

Abstract
Background
Deficiency of adenosine deaminase 2 (DADA2) is a syndrome with pleiotropic manifestations including vasculitis and hematologic compromise. A systematic definition of the relationship between ADA2 mutations and clinical phenotype remains unavailable.
Objective
We tested whether the impact of ADA2 mutations on enzyme function correlates with clinical presentation.
Methods
DADA2 patients with severe hematologic manifestations were compared with vasculitis-predominant patients. Enzymatic activity was assessed using expression constructs reflecting all 53 missense, nonsense, insertion and deletion genotypes from 152 patients across the DADA2 spectrum.
Results
We identified DADA2 patients presenting with pure red cell aplasia (PRCA, n = 5) or bone marrow failure syndrome (BMF, n = 10). Most patients did not exhibit features of vasculitis. Recurrent infection, hepatosplenomegaly and gingivitis were common in patients with BMF, of whom half died from infection. Unlike DADA2 patients with vasculitis, patients with PRCA and BMF proved largely refractory to tumor necrosis factor inhibitors. ADA2 variants associated with vasculitis predominantly reflected missense mutations with at least 3% residual enzymatic activity. By contrast, PRCA and BMF were associated with missense mutations with minimal residual enzyme activity, nonsense variants, and insertions / deletions resulting in complete loss of function.
Conclusion
Functional interrogation of ADA2 mutations reveals an association of subtotal function loss with vasculitis, typically responsive to TNF blockade, whereas more extensive loss is observed in hematologic disease which may be refractory to treatment. These findings establish a genotype-phenotype spectrum in DADA2.
Clinical implications
Genotype correlates with clinical phenotype and therapeutic response in DADA2.
Keywords: adenosine deaminase 2, DADA2, vasculitis, pure red cell aplasia, bone marrow failure
Capsule Summary
DADA2 is a monogenic disorder with multi-organ system manifestations. We present a cohort of DADA2 patients with severe hematologic defects and describe novel genotype-phenotype correlations based on functional analysis of 53 ADA2 mutations.
Graphical Abstract
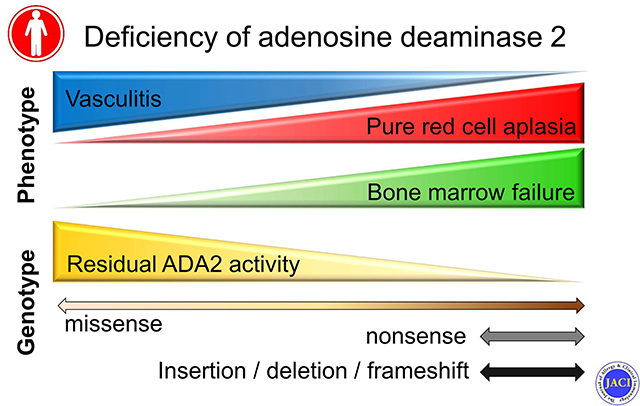
Introduction
Deficiency of adenosine deaminase 2 (DADA2) is a monogenic autoinflammatory disease initially characterized as a cause of stroke and systemic vasculitis in young children 1, 2. Since its initial description in 2014, the clinical spectrum of this condition has expanded considerably, and variable hematologic and immunologic abnormalities have been described in about half of DADA2 patients 3, 4 Primary presentations of the disease include pure red cell aplasia (PRCA) that mimics Diamond-Blackfan anemia and bone marrow failure (BMF) with variable cytopenia, even without vasculitis or systemic inflammation 5–7. The severity of these manifestations can result in transfusion dependency in patients with PRCA or a need for hematopoietic stem transplant (HSCT) in those with BMF 8–11. Some patients present with humoral immunodeficiency and recurrent infection, further complicating our understanding of DADA2 12, 13. How mutations in the same gene can present with different phenotypes is poorly understood.
ADA2 is an extracellular enzyme primarily secreted by monocytes and macrophages 14, 15. While ADA2 is capable of catalyzing the deaminase reaction that converts adenosine to inosine, its physiologic function is not known. Biallelic mutations in the encoding gene ADA2 (formerly known as CECR1) and very low levels of ADA2 enzymatic activity in the peripheral blood are diagnostic of DADA2 2. Missense variants are most common but nonsense mutations, insertions / deletions (indels) and splice site mutations have been described 4
A systematic analysis comparing ADA2 mutations associated with different clinical phenotypes is lacking. Previous studies have not been able to establish convincing genotype-phenotype correlations, in part due to a limited number of cases and preferential recruitment of patients with a specific phenotype based on the subspecialty of the investigators. Establishing genotype-phenotype correlations has important diagnostic and therapeutic implications. Whereas tumor necrosis factor inhibitors (TNFi) prevent strokes and improve manifestations of vasculitis in DADA2 16, their efficacy for PRCA and BMF is less clear. HSCT may be considered earlier for patients with severe hematologic presentations 9.
Here we report 15 new cases of DADA2 with PRCA or BMF as primary presentation. Based on the genetic findings we observed in these patients, we systematically studied ADA2 mutations from 152 published cases encompassing the different phenotypes by in silico analysis and functional assay. Our results provide strong evidence for genotype-phenotype correlations in DADA2 with potentially direct clinical relevance.
Methods
Patients
These studies were approved by the Institutional Review Boards at Boston Children’s Hospital and Brigham and Women’s Hospital. We performed retrospective chart review of 15 patients with DADA2 from 12 families. The patients were enrolled through a world-wide collaboration with approval by the local ethics committees. Research diagnostic testing was performed with written informed consent from the parent or guardian and assent when appropriate. Clinical and laboratory data for the cohort are described in Tables E1–E3 in Online Repository).
Literature search
Please see Supplemental methods in Online Repository for details of literature review and criteria for case selection. Cases selected from each publication and their phenotype are detailed in Table E4 in Online Repository. A complete list of mutations from the selected cases are displayed in Table E5 in Online Repository.
Analysis of ADA2 mutations
Construction of pcDNA3.1 plasmid for expression of wild-type ADA2 was as described 17. Site-directed mutagenesis was performed using the NEB Q5 mutagenesis kit (New England Biolabs, Ipswich, MA). The list of mutations and primer pairs used to generate mutant constructs are available in Tables E6 in Online Repository. Mutant constructs were purified using Purelink Quick Plasmid Miniprep kit (Thermo Fisher Scientific, Waltham, MA) and verified by sequencing. Plasmids were transfected into 293T cells using Fugene 6 (Promega, Madison, WI). Medium was collected after 72 hours and ADA2 activity was quantified using an established spectrophotometric assay that couples the release of ammonia from adenosine with the consumption of NADH 2,17. Each mutant was analyzed by three independent experiments and measurements were normalized to the activity of wildtype ADA2 from the same run.
Statistical analysis
The Kruskal-Wallis test was used for comparison of ADA2 activity between multiple mutation types and disease phenotypes. Chi-square was used for comparison of mutation types between clinical phenotypes. All tests were two-sided, and P < 0.05 was considered significant. Statistical analyses were performed using Prism 5.0 software (GraphPad Software, La Jolla, CA).
Results
A series of DADA2 patients with primary hematologic defects
We present an international cohort of 15 DADA2 patients from 12 families with PRCA (n = 5) or BMF (n = 10) as their primary presentation. Summarized data for the cohort are displayed in Table 1. Clinical manifestations and laboratory data for each patient are provided in Tables E1 and E2 in Online Repository, respectively. The age of onset for PRCA was very early (median 0.3 years, range 0.1 – 12 years); only 1 patient presented after 6 months of age (Table 1). The age of onset was more variable for the BMF group (median 2.2 years, range 0.1 – 13 years). Patients with PRCA displayed normocytic or microcytic anemia with very low reticulocyte count, consistent with defective erythrocyte production. Most patients with BMF had severe neutropenia and mild anemia, while 2 patients had pancytopenia. Consistent with previous studies, low immunoglobulin levels (IgG, IgM and/or IgA) were common in patients with DADA2 (Table 1). Cases of severe infection have been described in DADA2-associated BMF 8, 18, 19. Indeed, recurrent infection was more common in the BMF group (80% vs 20% in PRCA group). These patients experienced a variety of infections, and 5/10 patients ultimately succumbed to sepsis (Table E1 in Online Repository). It is noteworthy that two patients (K-1 and L-1) each had one sibling that died from severe infection before the discovery of DADA2, suggesting that mortality for this phenotype is even higher than estimated here.
Table 1.
Summary of clinical characteristics in DADA2 patients with PRCA or BMF
| PRCA | BMF | |
|---|---|---|
| Number of cases | 5 | 10 |
| Median age of onset (year) | 0.3 | 2.2 |
| Sex (% female) | 40 | 50 |
| Anemia (%) | 100 | 80 |
| Lymphopenia (%) | 0 | 40 |
| Neutropenia (%) | 0 | 90 |
| Thrombocytopenia (%) | 0 | 30 |
| Low IgG (%) | 20 | 30 |
| Low IgM (%) | 40 | 60 |
| Low IgA (%) | 60 | 50 |
| Recurrent infection (%) | 20 | 80 |
| Stroke (%) | 20 | 0 |
| Skin vasculitis (%) | 0 | 20 |
| Oral ulcers / Gingivitis (%) | 20 | 70 |
| Hepatosplenomegaly (%) | 40 | 90 |
| Death (%) | 20 | 50 |
Unlike patients from previous large series focused on DADA2 as a monogenic vasculitis 1, 2, 20–22, most DADA2 patients in this PRCA / BMF cohort (12/15, 80%) had no history of vasculitis. Two patients with BMF had cutaneous vasculitis and one patient with PRCA developed sudden-onset squinting and transient hemiparesis with MRI findings compatible with a small ischemic stroke. In the BMF group, almost all patients exhibited hepatosplenomegaly and half experienced severe gingivitis, a feature associated with neutropenia 23 that has not previously been reported in DADA2 (Table E1 in Online Repository). Treatment regimens for these patients include disease modifying anti-rheumatic drugs (DMARDs), biologics, intravenous immunoglobulin, granulocyte colony stimulating factor (GCSF) and HSCT. Unlike the success of TNFi therapy for prevention of stroke and treatment of vasculitis 16, most cases in this cohort did not respond to TNFi (Table E1 in Online Repository). In 10 patients that received TNFi, only one (patient C-1 with PRCA and stroke) showed sustained improvement of hematologic features. Three patients showed improvement of vasculitis and/or systemic inflammation but their cytopenia did not improve. One patient with BMF developed Pseudomonas aeruginosa sepsis soon after initiation of TNFi.
Patients in this cohort did not exhibit clinical features of autoimmunity. All patients with PRCA showed negative direct Coomb’s test. Four patients in the BMF group were found to have autoantibodies: 2 with low-titer anti-nuclear antibodies, 1 with anti-neutrophil antibodies, and 1 with non-specific anti-neutrophil cytoplasmic antibodies (Table E3 in Online Repository).
Biallelic mutations in ADA2 were confirmed in all patients (Table E1 in Online Repository). Nine unique ADA2 mutations were found in this cohort, and two (F212del and K449Nfs*2) were novel variants (Table 2). To confirm the pathogenicity of these mutations, we expressed these variants in 293T cells and measured ADA2 activity using an established spectrophotometric assay 2,24 All mutations from our patient cohort displayed minimal residual ADA2 activity (<2% of wildtype; Table 2). Interestingly, among the 7 previously-described mutations, 6 had been described in patients with severe hematologic manifestations without vasculitis 6, 8, 10, 13, 18, 25, 26. Moreover, whereas most vasculitis-associated ADA2 mutations in prior studies were missense variants 4, 6 patients in this cohort had homozygous indel mutations resulting in frameshift and early truncation. All siblings in one family (A-1, A-2 and A-3) exhibited PRCA while another pair of siblings had severe neutropenia (J1 and J2). These observations together raised the possibility of genotype differences among the clinical phenotypes in DADA2.
Table 2.
Characterization of ADA2 mutations
| Protein | cDNA | n | Phenotype | Type | Domain | ADA2 activity (%WT) | Published phenotype [ref#] |
|---|---|---|---|---|---|---|---|
| G47W | c.139G>T | 1 | BMF | missense | Dimerization | 0.3 ± 0.5 | Vasculitis * 25 |
| R49Afs*13 | c.137dupT | 4 | PRCA, BMF | frameshift | Dimerization | UD | Hemolytic anemia 8 |
| F178S | c.533T>C | 2 | BMF | missense | Catalytic | 0.8 ± 0.6 | PRCA 8 |
| F212del | c.634_636delTTC | 1 | BMF | deletion | Catalytic | 0.8 ± 1.2 | - |
| G321E | c.962G>A | 1 | PRCA | missense | Catalytic | 1.8 ± 1.0 | BMF 18 |
| G358R | c.1072G>A | 4 | BMF | missense | Catalytic | 1.7 ± 0.6 | PRCA 6 |
| K449Nfs*2 | c.1346_1347insTT | 1 | BMF | frameshift | Catalytic | UD | - |
| K466Tfs*2 | c.1397_1403AGGCTGAdel | 1 | BMF | frameshift | Catalytic | UD | PRCA 10 |
| V458D | c.1373T>A | 1 | BMF | missense | Catalytic | 2.4 ± 0.4 | BMF 13 Vasculitis * 26 |
Abbreviations: UD, undetectable.
This mutation was previously described in a patient with compound heterozygous mutations.
Genotype comparison of vasculitis and hematologic phenotypes in DADA2
To investigate possible genotype-phenotype correlations, we performed a literature review of published DADA2 cases with vasculitis, PRCA or BMF as the primary presentation. A list of included studies and details of case selection are provided in Methods and Table E4 in Online Repository. We reviewed 186 cases, of which 152 were selected for further investigation (Figure 1A). Details of case selection and exclusion are provided in Supplemental Method in Online Repository. Cases that appeared in multiple publications were analyzed only once and those with other phenotypes or incomplete data on ADA2 mutations were excluded (Table E4 in Online Repository). Because vasculitis is the most common presentation of DADA2, the vasculitis group (n = 100) was pooled from 11 major case series from around the world to minimize bias and regional differences. Including the 5 cases in our cohort, we identified 38 cases of DADA2 with PRCA as the primary manifestation. The BMF group consisted of 29 cases including the 10 patients from our cohort. Two patients with PRCA and 5 in the BMF group were described to have features of vasculitis.
Figure 1. Analysis of ADA2 mutations by patient phenotype.

A) Schematic of literature review and case selection for mutation analysis. B) Venn diagram of unique ADA2 mutations illustrating overlaps between disease phenotypes. C) Display of ADA2 gene structure illustrating the distribution of mutations associated with different phenotypes. Shared mutations are displayed by color coding. D) Circle charts illustrating the types of mutations associated with each phenotype. Analysis of individual alleles is displayed in the upper panel while analysis of homozygous individuals is shown in the lower panel.
One notable demographic difference between groups was age at presentation. In line with the observation in our cohort, DADA2 patients with PRCA presented very early in childhood [median age 0.5 years; interquartile range (IQR) 0.2 – 2.6] while those with vasculitis and BMF generally presented later (vasculitis: median 5.0 years, IQR 1.0 – 10.0 vs. BMF: median 5.0 years, IQR 2.0 – 14.0), including many cases diagnosed in adulthood (Kruskal-Wallis test, p < 0.001; Figure E1 in Online Repository).
Among the three groups (167 combined patients), 61 unique ADA2 mutations were found (Figure 1B). Surprisingly, only two of these mutations were shared by all three groups: H112Q and R169Q. The greatest overlap was found between PRCA and BMF groups, with 7 shared mutations not reported in the vasculitis group. Two mutations were shared by the vasculitis and PRCA groups, while another two were shared by the vasculitis and BMF groups. Plotting ADA2 mutations according to exon location, mutations associated with all three groups were scattered throughout the gene, without preferential concentration in specific domains (Figure 1C).
When the types of mutation were characterized (counting each allele individually), most mutations associated with vasculitis were missense variants (Figure 1D). Less than 10% of the mutant alleles in the vasculitis group belonged to other categories. In contrast, missense mutations accounted only for 53% of the variants in the PRCA group and 72% in the BMF group, respectively (Chi-square, p < 0.0001). Indels comprised the majority of remaining mutations for both groups (38% for PRCA and 16% for BMF; Figure 1D). When cases with compound heterozygous mutations were excluded, all 63 patients in the vasculitis group had homozygous missense mutations while more variable mutation types were found in the PRCA and BMF groups (Chi-square, p < 0.0001).
Using histograms to assess the most common ADA2 variants in each group, only a few overlapping mutations were found between the groups. R169Q was found in multiple patients in all three groups, while the most common mutation associated with vasculitis, G47R, was not seen in the other groups (Figure 2B). In contrast, G358R was seen in patients with PRCA and BMF, but not in those with vasculitis. R169Q and G358R were the only variants in the BMF group found in more than 2 cases.
Figure 2. Analysis of common mutations associated with each disease phenotype.

A) Histogram display of allelic count for the most common mutations associated with each disease phenotype. All cases in the current cohort and those selected from literature review were included. B) Phenotype distribution of the most common mutation association with vasculitis (G47R), PRCA (G358R) and BMF (R169Q).
Functional analysis of ADA2 mutations
The abundance of indels in the PRCA group suggests that the more detrimental mutations may be associated with this phenotype. However, missense mutations still accounted for more than 50% of variants. To understand whether functional differences exist among mutations groups, we created expression plasmids for each ADA2 mutant and transfected them into 293T cells. ADA2 enzymatic activity in the supernatant served as a functional readout for each mutation. Constructs for all 53 missense, nonsense, and indel variants from our patient cohort and published cases (Table E5 in Online Repository) were analyzed using this method. Splicing defects were not evaluated as the sequences for aberrantly-spliced complementary DNA are not available.
Our functional analysis confirmed that all mutations caused a reduction in ADA2 activity (Figure 3A). Not surprisingly, early translational termination caused by nonsense mutations and indels with frameshift completely abrogated ADA2 function. Missense variants, on the other hand, showed a wide spectrum of impact ranging from partial to complete loss of enzyme activity. Stratification by patient phenotype showed significantly greater residual ADA2 activity for mutations associated with vasculitis compared those associated with PRCA or BMF (Kruskal-Wallis test, p = 0.0002; Figure 3B). Examination of different cut-off levels revealed that a residual activity of >3% effectively segregated half of mutations associated with vasculitis from the other two groups (Chi-square, p < 0.0001; Figure 3C,D). All mutations associated with PRCA or BMF displayed residual activity under this threshold aside for Y353H, which demonstrated 4% residual activity.
Figure 3. Functional analysis of ADA2 mutations in vitro.

A) ADA2 enzyme activity of individual mutant constructs sorted by mutation type. B) ADA2 enzyme activity of individual mutant constructs sorted by disease phenotype. C) Stratification of mutations within each disease phenotype according to various cut-off values of residual ADA2 enzyme activity. D) Bar graph display of residual enzyme activity for individual mutations associated with each disease phenotype. Dotted line in all panels represent the cut-off value of 3% residual activity. For all panels, results are normalized as percentage of residual activity relative to wildtype (WT) ADA2. Each dot or bar represents the average of results from three independent experiments and error bar represents standard deviation.
To ensure that the statistics were not skewed by the greater number of nonsense and indel variants in the PRCA and BMF groups, we repeated the analysis including only missense mutations. A similar pattern was observed, as missense ADA2 mutations associated with vasculitis displayed significantly more residual enzymatic activity than those associated with the hematologic phenotypes (Kruskal-Wallis p < 0.0001; Figure E2A in Online Repository). We applied several in silico prediction algorithms to assess the pathogenicity of missense mutations associated with the three phenotypes. Consistent with our experimental data, analysis by SIFT 27 predicted that mutations associated with vasculitis would impair gene function significantly less than those associated with PRCA or BMF (Figure E2B in Online Repository). Such difference was not predicted by other algorithms (Polyphen2, MutationTaster and CADD; Figure E2C–F in Online Repository). Taken together, these findings suggest that the more deleterious ADA2 mutations are associated with severe hematologic phenotypes.
Establishing genotype-phenotype correlations in DADA2
Analysis of individual mutations cannot account for patients with compound heterozygous mutations. To evaluate whether the functional studies can be utilized to predict phenotype using actual mutation configurations from patients, we clustered ADA2 mutations into three categories using the 3% residual activity cut-off: type A, hypomorphic missense variants with ≥ 3% residual enzymatic activity compared to wildtype ADA2; type B, missense mutations with minimal (<3%) residual activity, and type C, indels and nonsense mutations with complete absence of enzyme activity. Based on the biallelic mutations identified, each patient was assigned to one of 6 groups (AA, AB, AC, BB, BC, CC) that reflected the predicted functional category of both mutations. For example, a patient with two type A mutations was assigned to group AA, while another patient with compound heterozygous type B and type C mutations was assigned to group BC. This method stratifies biallelic mutations for each patient into groups with a gradient of predicted residual ADA2 activity, where groups AA and CC have the highest and the lowest predicted activity, respectively. Patients with splice-site mutations (n = 7) were excluded from this analysis due to the lack of functional data to evaluate these variants.
For each phenotype, the percentage of patients assigned to each mutation group was plotted. Almost all DADA2 patients with vasculitis had at least one mutation with ≥ 3% residual ADA2 function and therefore were distributed to the AA, AB and AC groups (Figure 4). Most of the remaining patients, the majority of whom were homozygous for R169Q, were assigned to the BB group. By contrast, the majority of PRCA and BMF cases were found in the BB, BC, and CC groups, which have lower predicted residual ADA2 function (Chi-square, p < 0.0001). To reflect the actual number of cases in each category, all three groups were plotted together in Figure E3 in Online Repository. Accordingly, the prevalence of BMF and PRCA cases was greater in the genotype categories predicted to have lower residual ADA2 activity. These findings support the existence of genotype-phenotype correlations in DADA2, where missense mutations with greater residual enzymatic function favor the development of vasculitis while more detrimental missense mutations, indels and early-termination mutations causing more extensive disruption of protein function are associated with hematologic manifestations.
Figure 4. Genotype to phenotype analysis using patient mutation configurations.

Distribution of patients with vasculitis, PRCA or BMF phenotype in genotype categories assigned based on ADA2 mutation type and residual enzymatic activity of missense mutations (p < 0.0001, Chi-square test). Bars represent the percentage of patients of the given phenotype.
Discussion
DADA2 was first described as a form of monogenic vasculitis that mimics polyarteritis nodosa. Case reports and small case series have subsequently established severe hematologic defects as an alternate presentation of DADA2. The 15 new cases with PRCA / BMF described in this study represent the largest series to date for the severe hematologic phenotype of DADA2. We found that patients with PRCA tend to present very early in life and that those with BMF exhibit a high rate of mortality from recurrent infections. Extending the functional analysis of ADA2 mutations to include the more than 150 published cases in the literature, our work provides new evidence for genotype-phenotype correlations in DADA2. Mutations that are most detrimental to protein function, as measured by residual ADA2 activity, are enriched in patients with severe hematologic involvement.
Genotype-phenotype correlations in DADA2 have been difficult to establish due to incomplete penetrance and variable clinical manifestations in family members with identical mutations 21. Further, large case series have primarily characterized patients with vasculitis. A recent study comparing 12 patients with vasculitis and 10 with PRCA concluded that mutations in the dimerization domain of ADA2 were associated with vasculitis while those in the catalytic domain aligned with PRCA 11. However, almost all patients with vasculitis shared the G47R missense mutation. Our analysis of a broader range of variants showed that mutations associated with each phenotype were distributed throughout the gene without preferential location to specific domains.
The physiologic function of ADA2 is not fully elucidated and therefore it remains unclear whether the same mechanism underlies the development of vasculitis and hematologic defects. Based on our stratification of ADA2 mutations, it is possible that only a small amount of ADA2 is required to maintain normal hematopoiesis, since patients with the most detrimental mutations are most prone to developing PRCA and BMF. It remains unexplained why identical mutations result in PRCA in some patients and BMF in others. Whether ADA2 is produced by specific hematopoietic progenitor cells or acts differentially on these cells as a soluble factor warrants further investigation. Additional modifier genes and extrinsic factors in the bone marrow environment may also contribute to variable phenotype.
How ADA2 mutations with residual enzymatic function cause vasculitis remains to be determined. Carmona-Rivera et al. recently found that in the absence of ADA2, adenosine can trigger the formation of neutrophil extracellular traps (NET), which stimulates macrophages to produce TNF-α 28. While this mechanism may explain the basis of vasculitis and the effectiveness of TNFi therapy, it does not account for the observation that patients with the most detrimental mutations (e.g. nonsense and indels with frameshift) often do not present with vasculitis. It remains to be seen whether the same mechanism applies to the hematologic phenotype of DADA2, which seems less responsive to TNFi. Of the 10 patients that received TNFi in this case series, only one patient showed sustained improvement of PRCA after initiation of etanercept and corticosteroids. Three other patients showed improved fever and/or systemic inflammation without amelioration of baseline cytopenia. Recurrent infections are common in patients with BMF, and the addition of TNFi may further compromise immune defense. Additional studies are needed to address whether TNFi should be used routinely for all DADA2 patients.
Critically, the amount of ADA2 generated by our overexpression system is 20-fold higher than the average plasma ADA2 activity (>250 U/L for supernatant from 293T cells, compared to 12 U/L for healthy adult control plasma) 15. This overexpression increases the dynamic range of our measurements, enabling us to demonstrate that ADA2 mutations associated with vasculitis provide greater residual function than mutations associated with PRCA and BMF. In clinical practice, measurement of plasma ADA2 is limited by the inability of current techniques to resolve small differences in residual activity, which in most DADA2 patients therefore appears uniformly very low. We do not expect that the difference between levels of residual ADA2 function confirmed here in vitro will be observable in clinical samples, as would be required to extrapolate an expected clinical course from measured plasma ADA2 activity.
Although we here stratified each case into a phenotypic category based on primary manifestations, DADA2 phenotypes likely represent continua rather than distinct categories. DADA2 patients with vasculitis can develop anemia and leukopenia as part of their clinical course. Similarly, patients with severe hematologic defects remain susceptible to vasculitis. This potential overlap in phenotypic spectra is best illustrated by the R169Q missense variant, one of the most common pathogenic mutations in DADA2. The R169Q substitution renders minimal residual ADA2 activity (category B, <3%) and is found in all phenotypic categories in our analysis. Supporting this view, a wide spectrum of manifestation including stroke, red cell aplasia and profound cytopenias were reported in a cohort of patients with homozygous R169Q mutations 29. Inference from genotype is also complicated by high prevalence of compound heterozygosity in DADA2. Therefore, our ability to predict phenotype based on genotype alone remains limited and treatment decisions should be guided by clinical findings.
The spectrum of clinical manifestations in DADA2 extends beyond vasculitis, PRCA and BMF. Common variable immunodeficiency (CVID) has been recently described as a manifestation of the disease 13. It is unclear whether CVID represents a distinct phenotype in DADA2 because many of these patients also exhibited vasculitis and hematologic defects. The prevalence of hypogammaglobinemia in our cohort is similar to the general estimate for all patients 3. We suspect that humoral immunodeficiency with low immunoglobulin levels likely represents a common clinical feature of DADA2 regardless of the presenting phenotype. DADA2 can also manifest as autoimmunity (systemic lupus erythematosus and anti-phospholipid syndrome) and lymphoproliferative disease 30, 31. Patients in our cohort did not exhibit these features and autoantibodies were present only in a few patients. With only a limited number of cases reported, it is difficult to establish genotype correlations for these uncommon DADA2 phenotypes.
The broad clinical spectrum of DADA2 and variability in patient presentation are well recognized, but little is known about factors that influence disease phenotype. By characterizing a cohort of patients with PRCA or BMF, our work highlights the severity of hematologic manifestations and their associated morbidity and mortality in DADA2 patients. Systematic comparison of ADA2 mutations in patients with vasculitis, PRCA and BMF through functional analysis revealed a distinct correlation between mutation pathogenicity and disease phenotype. Further studies are needed to determine differences in the underlying pathophysiology of vasculitis and hematologic defects in DADA2.
Supplementary Material
Acknowledgements
We thank Drs. M. Hershfield, N. Ganson and S. Kelly for assistance with developing the spectrophotometric assay for quantitation of ADA2 activity, and S. Klayme and G. Debo for technical assistance.
Funding Sources: This work was supported by the National Institute of Health / National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) K08-AR074562 (P.Y.L.), R01-AR065538, R01-AR073201, R01-AR075906 and P30-AR070253 (P.A.N.), the Fundación Bechara (P.A.N.), the Arbuckle Family Foundation for Arthritis Research (P.A.N.), the Rheumatology Research Foundation Investigator Award (P.Y.L.), Boston Children’s Hospital Faculty Career Development Award (P.Y.L), the National Natural Science Foundation of China Grants 31771548 and 81971528 (Q.Z), and the Zhejiang Provincial Natural Science Foundation of China Grant LR19H100001 (Q.Z).
Abbreviations
- ADA2
adenosine deaminase 2
- DADA2
Deficiency of ADA2
- BMF
Bone marrow failure
- CADD
Combined Annotation Dependent Depletion
- CVID
combined variable immunodeficiency
- GCSF
granulocyte colony stimulating factor
- HSCT
Hematopoietic stem cell transplant
- PRCA
Pure red cell aplasia
- TNF
Tumor necrosis factor
- TNFi
TNF inhibitor
Footnotes
Conflict of Interest disclosure statement: The authors declare no conflict of interest relevant to the study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med 2014; 370:921–31. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Stone DL, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med 2014; 370:911–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee PY. Vasculopathy, Immunodeficiency, and Bone Marrow Failure: The Intriguing Syndrome Caused by Deficiency of Adenosine Deaminase 2. Front Pediatr 2018; 6:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyts I, Aksentijevich I. Deficiency of Adenosine Deaminase 2 (DADA2): Updates on the Phenotype, Genetics, Pathogenesis, and Treatment. J Clin Immunol 2018; 38:569–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghurye RR, Sundaram K, Smith F, Clark B, Simpson MA, Fairbanks L, et al. Novel ADA2 mutation presenting with neutropenia, lymphopenia and bone marrow failure in patients with deficiency in adenosine deaminase 2 (DADA2). Br J Haematol 2019; 186:e60–e4. [DOI] [PubMed] [Google Scholar]
- 6.Hashem H, Egler R, Dalal J. Refractory Pure Red Cell Aplasia Manifesting as Deficiency of Adenosine Deaminase 2. J Pediatr Hematol Oncol 2017; 39:e293–e6. [DOI] [PubMed] [Google Scholar]
- 7.Michniacki TF, Hannibal M, Ross CW, Frame DG, DuVall AS, Khoriaty R, et al. Hematologic Manifestations of Deficiency of Adenosine Deaminase 2 (DADA2) and Response to Tumor Necrosis Factor Inhibition in DADA2-Associated Bone Marrow Failure. J Clin Immunol 2018; 38:166–73. [DOI] [PubMed] [Google Scholar]
- 8.Ben-Ami T, Revel-Vilk S, Brooks R, Shaag A, Hershfield MS, Kelly SJ, et al. Extending the Clinical Phenotype of Adenosine Deaminase 2 Deficiency. J Pediatr 2016; 177:316–20. [DOI] [PubMed] [Google Scholar]
- 9.Hashem H, Kumar AR, Muller I, Babor F, Bredius R, Dalal J, et al. Hematopoietic stem cell transplantation rescues the hematological, immunological, and vascular phenotype in DADA2. Blood 2017; 130:2682–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ulirsch JC, Verboon JM, Kazerounian S, Guo MH, Yuan D, Ludwig LS, et al. The Genetic Landscape of Diamond-Blackfan Anemia. Am J Hum Genet 2018; 103:930–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ozen S, Batu ED, Taskiran EZ, Ozkara HA, Unal S, Guleray N, et al. A Monogenic Disease with a Variety of Phenotypes: Deficiency of Adenosine Deaminase 2. J Rheumatol 2019. [DOI] [PubMed] [Google Scholar]
- 12.Arts K, Bergerson JRE, Ombrello AK, Similuk M, Oler AJ, Agharahimi A, et al. Warts and DADA2: a Mere Coincidence? J Clin Immunol 2018; 38:836–43. [DOI] [PubMed] [Google Scholar]
- 13.Schepp J, Proietti M, Frede N, Buchta M, Hubscher K, Rojas Restrepo J, et al. Screening of 181 Patients With Antibody Deficiency for Deficiency of Adenosine Deaminase 2 Sheds New Light on the Disease in Adulthood. Arthritis Rheumatol 2017; 69:1689–700. [DOI] [PubMed] [Google Scholar]
- 14.Hashem H, Kelly SJ, Ganson NJ, Hershfield MS. Deficiency of Adenosine Deaminase 2 (DADA2), an Inherited Cause of Polyarteritis Nodosa and a Mimic of Other Systemic Rheumatologic Disorders. Curr Rheumatol Rep 2017; 19:70. [DOI] [PubMed] [Google Scholar]
- 15.Lee PY, Schulert GS, Canna SW, Huang Y, Sundel J, Li Y, et al. Adenosine deaminase 2 as a biomarker of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Ann Rheum Dis 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ombrello AK, Qin J, Hoffmann PM, Kumar P, Stone D, Jones A, et al. Treatment Strategies for Deficiency of Adenosine Deaminase 2. N Engl J Med 2019; 380:1582–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee PY, Huang Y, Zhou Q, Schnappauf O, Hershfield MS, Li Y, et al. Disrupted N-linked glycosylation as a disease mechanism in deficiency of ADA2. J Allergy Clin Immunol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cipe FE, Aydogmus C, Serwas NK, Keskindemirci G, Boztug K. Novel Mutation in CECR1 Leads to Deficiency of ADA2 with Associated Neutropenia. J Clin Immunol 2018; 38:273–7. [DOI] [PubMed] [Google Scholar]
- 19.Hsu AP, West RR, Calvo KR, Cuellar-Rodriguez J, Parta M, Kelly SJ, et al. Adenosine deaminase type 2 deficiency masquerading as GATA2 deficiency: Successful hematopoietic stem cell transplantation. J Allergy Clin Immunol 2016; 138:628–30 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caorsi R, Penco F, Grossi A, Insalaco A, Omenetti A, Alessio M, et al. ADA2 deficiency (DADA2) as an unrecognised cause of early onset polyarteritis nodosa and stroke: a multicentre national study. Ann Rheum Dis 2017; 76:1648–56. [DOI] [PubMed] [Google Scholar]
- 21.Nanthapisal S, Murphy C, Omoyinmi E, Hong Y, Standing A, Berg S, et al. Deficiency of Adenosine Deaminase Type 2: A Description of Phenotype and Genotype in Fifteen Cases. Arthritis Rheumatol 2016; 68:2314–22. [DOI] [PubMed] [Google Scholar]
- 22.Rama M, Duflos C, Melki I, Bessis D, Bonhomme A, Martin H, et al. A decision tree for the genetic diagnosis of deficiency of adenosine deaminase 2 (DADA2): a French reference centres experience. Eur J Hum Genet 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kyle RA, Linman JW. Gingivitis and chronic idiopathic neutropenia: report of two cases. Mayo Clin Proc 1970; 45:494–504. [PubMed] [Google Scholar]
- 24.Lee PY, Huang Y, Zhou Q, Schnappauf O, Hershfield MS, Li Y, et al. Disrupted N-linked glycosylation as a disease mechanism in deficiency of ADA2. J Allergy Clin Immunol 2018; 142:1363–5 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamprecht P, Humrich JY, Diebold I, Riemekasten G. Diagnosis of deficiency of adenosine deaminase 2 with early onset polyarteritis nodosa in an adult patient with a novel compound heterozygous CECR1 mutation. Clin Exp Rheumatol 2018; 36 Suppl 111:177. [PubMed] [Google Scholar]
- 26.Springer JM, Gierer SA, Jiang H, Kleiner D, Deuitch N, Ombrello AK, et al. Deficiency of Adenosine Deaminase 2 in Adult Siblings: Many Years of a Misdiagnosed Disease With Severe Consequences. Front Immunol 2018; 9:1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 2003; 31:3812–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carmona-Rivera C, Khaznadar SS, Shwin KW, Irizarry-Caro JA, O’Neil LJ, Liu Y, et al. Deficiency of adenosine deaminase 2 triggers adenosine-mediated NETosis and TNF production in patients with DADA2. Blood 2019; 134:395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Montfrans JM, Hartman EA, Braun KP, Hennekam EA, Hak EA, Nederkoorn PJ, et al. Phenotypic variability in patients with ADA2 deficiency due to identical homozygous R169Q mutations. Rheumatology (Oxford) 2016; 55:902–10. [DOI] [PubMed] [Google Scholar]
- 30.Skrabl-Baumgartner A, Plecko B, Schmidt WM, Konig N, Hershfield M, Gruber-Sedlmayr U, et al. Autoimmune phenotype with type I interferon signature in two brothers with ADA2 deficiency carrying a novel CECR1 mutation. Pediatr Rheumatol Online J 2017; 15:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trotta L, Martelius T, Siitonen T, Hautala T, Hamalainen S, Juntti H, et al. ADA2 deficiency: Clonal lymphoproliferation in a subset of patients. J Allergy Clin Immunol 2018; 141:1534–7 e8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.