

Abstract
Background & Aims
Microvillus inclusion disease (MVID) is caused by inactivating mutations in the myosin VB gene (MYO5B). MVID is a complex disorder characterized by chronic, watery, life-threatening diarrhea that usually begins in the first hours to days of life. We developed a large animal model of MVID to better understand its pathophysiology.
Methods
Pigs were cloned by transfer of chromatin from swine primary fetal fibroblasts, which were edited with TALENs and single strand oligonucleotide to introduce a P663 to L663 substitution in the endogenous swine MYO5B (corresponding to the P660L mutation in human MYO5B, associated with MVID) to fertilized oocytes. We analyzed duodenal tissues from patients with MVID (with the MYO5B P660L mutation) and without (controls), and from pigs, using immunohistochemistry. Enteroids were generated from pigs with MYO5B(P663L) and without the substitution (control pigs).
Results
Duodenal tissues from patients with MVID lacked MYO5B at the base of the apical membrane of intestinal cells; instead MYO5B was intracellular. Intestinal tissues and derived enteroids from MYO5B(P663L) piglets had reduced apical levels and diffuse sub-apical levels of NHE3 and SGLT1, which regulate transport of sodium, glucose, and water, compared with tissues from control piglets. However, intestinal tissues and derived enteroids from MYO5B(P663L) piglets maintained CFTR on apical membranes, like tissues from control pigs. Liver tissues from MYO5B(P663L) piglets had alterations in BSEP, a transporter that facilitates bile flow, which is normally expressed in the bile canaliculi in the liver.
Conclusions
We developed a large animal model of MVID that has many features of the human disease. Studies of this model could provide information about the functions of MYO5B and MVID pathogenesis, and might lead to new treatments.
Keywords: malabsorption, missense mutation, motor protein, plasma membrane
Lay Summary
The authors use gene editing to create pigs with the genetic mutation that causes microvillus inclusion disease. These pigs develop many features of the human disease and might be used to study how it develops and potential therapeutics.
Graphical Abstract
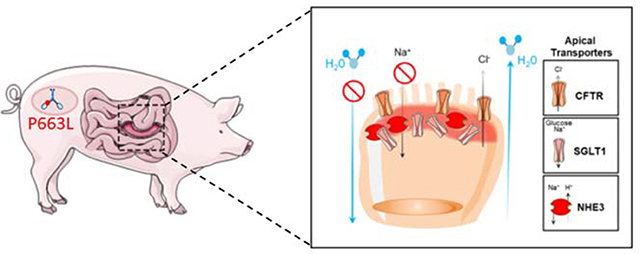
Introduction
Microvillus Inclusion Disease (MVID) is a congenital diarrhea disorder that results from inactivating mutations in the molecular motor, Myosin Vb (MYO5B)1–3. Among congenital diarrhea disorders, MVID is one of the most severe, with life-threatening diarrhea developing in the first week of life, requiring early management with administration of total parenteral nutrition (TPN)4. No definitive treatments exist for MVID outside of chronic TPN or small bowel transplantation. In a subset of individuals, treatment is also accompanied by liver transplantation5, 6. Whether TPN contributes to the liver cholestasis observed in individuals with MVID has been controversial7, 8.
While the prevalence of MVID in the general population is low (less than 200 known cases, orpha.net), there is an increased incidence of MVID in Turkey and in the Navajo Nation in the American Southwest9. In the Navajo tribe, the majority of individuals with MVID have a missense mutation (1979C>T p.Pro660Leu, exon 16 referred to as MYO5B(P660L)3, 10. Prior to 2015, MVID studies solely relied on in vitro cell lines and human tissue2, 11–13. Recently, mouse models have been generated to elucidate the pathogenesis of MVID by characterizing alterations in physiological function and intestinal structure resulting from loss of Myo5b in vivo14–17. While mouse models of human diseases are useful, complex diseases such as MVID may be better understood using large animal models that are closer to human physiology. To date, no large animal model has been generated for MVID. To better understand the pathogenesis of human MVID, we developed a porcine model of MVID using gene editing to express a mutation in MYO5B (P663L) homologous with the human MYO5B mutation found in the Navajo tribe (P660L). Piglets expressing the MYO5B(P663L) mutation demonstrated the presence of microvillus-lined inclusions, a hallmark of MVID, along the length of the small and large intestine. Alterations in sodium transporters (NHE3 and SGLT1) that facilitate enterocyte water absorption were present, with prominent subapical expression of these transporters in MYO5B(P663L) pig small and large intestine. The cystic fibrosis transmembrane conductance regulator (CFTR), which secretes chloride into the intestinal lumen was maintained on the apical membrane of enterocytes in MYO5B(P663L) enterocytes. Immunostaining of WT and MYO5B(P663L) liver showed alterations in bile salt export pump (BSEP) at the apical canalicular membrane of hepatocytes in MYO5B(P663L) pigs compared to WT. These results show that villus blunting as well as liver alterations that likely contribute to cholestasis occur early on in pigs with the Navajo mutation in MYO5B. Development of treatments that promote the proper delivery of sodium transporters to the apical membranes of enterocytes and BSEP to the canalicular membrane of hepatocytes may be efficacious in treating MVID.
Methods
TALEN design, assembly, and RNA synthesis
All TALENs were designed using the TAL Effector Nucleotide Targeter 2.0 software and assembles using standard methods 18–23. Linearized TALEN DNA was transcribed in vitro using the mMessage Machine T3 kit (Ambion). Synthesis reactions were assembled in a 20 μL reaction with 1 μg linearized plasmid DNA, 1× NTP/CAPs (Ambion), 1× reaction buffer (Ambion), and 2 μL enzyme mix (Ambion). Reactions were incubated for 2 h at 37°C, treated with Turbo DNase (Invitrogen), t hen cleaned up with the RNeasy Mini Kit (Qiagen).
Cell culture and transfection of swine embryonic fibroblasts
Fetal fibroblasts isolated from day 38–45 Landraces pig fetuses were cultured in 1× high-glucose Dulbecco’s modified eagle medium (DMEM) (Invitrogen) with 10% FBS (Atlas Biologicals), 2 mM L-glutamine (Corning), 10 mM HEPES buffer (Lonza), 1× penicillin/streptomycin solution (Corning), 5 μg/mL Apo-Transferrin (Sigma), 20 ng/mL recombinant human IGF-1 (R&D Systems), and 25 ng mL−1 recombinant human EGF (R&D Systems) and transfected using the Neon Transfection System (Thermo Fisher Scientific). Briefly, each transfection reaction included 600,000 fibroblasts, 1 μg of TALEN RNA, and 0.2 nmol HDR oligonucleotide, and the transfection reaction was pulsed once at 1800 V for 20 ms using the Neon™ transfection system (Thermo Fisher Scientific). Transfected cells were cultured 3 days at 30 °C, before splitting for restriction fragment length polymorphism (RFLP) analysis and plating for colony isolation at 38.5 °C. Individual colonies were collected in 10 cm dis hes, where 80–250 transfected cells were plated and allowed to grow for 10–14 days and individual colonies were aspirated under gentle trypsinization. The picked colonies were subsequently genotyped by RFLP and sequence analysis.
Detection and sequence validation of gene modification
Transfected cells harvested at day three were prepared for PCR analysis by pelleting and resuspending in PCR-safe lysis buffer (10 mm Tris·Cl, pH 8.0; 2 mM EDTA; 2.5% (vol/vol) Tween-20; 2.5% (vol/vol) Triton X-100; 100 μg mL−1 proteinase K) at ~1000 cells per μL, followed by incubation at 55 °C for 60 min and 95 °C for 15 min. Typically, 1 μL of prepared lysate was used in a 2× AccuStart II PCR SuperMix (QuantaBio); all other applications were according to the manufacturer’s protocol. Gene modification in individual colonies was detected by RFLP analysis and direct sequencing of PCR amplicons, characterized by TOPO cloning (Invitrogen) and sequencing.
Animal husbandry and cloning
MYO5B pigs were produced under license of chromatin transfer technology from Hematech to Cooperative Resources International Center for Biotechnology (CRI_ICB), Verona, WI). All animal work was performed in Recombinetics Inc. facilities under its Animal Welfare Assurance #A4728–01. All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC).
Three MYO5B(P663L) pigs were born alive via Caesarean section. However, one MYO5B(P663L) pig died upon delivery. The tissue harvested from this pig lacked well developed epithelial tissues when examined by hematoxylin and eosin staining (H&E) and intestinal epithelial markers were poorly detected. Two MYO5B(P663L) pigs were euthanized within 24 hours of delivery by intracardiac barbiturate injection following standard procedures of the Veterinary Diagnostic Laboratory of the University of Minnesota under its Animal Welfare Assurance #A3456–01. Three WT pigs were age matched from different litters and examined as controls. For all experiments n=2 MYO5B(P663L) pigs and 3 WT pigs.
Immunostaining
Paraffin
Tissue was excised and immediately fixed in 4% paraformaldehyde (PFA) or 10% neutral buffered formalin overnight at 4°C. Fix ed tissue was embedded in paraffin and 6-micron sections were used for immunostaining. Slides were heated prior to deparaffinization. Antigen retrieval was performed using Target Retrieval Solution citrate buffer pH 6 (Dako, Cat# S1699) using a pressure cooker set on high for 15 minutes. Slides were cooled in antigen retrieval on ice prior to blocking using protein block serum-free (Dako, Cat# X0909) for 1.5 hours at room temperature. Primary antibodies were added to antibody diluent with background reducing components (Dako, Cat# S3022) following the protein block and incubated overnight at 4°C. Sections were washed 3 times in 1× PBS for 5 minutes per wash. Secondary antibodies were diluted 1:200 using antibody diluent (Dako, Cat# S0809) and incubated for 1 hour in the dark at room temperature. Hoechst 33342 (Thermo Fisher Scientific, Cat# 62249; 10 mg/ml) in PBS was applied to slides for 5 minutes to stain the nuclei. Slides were washed 3 times in PBS for 5 minutes each wash. Prolong Gold Antifade (Thermo Fisher Scientific, Cat# P36934) was used to coverslip each slide. Slides were imaged using an Axio microscope or a LSM880 with Airyscan. Antibodies used are listed in Supplemental Table 1.
All authors had access to the study data and reviewed and approved the final manuscript.
Results
Generation of a MVID pig model by gene-editing
To generate a pig model of MVID, we mimicked a missense mutation p.Pro660Leu in the MYO5B gene identified in the Navajo population3, 10. MYO5BP660L lies within exon 28 of the swine MYO5B gene at position 663, which shares 100% amino acid identity with human exon 16 (Supplemental Figure 1A). TALENs flanking MYO5BP663L were designed and subsequently transfected into fetal Landrace fibroblasts with or without a homology directed repair (HDR) oligonucleotide containing the P663L mutation and a HindIII restriction fragment length polymorphism (RFLP) site (Supplemental Figure 1B). Cutting and homology repair efficiency of the TALENs was determined by Surveyor and RFLP assay, respectively (Supplemental Figure 1C and D). Colonies derived from single cells were isolated and genotyped for the MYO5BP663L mutation (data not shown). Owing to the low efficiency of the genome engineering tools, no homozygous colonies were isolated besides one semi-homozygous clone (clone 44) that had the intended P663L mutation on one allele, and a 70 base pair deletion on the other allele which removes a portion of exon 28 and intron 27, including the predicted splice receptor (Allele 1). The deletion on allele 1 is predicted to create a null mutation, as it leads to the incorporation of intron 27 and downstream stop codons. The semi-homozygous clone was subjected to chromatin transfer resulting in two viable pregnancies and 4 F0 male piglets (Supplemental Figure 1E). MYO5BP663L/KO, referred to as MYO5B(P663L), F0 and wildtype control pigs were genotyped by RFLP and sequence validated (Supplemental Figure 1F and G).
MYO5B(P663L) Pigs Display Intestinal Abnormalities Characteristic of MVID
The distribution of MYO5B in duodenal biopsy tissue from healthy individuals and Navajo patients with MVID (MYO5B(P660L)) was examined by immunostaining. In healthy control tissue, MYO5B appeared predominantly expressed at the base of the brush border. MVID patients with the MYO5B P660L mutation, lacked MYO5B near the apical membrane, instead MYO5B was observed intracellularly (Supplemental Figure 2A). The altered localization of MYO5B in Navajo MVID patients is consistent with previous findings showing that the P660L mutation creates a rigor disfunction in the motor, which can bind F-actin but cannot complete a motor stroke12.
Similar to healthy humans, the duodenum of WT pigs showed MYO5B primarily restricted to the base of the brush border as delineated by dipeptidyl peptidase 4 staining (DPPIV). MYO5B(P663L) pigs showed MYO5B primarily expressed near the Golgi complex (GM130) with little expression at the base of the apical membrane (Supplemental Figure 2B). Measurement of mean fluorescence intensity of MYO5B in WT and MYO5B(P663L) pigs demonstrated no difference, suggesting that protein levels are not significantly altered by the MYO5B(P663L) mutation, but that the MYO5B(P663L) mutation likely prevents proper function of MYO5B (Supplemental Figure 2C). The immunostaining of MYO5B in MYO5B(P663L) piglets resembled that of human MVID patients with the Navajo P660L mutation12.
H&E staining of the duodenum of WT and MYO5B(P663L) pigs showed large differences in villi size and structure, MYO5B(P663L) pigs exhibited stunted and thickened villi compared to WT (Figure 1A) consistent with reports demonstrating villus atrophy in patients with MVID24–26. Duodenal tissue of WT piglets showed well-structured villi and uniformed packing of microvilli (Figure 1B). MYO5B(P663L) duodenal tissue demonstrated profound cell rounding at the tips of villi and cells lacking organized microvilli reminiscent of findings reported by Cutz et al. demonstrating that proximal intestinal enterocytes of MVID patients appeared rounded and smoothed, lacking well-developed microvilli27.
Figure 1: Changes in small intestinal cell morphology resulting from MYO5B(P663L) mutation.

(A) H&E of WT and MYO5B(P663L) pig duodenum. Scale bars= 50 μm. (B) SEM of pig small intestine demonstrating uniform villi in WT pigs. The cells along the villi of WT pig duodenum appeared well aligned with uniform microvilli packing. MYO5B(P663L) pigs had broad, stunted villi with prominent cell rounding evident at the tips with disorganized microvilli packing. (C) TEM of the duodenum of neonatal WT and MYO5B(P663L) pigs demonstrated well-formed brush border in WT pig enterocytes. MYO5B(P663L) enterocytes had shortened microvilli, densely packed subapical vesicles, lateral microvilli in gaps between cells (indicated by yellow arrows) and the presence of intracellular microvillus lined inclusions. (D) Prominent F-actin positive inclusions were observed in the small intestine of MYO5B(P663L) pigs. No inclusions were observed in WT pigs. (E) DPPIV immunostaining of WT pig duodenum showed apical localization, in contrast MYO5B(P663L) pig duodenum had diffuse subapical DPPIV Scale bars= 2 μm.
Transmission electron microscopy (TEM) and F-actin staining confirmed changes in cellular morphology in MYO5B(P663L) piglet duodenal tissue, where large intracellular inclusions and lateral microvilli were observed that are characteristic of MVID24, 27 (Figure 1C&D). The presence of lateral microvilli in humans with MVID has not been fully recapitulated in mouse models of MVID28–30. MYO5B(P663L) pigs also exhibited a large accumulation of vesicles below the apical membrane that was not observed in WT pigs, consistent with data obtained from intestinal biopsies from MVID patients31.
Altered Expression of Apical Transporters in MYO5B(P663L) Pigs
The small intestine is the primary site of fluid and electrolyte absorption32. Loss of apical expression of transporters that promote water absorption and maintenance of chloride secretion through CFTR may be the primary cause of MVID associated diarrhea14. SGLT1 is the primary transporter for absorption of galactose and glucose and is responsible for Na+-dependent sugar transport, which is accompanied by water transport thus facilitating hydration32. Immunostaining of the duodenum of WT pigs showed SGLT1 on the apical membrane of enterocytes (Figure 2A). In MYO5B(P663L) pigs, SGLT1 expression was reduced on the apical membrane and subapical SGLT1 was observed. Confocal high magnification images revealed SGLT1 located below the brush border in MYO5B(P663L) pigs, consistent with data obtained from patients with MVID and Myo5b KO mice12, 14. The sodium hydrogen exchanger 3 (NHE3) also promotes small intestinal Na+ and water absorption. In patients with MVID and in Myo5b KO mouse models, NHE3 is mislocalized displaying predominantly intracellular expression14, 17, 31, 33. In the duodenum of WT pigs, NHE3 expression was restricted to the brush border as expected (Figure 2B). In contrast, MYO5B(P663L) pigs exhibited NHE3 in inclusions and diffusely below the apical membrane in the duodenum.
Figure 2: Localization of apical transporters in the duodenum of neonatal pigs.

(A) Confocal imaging demonstrated the presence of diffuse SGLT1 below the apical brush border in MYO5B(P663L) enterocytes compared to WT. (B) NHE3 was observed below the apical membrane in MYO5B(P663L) enterocytes with reduced apical localization compared to WT. (C) CFTR was observed on the apical membrane of MYO5B(P663L) enterocytes as in WT. (D) Alkaline phosphatase immunostaining showed intracellular localization of alkaline phosphatase in MYO5B(P663L) pigs compared to WT. Scale bars=50 and 2 μm in low and high magnification respectively.
The cystic fibrosis transmembrane conductance regulator (CFTR) is a chloride channel that is present on the apical membrane of enterocytes. CFTR is a target of cholera toxin resulting in profound secretory diarrhea by the active secretion of Cl− into the gut lumen, which draws water by osmosis resulting in diarrhea34, 35. In the duodenum of WT pigs, CFTR was localized to the apical membrane of enterocytes (Figure 2C). In MYO5B(P663L) pigs, CFTR was largely maintained on the apical membrane, in contrast to the subapical expression of SGLT1 and NHE3 in MYO5B(P663L) enterocytes. The observed pattern of CFTR expression in MYO5B(P663L) pigs closely mirrored findings in Myo5b KO mouse models and in human MVID tissue14, 33. The loss of apical Na+ transporters and the retention of CFTR in the brush border of enterocytes in MYO5B(P663L) pigs suggests that the inability to absorb Na+, coupled with functional Cl− secretion, may be the primary source of MVID-associated diarrhea.
Intestinal alkaline phosphatase is mislocalized in enterocytes of patients with MVID and in Myo5b KO mouse models14, 16, 31. Consistent with previous studies, MYO5B(P663L) pigs exhibited decreased apical alkaline phosphatase and large amounts of cytoplasmic alkaline phosphatase compared to WT pigs by immunofluorescence. Collectively, these observations suggest that the pig model of MVID shares critical characteristics with human MVID disease and supports a central role for MYO5B in trafficking NHE3, SGLT1 and alkaline phosphatase to the apical membrane of enterocytes.
Intestinal Abnormalities Persist Throughout the Small and Large Intestines of MYO5B(P663L) Piglets
Due to the limited availability of tissue biopsies from MVID patients, the impact of mutations in MYO5B along the length of the small intestine is not clear. Data from mouse models indicate that the duodenum is most affected by altered expression of MYO5B compared to the jejunum and ileum17. Germline Myo5b KO mice exhibit the greatest number of inclusions in the duodenum, with fewer inclusions present in the jejunum and fewer still in the ileum. In addition to the decreasing number of inclusions along the length of the small intestine, structural abnormalities of fused villi occurred at a higher frequency in the duodenum compared to the jejunum and ileum of germline Myo5b KO mice17. However, in humans it has been reported that major alterations exist along the entire small bowel in MVID patients36. WT pigs exhibited phosphorylated ezrin-radixin-moesin (P-ERM) expression on the apical membrane of enterocytes along the whole length of the small intestine. Inclusions positive for P-ERM were observed in the duodenum, jejunum and ileum at similar frequency in MYO5B(P663L) pigs (Figure 3A, C, E). Subapical expression of CD10, a brush border membrane associated peptidase, has been used to diagnose MVID36–38. Micrographs of CD10 immunostaining demonstrate that in WT pigs CD10 is localized to the brush border of enterocytes throughout the small intestine (Figure 3B, D, F). In MYO5B(P663L) pigs, subapical accumulation of CD10 was observed in the duodenum, jejunum and ileum. NHE3 expression along the small intestine showed a similar pattern to CD10 with decreased expression of NHE3 in the brush border and large pools of subapical NHE3 in the jejunum and ileum of MYO5B(P663L) pigs compared to WT pigs (Supplemental Figure 3).
Figure 3: Alterations in brush border markers persist along the length of the small intestine in MYO5B(P663L) pigs.

(A) Phosphorylated ezrin-radixin-moesin (P-ERM) immunostaining showed normal apical localization in WT enterocytes. In contrast, MYO5B(P663L) enterocytes had prominent inclusions that were P-ERM positive. P-ERM immunostaining also demonstrated decreased microvilli height in MYO5B(P663L) enterocytes compared to WT enterocytes. (B) CD10 has been used to diagnose MVID in patients. CD10 immunostaining had apical localization in WT enterocytes. In MYO5B(P663L) enterocytes, CD10 was observed diffusely below the apical membrane. (C & E) Immunofluorescence staining showed P-ERM positive inclusions throughout the jejunum and ileum in MYO5B(P663L) pigs. (D & F) CD10 localized subapically in MYO5B(P663L) enterocytes in the jejunum and ileum. Scale bars=50 and 2 μm in low and high magnification respectively.
Immunostaining of the proximal and distal colon of WT pigs showed apical localization of NHE3 and SGLT1. In contrast, MYO5B(P663L) pigs exhibited large amounts of subapical NHE3 and decreased apical localization of SGLT1 in the proximal and distal colon (Supplemental Figure 4A–D). Moreover, large intracellular inclusions defined by gamma actin immunofluorescence were observed in the proximal and distal colon of MYO5B(P663L) pigs (Supplemental Figure 4E), which was surprising since no inclusions were previously reported in the colons of Myo5b KO mice17. Collectively, these data suggest that MYO5B(P663L) pigs have altered expression of apical membrane components along the entire length of the small and large intestines.
Enteroids Generated from MYO5B(P663L) Pigs Recapitulate in vivo Findings
Enteroids were generated from WT and MYO5B(P663L) pigs to further elucidate enterocyte abnormalities resulting from mutation of MYO5B. Gross morphology of enteroids appeared similar between WT and MYO5B(P663L) enteroids (Figure 4A). However, immunostaining of enteroids showed a poorly developed brush border and mislocalized P-ERM in MYO5B(P663L) enteroids (Figure 4A). Additionally, MYO5B(P663L) enteroids had subapical inclusions consistent with in vivo findings (Figure 4B). To promote differentiation of enteroids, 3D enteroids were plated onto transwells to form a 2D monolayer and were cultured using an air liquid interface (Figure 4C). WT enteroid monolayers demonstrated a well-defined brush border marked by F-actin and apical NHE3 expression (Figure 4D). In contrast MYO5B(P663L) enteroids displayed large amounts of intracellular NHE3 in enterocytes. SGLT1 immunofluorescence was localized to the apical membrane of WT enterocytes, while MYO5B(P663L) enterocytes showed cytoplasmic expression of SGLT1 (Figure 4E). We observed similar levels of CFTR on the apical membrane of WT and MYO5B(P663L) enterocytes, although subapical expression of CFTR was present in WT and MYOB(P663L) enteroid monolayers (Figure 5A).
Figure 4: Pig MYO5B(P663L) enteroids mimic in vivo findings.

(A) Brightfield image of WT and MYO5B(P663L) 3D enteroids appeared similar. Immunofluorescence staining for P-ERM and p120 showed disorganized cell structure in MYO5B(P663L) enteroids and aberrant expression of P-ERM. (B) F-actin staining showed the presence of subapical inclusions in MYO5B(P663L) derived enteroids. (C) Schematic of the development of 2D enteroid monolayer cultured using air liquid interface. (D) Immunostaining of enteroid monolayers demonstrated well developed brush border in WT enterocytes that expressed NHE3 on the apical membrane. Cultured MYO5B(P663L) enterocytes appeared disorganized with subapical accumulation of NHE3. (E) SGLT1 immunostaining showed apical expression in many WT enterocytes. In MYO5B(P663L) enterocytes SGLT1 was observed diffusely below the apical membrane. Scale bars=50 μm in (A), 2 μm in (B), 10 μm in (C & D).
Figure 5: Functional CFTR in MYO5B(P663L) enteroids.

(A) In enteroid monolayers cultured using an air liquid interface CFTR was observed on the apical membrane of MYO5B(P663L) enterocytes. (B) Brightfield micrograph of differentiated WT and MYO5B(P663L) pig derived 3D enteroids before and 1 hour after administration of forskolin. (C) Forskolin swelling, measured as percent change in diameter, after 1 hour forskolin administration. MYO5B(P663L) swelled to a greater degree compared with WT enteroids. *P<0.05, n=155 WT, n=156 MYO5B(P663L) enteroids, performed in three separate experiments. (D) Fecal chloride measures from WT and MYO5B(P663L) piglets as determined by a chloride probe performed in triplicate.
To determine whether CFTR was functional in WT and MYO5B(P663L) enteroids, we used a forskolin swelling assay in 3D enteroids. Administration of forskolin increases the level of intracellular cyclic adenosine monophosphate resulting in activation of CFTR and swelling of enteroids39, 40. Measurement of enteroids before and after forskolin-induced swelling demonstrated that MYO5B(P663L) enteroids swelled to a greater degree than WT enteroids, suggesting that CFTR is functional in MYO5B(P663L) enteroids and may have greater activity than WT enteroids (Figure 5B, C). The increased swelling of MYO5B(P663L) enteroids could also be attributed to the decreased water absorption that results from improper apical localization of NHE3 and SGLT1. We postulate that CFTR activation and the decreased ability of MYO5B(P663L) enteroids to absorb water both contribute to the increased swelling observed in these enteroids.
The intestines of the MYO5B(P663L) piglets showed no formed stool and remarkably clear fluid in the lumen throughout. Fecal chloride levels were measured to determine whether chloride secretion was higher in MYO5B(P663L) piglets compared to WT in vivo (Figure 5D). Chloride concentration was significantly higher in the fecal content of MYO5B(P663L) pigs compared to WT suggesting that CFTR mediated chloride secretion was likely occurring in vivo and that MYO5B(P663L) pigs exhibited a chloride secretory diarrhea.
MYO5B(P663L) Pigs Exhibit Altered Expression of Apical Transporters in Hepatocytes
Studies have reported that many individuals with MVID manifest cholestasis and progressive liver disease41. Recent reports have also noted mutations in MYO5B that do not result in recurrent diarrhea (MVID) in humans, but are associated with low γ-glutamyltransferase cholestasis42, 43. Previously, alterations in liver function in patients with MVID were attributed to prolonged use of TPN7, 8. However, recent studies suggest that mutations in MYO5B are the direct cause of hepatocyte alterations that cause cholestasis41–43. Currently, no published data exists regarding alterations in liver following loss of Myo5b in animal models of MVID. H&E staining showed the presence of lipid droplets in hepatocytes from WT piglets, but no other gross alterations were observed in liver morphology (Figure 6A). Immunostaining for MYO5B in WT pig hepatocytes showed MYO5B closely associated with the canaliculi as defined by MRP2 immunostaining. In MYO5B(P663L) pig hepatocytes, MYO5B appeared in clusters farther away from the canaliculi and had decreased cytoplasmic expression of MYO5B compared to WT hepatocytes (Figure 6B & C). ABCB1 (MDR1) is normally expressed in the canaliculi of hepatocytes and is responsible for the secretion of xenobiotics across the bile canaliculi membrane44. MYO5B(P663L) pig hepatocytes showed decreased expression of ABCB1 at the apical membrane and more diffuse expression at the apical membrane compared to WT hepatocytes (Figure 6D).
Figure 6: Expression of MYO5B and MDR1 in WT and MYO5B(P663L) hepatocytes.

(A) H&E of WT and MYO5B(P663L) pig liver showed the presence of lipid droplets in WT hepatocytes but no other difference was observed. Scale bars=50 μm. (B & C) In WT pigs, MYO5B appeared closely associated with the canalicular membrane, delineated by MRP2 immunostaining. MYO5B also appeared throughout the cytoplasm of hepatocytes. In MYO5B(P663L) hepatocytes MYO5B appeared in dense clusters more distant from the canalicular membrane compared to WT hepatocytes, distance of MYO5B from MRP2 is indicated by arrows. Less cytoplasmic MYO5B was observed in MYO5B(P663L) pigs compared to WT pigs. Scale bars=50 and 5 μm, respectively. (D) MDR1 in WT and MYO5B(P663L) hepatocytes demonstrated decreased expression of MDR1 in MYO5B(P663L) hepatocytes compared with WT. MDR1 appeared diffusely below the canalicular membrane in MYO5B(P663L) hepatocytes.
To determine whether the pig MYO5B(P663L) mutation resulted in liver alterations of other canalicular membrane proteins, we immunostained liver sections for the bile salt export pump (BSEP). Previous investigations have noted a disrupted canalicular distribution of BSEP in patients with mutations in MYO5B41, 42. In MYO5B(P663L) liver hepatocytes, we observed thickened and irregular canalicular and cytoplasmic staining of BSEP compared to WT hepatocytes (Figure 7A). Immunostaining for MRP2 showed subtle alterations with increased thickness of the canalicular membrane in MYO5B(P663L) hepatocytes, but without the cytoplasmic localization seen for BSEP (Figure 7B). The localization of MRP2 in MYO5B(P663L) pigs is consistent with previous reports in patients with MVID who exhibited cholestasis41. Immunostaining for BSEP and MRP2 demonstrate alterations in canalicular morphology in MYO5B(P663L) pigs compared to WT, indicative of a role for MYO5B in the proper formation and delivery of BSEP to the canalicular membrane.
Figure 7: MYO5B(P663L) pigs exhibit alterations in BSEP localization in hepatocytes.

(A) Z-stack projections of WT liver showed well developed canaliculi with apical BSEP localization. MYO5B(P663L) presented canaliculi that appeared stunted and thickened with BSEP present below the canalicular membrane in hepatocytes. (B) MRP2 was observed on the canalicular membrane of WT and MYO5B(P663L) hepatocytes, however MYO5B(P663L) hepatocyte canaliculi appeared less developed. Scale bars=10 μm.
Discussion
A porcine model of MVID exhibits large changes in intestinal epithelium, which are accompanied by alterations in BSEP expression in the liver. We demonstrate that the P663L mutation in MYO5B results in altered localization of MYO5B in the intestine and liver. Proper localization of MYO5B is likely critical for the proper trafficking and anchoring of brush border components. Previous studies suggest that MYO5B serves as a processive anchor, localizing vesicles and their cargoes to the subapical brush border45. In MYO5B(P663L) pigs, MYO5B was predominantly localized in close proximity to the Golgi apparatus, with little sub-apical MYO5B. In human MVID patients with the Navajo MYO5B P660L mutation, a similar distribution of cytoplasmic MYO5B was observed, suggesting that this pig model may closely mimic human MVID. Our data in humans and pigs demonstrate that with the human P660L or pig P663L mutation in MYO5B, MYO5B is concentrated in the cytosol of enterocytes, a pattern distinct from WT MYO5B. In the liver, the altered location of MYO5B in pig hepatocytes is more subtle between WT and MYO5B(P663L) hepatocytes. In MYO5B(P663L) pig liver, MYO5B is still located relatively near the canalicular membrane, although it appears to be more focally concentrated in the cytoplasm compared to WT. This may in part explain why individuals with MVID present with profound intestinal abnormalities immediately after birth, while liver cholestasis manifests later in life.
MYO5B(P663L) pigs showed pronounced villus blunting and cell rounding in the small intestine by electron microscopy, suggesting that MYO5B is crucial for proper villi formation. Villus atrophy and villus blunting have been reported in individuals maintained without enteral feeding46–50 raising the possibility that in MVID biopsies, this observation may not be the direct result of MYO5B mutation. Neonatal mouse models of MVID have not reported pronounced cell rounding at the tips of villi or stunting of villi15–17. However, these intestinal aberrations have been well documented in patients with MVID24, 27, 28, 31. The cell rounding observed by SEM in patients with MVID reported by Cutz et al. is remarkably similar to our observations in MYO5B(P663L) pigs27. While germline Myo5b KO mice lack prominent cell rounding, the dominant negative mutation in the P660L (P663L in pigs) mutation may elicit a greater impact on intestinal epithelium than knockout of MYO5B. Our pig model of MVID supports a role for MYO5B in maintenance or development of intestinal villi structure. TEM showed large gaps existing between intestinal cells in MYO5B(P663L) duodenum compared to WT duodenum. Additionally, lateral microvilli were observed between intestinal cells in MYO5B(P663L) pigs by TEM. The presence of lateral microvilli has been reported in intestinal tissue from patients with MVID by TEM28, 30, 51. It has been contentious whether fluid absorbed by enterocytes flows back into the intestinal lumen through the paracellular pathway in MVID patients. Mouse models of MVID suggested that junctional changes between intestinal cells may be the result of TPN14. However, MYO5B(P663L) pig intestine suggests that profound changes in cell to cell adhesion that result from the P663L mutation early in development may contribute to MVID-associated diarrhea.
The germline Myo5b KO mouse developed in our lab showed the most profound intestinal abnormalities in the proximal small intestine, which included increased frequency of inclusions and fused villi17. Due to the limited availability of tissue samples from the different segments of the intestine from the same patient with MVID, it has been difficult to determine whether individuals with MVID have a less severe intestinal phenotype in the distal small intestine. Examination of the small intestine of MYO5B(P663L) pigs demonstrated similar numbers of inclusions along the whole length of the small intestine as well as subapical accumulation of NHE3 and CD10 in the ileum. The distal small intestine did not show any abatement of mislocalization of brush border components suggesting that inherent differences exist between mouse Myo5b KO models of MVID and the MYO5B(P663L) pig model of MVID. These data also suggest that patients with MVID likely have alterations in transporter localization that persist along the length of the small and large intestines.
The mislocalization of sodium transporters, which showed loss of apical expression in the brush border and intracellular accumulation, closely recapitulates in vivo findings in MVID mouse models as well as immunostaining reports in human MVID patients12, 14, 17, 33, 52. In MYO5B(P663L) pigs, SGLT1 was dramatically reduced on the apical membrane and large amounts of SGLT1 was observed below the brush border compared to WT pigs. SGLT1 is the primary transporter responsible for Na+-dependent sugar transport and is estimated to account for the absorption of 5 liters of water per day in the small intestine of adult humans53, 54. NHE3 also demonstrated decreased apical expression as well as intracellular accumulation in MYO5B(P663L) pigs compared to WT enterocytes. NHE3 is the major sodium hydrogen exchanger in the intestinal brush border and is responsible for the majority of intestinal Na+ absorption, which facilitates net fluid absorption. Recessive SLC9A3 mutations, which result in absent or mutated NHE3 protein, cause congenital sodium diarrhea in humans55. The decreased expression of both SGLT1 and NHE3, which are responsible for a large proportion of Na+-dependent water absorption in the small and large intestines, may explain the severe diarrhea observed in our pig model of MVID and in MVID patients. In support of this concept, MVID patients have high concentrations of fecal Na+ suggesting defects in sodium absorption36.
The mislocalization of NHE3 and SGLT1 likely stems from improper trafficking of NHE3 and SGLT1 to the apical membrane. While apical proteins are often present in inclusions, inclusions are the result of endocytosis of the apical membrane and are not the primary cause of MVID associated diarrhea56. This was demonstrated by inhibition of inclusion formation by knocking out Pacsin 2 (Syndapin 2) in vivo in Myo5b KO mice, which results in rare inclusions. Still, SGLT1 apical localization is severely decreased in these mice, which die early in development and have watery stool. Additionally, adult inducible intestine-specific VillinCreERT2;Myo5bflox/flox mice that lack Myo5b have few inclusions, but still have significantly decreased apical NHE3 and SGLT1 and severe diarrhea14. Therefore, the improper delivery of apical proteins resulting from mutations in MYO5B, rather than inclusions, is likely the primary contributor to decreased apical localization of NHE3 and SGLT1.
In contrast to the loss of apical sodium transporters, CFTR was largely maintained at the apical membrane of enterocytes and increased swelling was observed in MYO5B(P663L) enteroids in response to forskolin. Consistent with these findings, we previously reported that, in tamoxifen induced VillinCreERT2;Myo5bflox/flox mice, CFTR contributes to basal and stimulated Isc to a greater degree than in tamoxifen-treated control mice14. Rhoads et al. previously reported in human jejunal tissue from MVID patients that chloride secretion was near the maximal rate of healthy tissue57. T84 cells with knockdown of MYO5B did not affect CFTR ion transport and CFTR was expressed in the brush border of Caco2 cells with MYO5B knockdown31, 33. Furthermore, CFTR was observed on the apical membrane in human MVID intestinal tissue14, 33. Forteza and colleagues reported that MYO5B loss of function and exposure of intestinal epithelium to glucocorticoids at birth results in PKA stimulation that drives CFTR-mediated Cl− secretion and thus secretory diarrhea58. These data suggest that active chloride secretion mediated through CFTR may be occurring in MVID, further driving dehydration that likely results from mislocalization of sodium transporters.
Cholestasis in patients with MVID was previously presumed to result from long term TPN alimentation, which is a known risk factor in individuals with intestinal failure7, 8, 59, 60. Our large animal model of MVID suggests that early alterations in canalicular membrane protein expression (BSEP and MDR1) result from mutation in MYO5B. In support of these findings, two groups have recently reported that individuals with mutations in MYO5B that have never received parenteral nutrition and do not present recurrent diarrhea, develop intrahepatic cholestasis42, 43. Patients with cholestasis who harbor a mutation in MYO5B have an abnormal distribution of BSEP with subapical expression below the canalicular membrane42, 43. Gonzales et al. also reported thickened canalicular staining similar to our findings in MYO5B(P663L) hepatocytes43. In patients with MVID, cholestasis has been linked to inherent loss of function of MYO5B accompanied by decreased apical BSEP and not TPN alimentation41, 61. In MYO5B(P663L) pig liver, we observed altered expression of MYO5B, which has also been reported in hepatocytes of patients with isolated cholestasis, who have mutations in MYO5B, and in MVID patients who develop cholestasis41–43. Additionally, Overeem et al. has recently found that mutant MYO5B(P660L) causes the intracellular accumulation of bile canaliculi resident proteins in hepatocyte HEPG2 and HUES9 cells62.
In summary, we have examined the effects of the P663L mutation in MYO5B in genetically-engineered neonatal pigs. Our findings demonstrate that the Navajo mutation in MYO5B results in intestinal abnormalities that persist along the length of the small and large intestines and include villus atrophy, inclusions, alterations in apical transporters and mislocalization of MYO5B. Moreover, this large animal model of MVID documents early changes in hepatocyte canalicular membrane formation and expression of BSEP that likely contribute to the cholestasis associated with MVID. While the development of a pig model for MVID was expensive and required cloning of piglets from engineered fibroblasts, the close correlation of pathology in the pigs with those in Navajo MVID patients indicates that significant insights into human disease can be gleaned from pig models. Our findings suggest that development of therapeutic treatments that correct trafficking of sodium transporters to the apical membrane and inhibit chloride secretion through CFTR may help ameliorate MVID associated diarrhea.
Supplementary Material
What you need to know.
BACKGROUND AND CONTEXT
Microvillus inclusion disease (MVID) is caused by inactivating mutations in the myosin VB gene (MYO5B).
NEW FINDINGS
We used gene editing to develop a large animal model of MVID that has many features of the human disease.
LIMITATIONS
At the early postnatal stage analyzed in the MYO5B(P663L) piglets, the full spectrum of disease observed in patients with MVID may not be present.
IMPACT
Studies of this model could provide information about MVID pathogenesis and lead to development of treatments.
Acknowledgements
This work was supported by the National Institute of Health (NIH) grants R43 DK109820 to T.M. and J.R.G., R01 DK48370, R01 DK70856 and a gift from the Christine Volpe Fund to J.R.G. A.C.E was supported by NIH F32 DK111101 and KO1 DK12186901. This work was supported by core resources of the Vanderbilt Digestive Disease Center (P30 DK058404), the Vanderbilt-Ingram Cancer Center (P30 CA68485), and imaging supported by both the Vanderbilt Cell Imaging Shared Resource and the Vanderbilt Digital Histology Shared Resource (supported by a VA Shared Equipment Grant 1IS1BX003097). The authors thank Christopher R. Marino, University of Tennessee, Memphis for the CFTR antibody R3194 (listed in Supplemental Table 1).
Footnotes
The authors have declared that no conflict of interest exists.
Authors names in bold designate shared co-first authorship.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Muller T, Hess MW, Schiefermeier N, et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet 2008;40:1163–5. [DOI] [PubMed] [Google Scholar]
- 2.Ruemmele FM, Muller T, Schiefermeier N, et al. Loss-of-function of MYO5B is the main cause of microvillus inclusion disease: 15 novel mutations and a CaCo-2 RNAi cell model. Hum Mutat 2010;31:544–51. [DOI] [PubMed] [Google Scholar]
- 3.Erickson RP, Larson-Thome K, Valenzuela RK, et al. Navajo microvillous inclusion disease is due to a mutation in MYO5B. Am J Med Genet A 2008;146A:3117–9. [DOI] [PubMed] [Google Scholar]
- 4.Al-Sinani S, Sharef SW, Lakhtakia R, et al. Diagnosis of microvillous inclusion disease: a case report and literature review with significance for oman. Oman Med J 2012;27:497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perry A, Bensallah H, Martinez-Vinson C, et al. Microvillous atrophy: atypical presentations. J Pediatr Gastroenterol Nutr 2014;59:779–85. [DOI] [PubMed] [Google Scholar]
- 6.Siahanidou T, Koutsounaki E, Skiathitou AV, et al. Extraintestinal manifestations in an infant with microvillus inclusion disease: complications or features of the disease? Eur J Pediatr 2013;172:1271–5. [DOI] [PubMed] [Google Scholar]
- 7.Teitelbaum DH. Parenteral nutrition-associated cholestasis. Curr Opin Pediatr 1997;9:270–5. [DOI] [PubMed] [Google Scholar]
- 8.Farrell MK, Balistreri WF. Parenteral nutrition and hepatobiliary dysfunction. Clin Perinatol 1986;13:197–212. [PubMed] [Google Scholar]
- 9.Comegna M, Amato F, Liguori R, et al. Two cases of microvillous inclusion disease caused by novel mutations in MYO5B gene. Clin Case Rep 2018;6:2451–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pohl JF, Shub MD, Trevelline EE, et al. A cluster of microvillous inclusion disease in the Navajo population. J Pediatr 1999;134:103–6. [DOI] [PubMed] [Google Scholar]
- 11.Kravtsov D, Mashukova A, Forteza R, et al. Myosin 5b loss of function leads to defects in polarized signaling: implication for microvillus inclusion disease pathogenesis and treatment. Am J Physiol Gastrointest Liver Physiol 2014;307:G992–G1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knowles BC, Roland JT, Krishnan M, et al. Myosin Vb uncoupling from RAB8A and RAB11A elicits microvillus inclusion disease. J Clin Invest 2014;124:2947–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thoeni CE, Vogel GF, Tancevski I, et al. Microvillus inclusion disease: loss of Myosin vb disrupts intracellular traffic and cell polarity. Traffic 2014;15:22–42. [DOI] [PubMed] [Google Scholar]
- 14.Engevik AC, Kaji I, Engevik MA, et al. Loss of MYO5B Leads to Reductions in Na(+) Absorption With Maintenance of CFTR-Dependent Cl(−) Secretion in Enterocytes. Gastroenterology 2018;155:1883–1897 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carton-Garcia F, Overeem AW, Nieto R, et al. Myo5b knockout mice as a model of microvillus inclusion disease. Sci Rep 2015;5:12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneeberger K, Vogel GF, Teunissen H, et al. An inducible mouse model for microvillus inclusion disease reveals a role for myosin Vb in apical and basolateral trafficking. Proc Natl Acad Sci U S A 2015;112:12408–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weis VG, Knowles BC, Choi E, et al. Loss of MYO5B in mice recapitulates Microvillus Inclusion Disease and reveals an apical trafficking pathway distinct to neonatal duodenum. Cell Mol Gastroenterol Hepatol 2016;2:131–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doyle EL, Booher NJ, Standage DS, et al. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res 2012;40:W117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bedell VM, Wang Y, Campbell JM, et al. In vivo genome editing using a high-efficiency TALEN system. Nature 2012;491:114–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carlson DF, Tan W, Lillico SG, et al. Efficient TALEN-mediated gene knockout in livestock. Proc Natl Acad Sci U S A 2012;109:17382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan W, Carlson DF, Lancto CA, et al. Efficient nonmeiotic allele introgression in livestock using custom endonucleases. Proc Natl Acad Sci U S A 2013;110:16526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan WS, Carlson DF, Walton MW, et al. Precision editing of large animal genomes. Adv Genet 2012;80:37–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carlson DF, Fahrenkrug SC, Hackett PB. Targeting DNA With Fingers and TALENs. Mol Ther Nucleic Acids 2012;1:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davidson GP, Cutz E, Hamilton JR, et al. Familial enteropathy: a syndrome of protracted diarrhea from birth, failure to thrive, and hypoplastic villus atrophy. Gastroenterology 1978;75:783–90. [PubMed] [Google Scholar]
- 25.Groisman GM, Sabo E, Meir A, et al. Enterocyte apoptosis and proliferation are increased in microvillous inclusion disease (familial microvillous atrophy). Hum Pathol 2000;31:1404–10. [PubMed] [Google Scholar]
- 26.Lake BD. Microvillus inclusion disease: specific diagnostic features shown by alkaline phosphatase histochemistry. J Clin Pathol 1988;41:880–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cutz E, Rhoads JM, Drumm B, et al. Microvillus inclusion disease: an inherited defect of brush-border assembly and differentiation. N Engl J Med 1989;320:646–51. [DOI] [PubMed] [Google Scholar]
- 28.Phillips AD, Schmitz J. Familial microvillous atrophy: a clinicopathological survey of 23 cases. J Pediatr Gastroenterol Nutr 1992;14:380–96. [PubMed] [Google Scholar]
- 29.Croft NM, Howatson AG, Ling SC, et al. Microvillous inclusion disease: an evolving condition. J Pediatr Gastroenterol Nutr 2000;31:185–9. [DOI] [PubMed] [Google Scholar]
- 30.Iancu TC, Mahajnah M, Manov I, et al. Microvillous inclusion disease: ultrastructural variability. Ultrastruct Pathol 2007;31:173–88. [DOI] [PubMed] [Google Scholar]
- 31.Ameen NA, Salas PJ. Microvillus inclusion disease: a genetic defect affecting apical membrane protein traffic in intestinal epithelium. Traffic 2000;1:76–83. [DOI] [PubMed] [Google Scholar]
- 32.Kiela PR, Ghishan FK. Physiology of Intestinal Absorption and Secretion. Best Pract Res Clin Gastroenterol 2016;30:145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kravtsov DV, Ahsan MK, Kumari V, et al. Identification of intestinal ion transport defects in microvillus inclusion disease. Am J Physiol Gastrointest Liver Physiol 2016;311:G142–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gabriel SE, Brigman KN, Koller BH, et al. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science 1994;266:107–9. [DOI] [PubMed] [Google Scholar]
- 35.Li C, Dandridge KS, Di A, et al. Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J Exp Med 2005;202:975–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruemmele FM, Schmitz J, Goulet O. Microvillous inclusion disease (microvillous atrophy). Orphanet J Rare Dis 2006;1:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Youssef N, F MR, Goulet O, et al. [CD10 expression in a case of microvillous inclusion disease]. Ann Pathol 2004;24:624–7. [DOI] [PubMed] [Google Scholar]
- 38.Groisman GM, Amar M, Livne E. CD10: a valuable tool for the light microscopic diagnosis of microvillous inclusion disease (familial microvillous atrophy). Am J Surg Pathol 2002;26:902–7. [DOI] [PubMed] [Google Scholar]
- 39.Dekkers JF, Wiegerinck CL, de Jonge HR, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med 2013;19:939–45. [DOI] [PubMed] [Google Scholar]
- 40.Foulke-Abel J, In J, Kovbasnjuk O, et al. Human enteroids as an ex-vivo model of host-pathogen interactions in the gastrointestinal tract. Exp Biol Med (Maywood) 2014;239:1124–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Girard M, Lacaille F, Verkarre V, et al. MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillous inclusion disease. Hepatology 2014;60:301–10. [DOI] [PubMed] [Google Scholar]
- 42.Qiu YL, Gong JY, Feng JY, et al. Defects in myosin VB are associated with a spectrum of previously undiagnosed low gamma-glutamyltransferase cholestasis. Hepatology 2017;65:1655–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gonzales E, Taylor SA, Davit-Spraul A, et al. MYO5B mutations cause cholestasis with normal serum gamma-glutamyl transferase activity in children without microvillous inclusion disease. Hepatology 2017;65:164–173. [DOI] [PubMed] [Google Scholar]
- 44.Keppler D. Cholestasis and the role of basolateral efflux pumps. Z Gastroenterol 2011;49:1553–7. [DOI] [PubMed] [Google Scholar]
- 45.Schafer JC, Baetz NW, Lapierre LA, et al. Rab11-FIP2 interaction with MYO5B regulates movement of Rab11a-containing recycling vesicles. Traffic 2014;15:292–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng Y, Teitelbaum DH. Epidermal growth factor/TNF-alpha transactivation modulates epithelial cell proliferation and apoptosis in a mouse model of parenteral nutrition. Am J Physiol Gastrointest Liver Physiol 2012;302:G236–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buchman AL, Moukarzel AA, Bhuta S, et al. Parenteral nutrition is associated with intestinal morphologic and functional changes in humans. JPEN J Parenter Enteral Nutr 1995;19:453–60. [DOI] [PubMed] [Google Scholar]
- 48.Pironi L, Paganelli GM, Miglioli M, et al. Morphologic and cytoproliferative patterns of duodenal mucosa in two patients after long-term total parenteral nutrition: changes with oral refeeding and relation to intestinal resection. JPEN J Parenter Enteral Nutr 1994;18:351–4. [DOI] [PubMed] [Google Scholar]
- 49.Shaw D, Gohil K, Basson MD. Intestinal mucosal atrophy and adaptation. World J Gastroenterol 2012;18:6357–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ney DM. Effects of insulin-like growth factor-I and growth hormone in models of parenteral nutrition. JPEN J Parenter Enteral Nutr 1999;23:S184–9. [DOI] [PubMed] [Google Scholar]
- 51.Morroni M, Cangiotti AM, Guarino A, et al. Unusual ultrastructural features in microvillous inclusion disease: A report of two cases. Virchows Arch 2006;448:805–10. [DOI] [PubMed] [Google Scholar]
- 52.Vogel GF, Janecke AR, Krainer IM, et al. Abnormal Rab11-Rab8-vesicles cluster in enterocytes of patients with microvillus inclusion disease. Traffic 2017;18:453–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wright E, Loo D, Hirayama B, et al. Sugar Absorption Physiology of the Gastrointestinal Tract: Physiology of the gastrointestinal tract. 4th ed. New York, Elsevier; …, 2006. [Google Scholar]
- 54.Hirschhorn N, Kinzie JL, Sachar DB, et al. Decrease in net stool output in cholera during intestinal perfusion with glucose-containing solutions. N Engl J Med 1968;279:176–81. [DOI] [PubMed] [Google Scholar]
- 55.Janecke AR, Heinz-Erian P, Yin J, et al. Reduced sodium/proton exchanger NHE3 activity causes congenital sodium diarrhea. Hum Mol Genet 2015;24:6614–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Engevik AC, Kaji I, Postema MM, et al. Loss of myosin Vb promotes apical bulk endocytosis in neonatal enterocytes. J Cell Biol 2019;218:3647–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rhoads JM, Vogler RC, Lacey SR, et al. Microvillus inclusion disease. In vitro jejunal electrolyte transport. Gastroenterology 1991;100:811–7. [DOI] [PubMed] [Google Scholar]
- 58.Forteza R, Ahsan MK, Carton-Garcia F, et al. Glucocorticoids and Myosin5b loss-of-function induce heightened PKA signaling in addition to membrane traffic defects. Mol Biol Cell 2019:mbcE18070415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lauriti G, Zani A, Aufieri R, et al. Incidence, prevention, and treatment of parenteral nutrition-associated cholestasis and intestinal failure-associated liver disease in infants and children: a systematic review. JPEN J Parenter Enteral Nutr 2014;38:70–85. [DOI] [PubMed] [Google Scholar]
- 60.Rangel SJ, Calkins CM, Cowles RA, et al. Parenteral nutrition-associated cholestasis: an American Pediatric Surgical Association Outcomes and Clinical Trials Committee systematic review. J Pediatr Surg 2012;47:225–40. [DOI] [PubMed] [Google Scholar]
- 61.Schlegel C, Weis VG, Knowles BC, et al. Apical Membrane Alterations in Non-intestinal Organs in Microvillus Inclusion Disease. Dig Dis Sci 2018;63:356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Overeem AW, Li Q, Qiu YL, et al. A molecular mechanism underlying genotype-specific intrahepatic cholestasis resulting from MYO5B mutations. Hepatology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.