

Summary
The recent discovery of extracellular RNAs in blood, including RNAs in extracellular vesicles (EVs), combined with low-input RNA-sequencing advances have enabled scientists to investigate their role in human disease. To date, most studies have been focusing on small RNAs, and methodologies to optimize long RNAs measurement are lacking. We used plasma RNA to assess the performance of six long RNA sequencing methods, at two different sites, and we report their differences in reads (%) mapped to the genome/transcriptome, number of genes detected, long RNA transcript diversity, and reproducibility. Using the best performing method, we further compare the profile of long RNAs in the EV- and no-EV-enriched RNA plasma compartments. These results provide insights on the performance and reproducibility of commercially available kits in assessing the landscape of long RNAs in human plasma and different extracellular RNA carriers that may be exploited for biomarker discovery.
Subject Areas: Medical Laboratory Technology, Omics, Transcriptomics
Graphical Abstract
Highlights
-
•
Low-input RNA-sequencing kits were evaluated using plasma extracellular RNA
-
•
Thousands of unique and diverse long RNA transcripts were detected at >80% coverage
-
•
Extracellular vesicle-enriched fractions had a unique set of long RNA transcripts
Medical Laboratory Technology; Omics; Transcriptomics
Introduction
RNA is an essential biomolecule that plays an important role in diverse cellular functions. Due to this central role, RNA expression has been extensively studied in the context of diagnosis, prognosis, and treatment of complex diseases. Technological advances, especially on the development of RNA sequencing (RNA-seq), provide new opportunities for discoveries related not only to gene expression but also to differing transcript isoforms, splice variants, and gene fusions in an unbiased way (Byron et al., 2016). In fact, RNA-seq-based tests have already made it into clinical applications, such as the FoundationOne Heme test (Foundation Medicine) that employs RNA-seq toward gene fusion detection in blood cancers (Intlekofer et al., 2018), the GEM ExTra test (Ashion Analytics) that integrates exome sequencing and RNA-seq for clinical use (Borad et al., 2014, Nasser et al., 2015), and the ExoDx Prostate test (Exosome Diagnostics) that utilizes RNA-Seq data from extracellular vesicles (i.e., exosomes) isolated from urine (McKiernan et al., 2016). Therefore, the clinical potential for RNA-seq demands further methodological testing toward protocols that maximize efficiency, RNA species output, and can be performed on small sample volumes, and/or low inputs of RNA.
Blood remains the most commonly collected biofluid in the clinic and in most diseases, it represents an ideal source of accessible biological information from diverse tissues. Blood contains a range of biomarkers including proteins, metabolites, DNA, and RNA that can be measured and analyzed for the development of blood-based biomarkers in diverse disease types such as cancer, cardiovascular disease, and neurodegenerative diseases. Recently, multiple efforts have been focusing on the development of diagnostic and therapeutic applications that are based on extracellular RNA (exRNA), primarily RNA encapsulated in extracellular vesicles (EVs) or carried in other carrier subtypes (Srinivasan et al., 2019). EVs (i.e, exosomes and microvesicles) are typically 20–1000 nm vesicles that are released from cells into the blood circulation (or other biofluids) and contain proteins, lipids, DNA, and RNA molecules (reviewed in (Raposo and Stoorvogel, 2013, Yanez-Mo et al., 2015)). The discovery of RNA molecules in blood EVs and the proof that EVs provide protection to RNA molecules from being degraded by RNAses (Wang et al., 2010) lead to an increased interest in the profiling of RNAome in blood EVs under different conditions. In fact, EVs contain all the RNA species that are found in the cell as well (i.e., mRNAs, tRNAs, lncRNAs, rRNAs, miRNAs, etc.) (Murillo et al., 2019). However, electrophoretic analysis of EV RNAs reveal that apart from intact small RNAs, they also contain fragments of longer RNAs (Lasser et al., 2011, Skog et al., 2008), which can make standard sequencing more difficult. Numerous studies have shown that RNAs detected in blood circulating EVs are associated with disease prognosis, diagnosis, and progression and can be therefore used for the development of clinical tests (Ingenito et al., 2019, Liu et al., 2019, Quinn et al., 2015). However, successful development of such biomarkers requires standardized and reproducible RNA-Seq protocols that can be used to measure EV-containing RNAs from small volumes of blood and therefore low yields of RNA input.
Our group has previously focused on the development and optimization of RNA-Seq methods to study small RNAs, such as miRNAs, tRNAs, and piRNAs in biofluids (Murillo et al., 2019, Shah et al., 2017, Yeri et al., 2018). However, equivalent methodology to profile the longer RNAs and their fragments have not been well reported. In this study, we focus on methodology to profile fragments of protein-coding and long non-coding RNA transcripts (e.g., mRNAs, lncRNAs, and other long non-coding RNAs) in biofluids and extracellular RNA carriers. In brief, we took plasma from healthy volunteers and divided the plasma into two independent, uniform pools, extracted the RNA, and compared the RNA profiles obtained across six different RNA-sequencing library preparation kits and two different laboratory sites to determine optimal performance as measured by the number of reads mapping to the genome/transcriptome and RNA species diversity. Using the best-performing RNA-seq kit based on these two metrics, we examined the profile of RNAs in EV-enriched and no-EV plasma compartments isolated from both pooled plasma or plasma from individual human subjects.
Results
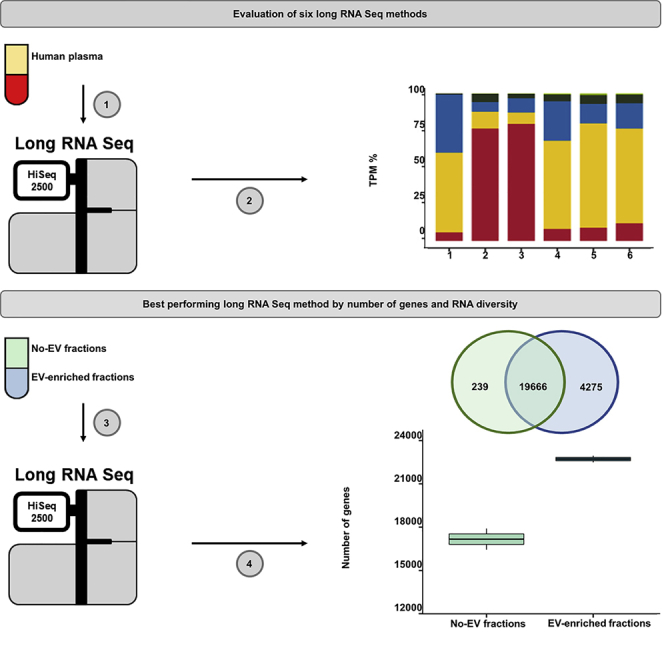
To allow for the systematic comparison of library construction kits/conditions, we employed total RNA from two independent pools of plasma samples. We divided the total RNA from both pools equally among the six different RNA sequencing kits/conditions and constructed libraries in duplicate at two independent sites. Following sample preparation, we performed long RNA sequencing using Illumina's HiSeq 2500 platform to assess genome and transcriptome mapping percentage (Figure 1). In order to standardize the number of input reads for downstream analysis and comparison across kits, FASTQs were randomly and uniformly down-sampled to 50 million read pairs prior to genome and transcriptome mapping.
Figure 1.

Total RNA from Pooled Plasma from Healthy Volunteers was Used to Prepare RNA-Sequencing Libraries to Evaluate Six Different Kits/Conditions. The total RNA was equally divided, and the libraries were constructed in duplicates. All libraries were sequenced on Illumina's HiSeq 2500 platform under the same conditions.
We found that all six tested kits/conditions tended to have similar percentages of reads mapped to the genome across pools, although Ovation SoLo (OS) showed the lowest percentage uniquely mapping reads in both pools (Figure 2A). The percentage of reads mapped to the transcriptome was higher in the TruSeq RNA Access kit (now called RNA Exome) with or without fragmentation, whereas the other kits had ∼50% or fewer of reads mapped to the transcriptome (Figure 2B). To measure the RNA biotypes captured across kits, we took the reads mapping to the transcriptome and displayed the percentage of reads assigned to each of four RNA biotypes defined by Ensembl/Havana/Vega (protein-coding, lncRNAs, ncRNAs, and pseudogenes) and calculated the percentage of counts (Figure 2C) and transcripts per kilobase million (TPM) (Figure 2D) represented by each biotype category. From this analysis, we found that the SMARTer Pico v2 and SMARTer/KapaHyper (Frag and FragRibo) kits had a higher proportion of lncRNA counts and TPMs across the pools. As expected, due to its capture probe design, the RNA Access kit showed the highest percentage of protein coding RNA when analyzed both by counts and TPMs (Figures 2C and 2D). However, when we looked at diversity of RNA species performance, the SMARTer Pico V2 with Fragmentation and Ribosomal RNA depletion (SMART_Pico_Frag) showed the highest number (154,942) of unique transcripts detected as compared with all other kits (Table 1). To assess reproducibility of each kit, we calculated the Spearman's correlation from DESeq2-normalized counts for all pools and replicates. The mean correlation for each pool, kit, and site combination listed is shown in Table S1. We found that all kits had a comparable reproducibility within and between site(s), except for the Ovation_SoLo_Frag and SMART_KAPA_FragRibo kits, which showed lower reproducibility.
Figure 2.

Assessment of Genome and Transcriptome (hg38) Mapping Percentage by RNA-Sequencing Library Preparation Kit/Condition and by RNA Species Using Total RNA from Pooled Plasma.
(A) Percentage of input reads for each RNA pool and kit/condition that were uniquely mapped (green), multimapped (black), or unmapped (gray) to the human genome using STAR.
(B) Percentage of input reads for each RNA pool and kit/condition that were quasimapped (green) or unmapped (gray) to the human transcriptome using salmon.
(C) Percentage of uniquely mapped read counts (light green bar in panel A) assigned to the human transcriptome using featureCounts for each RNA pool and kit/condition and represented by the following RNA species: protein coding (red), lncRNAs (yellow), non-coding RNAs (blue), pseudogenes (black), and other RNAs (including RNAs to be experimentally confirmed, immunoglobulin genes, and T cell receptor genes; green).
(D) Percentage of transcripts per kilobase million (TPM) quasimapped to the human transcriptome (light green bar in panel B) for each RNA pool and kit/condition and represented by the following RNA species: protein coding (red), lncRNAs (yellow), non-coding RNAs (blue), pseudogenes (black), and other RNAs (green).
Table 1.
Number of Long RNA Transcripts Detected in Human Plasma across Evaluated Kits.
| Kit | Mean | SD |
|---|---|---|
| Ovation_SoLo_Frag | 53,695 | 13,109 |
| RNA_Access_Frag | 43,215 | 3,568 |
| RNA_Access_noFrag | 45,961 | 7,756 |
| SMART_KAPA_Frag | 138,658 | 9,927 |
| SMART_KAPA_FragRibo | 113,303 | 34,667 |
| SMART_Pico_Frag | 154,942 | 14,618 |
Our main objective was to assess the long RNA profile of EV-enriched plasma fractions as per the traditional markers (i.e, CD9, CD63, and Alix) and whether is different than other fractions. EVs were isolated using iodixanol cushioned-density gradient centrifugation (C-DGUC), which enriches EVs based on their density (Witwer et al., 2013). Any isolation method will target slightly different EV populations and could give slightly different results regarding the associated exRNAs. However, the C-DGUC method is considered very stringent in separating EVs and lipoproteins (19) and is recommended in the MISEV guidelines to achieve better separation of EVs (20). All samples were dialyzed to remove iodixanol prior to downstream analyses (Figure 3A). As depicted in Figure S1, dialysis did not have any impact on the numbers of EVs; however, we observed a reduction in the average size of particles in both fraction pools as measured by NanoSight LM10. This may be due to the removal of iodixanol, which may cause the formation of larger aggregates. We next aimed to determine any differences in genome/transcriptome mapping percentage, representation of RNA species, and gene expression between fractions enriched in EVs and those that do not contain EVs. For this, we chose the SMARTer Pico V2 with Fragmentation and Ribosomal RNA depletion (SMART_Pico_Frag) protocol due to its overall performance and ability to capture the highest diversity of long RNA transcripts. As depicted in Figure 3B, fractions 6–10 were predominantly positive for the traditional EV markers CD9, CD63, and Alix (compared with fractions 1–5) and therefore we decided to proceed with two pools of fractions (1–5 and 6–10) for the RNA-seq. It is worth noting that fractions 6–10 were positive for other RNA carriers such as the Argonaute 2 (AGO2) protein and high-density lipoproteins (HDL), which was the only marker present in all fractions (Figure 3B). HDL generally segregates with denser fractions (as for small RNA carrier HDL, 21). ApoA1, used as a marker for lipoproteins, is not specific for HDL and may be exchanged between different classes of lipoproteins, including chylomicrons and VLDLs. We did not have control of the fasting state of the individuals whose plasma comprised the pool plasma used for analysis and the western blot. For the individual samples analyzed (subject 1 and 2), the samples were collected from fasting individuals. We recommend, for future studies, that researchers collect samples in the fasting state to eliminate this potential confounder.
Figure 3.

Isolation of Total RNA from EV- and No-EV-Enriched Plasma Fractions for RNA Sequencing
(A) Total RNA from extracellular vesicles (EVs) isolated from either pooled or individual plasma samples was used to compare gene expression between no-EV (fractions 1–5) and EV-enriched (fractions 6–10) plasma compartments. The different sizes of EVs were isolated by cushioned-density gradient ultracentrifugation using iodixanol and purified with dialysis prior to RNA extraction. RNA-sequencing libraries were constructed in duplicates using the SMARTer Pico v2 kit and subsequently sequenced on Illumina's HiSeq 2500 platform.
(B) Western blot analysis of CD9, CD63, and Alix markers for extracellular vesicles, AGO2 for Argonaute 2 proteins, and APOA1 for high-density lipoproteins in all fractions (1–12) retrieved from iodixanol cushioned-density gradient ultracentrifugation and dialysis of plasma samples.
RNA was isolated from fractions 1–5 and 6–10 using ExoRNeasy, and the RNA was quantified for each sample in triplicate using Quant-iT Ribogreen RNA Assay according to ThermoFisher's low-range Ribogreen protocol. Fractions 6–10 consistently had more RNA than fractions 1–5 (Table S2). Analysis of the RNA-seq data showed that fractions 6–10 tended to have higher percentages of reads mapped to the genome and transcriptome across both the pooled and individual plasma samples compared with fractions 1–5 (Figures 4A and 4B). As for the RNA species between the two fraction pools, we found that the percentage of counts and TPMs were similar across the pooled and individual plasma samples (Figures 4C and 4D). In addition, fractions 6–10 showed an increase in transcript diversity, with 67,297 and 74,716 transcripts detected in the pools and subjects, respectively, as compared with 45,057 and 34,664 transcripts detected in fractions 1–5 (Table 2).
Figure 4.

Assessment of Genome and Transcriptome (hg38) Mapping Percentage by RNA-Sequencing Library Preparation Kit/Condition and by RNA Species Using Total RNA from between no-EV (Fractions 1–5) and EV-Enriched (Fractions 6–10) Plasma Compartments.
The different fractions were isolated from either pooled plasma (i.e., Pool A and B) or plasma from individual subjects (i.e., Subject 1 and 2).
(A) Percentage of input reads for each RNA pool/subject and kit/condition by fractions that were uniquely mapped (green), multimapped (black), or unmapped (gray) to the human genome using STAR.
(B) Percentage of input reads for each RNA pool/subject and kit/condition by plasma compartment that were quasimapped (green) or unmapped (gray) to the human transcriptome using salmon.
(C) Percentage of uniquely mapped read counts (light green bar in panel A) assigned to the human transcriptome using featureCounts for each RNA pool/subject and kit/condition by plasma compartment and represented by the following RNA species: protein coding (red), lncRNAs (yellow), non-coding RNAs (blue), pseudogenes (black), and other RNAs (including RNAs to be experimentally confirmed, immunoglobulin genes, and T cell receptor genes; green).
(D) Percentage of transcripts per kilobase million (TPM) quasimapped to the human transcriptome (light green bar in panel B) for each RNA pool/subject and kit/condition by plasma compartment and represented by the following RNA species: protein coding (red), lncRNAs (yellow), non-coding RNAs (blue), pseudogenes (black), and other RNAs (green).
Table 2.
Number of Long RNA Transcripts Detected in No-EV (1–5) and EV-Enriched (6–10) Fractions from Pooled and Individual Plasma Samples.
| Sample Type | Fraction | Number of Transcripts (Mean)a |
|---|---|---|
| Pooled plasma | 1 to 5 | 45,057 |
| Pooled plasma | 6 to 10 | 67,297 |
| Individual plasma | 1 to 5 | 34,664 |
| Individual plasma | 6 to 10 | 74,717 |
Mean number of genes come from the duplicate runs using the SMARTer Stranded Total RNA-Seq Kit v2 - Pico Input Mammalian kit.
Lastly, we sought to examine any differences in the number of genes detected in fractions 1–5 and 6–10 in both the pooled and individual plasma samples. The total number of genes detected in pooled plasma was 24,180, which was very comparable to the number of genes (i.e., 24,613) detected in individual plasma samples. However, when comparing the two fraction pools (1–5 and 6–10), we found that the total number of genes detected in fractions 6–10 in both the pooled (23,941) and individual (24,513) samples was higher than the number of genes detected in the pooled (19,905) and individual (19,558) plasma samples in fractions 1–5 (Figure 5A). Although a comparable number of genes were commonly detected in both fraction pools in the pooled and individual plasma samples (19,666 and 19,458 respectively), a much higher number of genes was uniquely detected in fractions 6–10 as compared with fractions 1–5 in both the pooled (4,275 versus 239 genes) and individual samples (5,055 versus 100 genes) (Figures 5B and 5C). In pooled plasma samples, the uniquely expressed genes in fractions 6–10 represented 18% of the total number of genes, whereas in individual plasma samples, this number represented 21% of the total number of genes. Table 3 shows the number of genes and transcripts detected in fractions 1–5 and 6–10 for the pooled samples and the subject samples. GC content analysis revealed that the distribution pattern was similar for all samples. However, we observed a consistently higher number of reads in fractions 6–10 compared with fractions 1–5 in both the pooled and individual samples that was more apparent around the 50% mean GC content peak (Figure S2).
Figure 5.

Evaluation of Detected Genes in Extracellular Vesicles (EVs) from Pooled or Individual Plasma Samples Using the SMARTer Pico v2 Kit.
Genes were filtered to those with an average of 10 counts across all samples and then normalized using DESeq2.
(A) Box plots showing the total number of genes detected in EV-enriched and no-EV plasma compartments by sample type (pooled versus individual plasma).
(B) Venn diagram showing the number of commonly or uniquely detected genes in no-EV (fractions 1–5) and EV-enriched (fractions 6–10) compartments from pooled plasma samples.
(C) Venn diagram showing the number of commonly or uniquely detected genes in no-EV (fractions 1–5) and EV-enriched (fractions 6–10) compartments from individual plasma samples.
Table 3.
The Number of Genes (Detected at >10 Counts) and Transcripts (Detected at >1 TPM) for Each Pooled and Subject Fraction (1–5 and 6–10).
| Protein Coding | lncRNA | ncRNA | Pseudogene | Other | |
|---|---|---|---|---|---|
| Genes Detected Counts >10 | |||||
| Pool A 1-5 | 10,964 | 2,391 | 87 | 813 | 63 |
| Pool A 6-10 | 13,443 | 3,477 | 68 | 1,023 | 83 |
| Pool B 1-5 | 12,455 | 3,481 | 134 | 1,187 | 94 |
| Pool B 6-10 | 13,699 | 3,651 | 71 | 1,049 | 98 |
| Subject 1 1-5 | 9,160 | 1,528 | 57 | 592 | 33 |
| Subject 1 6-10 | 16,938 | 13,941 | 356 | 3,118 | 350 |
| Subject 2 1-5 | 12,133 | 4,698 | 137 | 1,212 | 106 |
| Subject 2 6-10 | 16,007 | 10,752 | 223 | 2,451 | 261 |
| Transcripts Detected TPM >1 | |||||
| Pool A 1-5 | 10,130 | 4,760 | 209 | 237 | 58 |
| Pool A 6-10 | 5,485 | 2,084 | 234 | 150 | 37 |
| Pool B 1-5 | 9,745 | 4,315 | 256 | 193 | 42 |
| Pool B 6-10 | 5,582 | 2,196 | 270 | 168 | 38 |
| Subject 1 1-5 | 8,442 | 4,164 | 125 | 189 | 42 |
| Subject 1 6-10 | 12,205 | 12,175 | 494 | 936 | 191 |
| Subject 2 1-5 | 9,904 | 5,983 | 260 | 343 | 69 |
| Subject 2 6-10 | 9,465 | 8,110 | 376 | 585 | 121 |
Total genes and transcripts are broken out by RNA biotype as in Figure 4.
One interesting question is whether or not we can detect full gene coverage or we detect only fragments. Figure 6A shows the transcript length distribution of RNA biotypes by abundance for each of the sample fractions, and Figure 6B shows the corresponding mean transcript length, in basepairs, of the transcript length distribution shown in Figure 6A. A large number of protein coding RNA and lncRNAs are detected across a range of RNA transcript lengths. Table 4 describes the number of genes detected at > 80% coverage for transcripts of different lengths. These data help describe and highlight the decreased genes counts and increased TPMs associated with lncRNAs in the gene and transcript plots for Figures 4C and 4D. The low number of gene counts reflects the low abundance of lncRNAs compared with mRNAs in these samples. However, the increased TPMs dedicated to lncRNAs in 4D is due to the length estimate that is included for Salmon outputs; RNA fragments are detected and divided by their length, which is slightly smaller for the detected lncRNAs than the mRNA lengths observed in the distribution of Figure 6.
Figure 6.

Distribution of Transcript Lengths by RNA Biotype for Each of the Sample Fractions.
(A) Histogram of the transcript lengths, by biotype, of transcripts detected in each sample type. The mean length for each biotype is displayed as a vertical dashed line.
(B) Mean transcript length, in basepairs, of transcripts detected in each sample type broken out by biotype.
Table 4.
Number of Transcripts Detected with >80% Coverage at Varying Transcript Lengths (bp).
| Fraction | 200–500 | 501–1,000 | 1,001–5,000 | 5,001–10,000 | >10,000 | |
|---|---|---|---|---|---|---|
| Pool A | 1 to 5 | 9,877 | 21,906 | 15,252 | 1,252 | 160 |
| Pool B | 1 to 5 | 13,067 | 30,275 | 22,624 | 2,140 | 327 |
| Pool A | 6 to 10 | 15,321 | 37,015 | 28,729 | 2,790 | 348 |
| Pool B | 6 to 10 | 14,853 | 34,982 | 26,306 | 2,429 | 309 |
| Subject 1 | 1 to 5 | 4,219 | 9,263 | 5,363 | 323 | 49 |
| Subject 2 | 1 to 5 | 8,039 | 17,035 | 11,668 | 880 | 148 |
| Subject 1 | 6 to 10 | 14,736 | 32,723 | 25,662 | 2,299 | 315 |
| Subject 2 | 6 to 10 | 12,924 | 27,737 | 20,481 | 1,679 | 231 |
Significant pathways from IPA pathway analysis of genes unique to fractions 6–10 include Glucocorticoid Biosynthesis, the Intrinsic Prothrombin Activation Pathway, multiple pathways related to thyroid hormone metabolism, and eNOS Signaling (Table 5). The complete list of pathways from IPA pathway analysis is shown in Table S3. Last, the full list of transcripts uniquely detected in fractions 1–5 or 6–10 is provided in Tables S4 and S5, respectively.
Table 5.
Ingenuity Pathway Analysis of Genes Uniquely Detected in EV-Enriched Fractions 6 to 10.
| Ingenuity Canonical Pathways | Ratio | Molecules | p Value |
|---|---|---|---|
| Glucocorticoid biosynthesis | 0.50 | CYP11B1,CYP11B2,CYP17A1,CYP21A2 | 0.0001 |
| Intrinsic prothrombin activation pathway | 0.17 | F12,FGA,FGB,KLK11,KLK13,KLK4,KLK5 | 0.0005 |
| Mineralocorticoid biosynthesis | 0.43 | CYP11B1,CYP11B2,CYP21A2 | 0.0013 |
| Phototransduction pathway | 0.14 | ARR3,GNAT1,GUCA1A,GUCY2D,GUCY2F,OPN4,RHO | 0.0018 |
| Thyronamine and iodothyronamine metabolism | 0.67 | DIO1,DIO3 | 0.0035 |
| Thyroid hormone metabolism I (via deiodination) | 0.67 | DIO1,DIO3 | 0.0035 |
| SPINK1 pancreatic cancer pathway | 0.11 | CELA2A,KLK11,KLK13,KLK4,KLK5,PRSS3 | 0.0117 |
| Extrinsic prothrombin activation pathway | 0.19 | F12,FGA,FGB | 0.0166 |
| TR/RXR activation | 0.08 | DIO1,DIO3,FGA,G6PC,SYT12,THRSP,TRH | 0.0269 |
| Maturity onset diabetes of Young (MODY) signalling | 0.15 | GCK,PDX1,SLC2A2 | 0.0309 |
| Coagulation system | 0.11 | F12,FGA,FGB,SERPIND1 | 0.0324 |
| Basal cell carcinoma signalling | 0.09 | BMP15,FZD10,FZD7,FZD9,KIF7,WNT4 | 0.0347 |
| eNOS signaling | 0.07 | AQP12A/AQP12B,AQP2,AQP5,AQP8,CCNA1,CHRM1,CHRM3,CHRNA3,CHRNB3,CNGA4 | 0.0398 |
| Glycine betaine degradation | 0.20 | BHMT2,SARDH | 0.0447 |
Discussion
RNA sequencing from blood offers the opportunity to develop biomarkers of health and disease using plasma or sub-compartments of plasma (e.g., extracellular vesicles), which is an easily available biofluid that can be obtained non-invasively. The lack of method standardization and reproducibility has hampered the growth of this emerging technology, especially in view of the small sample volumes typically available, different compartments in which the extracellular RNA is carried, and low quantities of RNA present in most biofluids. Previous studies, including by our group, had focused on small RNAs known to be most abundant in the extracellular compartment. However, recent studies suggest that circulating mRNA and other long RNAs may be specific disease markers. In this study, we used plasma exRNA to rigorously compare six RNA-seq library preparation kits tailored to longer (>200 nt) RNA sequences, and we present their differences in genome and transcriptome mapping percentage as well as long RNA species diversity. In addition, by using the kit with the greatest demonstrated gene diversity, SMARTer Pico V2, we showed that the EV-enriched fractions (i.e, fractions 6–10) yield different genome/transcriptome mapping percentages and have a distinct gene profile than the no-EV fractions (i.e., fractions 1–5). As the kit required may differ based on the aim of any given experiment, we hope this dataset provides a reference for genome mapping percentage and long RNA species diversity in a clinically relevant biofluid, plasma.
We firstly compared the genome/transcriptome mapping percentage and long RNA species diversity of six different library preparation kits/methods by using exRNA that was extracted from the same pool of plasma. Although the percentage of mapped reads varied modestly across kits, we found that the TruSeq RNA Access kit had a significantly higher percentage of reads mapped to the transcriptome, which was expected based on the design of the kit to enrich for mapping percentage of coding RNA sequences. We also found that fragmentation did not have any impact on the performance of this kit. Although the TruSeq RNA Access kit had higher percentages of counts and TPMs in protein coding genes, the SMARTer Pico v2 and SMARTer/KapaHyper kits tended to have more representation of non-coding RNAs and lncRNAs. Thus, experiments focused on protein-coding RNA versus those focused on non-coding RNA or increased RNA species diversity might choose differing library preparation kits.
The use of exRNA is an emerging research area toward the development of diagnostics and therapeutics in health tracking and disease states (Ingenito et al., 2019, Liu et al., 2019, Murillo et al., 2019, Quinn et al., 2015). Therefore, having shown that the library preparation kits vary in terms of transcriptome mapping percentage and RNA species diversity employing cell-free exRNA from pooled plasma samples, we next focused on the evaluation of exRNA from EV-enriched or no-EV fractions. For this part, we used exRNA from pooled and individual plasma samples isolated using C-DGUC. We observed that the concentration of RNA in the EV-enriched fractions (6–10) was higher in every case: 3.4–4.7 ng in the individuals samples and 10.1–10.8 ng in the pooled samples compared with 1.8–2.0 ng for the patient samples and 3.0–3.2 ng for the pooled samples (in fractions 1–5). EV-enriched fractions had increased percentage of mapping to the genome and transcriptome as compared with no-EV fractions that were negative for EV markers. From these data, we might surmise that the greatest amount and diversity of long RNA species are associated with the EV fractions compared with the EV-depleted (lipoprotein-rich) fractions.
Having demonstrated that the plasma exRNA in different compartments could impact the number of reads mapping to the genome/transcriptome, we next determined the impact on the number of genes detected. As most current RNA-based biomarkers rely on specific genes, profiling of the different blood exRNA compartments is of great importance. Overall, as described above, we detected more genes in EV-enriched fractions from both pooled and individual plasma samples compared with no-EV fractions. Although 19,666 and 19,458 genes were commonly detected between the two fraction pools (1–5 vs 6–10) from pooled and individual plasma samples respectively, there were ∼18x more uniquely detected genes in the EV-enriched fractions compared with the no-EV fractions from pooled samples (4,279 versus 239 genes) and ∼50x more in individual samples (5,055 versus 100 genes) (Figures 5B and 5C). Thus, not only a higher percentage of reads maps to the genome/transcriptome in EV-enriched fractions but a higher number of genes can be detected as well, which means higher RNA diversity. In a recent study, Wei et al. showed that different exRNA fractions isolated using ultrafiltration from human glioma stem cells had a distinctly different profile of both small and long RNAs (Wei et al., 2017). Although this study focused on exRNA from glioma stem cells and looked at different exRNA fractions than our study, it agrees with our findings that different fractions do exert different exRNA profiles and highlights the importance of the establishment of an exRNA roadmap based on the different fractions in biofluids.
RNA-seq will continue to play an integral role in the development of blood-based biomarkers. The meticulous, direct comparison of sequencing methods should provide justification based on desired RNA species detection (e.g. an unbiased RNA view versus a coding gene-only approach). This work demonstrates feasibility using different library prep kits to sequence RNA from plasma and shows that the aim of the study should dictate the choice of kit. Lastly, it demonstrates that different plasma exRNA compartments comprise of a unique RNA profile, which directly impacts detection of certain RNAs from blood circulation.
Limitations of the Study
Although we only used a small number of human samples, we were able to uniformly test each of the kits using the same starting material. We also then employed the best performing kit based on mapping percentage to genome/transcriptome and ability to detect the greatest diversity of RNA species on EV-enriched and no-EV plasma compartments to further demonstrate its performance. However, many more samples will need to be used to arrive at what should be expected from normal healthy subjects and how it varies in disease. Therefore, we are not able to recommend with high confidence a one-kit-fits-all for RNA-seq experiments in plasma. Based on our findings, each library preparation kit produces a varying genome/transcriptome mapping percentage and RNA species detection, which precludes us from doing so. We, however, recommend that different kits should be chosen depending on the goals and focus of each experiment using plasma. Furthermore, we were not able to sequence RNA from highly purified RNA carriers (e.g, CD9+ EVs versus AGO2 versus HDL) due to technical limitations and we, therefore, cannot comment on the RNA-seq performance and RNA cargo of each of the abovementioned RNA carriers. We do, however, encourage future studies to focus on the rigorous isolation of each RNA carrier and perform a systematic evaluation of RNA-seq protocols on plasma exRNA compartments for the creation of a comprehensive exRNA atlas by RNA carrier in blood.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Saumya Das MD, PhD (sdas@mgh.harvard.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The source code that generates the combined GENCODE and LNCipedia gene annotation can be accessed here: https://github.com/tgen/gencode-plus-lncipedia. The RNA sequencing data have been deposited in Dryad: https://doi.org/10.5061/dryad.kh1893236.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We acknowledge funding support from the NIH Extracellular RNA Communication Consortium Common Fund grants UH3TR000901 [SD], UH3TR000891 [KVKJ], UH3TR000906 and U01HL126494 [LCL], HL126497 [IG]. We also acknowledge funding support from the American Heart Association grant 16SFRN31280008 [SD] and NIH grants R01HL122547 [SD] and R01CA218500 [IG].
Author Contributions
Conceptualization, K.V.K.J., S.D.; Methodology, K.V.K.J., S.D., R.S.R., R.R.; Formal Analysis, E.H., A.Y.; Investigation, R.S.R., R.R., S.S.; Resources, K.V.K.J., S.D., L.C.L., I.G., M.G.S.; Data Curation, E.H., A.Y.; Writing—Original Draft, R.S.R., E.H., R.R.; Writing—Review & Editing, R.S.R., E.H., T.G.W., K.V.K.J., S.D., I.G., M.G.S., L.C.L.; Visualization, R.S.R., E.H., K.V.K.J., S.D.; Supervision, K.V.K.J., S.D.; Funding Acquisition, I.G., L.C.L., K.V.K.J., S.D.
Declaration of Interests
The authors declare no competing interests.
Published: June 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101182.
Contributor Information
Kendall Van Keuren-Jensen, Email: kjensen@tgen.com.
Saumya Das, Email: sdas@mgh.harvard.edu.
Supplemental Information
References
- Borad M.J., Champion M.D., Egan J.B., Liang W.S., Fonseca R., Bryce A.H., McCullough A.E., Barrett M.T., Hunt K., Patel M.D. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet. 2014;10:e1004135. doi: 10.1371/journal.pgen.1004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byron S.A., Van Keuren-Jensen K.R., Engelthaler D.M., Carpten J.D., Craig D.W. Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nat. Rev. Genet. 2016;17:257–271. doi: 10.1038/nrg.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingenito F., Roscigno G., Affinito A., Nuzzo S., Scognamiglio I., Quintavalle C., Condorelli G. The role of exo-miRNAs in cancer: a focus on therapeutic and diagnostic applications. Int. J. Mol. Sci. 2019;20:4687. doi: 10.3390/ijms20194687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer A.M., Joffe E., Batlevi C.L., Hilden P., He J., Seshan V.E., Zelenetz A.D., Palomba M.L., Moskowitz C.H., Portlock C. Integrated DNA/RNA targeted genomic profiling of diffuse large B-cell lymphoma using a clinical assay. Blood Cancer J. 2018;8:60. doi: 10.1038/s41408-018-0089-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasser C., Alikhani V.S., Ekstrom K., Eldh M., Paredes P.T., Bossios A., Sjostrand M., Gabrielsson S., Lotvall J., Valadi H. Human saliva, plasma and breast milk exosomes contain RNA: uptake by macrophages. J. Transl. Med. 2011;9:9. doi: 10.1186/1479-5876-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Bai X., Zhang A., Huang J., Xu S., Zhang J. Role of exosomes in central nervous system diseases. Front. Mol. Neurosci. 2019;12:240. doi: 10.3389/fnmol.2019.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKiernan J., Donovan M.J., O'Neill V., Bentink S., Noerholm M., Belzer S., Skog J., Kattan M.W., Partin A., Andriole G. A novel urine exosome gene expression assay to predict high-grade prostate cancer at initial biopsy. JAMA Oncol. 2016;2:882–889. doi: 10.1001/jamaoncol.2016.0097. [DOI] [PubMed] [Google Scholar]
- Murillo O.D., Thistlethwaite W., Rozowsky J., Subramanian S.L., Lucero R., Shah N., Jackson A.R., Srinivasan S., Chung A., Laurent C.D. exRNA atlas analysis reveals distinct extracellular RNA cargo types and their carriers present across human biofluids. Cell. 2019;177:463–477.e15. doi: 10.1016/j.cell.2019.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasser S., Kurdolgu A.A., Izatt T., Aldrich J., Russell M.L., Christoforides A., Tembe W., Keifer J.A., Corneveaux J.J., Byron S.A. An integrated framework for reporting clinically relevant biomarkers from paired tumor/normal genomic and transcriptomic sequencing data in support of clinical trials in personalized medicine. Pac. Symp. Biocomput. 2015:56–67. https://pubmed.ncbi.nlm.nih.gov/25592568/ [PubMed] [Google Scholar]
- Quinn J.F., Patel T., Wong D., Das S., Freedman J.E., Laurent L.C., Carter B.S., Hochberg F., Van Keuren-Jensen K., Huentelman M. Extracellular RNAs: development as biomarkers of human disease. J. Extracell. Vesicles. 2015;4:27495. doi: 10.3402/jev.v4.27495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G., Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah R., Yeri A., Das A., Courtright-Lim A., Ziegler O., Gervino E., Ocel J., Quintero-Pinzon P., Wooster L., Bailey C.S. Small RNA-seq during acute maximal exercise reveal RNAs involved in vascular inflammation and cardiometabolic health: brief report. Am. J. Physiol. Heart Circ. Physiol. 2017;313:H1162–H1167. doi: 10.1152/ajpheart.00500.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skog J., Wurdinger T., van Rijn S., Meijer D.H., Gainche L., Sena-Esteves M., Curry W.T., Jr., Carter B.S., Krichevsky A.M., Breakefield X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008;10:1470–1476. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S., Yeri A., Cheah P.S., Chung A., Danielson K., De Hoff P., Filant J., Laurent C.D., Laurent L.D., Magee R. Small RNA sequencing across diverse biofluids identifies optimal methods for exRNA isolation. Cell. 2019;177:446–462.e16. doi: 10.1016/j.cell.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Zhang S., Weber J., Baxter D., Galas D.J. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res. 2010;38:7248–7259. doi: 10.1093/nar/gkq601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z., Batagov A.O., Schinelli S., Wang J., Wang Y., El Fatimy R., Rabinovsky R., Balaj L., Chen C.C., Hochberg F. Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nat. Commun. 2017;8:1145. doi: 10.1038/s41467-017-01196-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witwer K.W., Buzas E.I., Bemis L.T., Bora A., Lasser C., Lotvall J., Nolte-’t Hoen E.N., Piper M.G., Sivaraman S., Skog J. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles. 2013;2 doi: 10.3402/jev.v2i0.20360. https://pubmed.ncbi.nlm.nih.gov/24009894/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanez-Mo M., Siljander P.R., Andreu Z., Zavec A.B., Borras F.E., Buzas E.I., Buzas K., Casal E., Cappello F., Carvalho J. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles. 2015;4:27066. doi: 10.3402/jev.v4.27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeri A., Courtright A., Danielson K., Hutchins E., Alsop E., Carlson E., Hsieh M., Ziegler O., Das A., Shah R.V. Evaluation of commercially available small RNASeq library preparation kits using low input RNA. BMC Genomics. 2018;19:331. doi: 10.1186/s12864-018-4726-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source code that generates the combined GENCODE and LNCipedia gene annotation can be accessed here: https://github.com/tgen/gencode-plus-lncipedia. The RNA sequencing data have been deposited in Dryad: https://doi.org/10.5061/dryad.kh1893236.