological function in the brain is not well described, although it is worth noting neuron mitochondria synthesize NAA (Baslow, 2003). NAA may contribute to lipid conversion to myelin, aspartate storage, or osmoregulation, although these roles remain speculative (Choi et al., 2007). NAA levels decrease in the AD brain, although NAA reduction does not differentiate AD from other neurodegenerative diseases. AD brains also display increased myo-inositol (mI). Little is known about the biological importance of mI changes (Kantarci et al., 2007; Martinez-Bisbal et al., 2004).
Lipid homeostasis is increasingly implicated in AD. Consistent correlations between sphingolipids and AD progression and severity are recognized (Varma et al., 2018). Ceramide accumulates in AD brains, which is of interest because ceramide can stimulate apoptosis (He et al., 2010). Lipid changes in AD blood or CSF may have the potential to serve as disease biomarkers (Wong et al., 2017).
3. Susceptibility to mitochondrial dysfunction and the AD COX defect
Mitochondria contribute to diverse cellular processes including apoptosis, cell growth and division, calcium storage, and lipid metabolism (Galluzzi et al., 2012). Deficient mitochondrial function is associated with numerous diseases (Wallace, 2005) and a driving role for mitochondrial dysfunction is recognized in multiple diseases (Niyazov et al., 2016). Primary mitochondrial disorders preferentially affect the brain and heart, organs known for their high energy demands. Based on these observations, the brain and heart are thought to have a lower threshold for tolerating mitochondrial dysfunction relative to other tissues (Wallace and Chalkia, 2013).
Changes in mitochondrial enzyme activity occur in AD (Swerdlow, 2012). Reduced cytochrome oxidase (COX) activity has consistently been observed across numerous tissues. Initial studies describe decreased COX activity in AD patient brains and platelets. The discovery of COX deficiency in non-degenerating tissues such as platelets suggests mitochondrial dysfunction occurs independently of neurodegenerative processes (Parker et al., 1990). Purified COX from AD brains showed anomalous kinetic behavior (Parker and Parks, 1995). COX activity reductions are easier to detect than complex I and II-III activity reductions (Mutisya et al., 1994).
Mitochondrial DNA (mtDNA) could contribute to reduced AD COX activity, and this possibility was addressed in studies utilizing cytoplasmic hybrids (cybrids). Generating cybrids involves repopulating mtDNA depleted cells (ρ0 cells) with exogenous mtDNA from human subject-derived platelets. Cybrid cell lines containing mitochondria transferred from AD patients, called AD cybrids, effectively model AD mitochondrial function on a stable nuclear background. This includes reduced COX activity and decreased mitochondrial oxygen consumption. AD cybrids also have decreased NAD+/NADH and increased ADP/ATP ratios (Silva et al., 2013). The collective cybrid evidence argues mtDNA contributes to AD mitochondrial dysfunction (Ghosh et al., 1999; Swerdlow et al., 1997).
Post-mortem analysis of AD brain tissue reveals decreases in COX subunits with disease progression (Kish et al., 1999). Elderly control subjects show decreased COX protein relative to young control subjects (Ojaimi et al., 1999). These findings suggest COX levels decline with age, and can surpass a threshold that associates with and possibly contributes to AD
AD hippocampi show increased numbers of COX deficient neurons. In one relevant study, the investigators performed an immunohistochemistry survey using antibodies to COX and complex II subunits. Neurons lacking COX staining in the presence of preserved complex II staining were designated COX deficient. This result suggests it is possible to selectively reduce COX subunits without proportionally eliminating other mitochondrial constituents (Cottrell et al., 2001). A subsequent study looked for correlations between COX deficiency and AD pathology, and found tangle-containing neurons are consistently COX-positive (Cottrell et al., 2002). The reasons for this are unclear but the non-randomness of the observation is consistent with the possibility of an NFT-mitochondria relationship.
4. Other AD mitochondrial defects
AD mitochondria show numerous changes (Swerdlow, 2012). Mitochondrial surface area decreases, while cristae structure is altered and increased variability in mitochondrial shape occurs (Baloyannis, 2006). Changes in the mitochondrial fission and fusion machinery are early AD features that may precipitate these changes. There are changes in fusion and fission proteins that appear to affect mitochondrial localization in AD neurons (Manczak et al., 2011; Wang et al., 2008). Indeed, neuronal cultures with AD-relevant fission/fusion protein alterations recapitulate AD-like changes in mitochondrial distribution (Wang et al., 2009).
Three-dimensional electron microscopy (3D EM) studies of AD brains revealed a novel mitochondrial morphology called “mitochondria on a string” (MOAS) (Zhang et al., 2016). The investigators who described this proposed MOAS formation occurs in response to AD bioenergetic stress. MOAS formation may inhibit mitophagy, thereby preserving a low level of mitochondrial function under extreme stress. Related findings suggested MOAS arises due to arrested mitochondrial fission.
Lysosomal alterations are potentially tied to AD mitochondrial defects. AD neurons attempt to increase lysosomal proteins early in disease progression, specifically upregulating cathepsin D mRNA and protein levels with concomitant increases in lysosomal structures (Cataldo et al., 1995). Dystrophic AD neurons experience disrupted axonal and dendritic autophagy, with an accompanying accumulation of partially digested cellular components (Nixon and Yang, 2011). Along these lines, AD neurons from one study reported increased COX protein and mtDNA levels despite morphometric analyses showing reduced mitochondrial mass (Hirai et al., 2001). Rather than signaling an increase in functional mitochondria, observed increases in COX and mtDNA in that study potentially reflected deficient mitochondrial degradation. In AD neurons containing increased mitochondrial components, those components tended to colocalize with lysosomal structures (Hirai et al., 2001).
Mitophagy impairment also occurs in the AD brain. A recent study found reductions in numerous elements of the mitophagy program in AD hippocampus, in induced pluripotent stem cell-derived human AD neurons, and in AD animal models. The authors concluded impaired mitophagy may contribute to AD and enhancing mitochondrial clearance may prove therapeutically useful (Fang et al., 2019).
The root cause for AD mitochondrial dysfunction remains controversial. Initially, researchers assumed mitochondrial defects were a consequence of neuronal degeneration, but considerable evidence argues this is unlikely to represent the sole cause. Aβ or tau pathology may also trigger mitochondrial defects (Caspersen et al., 2005; Devi et al., 2006), but cannot account for its entirety. If neurodegeneration, Aβ, or tau pathology do not entirely account for AD mitochondrial dysfunction, then what other possibilities warrant consideration?
5. Potential role of mtDNA
MtDNA inheritance and an accumulation of somatic mtDNA mutations influence mitochondrial function. These effects play out against the nuclear genome, certainly on a fundamental level and perhaps also in AD (Andrews et al., 2019). Studies implicate somatic mtDNA mutations in aging and AD progression (Corral-Debrinski et al., 1992; Corral-Debrinski et al., 1994; Cottrell et al., 2001).
Deletions represent one type of somatic mtDNA mutation that may contribute to neurodegeneration (Corral-Debrinski et al., 1992). The 4,997 base pair “common deletion” increases in the brain during normal aging. Brain areas with the highest metabolic activity display the greatest common deletion burden. One study that focused on AD COX-deficient neurons found an association between COX deficiency and increased aging-related mtDNA deletions (Krishnan et al., 2012), which suggests mtDNA deletions may contribute to AD bioenergetic defects. Early AD brains display increased common deletion burden compared to age-matched controls. Around age 75, though, the frequency of common deletion detection actually declines in the AD brain. This is in contrast to age-matched control brains, which accumulate the common deletion to a greater extent beyond age 75 (Corral-Debrinski et al., 1994; Hamblet and Castora, 1997).
Oxidative damage often associates with mitochondrial dysfunction and may contribute to mtDNA damage (Lovell and Markesbery, 2007). Levels of oxidative damage increase in the AD brain, especially in the parietal lobe (Nunomura et al., 2001; Nunomura et al., 1999; Perry et al., 2002). One study found a three-fold increase in the amount of an oxidized nucleoside related to AD brain mtDNA, while AD nuclear DNA displayed only a small increase in oxidative damage (Mecocci et al., 1994). This established that mtDNA experiences increased oxidative damage in AD and suggests mtDNA is more prone to damage by this mechanism than nuclear DNA. It is interesting to note that the region displaying the most mtDNA oxidative damage, the parietal lobe, also exhibits early reductions in glucose consumption during AD (Silverman et al., 2001), suggesting a possible link between mitochondrial dysfunction and brain hypometabolism.
Another study reported mtDNA control region point mutations accumulate to a greater extent in the AD brain (Coskun et al., 2004). Mutations in this mtDNA segment correlate with disrupted mtDNA transcription and replication. The AD brains in this study showed an approximate 50% reduction in their mtDNA/nuclear DNA ratio. Control region mutations, therefore, may also contribute to AD mitochondrial dysfunction.
More recent studies utilizing chip-based or next generation sequencing reveal increased mtDNA substitution variants in AD patients (Casoli et al., 2014; Casoli et al., 2015). In one study that assessed mtDNA integrity in early stage AD patient hippocampi, there was an observed dramatic increase in mtDNA mutations. Based on further analysis, the authors speculated this increase more likely reflected an enhanced accumulation of replication errors than a direct consequence of oxidative damage (Hoekstra et al., 2016).
MtDNA inheritance may also influence AD risk. Some studies suggest AD shows a maternal inheritance predominance (Edland et al., 1996), which is consistent with an mtDNA effect because mtDNA is maternally inherited. It is possible, though, that excess maternal inheritance reflects an artifact of enhanced female longevity or perhaps an inherently increased female AD risk (Heggeli et al., 2012). Other studies show a maternal AD history also associates with brain hypometabolism and lower COX activity in cognitively normal, middle aged adults (Mosconi et al., 2007; Mosconi et al., 2011). Furthermore, iIndividuals with a maternal AD history experience progressive brain hypometabolism in the same brain regions as AD patients (Mosconi et al., 2009). Epidemiology and endophenotype studies, therefore, are consistent with the possibility that mtDNA contributes to both AD risk and biomarker endpoints.
The AD cybrid data also suggest mtDNA contributes to AD pathogenesis (Swerdlow et al., 2017). As mentioned earlier, AD cybrids display reduced COX activity, while maintaining activity of other respiratory chain complexes (Ghosh et al., 1999). Beyond COX deficiency, AD cybrids produce more reactive oxygen species (ROS), have swollen mitochondria with reduced membrane potential, and recapitulate AD-like pathology features (Khan et al., 2000; Swerdlow et al., 1997; Trimmer et al., 2000). AD cybrids have elevated intracellular Aβ and secrete Aβ into the media at a greater rate (Khan et al., 2000; Onyango et al., 2010). Concomitantly, AD cybrids increase caspase-3 activity and cytochrome c release. These apoptotic responses may facilitate Aβ accumulation in AD cybrids (Khan et al., 2000). Aβ treatment in AD cybrids leads to an accentuated apoptotic response compared to age-matched control cybrids (Cardoso et al., 2004).
6. An AD mitochondrial cascade hypothesis
A mitochondrial cascade hypothesis argues AD neuropathology arises secondary to mitochondrial dysfunction (Swerdlow, 2018; Swerdlow et al., 2010, 2014; Swerdlow and Khan, 2004; Swerdlow and Khan, 2009). This view of AD does not attempt to discount the potential detrimental effects of aggregating proteins and tries to provide a rationale for why protein aggregation occurs in this disease.
The mitochondrial cascade hypothesis proposes an individual’s mtDNA and nuclear DNA inheritance determine their baseline mitochondrial function and durability. As the individual ages, their mitochondrial dysfunction declines; this decline may represent a consequence of, or occur independent of, somatic mtDNA mutation. Initially, compensatory responses can maintain adequate mitochondrial function in the face of diminished efficiency. At some point, though, the extent of mitochondrial function exceeds the limits of compensation and disease ensues (Figure 1). Insoluble protein aggregates may appear in response to changing mitochondrial function, either during the initial compensation phase or the subsequent uncompensated phase (Swerdlow et al., 2010).
Figure 1. Compensated versus uncompensated brain aging as predicted by the mitochondrial cascade hypothesis.
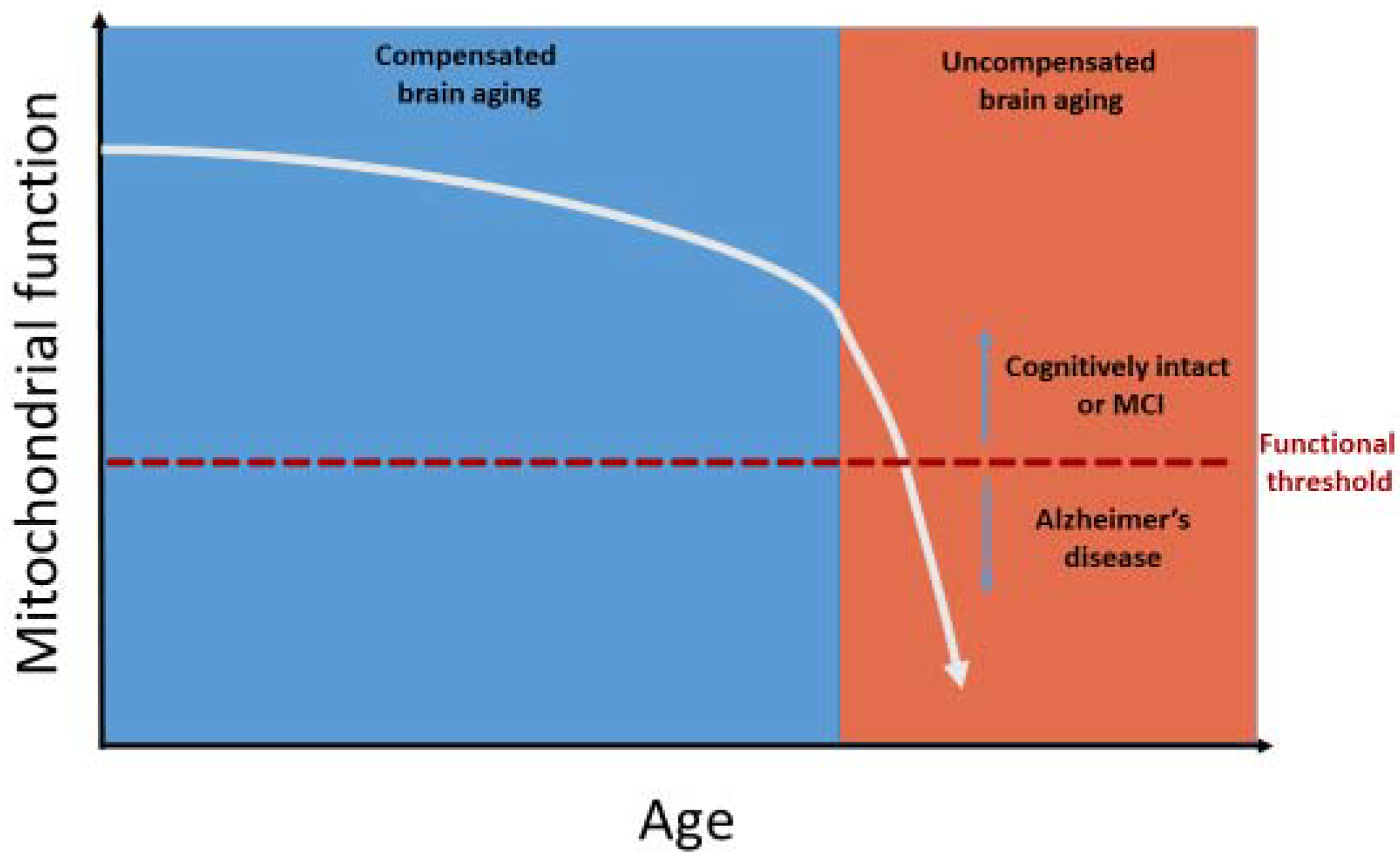
The sporadic AD mitochondrial cascade hypothesis proposes mitochondrial dysfunction progressively declines with advancing age, which initially prompts compensation (compensated brain aging). At some point, this decline reaches a point at which adequate compensation is no longer possible, and the brain transitions from compensated to uncompensated brain aging. Clinical symptoms and signs are most evident during the stage of uncompensated brain aging.
Threshold effects are pertinent to the mitochondrial cascade hypothesis. Each organ likely possesses its own threshold for tolerating mitochondrial dysfunction, beyond which normal function becomes impossible. Neurons are highly dependent on mitochondria for a variety of functions, rendering them particularly susceptible to mitochondrial dysfunction (Tomasi et al., 2013; Wallace and Chalkia, 2013). This could explain why AD presents primarily as a brain disease, albeit one that associates with subtle systemic phenotypes.
7. Connections between mitochondria and protein aggregation
Many studies report the effects of amyloid and tau species on mitochondrial function. Exposing cultured cells to Aβ peptides perturbs multiple mitochondrial endpoints, including mitochondrial membrane potential, respiratory chain activities, and oxygen consumption (Pereira et al., 1998). Isolated mitochondria, when incubated with Aβ, show decreased COX, alpha-ketoglutarate, and pyruvate dehydrogenase activities, which recapitulates observed reductions of these enzyme activities in tissues from AD subjects (Casley et al., 2002; Gibson et al., 1998). APP accumulates in mitochondrial translocases, where it appears to disrupt mitochondrial function (Anandatheerthavarada et al., 2003; Anandatheerthavarada and Devi, 2007; Devi et al., 2006). Aβ is found within mitochondria, where it interacts with a mitochondrial matrix alcohol dehydrogenase alternatively referred to as the Aβ-binding alcohol dehydrogenase (ABAD) enzyme. ABAD contributes to proper mitochondrial function and Aβ binding inhibits ABAD activity (Lustbader et al., 2004). Other studies interrogating the co-localization of mutant APP/Aβ with mitochondria show a physical interaction with cyclophilin D (Du et al., 2008), as well as accentuated mitochondrial free radical production and oxidative DNA damage (Manczak et al., 2006).
Studies of AD brain suggest mitochondrial transport throughout the cell is disturbed in AD neurons (Panchal and Tiwari, 2019). AD neurons contain reduced numbers of synapse mitochondria (Pickett et al., 2018). The reasons for this are not entirely clear, but one hypothesized cause is tau aberration. Tau contributes to microtubule stability, and dysfunctional tau may preclude the physiologic transport of mitochondria along microtubules.
Direct interactions between tau and mitochondria are also reported. Low-capacity runner (LCR) rats display metabolic defects along with hippocampal neurodegeneration. These rats accumulate hyperphosphorylated tau within their mitochondria. In this model, co-localization of tau with mitochondria is not an artifact of forced overexpression. The authors of this study speculated mitochondrial dysfunction in LCR rats altered tau physiology and contributed to neurodegeneration (Choi et al., 2014).
Additional studies reveal mitochondria-localized tau protein. In AD brains, hyperphosphorylated tau accumulates in voltage dependent anion channel 1 (VDAC1) structures present on the mitochondrial outer membrane (Manczak and Reddy, 2012). Phosphorylated tau also interacts with VDAC1 in APP, APP/PS1, and triple transgenic AD mice, where it appears to inhibit VDAC1 function (Manczak and Reddy, 2012).
Truncated tau species with the ability to impede mitochondrial function collect in AD neurons. Tau cleaved at aspartate 421 (Asp-421) increases in AD brains, and Aβ can promote this processing (Gamblin et al., 2003). Overexpressing this tau fragment in neuronal cell culture causes oxidative stress and mitochondrial fragmentation (Quintanilla et al., 2009). Overexpressing a different tau fragment, NH2-26-44, causes primary neuron death. N-terminal tau fragments cause mitochondrial dysfunction by disrupting adenine nucleotide transporter (ANT) function (Atlante et al., 2008).
Another AD related protein, apolipoprotein E (apoE), is pertinent to mitochondria. The APOE ε4 (APOE4) allele is the strongest known sporadic AD genetic risk factor. The apoE4 protein builds up in endosomal compartments and poorly stimulates cholesterol efflux relative to the apoE2 and apoE3 isoforms (Heeren et al., 2004). The apoE4 isoform appears particularly susceptible to c-terminal protease cleavage. Neuronal cells transfected with truncated apoE4 display increased NFT formation, suggesting apoE4 fragments can induce NFT formation (Huang et al., 2001). Subsequent studies revealed physical associations between apoE fragments and mitochondrial proteins. ApoE4 binds to mitochondrial complex III and IV subunits, and apoE4 fragments bind more strongly than full length apoE4. Expressing truncated apoE4 in Neuro-2a cells reduced complex III and IV activity compared to cells expressing full length apoE4. This line of research suggests apoE4-derived peptide fragments promote neurodegeneration by inhibiting mitochondrial function (Chang et al., 2005; Chen et al., 2011; Nakamura et al., 2009).
While proteins such as Aβ, APP, tau, and apoE can influence mitochondria, a reciprocal relationship in which mitochondria influence these proteins is also recognized (Figure 2). Fibroblasts treated with a mitochondrial uncoupler display increased tau phosphorylation at AD-relevant sites (Blass et al., 1990). This suggests the uncoupling of oxidative phosphorylation reported in the AD brain (Sims et al., 1987) may contribute to tau abnormalities. Studies with complex I inhibitors also demonstrate a relationship between mitochondrial function and tau. Two complex I inhibitors, annonacin and 1-methyl-4-phenylpyridinium (MPP+), alter tau splicing in human neurons, which leads to a predominance of 4R tau (Bruch et al., 2014). In rats, chronic rotenone treatment increases brain tau hyperphosphorylation and aggregation, as well as alpha-synuclein deposits (Hoglinger et al., 2005).
Figure 2. Mitochondria and AD-relevant proteins influence each other.
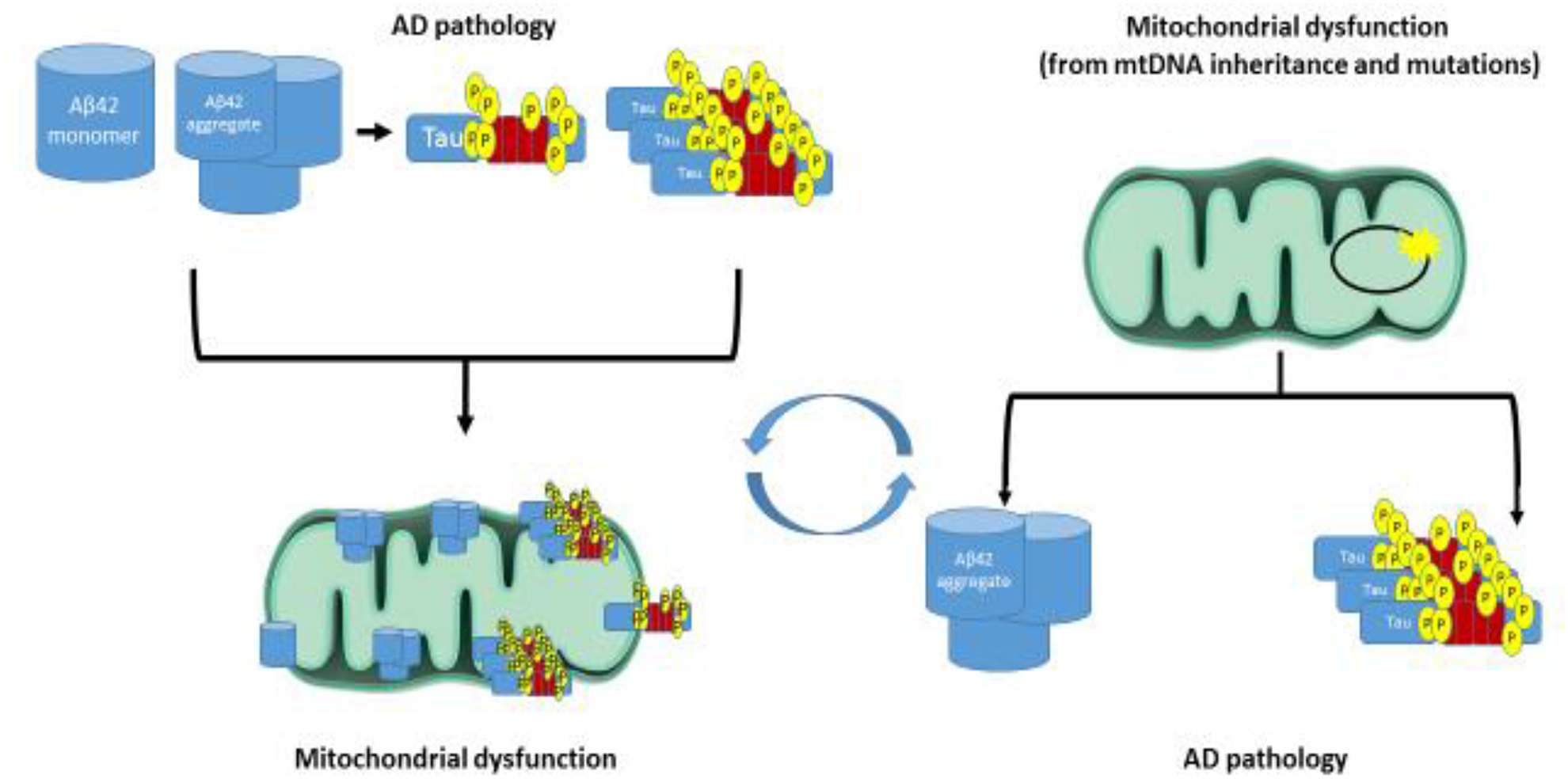
Amyloid and tau pathology could drive AD mitochondrial dysfunction. Alternatively, AD mitochondrial dysfunction could exist independent of these proteins, and may in fact set the stage for amyloid plaque and NFT formation.
8. Protein stress responses, mitochondria, and cell proteostasis
Unfolded protein responses (UPRs) help maintain cell proteostasis. Well described mechanisms for responding to unfolded proteins exist for the endoplasmic reticulum (ER) and the cytosol. The UPR in the ER (erUPR) utilizes numerous strategies to maintain cell proteostasis in the face of diverse stressors. One involves increasing ER chaperone protein expression to restore proper protein folding and prevent aggregation. Simultaneously, ER stress may initiate eukaryotic initiation factor 2α (eIF2α) phosphorylation, which inhibits general protein translation to reduce the cell protein load. The erUPR also includes a process called ER-associated degradation (ERAD). ERAD components degrade misfolded and accumulated proteins (Bravo et al., 2013). Together, these programs identify and rectify protein misfolding and aggregation in the ER to maintain homeostasis. Prolonged erUPR activation, however, promotes inflammatory responses and apoptosis (Fribley et al., 2009; Grootjans et al., 2016).
The cytosol also senses and responds to misfolded and aggregating proteins. Exposed hydrophobic residues act as markers of peptide misfolding. Cytosolic chaperones, including heat shock protein 70 (Hsp70) and heat shock protein 90 (Hsp90) family members, sense misfolding and refold proteins. The ubiquitin-proteasome system (UPS) degrades unsalvageable damaged or unfolded proteins. Members of the E3 ubiquitin ligase family tag misfolded proteins and designate them for destruction by the proteasome. ATP-independent small heat shock proteins (sHsps) help solubilize and prevent protein aggregates through direct binding (Buchberger et al., 2010).
Aggresomes additionally mediate cytosolic proteostasis. Aggresomes form when misfolded proteins accumulate into large inclusion bodies, which serve as misfolded protein repositories. Proteasomal components and chaperones move to aggresomes and attempt to degrade them (Kawaguchi et al., 2003). Autophagy-lysosome machinery also contributes to aggresome removal (Zaarur et al., 2014). Aggresome formation typically follows proteasome failure (Johnston et al., 1998).
Increasing evidence reveals an important role for mitochondria in proteostasis. Mitochondria possess intrinsic chaperones and proteases that handle internally misfolded proteins and aggregation. The presence of unfolded protein within mitochondria triggers a mitochondrial unfolded protein response (mtUPR), which upregulates the mitochondria’s resident chaperones and proteases (Jovaisaite et al., 2014). Most evidence for the mtUPR comes from studies in Caenorhabditis elegans (C. elegans). Diverse mitochondrial insults stimulate the mtUPR in C. elegans, including decreases in mitochondrial ribosomes, mtDNA depletion, and impaired mitochondrial translation. Respiratory chain inhibition and knockdown of mitochondrial chaperones and proteases also initiate the mtUPR. The mtUPR activates and operates independent of cytosolic heat shock and erUPR stress responses (Yoneda et al., 2004).
In C. elegans, activating transcription factor associated with stress-1 (ATFS1) mediates the mtUPR. Under non-stress conditions, mitochondria import ATFS-1 where it undergoes degradation. Mitochondrial impairment or stress impairs ATFS-1 import, which redirects it to the nucleus. Nuclear ATFS-1 upregulates the expression of mitochondrial chaperones and proteases (Nargund et al., 2012). In mammalian cells, activating transcription factor 5 (ATF5) plays a similar role to ATFS-1, mediating the mtUPR under select conditions. However, on some parameters the mammalian and C. elegans mtUPRs diverge, as disrupting mitochondrial translation, respiration, protein import, and membrane potential fail to activate the mammalian mtUPR (Fiorese et al., 2016).
Although these mitochondrial insults fail to engage the mammalian mtUPR, they do activate other proteostasis mechanisms, including the integrated stress response (ISR) (Figure 3). Mitochondrial stress inhibits cytosolic translation by increasing eIF2α phosphorylation. While eIF2α phosphorylation represses general cell translation, the translation of certain stress response factors, including activating transcription factor 4 (ATF4), increases. ATF4 knockdown impedes cell proliferation and slows recovery following ethidium bromide-induced mtDNA depletion (Quirós et al., 2017). Another study found respiration inhibitors induce an ATF4-dependent stress response (Garaeva et al., 2016).
Figure 3. Mitochondrial stress impacts cellular proteostasis.
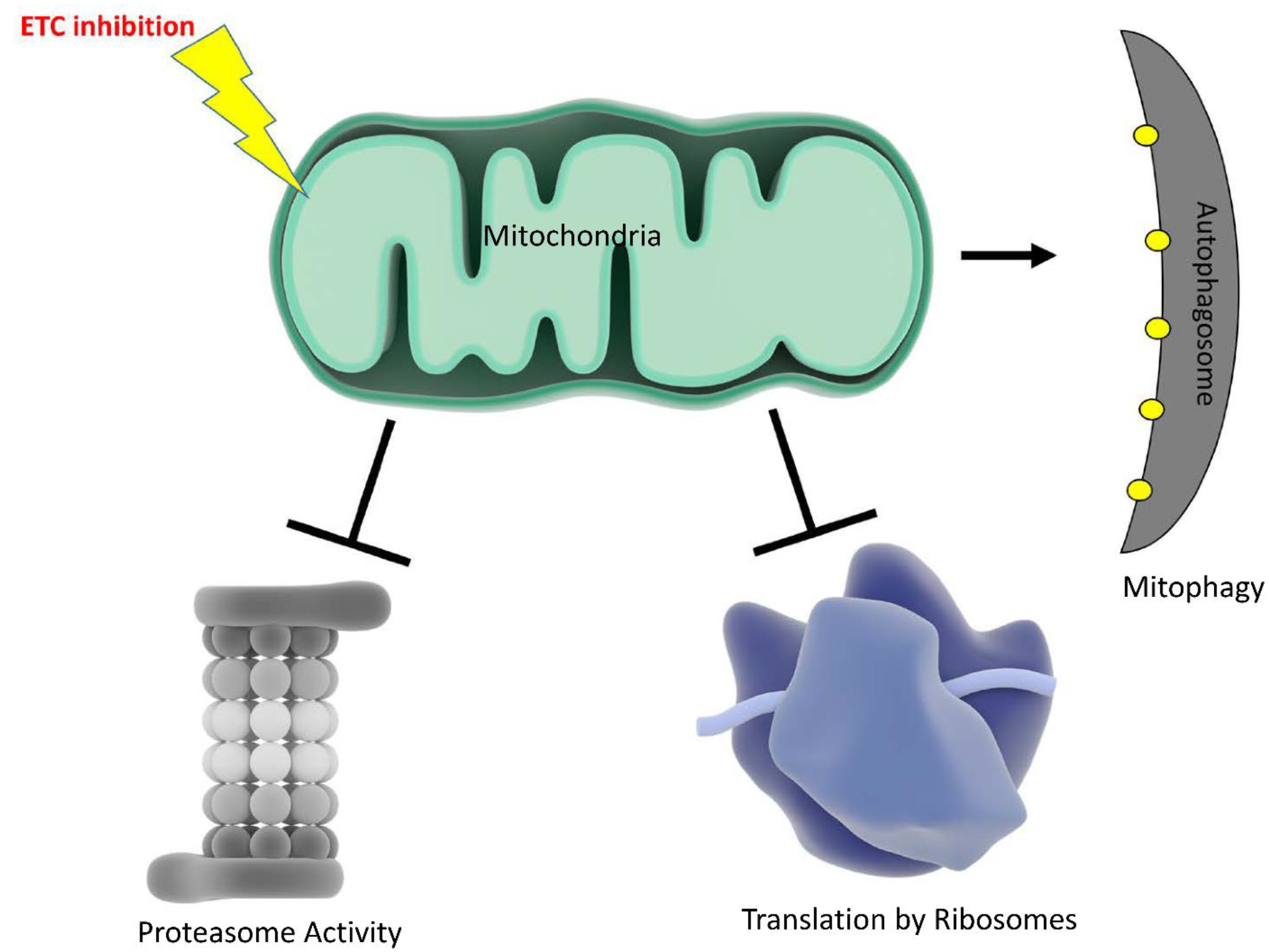
ETC inhibition decreases proteasome activity, inhibits protein translation, and increases mitophagy.
Studies performed in yeast, C. elegans, and mammalian cells reveal a relationship between the electron transport chain (ETC) and proteasome activity, in which ETC impairment associates with reduced proteasome function. Reactive oxygen species (ROS), which favor proteasome disassembly, may mediate this relationship as antioxidant treatment reverses proteasome disassembly. ATP levels and proteasome function also correlate, which implies bioenergetic status may also link mitochondrial function to the UPS (D’Amico et al., 2017).
Very specific types of mitochondrial stress can alternatively activate the proteasome. Defective mitochondrial protein import causes mitochondrial protein precursors to accumulate in the cytosol, which the cell responds to by increasing proteasome activity and decreasing protein translation. This response to the cytosolic accumulation of mitochondrial proteins is called the UPR activated by mistargeting of proteins (UPRam) (Wrobel et al., 2015).
In C. elegans there is also a mitochondrial stress response pathway that depends on lipid biosynthesis. Knockdown of a mitochondrial chaperone, mtHSP70, triggers cytosolic lipid accumulation which activates transcription factors that increase the expression of lipid metabolism and cytosolic chaperone genes (Kim et al., 2016). Further studies need to determine the relevancy of these pathways in mammalian cells.
9. Mitochondrial protein clearance pathways
Studies in yeast suggest mitochondria actively degrade aggregation prone-proteins. Disrupting this process by interfering with mitochondrial protein import and protease function promotes the formation of cytosolic protein aggregates (Ruan et al., 2017). As part of a related phenomenon, defective cytosolic chaperone function causes protein aggregates to accumulate within mitochondria. While aggregating proteins move into the mitochondria most robustly under heat shock stress conditions, under basal conditions a limited stream of aggregation-prone proteins continue to access the mitochondria, where they undergo degradation. Human cells appear to display a similar mechanism (Ruan et al., 2017).
In addition to the protease-mediated removal of mitochondria-internalized peptides, the lysosomal elimination of mitochondria through mitophagy also supports proteostasis. In APP transgenic mice, PTEN-induced putative kinase 1 (PINK1) knock-down impairs mitophagy and increases plaque deposition, while PINK1 overexpression enhances mitophagy and reduces plaque deposition. PINK1 overexpression also reduces synaptic loss and preserves memory task performance in these mice (Du et al., 2017).
Mitochondria can relegate protein aggregates to discrete regions. The contaminated sections specifically recruit parkin and undergo fission-mediated removal from the rest of the organelle. Under conditions of fission inhibition, parkin tagging of mitochondria transforms from a focal to diffuse pattern, which leads to a generalized mitophagy response. Fission, therefore, regulates a balance between selective degradation of mitochondrial protein aggregates and their less specific removal through mitophagy (Burman et al., 2017).
10. Concluding remarks
Mitochondrial function is altered in AD. Multiple factors may contribute to this, including genetics, protein stress, and stress responses. Accumulating evidence argues mitochondria play an important role in AD and may even contribute directly to its classic plaque and tangle histology. A growing body of experimental data firmly link mitochondrial dysfunction to Aβ and tau homeostasis. Some studies focus on the ability of Aβ and tau to alter mitochondrial function. Others show mitochondria influence Aβ and tau homeostasis, as well as their aggregation. The connections between mitochondria and cell proteostasis are extensive and reflect fundamental cell biology. Mitochondria modify protein translation and degradation infrastructure, and in the case of protein degradation serve as part of that infrastructure. Targeting mitochondria to mitigate protein aggregation in AD, and ideally to provide clinical relief, seems reasonable.
Acknowledgements
RHS is supported by the National Institute on Aging (P30 AG035982, R01 AG060733, R01 AG061194) and the Department of Defense (AZ 170111).
Abbreviations:
- Aβ
beta amyloid
- ABAD
Aβ-binding alcohol dehydrogenase
- AD
Alzheimer’s disease
- ANT
adenine nucleotide transporter
- ApoE
apolipoprotein E
- APP
amyloid precursor protein
- ATF
activating transcription factor
- ATFS1
ATF associated with stress-1
- BDNF
brain derived neurotrophic factor
- COX
cytochrome oxidase
- Cybrid
cytoplasmic hybrid
- eIF2α
eukaryotic initiation factor 2α
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- ETC
electron transport chain
- FDG-PET
(18F)-2-fluoro-deoxy-D-glucose positron emission tomography
- Hsp
heat shock protein
- ISR
integrated stress response
- LCR
low capacity runner
- MOAS
mitochondria on a string; mI, myo-inositol
- MPP+
1-methyl-4-phenylpyridinium
- mtDNA
mitochondrial DNA
- NAA
N-acetylaspartate
- NFTs
neurofibrillary tangles
- PINK1
PTEN-induced putative kinase 1
- PS
presenilin
- ROS
reactive oxygen species
- UPR
unfolded protein response
- UPRam
UPR activated by mistargeting of proteins
- UPS
ubiquitin-proteasome system
- VDAC
voltage dependent anion channel
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors have no conflict of interests with any organizations or entity mentioned here.
References
- An Y, Varma VR, Varma S, Casanova R, Dammer E, Pletnikova O, Chia CW, Egan JM, Ferrucci L, Troncoso J, Levey AI, Lah J, Seyfried NT, Legido-Quigley C, O’Brien R, Thambisetty M, 2018. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 14, 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG, 2003. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. The Journal of cell biology 161, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandatheerthavarada HK, Devi L, 2007. Amyloid precursor protein and mitochondrial dysfunction in Alzheimer’s disease. Neuroscientist 13, 626–638. [DOI] [PubMed] [Google Scholar]
- Andrews SJ, Fulton-Howard B, Patterson C, McFall GP, Gross A, Michaelis EK, Goate A, Swerdlow RH, Pa J, 2019. Mitonuclear interactions influence Alzheimer’s disease risk. Neurobiology of aging. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlante A, Amadoro G, Bobba A, de Bari L, Corsetti V, Pappalardo G, Marra E, Calissano P, Passarella S, 2008. A peptide containing residues 26–44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim Biophys Acta 1777, 1289–1300. [DOI] [PubMed] [Google Scholar]
- Baloyannis SJ, 2006. Mitochondrial alterations in Alzheimer’s disease. Journal of Alzheimer’s disease : JAD 9, 119–126. [DOI] [PubMed] [Google Scholar]
- Barba I, Fernandez-Montesinos R, Garcia-Dorado D, Pozo D, 2008. Alzheimer’s disease beyond the genomic era: nuclear magnetic resonance (NMR) spectroscopy-based metabolomics. Journal of cellular and molecular medicine 12, 1477–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baslow MH, 2003. N-acetylaspartate in the vertebrate brain: metabolism and function. Neurochemical research 28, 941–953. [DOI] [PubMed] [Google Scholar]
- Blass JP, Baker AC, Ko L, Black RS, 1990. Induction of Alzheimer antigens by an uncoupler of oxidative phosphorylation. Archives of neurology 47, 864–869. [DOI] [PubMed] [Google Scholar]
- Bravo R, Parra V, Gatica D, Rodriguez AE, Torrealba N, Paredes F, Wang ZV, Zorzano A, Hill JA, Jaimovich E, Quest AFG, Lavandero S, 2013. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol 301, 215–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruch J, Xu H, De Andrade A, Hoglinger G, 2014. Mitochondrial complex 1 inhibition increases 4-repeat isoform tau by SRSF2 upregulation. PLoS One 9, e113070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A, Bukau B, Sommer T, 2010. Protein Quality Control in the Cytosol and the Endoplasmic Reticulum: Brothers in Arms. Molecular Cell 40, 238–252. [DOI] [PubMed] [Google Scholar]
- Burman JL, Pickles S, Wang C, Sekine S, Vargas JNS, Zhang Z, Youle AM, Nezich CL, Wu X, Hammer JA, Youle RJ, 2017. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. The Journal of Cell Biology 216, 3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso SM, Santana I, Swerdlow RH, Oliveira CR, 2004. Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Abeta toxicity. Journal of neurochemistry 89, 1417–1426. [DOI] [PubMed] [Google Scholar]
- Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA, 2002. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. Journal of neurochemistry 80, 91–100. [DOI] [PubMed] [Google Scholar]
- Casoli T, Di Stefano G, Spazzafumo L, Balietti M, Giorgetti B, Giuli C, Postacchini D, Fattoretti P, Conti F, 2014. Contribution of non-reference alleles in mtDNA of Alzheimer’s disease patients. Annals of clinical and translational neurology 1, 284–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casoli T, Spazzafumo L, Di Stefano G, Conti F, 2015. Role of diffuse low-level heteroplasmy of mitochondrial DNA in Alzheimer’s disease neurodegeneration. Frontiers in aging neuroscience 7, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD, 2005. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 19, 2040–2041. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Barnett JL, Berman SA, Li J, Quarless S, Bursztajn S, Lippa C, Nixon RA, 1995. Gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron 14, 671–680. [DOI] [PubMed] [Google Scholar]
- Chang S, ran Ma T, Miranda RD, Balestra ME, Mahley RW, Huang Y, 2005. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proceedings of the National Academy of Sciences of the United States of America 102, 18694–18699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HK, Ji ZS, Dodson SE, Miranda RD, Rosenblum CI, Reynolds IJ, Freedman SB, Weisgraber KH, Huang Y, Mahley RW, 2011. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. The Journal of biological chemistry 286, 5215–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Chandrasekaran K, Demarest TG, Kristian T, Xu S, Vijaykumar K, Dsouza KG, Qi NR, Yarowsky PJ, Gallipoli R, Koch LG, Fiskum GM, Britton SL, Russell JW, 2014. Brain diabetic neurodegeneration segregates with low intrinsic aerobic capacity. Ann Clin Transl Neurol 1, 589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JK, Dedeoglu A, Jenkins BG, 2007. Application of MRS to mouse models of neurodegenerative illness. NMR in biomedicine 20, 216–237. [DOI] [PubMed] [Google Scholar]
- Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF, Wallace DC, 1992. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nature genetics 2, 324–329. [DOI] [PubMed] [Google Scholar]
- Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, McKee AC, Beal MF, Graham BH, Wallace DC, 1994. Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics 23, 471–476. [DOI] [PubMed] [Google Scholar]
- Coskun PE, Beal MF, Wallace DC, 2004. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proceedings of the National Academy of Sciences of the United States of America 101, 10726–10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell DA, Blakely EL, Johnson MA, Ince PG, Turnbull DM, 2001. Mitochondrial enzyme-deficient hippocampal neurons and choroidal cells in AD. Neurology 57, 260–264. [DOI] [PubMed] [Google Scholar]
- Cottrell DA, Borthwick GM, Johnson MA, Ince PG, Turnbull DM, 2002. The role of cytochrome c oxidase deficient hippocampal neurones in Alzheimer’s disease. Neuropathology and Applied Neurobiology 28, 390–396. [DOI] [PubMed] [Google Scholar]
- D’Amico D, Sorrentino V, Auwerx J, 2017. Cytosolic Proteostasis Networks of the Mitochondrial Stress Response. Trends in biochemical sciences 42, 712–725. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW, 1990. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Annals of neurology 27, 457–464. [DOI] [PubMed] [Google Scholar]
- Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK, 2006. Accumulation of Amyloid Precursor Protein in the Mitochondrial Import Channels of Human Alzheimer’s Disease Brain Is Associated with Mitochondrial Dysfunction. The Journal of Neuroscience 26, 9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du F, Yu Q, Yan S, Hu G, Lue L-F, Walker DG, Wu L, Yan SF, Tieu K, Yan SS, 2017. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain 140, 3233–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD, 2008. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med 14, 1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edland SD, Silverman JM, Peskind ER, Tsuang D, Wijsman E, Morris JC, 1996. Increased risk of dementia in mothers of Alzheimer’s disease cases: evidence for maternal inheritance. Neurology 47, 254–256. [DOI] [PubMed] [Google Scholar]
- Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X, Rocktaschel P, Croteau DL, Akbari M, Greig NH, Fladby T, Nilsen H, Cader MZ, Mattson MP, Tavernarakis N, Bohr VA, 2019. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nature neuroscience 22, 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM, 2016. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Current biology : CB 26, 2037–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fribley A, Zhang K, Kaufman RJ, 2009. Regulation of apoptosis by the unfolded protein response. Methods in molecular biology (Clifton, N.J.) 559, 191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, Muschner D, Thalheimer A, Turk A, Hoyer S, Zochling R, Boissl KW, Jellinger K, Riederer P, 1998. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. Journal of neural transmission (Vienna, Austria : 1996) 105, 423–438. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G, 2012. Mitochondrial control of cellular life, stress, and death. Circulation research 111, 1198–1207. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N, Miller R, Berry RW, Binder LI, Cryns VL, 2003. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America 100, 10032–10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaeva AA, Kovaleva IE, Chumakov PM, Evstafieva AG, 2016. Mitochondrial dysfunction induces SESN2 gene expression through Activating Transcription Factor 4. Cell Cycle 15, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh SS, Swerdlow RH, Miller SW, Sheeman B, Parker WD Jr., Davis RE, 1999. Use of cytoplasmic hybrid cell lines for elucidating the role of mitochondrial dysfunction in Alzheimer’s disease and Parkinson’s disease. Ann N Y Acad Sci 893, 176–191. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Sheu KF, Blass JP, 1998. Abnormalities of mitochondrial enzymes in Alzheimer disease. J Neural Transm 105, 855–870. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. , 1991. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706. [DOI] [PubMed] [Google Scholar]
- Grootjans J, Kaser A, Kaufman RJ, Blumberg RS, 2016. The unfolded protein response in immunity and inflammation. Nat Rev Immunol 16, 469–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossberg GT, Tong G, Burke AD, Tariot PN, 2019. Present Algorithms and Future Treatments for Alzheimer’s Disease. J Alzheimers Dis 67, 1157–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblet NS, Castora FJ, 1997. Elevated levels of the Kearns-Sayre syndrome mitochondrial DNA deletion in temporal cortex of Alzheimer’s patients. Mutation research 379, 253–262. [DOI] [PubMed] [Google Scholar]
- He X, Huang Y, Li B, Gong CX, Schuchman EH, 2010. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol Aging 31, 398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeren J, Grewal T, Laatsch A, Becker N, Rinninger F, Rye KA, Beisiegel U, 2004. Impaired recycling of apolipoprotein E4 is associated with intracellular cholesterol accumulation. The Journal of biological chemistry 279, 55483–55492. [DOI] [PubMed] [Google Scholar]
- Heggeli KA, Crook J, Thomas C, Graff-Radford N, 2012. Maternal transmission of Alzheimer disease. Alzheimer Dis Assoc Disord 26, 364–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA, 2001. Mitochondrial abnormalities in Alzheimer’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 21, 3017–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra JG, Hipp MJ, Montine TJ, Kennedy SR, 2016. Mitochondrial DNA mutations increase in early stage Alzheimer disease and are inconsistent with oxidative damage. Annals of neurology 80, 301–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman JM, Welsh-Bohmer KA, Hanson M, Crain B, Hulette C, Earl N, Coleman RE, 2000. FDG PET imaging in patients with pathologically verified dementia. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 41, 1920–1928. [PubMed] [Google Scholar]
- Hoglinger GU, Lannuzel A, Khondiker ME, Michel PP, Duyckaerts C, Feger J, Champy P, Prigent A, Medja F, Lombes A, Oertel WH, Ruberg M, Hirsch EC, 2005. The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. Journal of neurochemistry 95, 930–939. [DOI] [PubMed] [Google Scholar]
- Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, Mahley RW, 2001. Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proceedings of the National Academy of Sciences of the United States of America 98, 8838–8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR, 1998. Aggresomes: A Cellular Response to Misfolded Proteins. The Journal of Cell Biology 143, 1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovaisaite V, Mouchiroud L, Auwerx J, 2014. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. The Journal of Experimental Biology 217, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, Weigand SD, Petersen RC, Boeve BF, Knopman DS, Gunter J, Reyes D, Shiung M, O’Brien PC, Smith GE, Ivnik RJ, Tangalos EG, Jack CR Jr., 2007. Longitudinal 1H MRS changes in mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging 28, 1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao T-P, 2003. The Deacetylase HDAC6 Regulates Aggresome Formation and Cell Viability in Response to Misfolded Protein Stress. Cell 115, 727–738. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ 3rd, Shen J, 2017. Presenilin-1 mutations and Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America 114, 629–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SM, Cassarino DS, Abramova NN, Keeney PM, Borland MK, Trimmer PA, Krebs CT, Bennett JC, Parks JK, Swerdlow RH, Parker WD Jr., Bennett JP Jr., 2000. Alzheimer’s disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Annals of neurology 48, 148–155. [PubMed] [Google Scholar]
- Kim H-E, Grant AR, Simic MS, Kohnz RA, Nomura DK, Durieux J, Riera CE, Sanchez M, Kapernick E, Wolff S, Dillin A, 2016. Lipid Biosynthesis Coordinates a Mitochondrial-to-Cytosolic Stress Response. Cell 166, 1539–1552.e1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kish SJ, Mastrogiacomo F, Guttman M, Furukawa Y, Taanman JW, Dozic S, Pandolfo M, Lamarche J, DiStefano L, Chang LJ, 1999. Decreased brain protein levels of cytochrome oxidase subunits in Alzheimer’s disease and in hereditary spinocerebellar ataxia disorders: a nonspecific change? Journal of neurochemistry 72, 700–707. [DOI] [PubMed] [Google Scholar]
- Krishnan KJ, Ratnaike TE, De Gruyter HLM, Jaros E, Turnbull DM, 2012. itochondrial DNA deletions cause the biochemical defect observed in Alzheimer’s disease. Neurobiology of Aging 33, 2210–2214. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Markesbery WR, 2007. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res 35, 7497–7504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue L-F, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H, 2004. ABAD Directly Links Aß to Mitochondrial Toxicity in Alzheimer’s Disease. Science 304, 448. [DOI] [PubMed] [Google Scholar]
- Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH, 2006. Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15, 1437–1449. [DOI] [PubMed] [Google Scholar]
- Manczak M, Calkins MJ, Reddy PH, 2011. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Human molecular genetics 20, 2495–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Reddy PH, 2012. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum Mol Genet 21, 5131–5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Bisbal MC, Arana E, Marti-Bonmati L, Molla E, Celda B, 2004. Cognitive impairment: classification by 1H magnetic resonance spectroscopy. European journal of neurology 11, 187–193. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH, 2011. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecocci P, MacGarvey U, Beal MF, 1994. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Annals of neurology 36, 747–751. [DOI] [PubMed] [Google Scholar]
- Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE, 1997. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Annals of neurology 42, 85–94. [DOI] [PubMed] [Google Scholar]
- Mosconi L, Brys M, Switalski R, Mistur R, Glodzik L, Pirraglia E, Tsui W, De Santi S, de Leon MJ, 2007. Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proceedings of the National Academy of Sciences of the United States of America 104, 19067–19072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, de Leon M, Murray J, E L, Lu J, Javier E, McHugh P, Swerdlow RH, 2011. Reduced mitochondria cytochrome oxidase activity in adult children of mothers with Alzheimer’s disease. Journal of Alzheimer’s disease : JAD 27, 483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Mistur R, Switalski R, Brys M, Glodzik L, Rich K, Pirraglia E, Tsui W, De Santi S, de Leon MJ, 2009. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology 72, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Pupi A, De Leon MJ, 2008a. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci 1147, 180–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Tsui WH, Herholz K, Pupi A, Drzezga A, Lucignani G, Reiman EM, Holthoff V, Kalbe E, Sorbi S, Diehl-Schmid J, Perneczky R, Clerici F, Caselli R, Beuthien-Baumann B, Kurz A, Minoshima S, de Leon MJ, 2008b. Multicenter standardized 18F-FDG PET diagnosis of mild cognitive impairment, Alzheimer’s disease, and other dementias. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 49, 390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell J, Farlow M, Ghetti B, Benson MD, 1991. A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science 254, 97–99. [DOI] [PubMed] [Google Scholar]
- Mutisya EM, Bowling AC, Beal MF, 1994. Cortical cytochrome oxidase activity is reduced in Alzheimer’s disease. Journal of neurochemistry 63, 2179–2184. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Watanabe A, Fujino T, Hosono T, Michikawa M, 2009. Apolipoprotein E4 (1–272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Molecular neurodegeneration 4, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM, 2012. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestor PJ, Fryer TD, Smielewski P, Hodges JR, 2003. Limbic hypometabolism in Alzheimer’s disease and mild cognitive impairment. Annals of neurology 54, 343–351. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Yang D-S, 2011. Autophagy failure in Alzheimer’s disease--locating the primary defect. Neurobiol Dis 43, 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niyazov DM, Kahler SG, Frye RE, 2016. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment. Molecular syndromology 7, 122–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA, 2001. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60, 759–767. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, Smith MA, 1999. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 19, 1959–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojaimi J, Masters CL, McLean C, Opeskin K, McKelvie P, Byrne E, 1999. Irregular distribution of cytochrome c oxidase protein subunits in aging and Alzheimer’s disease. Annals of neurology 46, 656–660. [PubMed] [Google Scholar]
- Onyango IG, Ahn JY, Tuttle JB, Bennett JP Jr., Swerdlow RH, 2010. Nerve growth factor attenuates oxidant-induced beta-amyloid neurotoxicity in sporadic Alzheimer’s disease cybrids. Journal of neurochemistry 114, 1605–1618. [DOI] [PubMed] [Google Scholar]
- Panchal K, Tiwari AK, 2019. Mitochondrial dynamics, a key executioner in neurodegenerative diseases. Mitochondrion 47, 151–173. [DOI] [PubMed] [Google Scholar]
- Parker WD Jr., Filley CM, Parks JK, 1990. Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology 40, 1302–1303. [DOI] [PubMed] [Google Scholar]
- Parker WD Jr., Parks JK, 1995. Cytochrome c oxidase in Alzheimer’s disease brain: purification and characterization. Neurology 45, 482–486. [DOI] [PubMed] [Google Scholar]
- Pereira C, Santos MS, Oliveira C, 1998. Mitochondrial function impairment induced by amyloid beta-peptide on PC12 cells. Neuroreport 9, 1749–1755. [DOI] [PubMed] [Google Scholar]
- Perry G, Nunomura A, Hirai K, Zhu X, Perez M, Avila J, Castellani RJ, Atwood CS, Aliev G, Sayre LM, Takeda A, Smith MA, 2002. Is oxidative damage the fundamental pathogenic mechanism of Alzheimer’s and other neurodegenerative diseases? Free radical biology & medicine 33, 1475–1479. [DOI] [PubMed] [Google Scholar]
- Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW, 1991. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 7, 695–702. [DOI] [PubMed] [Google Scholar]
- Pickett EK, Rose J, McCrory C, McKenzie C-A, King D, Smith C, Gillingwater TH, Henstridge CM, Spires-Jones TL, 2018. Region-specific depletion of synaptic mitochondria in the brains of patients with Alzheimer’s disease. Acta neuropathologica 136, 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla RA, Matthews-Roberson TA, Dolan PJ, Johnson GV, 2009. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. The Journal of biological chemistry 284, 18754–18766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirós PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D, Gygi SP, Auwerx J, 2017. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. The Journal of Cell Biology 216, 2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, Thibodeau SN, Osborne D, 1996. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. The New England journal of medicine 334, 752–758. [DOI] [PubMed] [Google Scholar]
- Risacher SL, Anderson WH, Charil A, Castelluccio PF, Shcherbinin S, Saykin AJ, Schwarz AJ, Alzheimer’s Disease Neuroimaging I, 2017. Alzheimer disease brain atrophy subtypes are associated with cognition and rate of decline. Neurology 89, 2176–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosendorff C, Beeri MS, Silverman JM, 2007. Cardiovascular risk factors for Alzheimer’s disease. The American journal of geriatric cardiology 16, 143–149. [DOI] [PubMed] [Google Scholar]
- Ruan L, Zhou C, Jin E, Kucharavy A, Zhang Y, Wen Z, Florens L, Li R, 2017. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 543, 443–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrijvers EMC, Witteman JCM, Sijbrands EJG, Hofman A, Koudstaal PJ, Breteler MMB, 2010. Insulin metabolism and the risk of Alzheimer disease: the Rotterdam Study. Neurology 75, 1982–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva DF, Selfridge JE, Lu J, E L, Roy N, Hutfles L, Burns JM, Michaelis EK, Yan S, Cardoso SM, Swerdlow RH, 2013. Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum Mol Genet 22, 3931–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman DHS, Small GW, Chang CY, Lu CS, de Aburto MAK, Chen W, Czernin J, Rapoport SI, Pietrini P, Alexander GE, Schapiro MB, Jagust WJ, Hoffman JM, Welsh-Bohmer KA, Alavi A, Clark CM, Salmon E, de Leon MJ, Mielke R, Cummings JL, Kowell AP, Gambhir SS, Hoh CK, Phelps ME, 2001. Positron Emission Tomography in Evaluation of DementiaRegional Brain Metabolism and Long-term Outcome. JAMA 286, 2120–2127. [DOI] [PubMed] [Google Scholar]
- Simpson IA, Chundu KR, Davies-Hill T, Honer WG, Davies P, 1994. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Annals of neurology 35, 546–551. [DOI] [PubMed] [Google Scholar]
- Sims NR, Finegan JM, Blass JP, Bowen DM, Neary D, 1987. Mitochondrial function in brain tissue in primary degenerative dementia. Brain research 436, 30–38. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH, 2007. Is aging part of Alzheimer’s disease, or is Alzheimer’s disease part of aging? Neurobiology of aging 28, 1465–1480. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH, 2012. Mitochondria and cell bioenergetics: increasingly recognized components and a possible etiologic cause of Alzheimer’s disease. Antioxid Redox Signal 16, 1434–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH, 2018. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. Journal of Alzheimer’s disease : JAD 62, 1403–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH, Burns JM, Khan SM, 2010. The Alzheimer’s disease mitochondrial cascade hypothesis. Journal of Alzheimer’s disease : JAD 20 Suppl 2, S265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH, Burns JM, Khan SM, 2014. The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochimica et biophysica acta 1842, 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH, Khan SM, 2004. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Medical Hypotheses 63, 8–20. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH, Khan SM, 2009. The Alzheimer’s disease mitochondrial cascade hypothesis: an update. Experimental neurology 218, 308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH, Koppel S, Weidling I, Hayley C, Ji Y, Wilkins HM, 2017. Mitochondria, Cybrids, Aging, and Alzheimer’s Disease. Progress in molecular biology and translational science 146, 259–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH, Parks JK, Cassarino DS, Maguire DJ, Maguire RS, Bennett JP Jr., Davis RE, Parker WD Jr., 1997. Cybrids in Alzheimer’s disease: a cellular model of the disease? Neurology 49, 918–925. [DOI] [PubMed] [Google Scholar]
- Thies W, Bleiler L, 2011. 2011 Alzheimer’s disease facts and figures. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 7, 208–244. [DOI] [PubMed] [Google Scholar]
- Tomasi D, Wang G-J, Volkow ND, 2013. Energetic cost of brain functional connectivity. Proceedings of the National Academy of Sciences 110, 13642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer PA, Swerdlow RH, Parks JK, Keeney P, Bennett JP, Miller SW, Davis RE, Parker WD, 2000. Abnormal Mitochondrial Morphology in Sporadic Parkinson’s and Alzheimer’s Disease Cybrid Cell Lines. Experimental Neurology 162, 37–50. [DOI] [PubMed] [Google Scholar]
- Varma VR, Oommen AM, Varma S, Casanova R, An Y, Andrews RM, O’Brien R, Pletnikova O, Troncoso JC, Toledo J, Baillie R, Arnold M, Kastenmueller G, Nho K, Doraiswamy PM, Saykin AJ, Kaddurah-Daouk R, Legido-Quigley C, Thambisetty M, 2018. Brain and blood metabolite signatures of pathology and progression in Alzheimer disease: A targeted metabolomics study. PLoS Med 15, e1002482–e1002482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, 2005. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39, 359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Chalkia D, 2013. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol 5, a021220–a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Lee H. g., Li X, Perry G, Smith MA, Zhu X, 2009. Impaired Balance of Mitochondrial Fission and Fusion in Alzheimer’s Disease. The Journal of Neuroscience 29, 9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X, 2008. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proceedings of the National Academy of Sciences of the United States of America 105, 19318–19323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmer RA, Gunderson EP, Quesenberry CP Jr., Zhou J, Yaffe K, 2007. Body mass index in midlife and risk of Alzheimer disease and vascular dementia. Current Alzheimer research 4, 103–109. [DOI] [PubMed] [Google Scholar]
- Wong MW, Braidy N, Poljak A, Pickford R, Thambisetty M, Sachdev PS, 2017. Dysregulation of lipids in Alzheimer’s disease and their role as potential biomarkers. Alzheimers Dement 13, 810–827. [DOI] [PubMed] [Google Scholar]
- Wrobel L, Topf U, Bragoszewski P, Wiese S, Sztolsztener ME, Oeljeklaus S, Varabyova A, Lirski M, Chroscicki P, Mroczek S, Januszewicz E, Dziembowski A, Koblowska M, Warscheid B, Chacinska A, 2015. Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature 524, 485. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D, 2004. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. Journal of cell science 117, 4055–4066. [DOI] [PubMed] [Google Scholar]
- Zaarur N, Meriin AB, Bejarano E, Xu X, Gabai VL, Cuervo AM, Sherman MY, 2014. Proteasome failure promotes positioning of lysosomes around the aggresome via local block of microtubule-dependent transport. Mol Cell Biol 34, 1336–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Trushin S, Christensen TA, Bachmeier BV, Gateno B, Schroeder A, Yao J, Itoh K, Sesaki H, Poon WW, Gylys KH, Patterson ER, Parisi JE, Diaz Brinton R, Salisbury JL, Trushina E, 2016. Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s Disease. Scientific Reports 6, 18725. [DOI] [PMC free article] [PubMed] [Google Scholar]