

Abstract
Cardiac arrest (CA) afflicts ~ 550,000 people each year in the USA. A small fraction of CA sufferers survive with a majority of these survivors emerging in a comatose state. Many CA survivors suffer devastating global brain injury with some remaining indefinitely in a comatose state. The pathogenesis of global brain injury secondary to CA is complex. Mechanisms of CA-induced brain injury include ischemia, hypoxia, cytotoxicity, inflammation, and ultimately, irreversible neuronal damage. Due to this complexity, it is critical for clinicians to have access as early as possible to quantitative metrics for diagnosing injury severity, accurately predicting outcome, and informing patient care. Current recommendations involve using multiple modalities including clinical exam, electrophysiology, brain imaging, and molecular biomarkers. This multi-faceted approach is designed to improve prognostication to avoid “self-fulfilling” prophecy and early withdrawal of life-sustaining treatments. Incorporation of emerging dynamic monitoring tools such as diffuse optical technologies may provide improved diagnosis and early prognostication to better inform treatment. Currently, targeted temperature management (TTM) is the leading treatment, with the number of patients needed to treat being ~ 6 in order to improve outcome for one patient. Future avenues of treatment, which may potentially be combined with TTM, include pharmacotherapy, perfusion/oxygenation targets, and pre/postconditioning. In this review, we provide a bench to bedside approach to delineate the pathophysiology, prognostication methods, current targeted therapies, and future directions of research surrounding hypoxic–ischemic brain injury (HIBI) secondary to CA.
Electronic supplementary material
The online version of this article (10.1007/s13311-020-00856-z) contains supplementary material, which is available to authorized users.
Key Words: Global brain injury, hypoxic–ischemic brain injury, cardiac arrest, neuroprognostication, diffuse optical spectroscopy, targeted temperature management
Introduction
Roughly 550,000 people in the USA suffer cardiac arrest (CA) annually [1]. Despite improvements in advanced cardiac life support (ACLS), basic cardiac life support (BCLS), resuscitation protocols, and training, only 11.5% of out-of-hospital cardiac arrest (OHCA) patients treated by emergency medical services (EMS) survive until hospital discharge [1]. Of those who survive OHCA, fewer than 9% are discharged from the hospital with a diagnosis of “good functional status” [1].
Poor neurological outcome is common among CA survivors because CA causes hypoxic–ischemic brain injury (HIBI). HIBI is a complex pathophysiological mechanism of brain damage caused by oxygen deprivation, reduction in cerebral perfusion, and cytotoxicity. It is important to use the term “hypoxic” instead of “anoxic” to describe HIBI because 1) complete loss of oxygen is not necessary to cause HIBI, and 2) the ischemic component of HIBI should not be overlooked [2].
Over the past decade, there have been multiple modifications to monitoring and treatment guidelines for HIBI secondary to CA (Fig. 1). In 2006, the American Academy of Neurology (AAN) proposed a set of prognostic guidelines for CA-induced HIBI, consisting of brain stem reflex assessment, myoclonus status epilepticus 24 h after CA, somatosensory evoked potential (SSEP) 24–72 h after CA, and neuron-specific enolase (NSE) within 24–72 h after CA [3]. In 2015, the European Resuscitation Council/European Society of Intensive Care Medicine (ERC/ESICM) also adopted a multimodal approach and expanded its guidelines to include targeted temperature management (TTM), sedative clearance, and waiting at least 72 h prior to prognostication to account for delayed neurological recovery [4]. Despite these revised guidelines, development of improved treatments for global cerebral ischemia induced by CA is still identified internationally by experts as a critical area of unmet clinical need [5].
Fig. 1.

Recent evolution of guidelines for post-CA patient care. This timeline compares changes in guidelines from three recent meetings (AAN, 2006; ERC/ESICM, 2014; AHA, 2015) emphasizing a multimodal approach to patient care following CA. Also included are gaps in the standard of care for CA patients, as identified during a more recent collaborative meeting (ANIM, 2018). AAN = American Academy of Neurology; ERC/ESICM = European Resuscitation Council/European Society of Intensive Care Medicine; ANIM = Arbeitstagung NeuroIntensive Medizin; NSE = neuron-specific enolase, PLR = pupillary light reflex; SSEP = somatosensory evoked potential; MRI = magnetic resonance imaging, CT = computerized tomography; WLST = withdrawal of life-sustaining therapy
Accurate prognostication of neurological outcome for CA patients as early as possible is critical for helping clinicians make the most well-informed treatment and patient care decisions. Indeed, deleterious metabolic changes occur within seconds of injury [6] and hemodynamic dysregulation occurs within minutes [7]. However, despite the rapid dynamics of HIBI, many current clinical techniques for brain injury assessment and prognosis are not performed until 24–72 h after CA [3, 8].
Continuous monitoring of cerebral changes during CA-induced HIBI, beginning immediately after injury, has potential to improve prognostic accuracy and patient care. For instance, identifying poor prognosis as early as possible could be vital for informing prompt personalized treatment to improve outcome by ameliorating physiologic insults in real time as they are developing. In this review, we will discuss molecular markers, imaging techniques, and optical technologies that, when performed within the first 24 h after return of spontaneous circulation (ROSC), may provide the potential to improve short- term and long-term prognosis and open avenues for novel therapeutic intervention. We will conclude by discussing the current state of treatment, beginning with TTM and expanding to include pharmacological therapies and hemodynamic interventions.
Mechanisms of Brain Injury Following Global Ischemia and Reperfusion
Causes of CA
Sudden CA can be dichotomized to shockable and nonshockable presenting rhythms and whether the occurrence is in-hospital or out-of-the-hospital. Etiologies of shockable rhythms (ventricular fibrillation (VF) and pulseless ventricular tachycardia (VT)) are typically cardiac in origin and consist of dysrhythmic events that cause the heart to suddenly stop. Although shockable rhythms have usually been the predominant cause of CA in adults, their prevalence has recently declined [9, 10]. This trend has been linked to an increase in interventions such as coronary stents, pacemakers, implanted defibrillators, anti-platelet medication, and coronary artery bypass graft (CABG) surgery [9]. Shockable rhythms have been associated with better survival rates than nonshockable rhythms (pulseless electrical activity (PEA) and ventricular asystole) [11–13].
Causes of nonshockable CA typically include respiratory failure (e.g., drowning, drug overdose, choking, suffocation, hanging). Respiratory failure leading to nonshockable CA is characterized by a prolonged prearrest period of hypoxia and hypercapnia contributing to the dysfunction of the cardiopulmonary system. Progression of such rapid, untreated respiratory failure can eventually lead to hypotension, bradycardia, and typically the development of pulseless electrical activity or asystole. Meanwhile, death by hanging can induce a state of respiratory failure by the compression to the airway or fracture to the hyoid bone but typically is a result of interruption of venous and/or arterial cerebral blood flow by different degrees of compression of these vessels [14].
In a canine model, ischemic brain damage was observed to be greater following 7 min of asphyxial CA than after 10 min of VF CA [15]. This result suggests that shockable and nonshockable CA involve different mechanisms of brain injury that may require different types of treatment. More recently, a study comparing shockable (VF) with nonshockable (asphyxia) CA in rats (n = 25) and humans (n = 1007) found that VF CA was associated with less severe brain injury (p < 0.001) and more favorable outcome after ROSC [16]. Among nonshockable rhythms, a Swedish registry of cardiopulmonary resuscitation reported greater survival rates in patients with PEA than those with asystole, suggesting that resuscitative care may benefit from differentiating different types of nonshockable rhythms to better understand and possibly improve neurological outcome in a more patient-specific manner [17].
Hemodynamic Changes
CA leads to reduced cerebral blood flow (CBF), initiating a cascade of primary metabolic dysfunction in the neurons and secondary reperfusion injuries to the brain (Fig. 2) [18]. Cerebral perfusion after CA and resuscitation can be characterized by a gradual, multi-phasic (Table 1) restoration of CBF and cerebral autoregulation over the first 72 h [19, 20]. The initial post-resuscitation increase in blood pressure and CBF is termed reactive hyperemia. During this period, the ratio of CBF to cerebral metabolic rate of oxygen (CMRO2) will often exceed its normal range. Post-ischemia/reperfusion rises in CBF to CMRO2, whether acute or delayed, have been referred to as “luxury perfusion,” which has been linked to a loss in vascular tone and vasodilation secondary to acidosis [21–23].
Fig. 2.
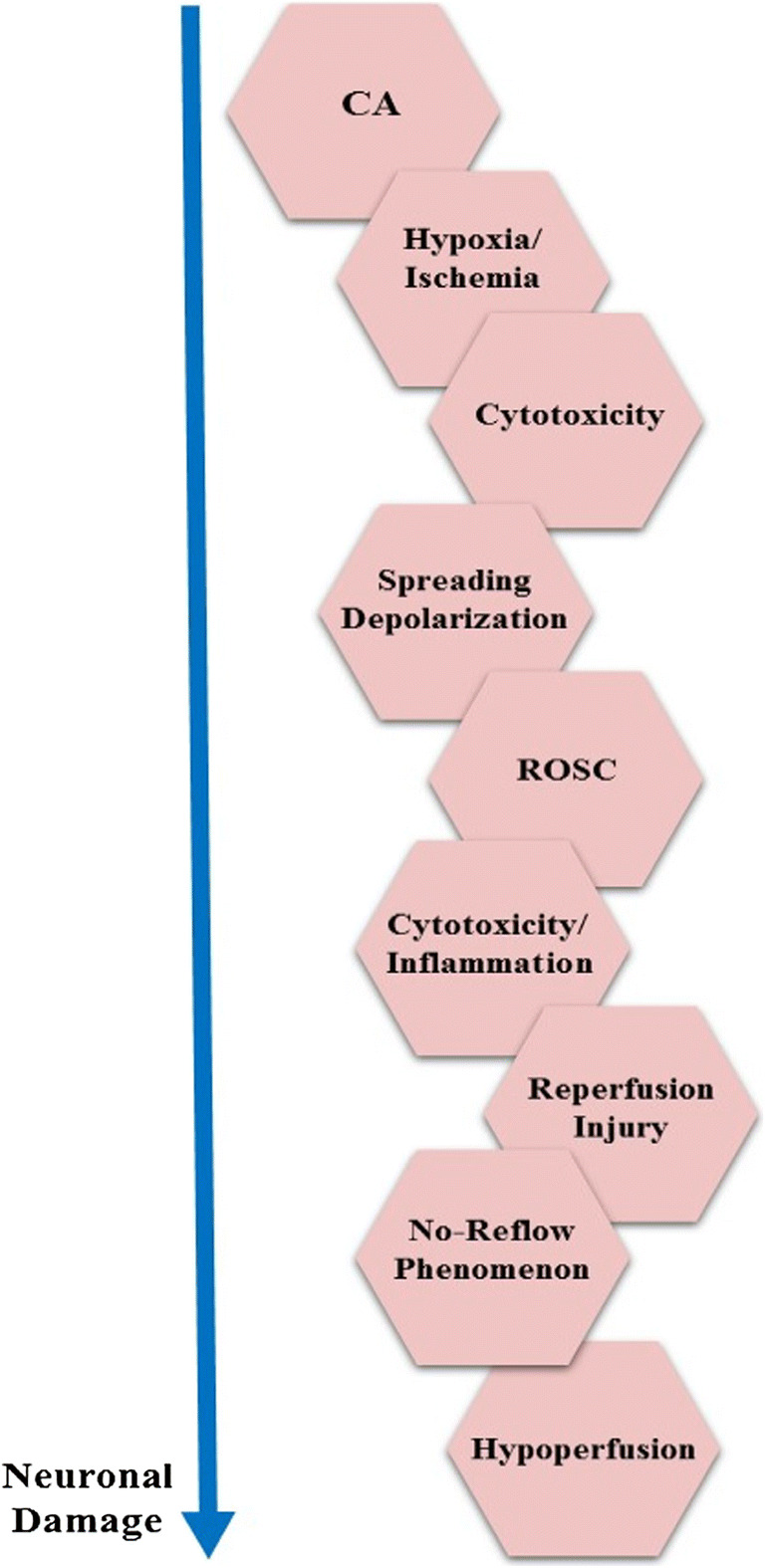
Mechanisms of post-CA neuronal damage. CA causes hypoxic–ischemic damage, leading to cytotoxicity and ultimately triggering cellular shutdown and spreading depolarization. Following ROSC, CBF is restored but mismatches between perfusion and metabolism can lead to ischemic injury (no-reflow) or luxury perfusion (reperfusion injury)
Table 1.
Three hemodynamic stages following resuscitation
| Hyperemia (0–30 min) | Hypoperfusion (30 min–12 h) | Restoration of blood flow (12–72 h) |
|---|---|---|
|
• Hyperemia linked to cerebral vasoparalysis [35] • Vasoparalysis linked to increasing lactic acid from anaerobic respiration [36], NO [37], and adenosine [38] • Loss of vascular tone is independent of changes in BP or reactivity to CO2, which is preserved [19, 21] |
• CBF decreases by 50% [39, 40] • Hypoperfusion linked to intravascular stasis (no-reflow phenomenon) [24] and vasoconstriction [27] • Imbalance of vasodilators (e.g., NO) [41] and vasoconstrictors (e.g., endothelin-1) [28] contributes to hypoperfusion |
• In survivors, large vessel blood flow exhibits gradual return to near-baseline, along with increased metabolism [42] • Increases in large vessel blood flow observed in nonsurvivors [29] |
Following resuscitation, three stages of hemodynamic alteration have been observed in patients and preclinical models. The first stage (hyperemia) is marked by increased blood flow. The second phase (hypoperfusion) is characterized by a reduction in blood flow. The third stage involves the recovery of near-normal CBF. Each box highlights specific pathophysiologic mechanisms linked to these hemodynamic changes
NO = nitric oxide, BP = blood pressure, CBF = cerebral blood flow
Reactive hyperemia is followed by hypoperfusion, a decrease in CBF that can exacerbate secondary injury. Hypoperfusion can arise as the result of the no-reflow phenomenon (incomplete microvascular filling during reperfusion) [24–26] and the actions of vasoconstrictors such as 20-hydroxyeicosatetraenoic acid (20-HETE) and endothelin-1 [27, 28]. As CBF is restored, the mean flow velocity in the middle cerebral artery (MFVMCA) increases toward normal in CA survivors, while MFVMCA is abnormally high in nonsurvivors [29], supporting a link between “luxury perfusion” and poor outcome. If vascular-specific markers can be actively monitored following resuscitation, CBF-targeted interventions may be targeted to ameliorate secondary injury [27].
CA-induced HIBI is linked to the disruption of normal cerebrovascular autoregulation, which protects the healthy brain from hemodynamic disturbances. Cerebrovascular autoregulation maintains constant CBF over a range of mean arterial pressure (MAP) (usually 50–150 mmHg). Brain injury after resuscitation has been linked to a right shift (higher MAP maximum) and narrowing of MAP on the autoregulation curve [30–33]. In pig models, hypothermia after hypoxic–asphyxial CA has been associated with a decrease in the right shift and a decrease in CBF [32].
Despite the well-known relationship between MAP and CBF, there is a lack of knowledge about optimal blood pressure targets after resuscitation for CA patients [33, 34].
Current guidelines suggest avoiding hypotension [4, 43, 44]. More recent studies suggest maintaining elevated MAP following CA to adequately perfuse the brain and maintain optimal CBF in the setting of cerebral dysregulation [44–46]. In one recent study, good neurological outcome (modified Rankin Scale ≤ 3) upon discharge from the hospital was associated with MAP > 90 mmHg in the first 6 h after resuscitation [44]. A second recent study, focused on CA patients with initial shockable rhythms, suggested that MAP targets should be > 75 mmHg to improve neurological outcome in that patient population [46].
Understanding cerebral autoregulation is critical for informing optimal MAP targets for CA patients. For example, patients with hypertension exhibit a cerebral autoregulation curve that is shifted to the right, suggesting that hypertensive patients may particularly benefit from higher blood pressure targets after CA [30]. Recently, methods for more individualized management of post-CA HIBI have been investigated. For instance, jugular bulb microdialysis is currently being used to assess cerebral oxidative metabolism by measuring lactate-to-pyruvate (LP) ratio, in which a high LP ratio is associated with anaerobic metabolism, alongside neurological outcome in a randomized control trial (n=66) comparing low (63 mmHg) versus high (77 mmHg) MAP following CA [45].
Metabolic Stress
Intracellular metabolic and ionic changes are hallmarks of ischemic injury that play an important role in homeostasis and neuronal survival. The cellular mechanisms of post-CA cerebral damage can be divided into two phases: primary and secondary injury.
The primary injury phase of HIBI is characterized by cellular excitotoxicity leading to neuronal and glial cell death. Oxygen is depleted within 15–20 s [6], causing loss of ATP production within 5 min [47–50], followed by anaerobic glycolysis for 4–5 min [51]. Inactivation of ATP dependent channels leads to cellular swelling (cytotoxic edema), accumulation of lactic acid, and increases in intracellular Ca2+. Increases in calcium also lead to membrane depolarization and release of glutamate largely mediated by NMDA receptors, which further increase the concentration of Ca2+. In vivo studies have shown that the concentration of Ca2+ in the hippocampus is substantially elevated during ischemia, and the reversal of these alterations during reperfusion has different dynamics in area CA3 than area CA1 [52]. Calcium activates apoptotic cascades involving caspases, calpains, proteases, phospholipases, and oxygen free radicals. Despite NMDA’s large role in Ca2+ influx and “excitotoxicity”, clinical trials using NMDA receptor antagonists were considered unsuccessful [53, 54].
Management of HIBI is mainly focused on limiting secondary injury [55]. Secondary injury involves a physiologic response to the imbalance of oxygen demand and supply. Immediately following resuscitation, reperfusion injury causes microvascular dysfunction, hyperoxia, cytotoxicity, inflammatory processes, and cerebral edema. Reperfusion exacerbates cellular excitotoxicity by delivering oxygen as a substrate for reactive oxygen species (ROS) and enzymatic reactions [56]. Endothelial dysfunction from ROS is responsible for impaired vasomotor regulation, decreased endothelial anticoagulation causing microthrombi [57, 58], and impaired blood-brain-barrier (BBB) permeability [59, 60]. Activation of lytic enzymes disrupts mitochondrial function and further contributes to loss of homeostasis and perpetuates necrosis [18, 56, 61]. Mitochondria are responsible for buffering cytosolic Ca2+ concentrations. Mitochondrial Ca2+ uniporters can be genetically deleted, leading to some neuroprotection during ischemia models, suggesting that they may be potential therapeutic targets in global ischemia [62, 63]. Also, we and others have recently shown that mitochondrial Zn2+ accumulation appears to precede and contribute to this mitochondrial Ca2+ dysregulation that eventually leads to neurotoxicity in selective CA1 pyramidal neurons, highlighting the potential therapeutic benefits of controlling such upstream mitochondrial Zn2+ dysregulation [64, 65]. Ischemic changes and reperfusion injuries are more deleterious to selective regions of the brain like the hippocampus (CA1 in particular), frontoparietal cortex (layers III, V, VI), basal ganglia, and Purkinje cells of the cerebellum [56]. Activation of lytic enzymes, apoptotic signaling cascade, and dysfunctional mitochondria all enhance excitotoxicity and lead to further irreversible brain damage [56].
Inflammation
The immune response following CA is orchestrated by a number of different inflammatory cells and markers. The process is complex and discussing it in totality would be beyond the scope of this review. Here, we summarize recent research into the role of microglia and briefly discuss inflammatory processes involving astrocytes and T lymphocytes. The inflammatory response has two major phases: 1) activation of resident macrophages and endothelial cells [56, 66]; 2) influx of peripheral inflammatory cells [56, 67].
Microglia, resident macrophages of the central nervous system (CNS), are activated within minutes of insult and will persist for days [68]. Microglia have long been thought to exacerbate ischemic injury, but current research highlights their “biphasic” or “dual” role after cerebral ischemia [60, 69]. Specifically, microglia not only release ROS and produce inflammatory cytokines and proteases [60, 70], they also clear necrotic debris and produce anti-inflammatory cytokines and growth factors [60, 71]. New therapeutic targets aim to control the regulatory mechanisms for microglial activation and polarization [69]. Clinical trials with stem cells and microRNA have shown potential for modulating microglia at different stages of ischemic stroke [60, 69, 72], and may hold promise for global ischemia.
Microglia, endothelial activation, astrocytes, and matrix metalloproteinases (MMPs) contribute to the breakdown of the BBB. Disruption of the BBB is associated with cerebral vasogenic edema, hemorrhagic transformation, leukocyte infiltration, and leakage of toxins [60]. Temporary BBB disruption during “no-flow” periods has also been observed in rodents via MRI variance imaging, with recovery of BBB 48 h later [68, 73]. Interestingly, microglia have also been shown to release progranulin, a growth factor associated with neurovascular protection and attenuation of BBB disruption after ischemic stroke [74–77]. In astrocytes, aquaporin 4 (AQP4) is upregulated after ROSC and associated with early vasogenic edema following CA [78]. In rodents, AQP4 inhibitors such as AER-271 mitigate early edema 3 h after ROSC [79]. Furthermore, loss of ATP results in impaired function of Na+/K+ pumps, leading to neuronal and astrocyte swelling due to loss of the membrane potential. Inability of neurotransmitter reuptake causes an increase in extracellular glutamate, resulting in excitotoxicity. Lactic acid, free radicals, and arachidonic acid build up much like in sepsis.
Cerebral edema is recognized as a robust factor in prognostication for neurological outcome, and one key goal of targeted therapy is to minimize edema following CA [56, 80, 81]. Cellular edema compresses surrounding capillaries, further reducing blood flow, increasing intracranial pressure and increasing Ca2+ influx. Leaks in the BBB result in the elevated influx of T-lymphocytes, particularly cytotoxic T Cells (CD8+), at 3 h after ROSC [68]. In fact, a recent preclinical study showed that mice lacking functional T-cells exhibited significantly reduced ischemic damage to neurons in the CA1 region of the hippocampus [82]. Recent preclinical studies have also shown mitigation of post-CA cerebral edema by SUR1-TRPM4 channel inhibitors, such as glibenclamide (glyburide), leading to improved outcome [83, 84], warranting their testing in clinical studies as has been done in large hemispheric stroke.
Temperature
Figure 3 summarizes key effects of temperature on the brain following CA-induced HIBI. Hyperthermia is associated with worse neurological outcome following HIBI [18, 85, 86]. Preclinical studies have demonstrated an increase in infarction size by 43.4% (95% CI, 29.8-56.9%) as a function of elevated temperature following stroke, and hyperthermia was shown to be even more harmful if present for over 2 h [87]. Thermotoxicity has been linked to pathophysiological mechanisms that further exacerbate ischemic and reperfusion injuries. For example, increased temperature (38–39 °C) is associated with greater BBB permeability [88], which is associated with cerebral vasogenic edema and altered intracranial pressure (ICP) [18]. Hyperthermia has also been linked to increases in glutamate production, which adds to Ca2+ influx, activation of metabolic pathways, and perpetuation of this vicious cycle ending in neuronal death [89–91]. Hyperthermia may also be associated with impaired autoregulation in CA patients [18]. Additionally, hyperthermia decreases the threshold for seizures [18, 55], which are often indicative of poor neurological recovery [92, 93]. On the other hand, different degrees of hypothermia seem to have little effect on seizures as there were no significant difference in the prevalence of seizures, or the ability of seizures to prognose outcome, in patients whose temperature was lowered to 33 °C versus 36 °C [93].
Fig. 3.

Effects of temperature on the post-CA brain. Core temperatures of 37.5 °C and above have been associated with deleterious changes in the brain after CA. However, if core temperature is temporarily reduced, therapeutic effects can be achieved. However, hypothermia also increases risk of complications such as bradycardia and impaired glycemic control. CBF = cerebral blood flow, BBB = blood–brain barrier
Experimental findings have strongly supported immediate cooling by 3–5 °C following resuscitation and continuing for approximately 48–72 h for optimal neuroprotection against secondary injury [94, 95]. Immediate therapeutic hypothermia in rat models improves cerebral blood flow (CBF) restoration and maintenance following ROSC and results in better neurological outcome [96]. Cerebral cooling following global ischemia was associated with fewer and shorter seizures in piglets [97, 98]. Furthermore, hypothermia has been associated with reductions in cerebral metabolism, excitotoxicity, free radical production, and BBB permeability [95, 99, 100]. In fact, cerebral metabolism is reduced at a rate of 5-7% for every 1 degree Celsius drop in temperature, emphasizing the impact of even a small change in temperature [95, 99, 101, 102]. In rodent models of cerebral ischemia, hypothermia was linked to reductions in lactate dehydrogenase (LDH) activity [103], free fatty acid accumulation [104], and excitatory amino acid (EAA) production [105, 106]. Indeed, for rats maintained at 33 °C and 30 °C, glutamate release was inhibited completely and dopamine release was reduced [104]. Such reduction of EAA helps to delay onset of, and damage due to, anoxic cell depolarization [107]. Moreover, hypothermia was shown to suppress nitric oxide and superoxide species in hippocampal slice cultures [108] and during ischemia and reperfusion in gerbils [109]. Also, the rapidity of temperature change after CA is important, as shown by one study demonstrating cooling within 1 h of global ischemia in a rodent model was associated with reduction in BBB leakiness and brain edema 24 h later [110].
Cooling is also known to be involved with inhibition of neuro-inflammatory and apoptotic pathways [95, 100, 111]. Furthermore, hypothermia has been associated with decreased microglial and nuclear NFkB activation [112]. Numerous studies have demonstrated hypothermic disruption of both the intrinsic and extrinsic pathways of apoptosis [100]. For instance, hypothermia has been linked to activation of anti-apoptotic Bcl-2 and suppression of pro-apoptotic factor BAX [113]. Reductions in pro-inflammatory cytokines (IL-1b, IL-6, IL-18, and TNF) [114–116] and increases in anti-inflammatory cytokines (IL-10) [117] have both been observed with cooling.
It is important to note that therapeutic hypothermia carries several well documented clinical risks. Hypothermia has been associated with a decrease in cardiac output of about 25-40% secondary to temperature induced bradycardia [118, 119]. Hypothermia has also been linked to vasodilation of coronary arteries in healthy adults, as well as vasoconstriction of coronary arteries in adults with severe atherosclerotic disease [120]. Other risks of hypothermia include immunosuppression, risk of infection, electrolyte disturbances, insulin resistance, impaired drug clearance, mild coagulopathy, and cold diuresis [95].
Techniques for Injury Assessment, Monitoring, and Prognosis
Accurate assessment of HIBI secondary to CA, continuous monitoring of CA-induced cerebral alterations, and improved prognosis of neurological recovery require multimodal approaches. Clinical guidelines for assessment of HIBI patients include testing brainstem reflexes and assessing EEG activity. When possible, it is also recommended to measure somatosensory evoked potential (SSEP), the EEG recorded activity induced by a stimulus (usually electrical activity to the median nerve). Additional modalities include brain imaging (e.g., computed tomography; magnetic resonance imaging; near-infrared spectroscopy) and biomarker assays (e.g., neuron-specific enolase).
A retrospective cohort study of 226 unconscious post-CA patients at a single tertiary care center from 2011–2017 compared the false predictive rate (FPR) of this multimodal approach for three different sets of guidelines. The ERC/ESICM (2014) guidelines proved more predictive of poor neurological outcome at 6 months (FPR < 0%) than the American Academy of Neurology (AAN) guidelines (FPR < 8%) or the American Heart Association (AHA) guidelines (FPR < 12%) [121]. The most robust predictors of poor neurological outcome using the ERC/ESICM guidelines were absence of N20 SSEP and/or absence of pupillary and corneal reflexes [121].
When using a multimodal approach, it is possible to minimize the degree of “self-fulfilling prophecy” and premature withdrawal of life-sustaining therapy (WLST) since prognostic limitations of relying on any one single approach can be mitigated [121–124]. However, some of the aforementioned tools and tests are expensive and require highly-trained personnel to interpret the results [122, 125].
Clinical Examination
Two landmark studies in 1974 [126] and 1977 [127] demonstrated the association between with neurological outcome of comatose CA survivors and parameters measured via initial clinical examination. In 1985, a multicenter study evaluated 210 comatose patients and established the Levy Criteria, the first set of guidelines to predict long-term neurological outcome [128]. Subsequently, these criteria evolved into the currently used Cerebral Performance Categories (CPC) to measure neurological outcome. CPC 1 represents the best possible outcome, with minor cranial nerve abnormalities and other mild neurological and psychological deficits but overall ability to lead a normal life. CPC 5 represents death. Prior to 2006, neurological outcome was dichotomized between “good” (CPC 1–3) and “poor” (CPC 4–5). In 2015, the Utstein Recommendations were to include CPC score of 3 and under as “poor” due to the patient’s limited cognition and severe neurological deficits [129, 130]. Alternative measurements include the Glasgow Outcome Scale (GOS) and modified Rankin Scale (mRS).
An absent pupillary light reflex (PLR) 72 h after CA has been shown in many studies to be prognostic of poor outcome (CPC score of 3–5 at discharge) [3, 128, 131]. Although PLR is often dichotomized as either absent or present, recent studies have used automated pupillometry to quantify the degree of pupillary constriction. In one such study employing pupillometry, 55 adult patients were assessed at the start of TTM, approximately 6 h after ROSC, and throughout 24 h of TTM, finding that NPi < 0.37 (Neurological Pupil index). By 6 h after ROSC, quantitative pupillometry was highly predictive of neurological outcome at discharge [131]. Other aspects of the clinical exam are focused on the brain stem response, including corneal reflex and motor response to pain. The main limitation in examining brain reflexes is their limited sensitivity in predicting good recovery as well as suppressive effects of sedation and neuromuscular blocking drugs [124, 132]. Either completely absent motor response or the presence of extensor (decerebrate) posturing to pain is defined as motor score (M) = 1 or 2, respectively, on the Glasgow Coma Scale. The ERC-ESICM recommends testing corneal reflex and PLR at 72 h while cautioning the interpretation of a low motor score of 1 or 2 on the Glasgow Coma Scale due to low specificity [124]. Neverthelesss, motor response has a high sensitivity and is best used in conjunction with other neuroprognosticators. Current research is also investigating the accuracy of optic nerve sheath diameter in determining neurological outcome (NCT03195881).
Patients presenting with myoclonic status epilepticus (MSE) have traditionally been thought to have a poor prognosis; however, exceptions to this have also been reported, especially in the era of TTM [43]. More invasive technologies that include intracranial monitoring have shown to be useful in select patients, but these techniques can carry risks, especially in patients with coagulopathy, and larger studies are needed for further validation [123].
Brain Imaging
The ratio of gray matter to white matter, measured with computed tomography (CT) within the first 48 h after resuscitation, has been shown to predict outcome following CA [133–136]. Lower values of the gray/white matter ratio can indicate increased edema, which reduces the ability of the gray matter to attenuate the CT source [136]. Combining CT data measured within 3 days of resuscitation with Glasgow Coma Scale data acquired 3 days after resuscitation showed potential for improving prognostication of recovery 6 months after resuscitation, relative to using only the Glasgow scale [137]. Combining CT data with neuron-specific enolase (NSE) testing (see Section 3.4) was shown to improve prognostication of patient recovery from CA, relative to either method individually [138].
Obtaining quantitative information about changes in gray matter and white matter independently is critical for assessing hypoxic–ischemic brain injury, since gray matter is more vulnerable to hypoxia than white matter [55] and CA-induced changes in gray matter in the cortex are different than those in the subcortex [139]. Recently, advances in MRI data processing for magnetic resonance imaging (MRI) have increased the precision with which hypoxia-related structural alterations in gray matter can be quantified. This has led to the creation of multivariate models combining changes in cortical thickness with alterations in gray matter volume (both measured with conventional T1-weighted MRI) to predict brain recovery 1 year after CA in patients [139]. Diffusion-weighted MRI has shown high sensitivity for predicting post-CA recovery, but it can suffer from poor specificity because patients who are younger, healthier, or underwent less-severe injury can often have good long-term prognosis despite initially showing abnormalities on MRI [140–142].
Recently, techniques such as diffusion-weighting [143], phase analysis [144], and connectivity mapping [145] have been used to obtain additional quantitative information from MRI to better characterize ischemic damage in the brain. MRI data has also been used in tandem with CT data to improve prediction of neurological recovery, showing that the combination of these two modalities provided better prognostic accuracy than either of the two individually[146]. Furthermore, positron emission tomography (PET) has been used to measure CBF and brain oxygen metabolism in a preclinical CA model, revealing oligemia and reduced oxygen extraction in the cortex concomitant with excess CBF relative to oxygen extraction in the cerebellum [147].
Electrophysiological Tools: EEG, SSEP, and BIS
Electroencephalography (EEG) is a technique that measures the changes in electrical potential from brain activity. Different EEG waveforms are observed in healthy brains versus disease states such as coma, seizure, or dementia. After HIBI, various EEG patterns have been identified as being benign or malignant. Benign EEG activity includes theta dominant or frontal rhythmic delta, whereas malignant activity includes isoelectric (silent), low amplitude delta, alpha coma or burst suppression [148]. Malignant EEG patterns have demonstrated low sensitivity but high specificity for identifying poor outcome [149]. This result may seem counterintuitive, but the presence of benign EEG is encountered more commonly than expected in post-CA patients, yielding low specificity.
Another method for prognostication is to apply electrical current to peripheral nerves to induce EEG patterns, commonly referred to as somatosensory evoked potentials (SSEP). For example, the absence of the N20 cortical wave during median nerve stimulation has provided 100% specificity for identifying poor outcomes in multiple studies [3]. The explanation for these findings was that cortical tissue is much more prone to anoxic brain injury, so by the time brainstem reflexes are absent, the cortex is irreversibly damaged. A prospective study [150] examined whether using combinations of 2 or more variables (e.g., bilaterally absent SSEP, unreactive EEG background, early myoclonus, incomplete recovery of brainstem reflexes) predicted survivability following anoxic brain injury. When 2 or more of the aforementioned variables were used, a specificity of 1, sensitivity of 0.72 and an unweighted accuracy of 0.9 were reported. However, a limitation in this study was that during post-CA care, administration of antiepileptics or sedatives could cause exogenous changes to EEGs. Despite the high specificity of SSEPs in identifying poor outcome, it can often be difficult to obtain them in a timely manner in the intensive care unit setting due to the need for someone experienced in the technique.
The bispectral index (BIS), a quantity obtained from EEG that has historically been used to characterize depth of anesthesia, is another potential prognostication method for assessing neurologic function in CA patients during TTM [151]. In an initial exploratory study, it was discovered that low BIS scores during therapeutic hypothermia in CA patients predicted poor neurologic outcome after discharge, and that decrease in BIS was associated with an increase in the odds of poor outcome [151]. In a subsequent study measuring CA patients undergoing TTM, the “good outcome” (cerebral performance category score of 1–2) patient group had a significantly higher mean BIS value after ROSC compared to the “poor outcome” (cerebral performance category score of 3–5) patient group [152]. Although these studies show promising uses for BIS, which is far easier to implement in the intensive care setting compared to SSEPs, several limitations exist with the usage of BIS. For instance, since BIS only outputs a processed EEG signal, BIS signal interference from confounding factors such as extraneous EMG activity or electrical devices such as pacemakers cannot easily be detected [153]. More studies concomitantly measuring BIS and EEG signals are needed to confirm the accuracy of BIS prediction of poor neurologic outcome. Nevertheless, EEG-based prognostication techniques show significant promise for predicting poor post-CA outcomes and thus can be of great use to inform decisions about withdrawal of care. However, there remains an unmet need for a reliable positive prognosticator to aid clinical decisions beyond withdrawal of care.
Molecular Biomarkers
Neuron-specific enolase (NSE) and S100B are neuro-injury biomarkers that may be used as strong early prognostic indicators of poor neurological outcome [154]. Unlike many neuro-prognostic markers, these markers are not significantly influenced by sedatives, neuromuscular relaxants [155, 156], temperature changes (33 vs 36 °C), or prolonged hypothermia during standard TTM management [157]. While traditionally, NSE values above 33 μg/L at 48 h were considered highly prognostic of death or poor neurological outcome, in the more recent TTM era, recent studies suggest that a higher cutoff may be needed to maintain a low false positive rate and that serial measurements may provide added benefit [155]. Another recent study on TTM patients further supported stronger prognostication of NSE values at 48 h as compared to 24 h, whereas minimal difference was found between values drawn at 48 versus 72 h [158]. NSE is a more robust prognosticator than S100B for poor outcome (CPC 3–5) at 6 months [155, 156]. However, studies have also shown that combined S100B and procalcitonin measurements at 24 h after CA are good neuro-prognosticators of poor neurological outcome (CPC 3–5). Combining S100B and procalcitonin biomarkers improved sensitivity from 83.3% and 70.2% (measuring S100B and procalcitonin alone, respectively) to 85.1% [159]. Currently, in the TTM era, there is no clear consensus for the optimal threshold that should be placed on NSE value for best prognostication [132, 154, 155]. However, numerous studies and recent expert recommendations support the use of NSE within a multimodal approach in combination with clinical, imaging, and electrophysiological data, rather than in isolation, to maximize sensitivity and specificity of prognostication [43, 155, 158, 160]. More recent studies have also analyzed miRNA, which are smaller molecules that can cross the blood–brain barrier and may be detectable earlier than NSE [161].
Emerging Tools: Diffuse Optical Spectroscopy and Imaging and Spreading Depolarization
Over the past two decades, techniques involving diffusely scattered light have become increasingly investigated for characterizing the complex changes in brain tissue resulting from global cerebral ischemia. These methods, collectively known as diffuse optical spectroscopy and Imaging (DOS(I)), and including Near-Infrared Spectroscopy (NIRS), shine visible-to-near-infrared (VIS-NIR) light onto the tissue and use a fiber-optic probe or camera to detect the light that is scattered back to the surface by the tissue [162]. Some of the light is absorbed by tissue constituents such as oxygenated hemoglobin, deoxygenated hemoglobin, water, and lipid, causing wavelength-dependent attenuation in the detected signal. Therefore, by performing DOS(I) at multiple wavelengths, the absorption coefficient (and thus, the concentration) of each individual chromophore in the tissue can be measured.
Early uses of DOS(I) techniques to characterize cerebral ischemia included imaging hemoglobin oxygenation changes in a neonatal piglet hemorrhage model [163] and identifying secondary energy failure via NIRS measurements of Cytochrome aa3 [164]. More recently, NIRS oximetry devices have become more widely incorporated into studies trying to improve prognostication following CA [165–169]. Some applications of these devices include examining the effect of TTM in preclinical models, [170, 171], using NIRS to inform CPR quality and detect ROSC [172], and testing the potential of NIRS for initial “prehospital” assessment of traumatic brain injury severity [173]. However, it is important to note that tissue oxygenation values measured with commercial NIRS systems may not strictly represent cerebral oxygen consumption [174], as perturbations to CBF and oxygen extraction will both impact the measured tissue oxygenation values. Therefore, it is necessary to incorporate multi-modal approaches for more accurate quantitative monitoring of tissue perfusion, oxygenation, and metabolism.
Over the past decade, numerous hybrid technologies, incorporating multiple modalities of diffuse light, have been investigated for assessing the complex dynamic relationship between cerebral blood flow changes, alterations in brain oxygenation, and deficits in tissue metabolism in response to global cerebral ischemia. Typically, one of the technologies in such a hybrid setup quantifies cerebral blood flow, and the other measures parameters related to brain oxygenation. Using these measurements in tandem provides metrics for characterizing changes in flow-metabolism coupling and cerebral autoregulation. For example, NIRS has been combined with diffuse correlation spectroscopy (DCS) in the neonate brain for quantifying perfusion and metabolism changes associated with hypothermic interventions during cardiopulmonary bypass and encephalopathy [175, 176]. Furthermore, a hybrid NIRS/laser Doppler approach has been used to quantify limits of autoregulation in a preclinical model of CA + CPR with TTM [32]. Also, a combination of laser Doppler and jugular catheter oxygen measurement has been performed to assess the relationship between CBF and cerebral oxygen extraction in the first 72 h after resuscitation [20]. Utilizing a multimodal approach, NIRS along with mean arterial pressure (MAP) have been used simultaneously for assessing differences between cerebral and peripheral perfusion to characterize autoregulation [177]. Our group has also examined differences between cerebral perfusion and peripheral perfusion in a preclinical CA model using multiple optical modalities and have found a dissociation between the cerebral and peripheral pulsatility [178]. Moreover, DCS has been used along with oximetry to monitor the potential for preconditioning due to repeated occlusion and reperfusion [179]. Camera-based diffuse optical techniques have also been used for measuring CBF and intrinsic optical signal (IOS; a parameter related to tissue absorption and scattering) along with a voltage-sensitive dye to quantify neurovascular coupling in a preclinical CA model [180, 181]. Recently, in neonates that experienced hypoxic–ischemic encephalopathy shortly after birth, wavelet techniques were employed using NIRS data in combination with blood pressure to quantify autoregulation and using NIRS in tandem with EEG to assess neurovascular coupling [182].
In the future, it may be possible to enhance contrast in diffuse optical hemodynamic measurements by incorporating techniques such as photoacoustic tomography [183] and improve localized detection of cerebral hemodynamic changes using diffuse optical tomography [184] or “whole-head” NIRS with multiple source-detector separations and principal component analysis to separate contributions from different regions of the brain [185]. Additionally, characterizing the biophysical meaning of the tissue scattering changes during cerebral ischemia and reperfusion shows significant potential as a new marker of cytotoxic edema, metabolic compromise, and neuronal injury [186–188]. In fact, our group has recently found in a preclinical model that changes in CBF, tissue oxygenation, cerebral metabolism, and tissue scattering precede, and may be predictive of, EEG recovery in the ultra-early period (first 1–5 min) after reperfusion [187, 189]. Furthermore, we show that the flow–metabolism ratio (CBF/CMRO2) in the brain at ultra-early time points (1–2 min after ROSC) may be diagnostic of CA severity (duration) and predict bursting, and this metric appears robust across different ischemia durations [190]. Additionally, we recently demonstrated that combining CBF and tissue oxygenation data shows promise for continuously measuring CMRO2 on an absolute scale (e.g., μM O2/min), which has traditionally been more challenging to measure but potentially very important [191]. Absolute CMRO2 measurement enables direct quantitative comparison between cerebral metabolism of multiple subjects or a single subject on multiple days, without the need for baseline measurements or perturbations such as hypercapnia.
One area of rapidly growing interest for both prognostication and potential targeted intervention is the spreading depolarization (SD) theory of neuronal injury from various insults including global ischemia. SD occurs when an insult (e.g., anoxia) causes neurons to depolarize in a wave that propagates and is followed by severe suppression of neuronal electrical activity [192–194]. The extremely energy-demanding strain placed on individual cells during this phenomenon induces physical changes (cell swelling, beading, energy store depletions, and loss of local blood flow) to brain tissue that accompany the electrical alterations. The area surrounding the brain necrosis, known as the penumbra, experiences the SD in waves emanating from the core [195]. Spreading depolarizations occur in conjunction with cytotoxic edema [196, 197]. SD has recently become the topic of intense preclinical and clinical research due to its interest as a potential target for therapeutic intervention [197–200]. Our group has recently shown that analysis of SD and repolarization during CA may play a potential role in very early prognostication of neurological outcome [188].
Targeted Temperature Management
In 2002, significant improvement in post-CA survival and neurologic function after shockable rhythm was reported in patients treated with induced hypothermia from 32 to 34 °C, a method that became widely adopted in hospitals [201–203]. However, there has been disagreement about the underlying therapeutic mechanisms associated with this temperature range. For example, there has been debate over whether the improved clinical outcomes were associated with the temperature being dropped to 32–34 °C or whether the effect was simply due to the prevention of fever, since fever has been associated with poorer post-CA outcomes [204–206]. In 2013, a landmark trial researched the differences in all-cause mortality and neurologic function between patients randomly assigned to TTM at 33 °C versus 36 °C, showing that hypothermia at 33 °C did not demonstrate a benefit compared to 36 °C [207]. The primary outcome, all-cause mortality at the end of this TTM trial, did not differ significantly between the two temperature groups, and the secondary outcomes, poor neurologic function or death at 180 days, also did not show a significant difference.
Further sub-studies stemming from this TTM trial have compared the 33 °C and 36 °C treatment groups within different subpopulations, including patients with nonshockable rhythm (NSR). Current guidelines recommend hypothermia for all resuscitated patients regardless of initial rhythm, but evidence for TTM in patients with NSR is limited, and management remains controversial [94, 208, 209]. In 2019, another landmark multicenter study was reported showing that TTM targeting 33 °C after nonshockable rhythm leads to improved neurological outcome in survivors by 90 days after CA [210]. Prior to this, a recent retrospective cohort study had linked TTM (32–34 °C within 6 h of ROSC) to decreased mortality and improved neurological outcomes in patients with NSR [209]. Moreover, another recent study found that there was no difference between patient outcomes for TTM at 33 °C versus TTM at 36 °C for NSR patients [211]. Thus, evidence for TTM’s benefit in nonshockable rhythm has been significantly strengthened in recent years.
The effects of bradycardia on mortality and neurologic outcome in CA patients were also studied in the patients from the 2013 TTM trial [212]. A subset analysis of the 33 °C group showed that bradycardia less than 50 beats/min was independently associated with lower 180-day mortality and lower odds of unfavorable neurologic outcome, and that the 33 °C group of patients experienced a more pronounced heart rate-lowering effect from the TTM as compared to the 36 °C group. This effect corroborated the previously known trend that core temperatures lower than 35.5 °C may lead to the development of sinus bradycardia [95]. However, these independent associations do not definitively prove that TTM at 33 °C can decrease mortality and improve neurologic outcome as compared to at 36 °C. Two possible confounding factors, including sedation and differences in medication use, were not controlled for in this study. Therefore, further investigation is needed to research the interactions between sedation, medication regimens, and heart rate in CA patients treated with TTM.
The optimal duration of cooling and rewarming is also a controversial topic in TTM. A recent randomized clinical trial compared 24 h versus 48 h of 33 °C TTM in CA patients suffering from shockable versus nonshockable rhythms. Better neurological outcome (CPC 1–2) was observed in the 48-h group than in the 24-h group. However, study results were not statistically significant, requiring larger randomized trials [213]. Furthermore, adverse events were also more common in the 48-h group [214]. Following TTM, slow rewarming is recommended [43, 94, 215] to avoid the risk of rebound fever over the initial 24 h following rewarming [216]. However, a recent retrospective observational study compared rewarming at 0.15 °C/h versus 0.25 °C/h and did not find significant differences in neurological outcome [215]. A current clinical trial (NCT02555254) is further investigating optimal rates of rewarming.
Current standards and guidelines for TTM do not recommend a particular method for modifying core temperature, and research comparing different TTM methods is limited [217, 218]. One recent retrospective study found endovascular cooling to be more precise than surface cooling, but the neurological outcomes in the two groups were similar [218], while endovascular cooling also requires placing central venous catheters and thus carries its associated risks [94]. Recently, esophageal cooling catheters have been safely implemented during TTM and are increasingly being used [219]. More innovative methods of targeted temperature management are currently being tested. For instance, the PRINCESS Clinical Trial (n=677) randomized patients to either trans-nasal evaporative intra-arrest cooling or to standard of care. Both groups received systemic therapeutic hypothermia (32 °C or 34 °C) for 24 h. The results showed that trans-nasal cooling did not significantly improve 90-day neurological outcome [220]. Furthermore, patients receiving TTM must be carefully monitored to minimize complications related to ventilation, oxygenation, carbon dioxide, bradycardia, hypotension, insulin resistance, infection, fever, acute respiratory disease, shivering, and over-sedation [94].
In a study focusing on quality of life and cognitive function after CA, patients from the 2013 TTM trial were invited to complete a follow-up assessment 6 months after discharge from the hospital [221]. Quality of life and cognitive function were assessed via four outcome measures: clinician-reported, performance, observer-reported, and patient-reported. No significant difference was found between the 33 °C and 36 °C groups on MMSE, IQCODE, Two Simple Questions, MCS, and PCS scores (all tests for cognitive function, level of daily function and mental recovery, and overall measures of health). The finding that cognitive function did not differ between the two TTM groups was confirmed by a second study [222]. Current research is ongoing to further investigate TTM mechanisms and their effects on quality of life, mortality, and neurological function. For instance, the TTM2 trial is an international multicenter randomized superiority trial, comparing 33 °C after CA to early treatment of temperatures greater than 37.8 °C (NCT02908308).
Emerging Treatments
Pharmacotherapy
Pharmacotherapies in cerebral ischemia continue to be explored and the current research has grown extensively. Here, we will introduce several pharmacotherapies and their accompanying pathways, including SUR1/glyburide, orexin, eicosanoids, prolyl hydroxylase, and ROS inhibitors.
SUR1 is a subunit that regulates intracellular Ca2+-activated nonselective cation channels that are opened by ATP depletion [223]. Typically, SUR1-regulated ion channels are not constitutively expressed in the CNS and are only activated during nerve damage caused by events such as ischemia, trauma, and hemorrhage. Sustained channel opening leads to sodium rushing into the cell, causing water to follow in, precipitating cell swelling and cell death. Glyburide, which has historically been used to treat diabetes mellitus type 2, is currently undergoing clinical trials (NCT02864953) to treat stroke patients by its mechanism of targeting the SUR1-TRPM4 channel complex and inhibiting its overactivity, thus decreasing edema formation [224, 225]. Indeed, glyburide has been shown to reduce post-CA cerebral edema and improve outcome in animal models [83, 84], suggesting its possible benefit in humans also. The completion of ongoing clinical trials is warranted to investigate this exciting possibility.
Orexin, a hypothalamic excitatory neuropeptide responsible for arousal, also plays an important role in neurological recovery after CA [226, 227]. Using a rodent model, our group has demonstrated a decline in orexin-A 4 h after resuscitation with a gradual increase to normal levels that temporally coincides with arousal [226]. An intranasal orexin-A treatment administered to rats 30-min after resuscitation was linked to significant reductions in neuroinflammatory markers (IL1b, iNOS, TNF-a, GFAP, CD11b) and accelerated neurological recovery [227]. In further support of a likely important role for orexin in post-CA recovery, our group showed that inhibition of orexin in rats after resuscitation is linked to worse neurological outcome, underscoring a causal relationship between orexinergic activity and arousal level after CA [226].
Eicosanoids, metabolites of arachidonic acid, are produced by cytochrome P450 (CYP) and play a role in vasodilation and vasoconstriction. 20-hydroxyeicosatetraenoic acid (20-HETE), an eicosanoid producing CYP enzyme, is an important vasoconstrictor. Inhibition of 20-HETE in a preclinical CA model was associated with improved cortical perfusion and better short-term outcomes [228].
Prolyl hydroxylase domain proteins (PHDs) hydroxylate targets such as hypoxia-inducible transcription factors (HIF). These proteins are upregulated in the presence of oxygen, marking HIF for proteasome degradation. In a murine model, HD1 inhibition was protective against cerebral ischemia [229].
Animal models have played a vital role in improving our understanding of the pathophysiology, prognosis, and potential treatments for HIBI after CA. A recent groundbreaking swine experiment [230] has questioned our understanding of death [231]. Irreversible cellular injury is the ultimate endpoint of HIBI and brain death. Interestingly, Vrsejla et al. have demonstrated a pig brain’s ability to resume microcirculation and molecular and cellular activity up to 4 h postmortem (after decapitation) and to maintain such activity up to 10 h postmortem during their tests. The cocktail of pharmacologic agents used in their perfusate that was injected in the pig brains to restore brain activity could potentially be key components that provide neuroprotective benefits following CA. These animal experiments have major implications for humans, both in advancement of treatments and prognostication as well as the fundamental definitions we carry for death.
More clinical trials are needed to test proven preclinical pharmacotherapies in humans. Indeed, ongoing clinical trials are investigating gas therapy, including Xenon versus oxygen (NCT0317618), as well as repurposing of compounds such as vitamin C (NCT03509662) following CA. In short, the future of treatment for the globally ischemic brain holds promise as more pharmacologic approaches are tested in the clinical setting.
ROS Inhibitors
Reactive oxygen species (ROS), the byproducts of metabolic enzymatic reactions, are involved in apoptosis, cell signaling, the immune response, and protein expression [232, 233]. Under normal conditions, the ROS produced in the brain are cleared by antioxidative enzymes to prevent an over-proliferation of ROS. However, during ischemic conditions, antioxidative enzymes are unable to clear the ROS as fast as they are produced, due to increased mitochondrial metabolism of succinate and increased activity of NADH oxidase and xanthine oxidase, among other enzymes. This overproduction of ROS can lead to brain tissue damage, protein degradation or aggregation, lipid peroxidation, DNA damage, and inactivation of key metabolic enzymes.
Therefore, therapies either targeting the generation of ROS or mitigating the effects of formed ROS have been developed to combat excessive ROS proliferation after ischemia. Treatments preventing the formation of ROS include mitochondrial electron transport chain uncouplers, NADPH oxidase inhibitors, and xanthine oxidase inhibitors, whereas treatments mitigating the effects of formed ROS involve ROS scavengers and ROS degradation. Vitamins A, C, and E and lipoic acid are antioxidants used as ROS scavengers, but clinical trials have shown no definitive benefit of using vitamins or supplements to treat ischemic stroke [232, 234, 235]. Use of other free radical scavengers such as NXY-059 and citicoline have not shown demonstrative benefit for patients [232, 236, 237]. These findings of ROS scavengers may be attributable to the fact that blanket scavenging may disrupt the physiological effects of ROS, causing a host of other issues [233]. Therefore, more focused scavenging may prove effective at limiting the damage from ROS. Preliminary studies have shown that the other side of the equation, preventing excessive ROS production, may prove beneficial [233, 238], but more studies are needed to confirm the efficacy of inhibiting the generation of ROS.
Perfusion and Oxygenation Targets
Over the past 10-15 years, a number of studies [7, 34, 44, 239–243] have investigated the ideal targets for blood pressure, CBF, and brain oxygenation during and immediately after CPR. Hyperoxia has long been the standard of care during resuscitation from CA, but this practice is currently a topic of significant debate, as the clinical literature is not consistent on whether hyperoxia or normoxia is better for improving neurological recovery following CA [241]. In a recent cohort study [34], a link was shown between hyperoxia in the first 6 h after resuscitation and impaired neurological recovery upon discharge from the hospital. Additionally, multiple preclinical studies going back several decades [239, 244, 245] have suggested that normoxia may facilitate better neurological recovery than hyperoxia. However, to properly interpret the effect of inspired oxygen on neurological recovery, it is critical to also understand the relationship between brain oxygenation and other crucial hemodynamic parameters such as MAP and CO2, as was done in a recently completed randomized control trial [242, 246]. This study, known as the COMACARE trial, showed that although elevated target values of oxygen and CO2 produced an increase in brain oxygenation (as measured by NIRS), these interventions did not ultimately lead to a significant change in neuron-specific enolase (NSE) concentration measured 48 h after resuscitation. Therefore, the optimization of target oxygen level during and immediately after ROSC continues to be an area of active, crucial research. Recently, our group has demonstrated in a preclinical model that it may be critical to target not just oxygenation, but the ratio of cerebral perfusion to oxygen metabolism (CBF/CMRO2), and that the window for optimizing this value may be quite transient (e.g., within the first several min after ROSC) [190]. We anticipate that future work in this area will continue to emphasize multimodal monitoring and multivariate optimization of cerebral hemodynamic parameters to facilitate improved outcome following CA and CPR.
Recently, several groups (including our own) have investigated the impact of peripheral blood pressure, perfusion, and pulsatility on neurological recovery following global cerebral ischemia. In the cohort study described in [34], it was also found that raising blood pressure within the first 6 h after ROSC may help improve neurological outcome after CA [44]. However, it has also been shown that the optimal levels of perfusion to target can vary from patient-to-patient, because these values are dependent on the MAP-CBF autoregulation curve, which can differ among patients [243]. This result emphasizes the need for a personalized-medicine approach to treatment, guided by continuous multimodal monitoring of cerebral autoregulation. New techniques, such as stimulating the vagus nerve [247] may be possible to employ in this context to optimize perfusion as needed for individual patients. Furthermore, targeting specific values of perfusion pressure [248] or even inducing variations in blood pressure [7] may help to facilitate improved neurological recovery. Additionally, our group has shown that in a preclinical model of asphyxial CA and resuscitation, increased peripheral blood pressure pulsatility (e.g., pulse pressure) after resuscitation is associated with worse short-term neurological recovery [178]. This finding, in conjunction with other studies, suggests that monitoring and targeting specific mean arterial pressure as well as the pulse pressure in the periphery and the brain may play an important role in post-resuscitation neurological recovery in patients.
Preconditioning and Postconditioning
Preconditioning and postconditioning are defined as exposing an organ to sub-lethal injuries before, during, or after a hypoxic insult to build tolerance and mitigate damage. Ischemic tolerance from these procedures has been thoroughly described in other organs such as the heart and kidneys. TTM can be thought of as preconditioning and postconditioning for the brain. This section does not aim to provide a comprehensive review but rather to make the reader aware of the breadth of work in this area and where temperature control may fit into the picture.
Neuroprotective potential is dependent on the type, severity, and duration of the insult. A stimulus that is too great will permanently damage brain tissue, while a stimulus that is too small will result in no neuroprotective effect. This invokes a continuum of the same insult at different intensities: no effect, protective, apoptogenic, and neurogenic. Preconditioning stimuli that are neuroprotective for global ischemia can be produced via a variety of insults. These sublethal conditions include, for example, hypoxia, ischemia, NMDA agonists, ethanol, thrombin, LPS, hyperbaric oxygen, anesthetics, and opioid receptor antagonists. Also, insults can be either global or regional/remote (i.e. induction of a milder ischemia to a peripheral limb) and still provide neuroprotection [249, 250]. All of these mechanisms utilize a few common pathways described below. All of their effects repeatedly have been shown to reduce (at different timepoints and effect) the loss of neurons during an ischemic event. Clinical trials for preconditioning in CA have been initiated to evaluate therapeutic benefit in humans (NCT03093948).
Preconditioning and postconditioning both induce a short-term weak response and a long-term robust response. The short-term response is experienced within minutes and may last around 2 h. The short-term response relies on membrane receptors, adenosine receptors, and electrical potentials and their effects on KATP channels [251, 252]. Another mechanism of short-term ischemic preconditioning may include the expression of miRNA that targets mRNA responsible for inflammation, ionic balance, oxidative stress, and transcription, as shown in an animal study [253].
The long-term robust response peaks around 3 days (with a protective window of ~ 1–7 days) and has a greater protective effect than the short-term response. This response takes time and results in greater protection because of changes in protein expression. These changes fall into the broad categories of inflammatory signaling, heat–shock proteins, nitrous oxide, free radical sensors, adenosine and BCL/caspase, Nf-KB, and various other cell survival pathways. Many studies have described these protective mechanisms via signal transduction in greater length and detail. Most of these previous reports describe a general downstream effect of reducing cell death through mechanisms related to KATP channels described above as well as by inhibition of the mitochondrial permeability transition pore (MPTP) and thereby blocking apoptosis or necrosis [254]. It has also been shown that mitigation of excitotoxicity can occur via NMDA regulation [255].
A commonly cited problem in ischemic preconditioning is the lack of clinical application due to the random nature of events such as focal stroke or CA. Therefore, while preconditioning may be a useful preventative measure, its practicality is clearly a challenge. Postconditioning, however, does not suffer from this limitation. Ischemic postconditioning also entails inducing sublethal injuries to the brain parenchyma, but it takes place after the initial insult. Ischemic postconditioning can also be categorized as either classic or remote, where classic is intermittent occlusion of the same vessels that caused the injury and remote is intermittent occlusion of vessels on an extremity. The underlying molecular mechanisms for postconditioning appear to have some similarities to those for preconditioning. One preclinical study showed the possible utility of intermittent ischemic postconditioning in reduction of infarct size [256]. The group performed three cycles of 30-s occlusion/10-s release of the bilateral CCA after reperfusion in rats, robustly reducing infarct size in focal ischemia. In 2009, this group used the same focal ischemic model, but rather than classic postconditioning, they used remote (on a limb) postconditioning to show a similar effect [257] via many of the same pathways described above. Recently, a clinical study showed a decrease in stroke incidence in intracerebral artery stenosis patients using remote ischemic preconditioning by inflating a pneumatic cuff around patients’ arms [258]. In this study, the incidence of recurrent stroke at 300 days was 26.7% in the control while the preconditioned group demonstrated a reduction in the incidence of recurrent stroke to 7.9% at 300 days.
Other methods of preconditioning are being investigated to mitigate ischemic brain injury without the use of ischemia itself as the preconditioning insult. Indeed, hypothermia itself has been shown in an animal model to provide a preconditioning type of neuroprotection [259]. Moreover, multiple preclinical studies have shown that partial caloric restriction or ketogenic diet appears to also induce a preconditioning-type of neuroprotection from ischemic injury [260, 261]. In fact, our group has recently shown in an animal model that ultrashort (overnight) caloric restriction before asphyxial CA induces mild ketosis and provides significant neuroprotection that may signify a preconditioning-type of effect [262].
Conclusion
HIBI caused by CA is a complex mechanism of brain damage caused by insults including oxygen deprivation, reduction in cerebral perfusion, and cytotoxicity [2, 263]. For decades the clinical exam was the principal method for determining injury severity and predicting neurological outcome. Electrophysiology, brain imaging, and molecular biomarkers are beginning to be integrated together to increase diagnostic and prognostic accuracy. Clinicians are often conflicted about when to discuss early predictions of outcome for neurologically impaired and comatose patients with family members to elucidate treatment plans [264]. Therefore, multimodal monitoring approaches are highly recommended as potential means of minimizing “self-fulfilling prophecy” and avoiding premature withdrawal of life-sustaining therapy [121, 130]. Emerging monitoring tools such as diffuse optical technologies, which can provide continuous monitoring of HIBI-related perturbations to the brain, may provide earlier diagnosis/prognosis, more accurate assessment of injury, lower false predictive rates, and better-informed patient treatment. Identifying poor prognosis early on with novel monitoring tools could be vital to either provide prompt treatment or improve outcome by ameliorating physiologic insults as they are occurring or having critical discussions with family members about goals of care.
There have been few recent therapeutic advancements in CA patient treatment, and while existing therapies (e.g., TTM) provide some benefit, there is an urgent need to further reduce devastating outcomes for patients and their families [5, 68]. For TTM, the number of patients needed to treat in order to improve recovery for one patient is ~ 6, making us turn toward the investigation of other potential treatments to further reduce morbidity and mortality [265]. Emerging therapies targeting metabolic factors, pharmacologic therapies, ROS scavengers, preconditioning, and optimizing peripheral blood pressure, CBF, and brain oxygenation may be integrated with the current standards of treatment early on to improve patient outcomes (Fig. 4). Our group anticipates that future therapies will potentially incorporate advanced monitoring tools to collect continuous, quantitative cerebral hemodynamic information at the bedside that may provide real-time feedback to inform dynamic, patient-specific, brain-centered treatment plans and address physiologic changes within minutes of cardiac arrest.
Fig. 4.

Targeted therapies for post-CA patient care. The base of the pyramid highlights the currently used treatment for CA patients (TTM), with bullet points identifying TTM parameters that remain topics of debate, interest, or investigation. Upon this foundation, new treatment mechanisms (upper levels of the pyramid) are being introduced. 20-HETE = 20-hydroxyeicosatetraenoic acid; AQP4 = aquaporin 4; ROS = reactive oxygen species; NMDA = LPS = lipopolysaccharide; MAP = mean arterial pressure; CBF = cerebral blood flow; CMRO2 = cerebral metabolic rate of oxygen; StO2 = tissue oxygenation
Electronic supplementary material
(PDF 173 kb)
Acknowledgments
Support for this work was provided by the Roneet Carmell Memorial Endowment Fund to Y.A., the Arnold and Mabel Beckman Foundation, the National Science Foundation Graduate Research Fellowship Program (DGE-1321846, to C.C.), the United States National Institutes of Health (P41EB015890), and the National Center for Research Resources and National Center for Advancing Translational Sciences, National Institutes of Health, through the following grants: TL1 TR001415-01 to R. H.W., R21 EB024793 to Y. A., KL2 TR001416 to Y.A. via UL1 TR001414, and a CTSA pilot grant to Y.A. via UL1 TR001414. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Abbreviations
- 20-HETE
hydroxyeicosatetraenoic acid
- AAN
American Academy of Neurology
- ACLS
advanced cardiac life support
- AQP4
aquaporin 4
- AHA
American Heart Association
- ATP
adenosine triphosphate
- BBB
blood–brain barrier
- BCLS
basic cardiac life support
- BIS
bispectral index
- BP
blood pressure
- CA
cardiac arrest
- CABG
coronary artery bypass graft
- CBF
cerebral blood flow
- CCA
common carotid artery
- CMRO2
cerebral metabolic rate of oxygen
- CNS
central nervous system
- CO2
carbon dioxide
- CPC
cerebral performance category
- CT
computed tomography
- DCS
diffuse correlation spectroscopy
- DOS(I)
diffuse optical spectroscopy and imaging
- EAA
excitatory amino acid
- EEG
electroencephalography
- EMG
electromyography
- EMS
emergency medical services
- ERC
European Resuscitation Council
- ESICM
European Society of Intensive Care Medicine
- GOS
Glasgow Outcome Scale
- HIBI
hypoxic–ischemic brain injury
- HIF
hypoxia-inducible transcription factors
- ICP
intracranial pressure
- LDH
lactate dehydrogenase
- LP
lactate-to-pyruvate
- LPS
lipopolysaccharides
- MAP
mean arterial pressure
- MCA
middle cerebral artery
- MFV
mean flow velocity
- MCS
mental health composite score
- MMP
matrix metalloproteinase
- MMSE
mini-mental state exam
- MPTP
mitochondrial permeability transition pore
- MRI
magnetic resonance imaging
- mRS
modified Rankin Scale
- MSE
myoclonus status epilepticus
- NADH
nicotinamide adenine dinucleotide
- NIRS
near-infrared spectroscopy
- NMDA
N-methyl-d-aspartate
- NO
nitric oxide
- NPi
Neurological Pupil index
- NSE
neuron-specific enolase
- NSR
nonshockable rhythm
- OHCA
out-of-hospital cardiac arrest
- PCS
physical health composite score
- PEA
pulseless electrical activity
- PET
positron emission tomography
- PHDs
prolyl hydroxylase domain proteins
- PLR
pupillary light reflex
- ROS
reactive oxygen species
- ROSC
return of spontaneous circulation
- SD
spreading depolarization
- SSEP
somatosensory evoked potential
- SUR
sulfonylurea receptor
- TTM
targeted temperature management
- VF
ventricular fibrillation
- VIS–NIR
visible to near-infrared
- VT
ventricular tachycardia
- WLST
withdrawal of life-sustaining therapy.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Melika Hosseini and Robert H. Wilson contributed equally to this work.
References
- 1.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart Disease and Stroke Statistics—2018 Update: A Report From the American Heart Association. Circulation 2018;137. 10.1161/CIR.0000000000000558. [DOI] [PubMed]
- 2.Arciniegas DB. Hypoxic-ischemic brain injury: Addressing the disconnect between pathophysiology and public policy. NeuroRehabilitation. 2010;26:1–4. doi: 10.3233/NRE-2010-0530. [DOI] [PubMed] [Google Scholar]
- 3.Wijdicks EFM, Hijdra A, Young GB, Bassetti CL, Wiebe S. Practice Parameter: Prediction of outcome in comatose survivors after cardiopulmonary resuscitation (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006;67:203–10. doi: 10.1212/01.wnl.0000227183.21314.cd. [DOI] [PubMed] [Google Scholar]
- 4.Nolan JP, Soar J, Cariou A, Cronberg T, Moulaert VRM, Deakin CD, et al. European Resuscitation Council and European Society of Intensive Care Medicine Guidelines for Post-resuscitation Care 2015. Section 5 of the European Resuscitation Council Guidelines for Resuscitation 2015. Resuscitation. 2015;95:202–22. doi: 10.1016/j.resuscitation.2015.07.018. [DOI] [PubMed] [Google Scholar]
- 5.Wartenberg KE, Hwang DY, Haeusler KG, Muehlschlegel S, Sakowitz OW, Madžar D, et al. Gap Analysis Regarding Prognostication in Neurocritical Care: A Joint Statement from the German Neurocritical Care Society and the Neurocritical Care Society. Neurocrit Care. 2019;31:231–44. doi: 10.1007/s12028-019-00769-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siesjö BK. Cell damage in the brain: a speculative synthesis. J Cereb Blood Flow Metab. 1981;1:155–85. doi: 10.1038/jcbfm.1981.18. [DOI] [PubMed] [Google Scholar]
- 7.Van Den Brule JMD, Van Der Hoeven JG, Hoedemaekers CWE. Cerebral perfusion and cerebral autoregulation after cardiac arrest. Biomed Res Int 2018;2018. 10.1155/2018/4143636. [DOI] [PMC free article] [PubMed]
- 8.Rossetti AO, Tovar Quiroga DF, Juan E, Novy J, White RD, Ben-Hamouda N, et al. Electroencephalography Predicts Poor and Good Outcomes after Cardiac Arrest: A Two-Center Study. Crit Care Med. 2017;45:e674–82. doi: 10.1097/CCM.0000000000002337. [DOI] [PubMed] [Google Scholar]
- 9.Keller SP, Halperin HR. Cardiac Arrest: the Changing Incidence of Ventricular Fibrillation. Curr Treat Options Cardiovasc Med 2015;17. 10.1007/s11936-015-0392-z. [DOI] [PMC free article] [PubMed]
- 10.Meaney PA, Nadkarni VM, Kern KB, Indik JH, Halperin HR, Berg RA. Rhythms and outcomes of adult in-hospital cardiac arrest. Crit Care Med. 2010;38:101–8. doi: 10.1097/CCM.0b013e3181b43282. [DOI] [PubMed] [Google Scholar]
- 11.Andrew E, Nehme Z, Lijovic M, Bernard S, Smith K. Outcomes following out-of-hospital cardiac arrest with an initial cardiac rhythm of asystole or pulseless electrical activity in Victoria, Australia. Resuscitation. 2014;85:1633–9. doi: 10.1016/j.resuscitation.2014.07.015. [DOI] [PubMed] [Google Scholar]
- 12.Strömsöe A, Svensson L, Axelsson ÅB, Claesson A, Göransson KE, Nordberg P, et al. Improved outcome in Sweden after out-of-hospital cardiac arrest and possible association with improvements in every link in the chain of survival. Eur Heart J. 2015;36:863–71. doi: 10.1093/eurheartj/ehu240. [DOI] [PubMed] [Google Scholar]
- 13.Wissenberg M, Lippert FK, Folke F, Weeke P, Hansen CM, Christensen EF, et al. Association of national initiatives to improve cardiac arrest management with rates of bystander intervention and patient survival after out-of-hospital cardiac arrest. JAMA - J Am Med Assoc. 2013;310:1377–84. doi: 10.1001/jama.2013.278483. [DOI] [PubMed] [Google Scholar]
- 14.McHugh TP, Stout M. Near-hanging injury. Ann Emerg Med. 1983;12:774–6. doi: 10.1016/S0196-0644(83)80256-X. [DOI] [PubMed] [Google Scholar]
- 15.Vaagenes P, Safar P, Moossy J, Rao G, Diven W, Ravi C, et al. Asphyxiation versus ventricular fibrillation cardiac arrest in dogs. Resuscitation. 1997;35:41–52. doi: 10.1016/S0300-9572(97)01108-8. [DOI] [PubMed] [Google Scholar]
- 16.Uray T, Lamade A, Elmer J, Drabek T, Stezoski JP, Missé A, et al. Phenotyping Cardiac Arrest. Crit Care Med. 2018;46:e508–15. doi: 10.1097/CCM.0000000000003070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bergström M, Schmidbauer S, Herlitz J, Rawshani A, Friberg H. Pulseless electrical activity is associated with improved survival in out-of-hospital cardiac arrest with initial non-shockable rhythm. Resuscitation. 2018;133:147–52. doi: 10.1016/j.resuscitation.2018.10.018. [DOI] [PubMed] [Google Scholar]
- 18.Sekhon MS, Ainslie PN, Griesdale DE. Clinical pathophysiology of hypoxic ischemic brain injury after cardiac arrest: A “two-hit” model. Crit Care. 2017;21:1–10. doi: 10.1186/s13054-017-1670-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buunk G, van der Hoeven JG, Meinders AE. Cerebrovascular reactivity in comatose patients resuscitated from a cardiac arrest. Stroke. 1997;28:1569–73. doi: 10.1161/01.str.28.8.1569. [DOI] [PubMed] [Google Scholar]
- 20.Lemiale V, Huet O, Vigué B, Mathonnet A, Spaulding C, Mira J, et al. Changes in cerebral blood flow and oxygen extraction during post-resuscitation syndrome. Resuscitation. 2008;76:17–24. doi: 10.1016/j.resuscitation.2007.06.028. [DOI] [PubMed] [Google Scholar]
- 21.Lassen NA. The luxury-perfusion syndrome and its possible relation to acute metabolic acidosis localised within the brain. Lancet. 1966;288:1113–5. doi: 10.1016/s0140-6736(66)92199-4. [DOI] [PubMed] [Google Scholar]
- 22.Beckstead JE, Tweed WA, Lee J, MacKeen WL. Cerebral blood flow and metabolism in man following cardiac arrest. Stroke. 1978;9:569–73. doi: 10.1161/01.STR.9.6.569. [DOI] [PubMed] [Google Scholar]
- 23.Iordanova B, Li L, Clark RSB, Manole MD. Alterations in Cerebral Blood Flow after Resuscitation from Cardiac Arrest. Front Pediatr 2017;5. 10.3389/fped.2017.00174. [DOI] [PMC free article] [PubMed]
- 24.Ames A, Wright RL, Kowada M, Thurston JM, Majno G. Cerebral ischemia. II. The no-reflow phenomenon. Am J Pathol. 1968;52:437–53. [PMC free article] [PubMed] [Google Scholar]
- 25.Fischer M, Hossmann KA. No-reflow after cardiac arrest. Intensive Care Med. 1995;21:132–41. doi: 10.1007/BF01726536. [DOI] [PubMed] [Google Scholar]
- 26.Hossmann K-AA. Reperfusion of the brain after global ischemia: hemodynamic disturbances. Shock. 1997;8:95–101. doi: 10.1097/00024382-199708000-00004. [DOI] [PubMed] [Google Scholar]
- 27.Li L, Poloyac SM, Watkins SC, St. Croix CM, Alexander H, Gibson GA, et al. Cerebral microcirculatory alterations and the no-reflow phenomenon in vivo after experimental pediatric cardiac arrest. J Cereb Blood Flow Metab. 2019;39:913–25. doi: 10.1177/0271678X17744717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bacic F, Uematsu S, McCarron RM, Spatz M. Secretion of Immunoreactive endothelin-1 by capillary and microvascular endothelium of human brain. Neurochem Res. 1992;17:699–702. doi: 10.1007/BF00968008. [DOI] [PubMed] [Google Scholar]
- 29.van den Brule JMD, Vinke E, van Loon LM, van der Hoeven JG, Hoedemaekers CWE. Middle cerebral artery flow, the critical closing pressure, and the optimal mean arterial pressure in comatose cardiac arrest survivors—An observational study. Resuscitation. 2017;110:85–9. doi: 10.1016/j.resuscitation.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 30.Strandgaard S. Autoregulation of cerebral blood flow in hypertensive patients. The modifying influence of prolonged antihypertensive treatment on the tolerance to acute, drug induced hypotension. Circulation. 1976;53:720–7. doi: 10.1161/01.CIR.53.4.720. [DOI] [PubMed] [Google Scholar]
- 31.Sundgreen C, Larsen FS, Herzog TM, Knudsen GM, Boesgaard S, Aldershvile J. Autoregulation of cerebral blood flow in patients resuscitated from cardiac arrest. Stroke. 2001;32:128–32. doi: 10.1161/01.STR.32.1.128. [DOI] [PubMed] [Google Scholar]
- 32.Lee JK, Brady KM, Mytar JO, Kibler KK, Carter EL, Hirsch KG, et al. Cerebral blood flow and cerebrovascular autoregulation in a swine model of pediatric cardiac arrest and hypothermia*. Crit Care Med. 2011;39:2337–45. doi: 10.1097/CCM.0b013e318223b910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhate TD, McDonald B, Sekhon MS, Griesdale DEG. Association between blood pressure and outcomes in patients after cardiac arrest: A systematic review. Resuscitation. 2015;97:1–6. doi: 10.1016/j.resuscitation.2015.08.023. [DOI] [PubMed] [Google Scholar]
- 34.Roberts BW, Hope Kilgannon J, Hunter BR, Puskarich MA, Pierce L, Donnino M, et al. Association between early hyperoxia exposure after resuscitation from cardiac arrest and neurological disability: Prospective multicenter protocol-directed cohort study. Circulation. 2018;137:2114–24. doi: 10.1161/CIRCULATIONAHA.117.032054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takagi S, Cocito L, Hossmann KA. Blood recirculation and pharmacological responsiveness of the cerebral vasculature following prolonged ischemia of cat brain. Stroke. 1977;8:707–12. doi: 10.1161/01.STR.8.6.707. [DOI] [PubMed] [Google Scholar]
- 36.Siesjö BK, Plum F. Cerebral Energy Metabolism in Normoxia and in Hypoxia. Acta Anaesthesiol Scand. 1971;15:81–101. doi: 10.1111/j.1399-6576.1971.tb00662.x. [DOI] [PubMed] [Google Scholar]
- 37.Humphreys SA, Koss MC. Role of nitric oxide in post-ischemic cerebral hyperemia in anesthetized rats. Eur J Pharmacol. 1998;347:223–9. doi: 10.1016/S0014-2999(98)00100-9. [DOI] [PubMed] [Google Scholar]
- 38.Sciotti VM, Van Wylen DGL. Increases in interstitial adenosine and cerebral blood flow with inhibition of adenosine kinase and adenosine deaminase. J Cereb Blood Flow Metab. 1993;13:201–7. doi: 10.1038/jcbfm.1993.24. [DOI] [PubMed] [Google Scholar]
- 39.Kågström E, Smith M-LL, Siesjö BK, Kagstrom E, Smith M-LL, Siesjo BK. Local cerebral blood flow in the recovery period following complete cerebral ischemia in the rat. J Cereb Blood Flow Metab. 1983;3:170–82. doi: 10.1038/jcbfm.1983.24. [DOI] [PubMed] [Google Scholar]
- 40.Safar P, Stezoski W, Nemoto EM. Amelioration of Brain Damage After 12 Minutes’ Cardiac Arrest in Dogs. Arch Neurol. 1976;33:91–5. doi: 10.1001/archneur.1976.00500020019004. [DOI] [PubMed] [Google Scholar]
- 41.Iadecola C, Pelligrino DA, Moskowitz MA, Lassen NA. Nitric oxide synthase inhibition and cerebrovascular regulation. J Cereb Blood Flow Metab. 1994;14:175–92. doi: 10.1038/jcbfm.1994.25. [DOI] [PubMed] [Google Scholar]
- 42.Bisschops LLA, Hoedemaekers CWE, Simons KS, Van Der Hoeven JG. Preserved metabolic coupling and cerebrovascular reactivity during mild hypothermia after cardiac arrest. Crit Care Med. 2010;38:1542–7. doi: 10.1097/CCM.0b013e3181e2cc1e. [DOI] [PubMed] [Google Scholar]
- 43.Callaway CW, Donnino MW, Fink EL, Geocadin RG, Golan E, Kern KB, et al. Part 8: Post-cardiac arrest care: 2015 American Heart Association guidelines update for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2015;132:S465–82. doi: 10.1161/CIR.0000000000000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roberts BW, Kilgannon JH, Hunter BR, Puskarich MA, Shea L, Donnino M, et al. Association Between Elevated Mean Arterial Blood Pressure and Neurologic Outcome After Resuscitation From Cardiac Arrest: Results From a Multicenter Prospective Cohort Study. Crit Care Med. 2019;47:93–100. doi: 10.1097/CCM.0000000000003474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mölström S, Nielsen TH, Nordström CH, Hassager C, Møller JE, Kjærgaard J, et al. Design paper of the “blood pressure targets in post-resuscitation care and bedside monitoring of cerebral energy state: A randomized clinical trial” Trials 2019;20. 10.1186/s13063-019-3397-1. [DOI] [PMC free article] [PubMed]
- 46.Russo JJ, Di Santo P, Simard T, James TE, Hibbert B, Couture E, et al. Optimal mean arterial pressure in comatose survivors of out-of-hospital cardiac arrest: An analysis of area below blood pressure thresholds. Resuscitation. 2018;128:175–80. doi: 10.1016/j.resuscitation.2018.04.028. [DOI] [PubMed] [Google Scholar]
- 47.Wagner SR, Lanier WL. Metabolism of glucose, glycogen, and high-energy phosphates during complete cerebral ischemia. A comparison of normoglycemic, chronically hyperglycemic diabetic, and acutely hyperglycemic nondiabetic rats. Anesthesiology. 1994;81:1516–26. doi: 10.1097/00000542-199412000-00028. [DOI] [PubMed] [Google Scholar]
- 48.Hoxworth JM, Xu K, Zhou Y, David Lust WD, Lamanna JC. Cerebral metabolic profile, selective neuron loss, and survival of acute and chronic hyperglycemic rats following cardiac arrest and resuscitation. Brain Res 1999; 821(2):467–79. 10.1016/S0006-8993(98)01332-8. [DOI] [PubMed]
- 49.Greer DM. Mechanisms of injury in hypoxic-ischemic encephalopathy: Implications to therapy. Semin Neurol. 2006;26:373–9. doi: 10.1055/s-2006-948317. [DOI] [PubMed] [Google Scholar]
- 50.Holzer M. Targeted temperature management for comatose survivors of cardiac arrest. N Engl J Med. 2010;363:1256–64. doi: 10.1056/NEJMct1002402. [DOI] [PubMed] [Google Scholar]
- 51.Michenfelder JD, Theye RA. The effects of anesthesia and hypothermia on canine cerebral ATP and lactate during anoxia produced by decapitation. Anesthesiology. 1970;33:430–9. doi: 10.1097/00000542-197010000-00013. [DOI] [PubMed] [Google Scholar]
- 52.Silver IA, Erecińska M. Ion homeostasis in rat brain in vivo: Intra- and extracellular [Ca2+] and [H+] in the hippocampus during recovery from short-term, transient ischemia. J Cereb Blood Flow Metab. 1992;12:759–72. doi: 10.1038/jcbfm.1992.107. [DOI] [PubMed] [Google Scholar]
- 53.Karen Birmingham. Future of neuroprotective drugs in doubt. Nat Med 2002;8(1):5–5. 10.1038/nm0102-5a. [DOI] [PubMed]
- 54.Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1:383–6. doi: 10.1016/S1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 55.Nolan JP, Neumar RW, Adrie C, Aibiki M, Berg RA, Böttiger BW, et al. Post-cardiac arrest syndrome: Epidemiology, pathophysiology, treatment, and prognostication. A Scientific Statement from the International Liaison Committee on Resuscitation; the American Heart Association Emergency Cardiovascular Care Committee; the Coun. Resuscitation. 2008;79:350–79. doi: 10.1016/j.resuscitation.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 56.Sanganalmath SK, Gopal P, Parker JCJRJCJR, Downs RK, Parker JCJRJCJR, Dawn B. Global cerebral ischemia due to circulatory arrest: insights into cellular pathophysiology and diagnostic modalities. Mol Cell Biochem. 2017;426:111–27. doi: 10.1007/s11010-016-2885-9. [DOI] [PubMed] [Google Scholar]
- 57.Adams JA. Endothelium and cardiopulmonary resuscitation. Crit. Care Med., vol. 34, 2006. 10.1097/01.CCM.0000246012.68479.49. [DOI] [PubMed]
- 58.Fischer M, Böttiger BW, Popov-Cenic S, Hossmann KA. Thrombolysis using plasminogen activator and heparin reduces cerebral no-reflow after resuscitation from cardiac arrest: An experimental study in the cat. Intensive Care Med. 1996;22:1214–23. doi: 10.1007/BF01709339. [DOI] [PubMed] [Google Scholar]
- 59.Kacimi R, Giffard RG, Yenari MA. Endotoxin-activated microglia injure brain derived endothelial cells via NF-κB, JAK-STAT and JNK stress kinase pathways. J Inflamm 2011;8. 10.1186/1476-9255-8-7. [DOI] [PMC free article] [PubMed]
- 60.Ma Y, Wang J, Wang Y, Yang GY. The biphasic function of microglia in ischemic stroke. Prog Neurobiol. 2017;157:247–72. doi: 10.1016/j.pneurobio.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 61.Cao W, Carney JM, Duchon A, Floyd RA, Chevion M. Oxygen free radical involvement in ischemia and reperfusion injury to brain. Neurosci Lett. 1988;88:233–8. doi: 10.1016/0304-3940(88)90132-2. [DOI] [PubMed] [Google Scholar]
- 62.Hamilton J, Brustovetsky T, Rysted JE, Lin Z, Usachev YM, Brustovetsky N. Deletion of mitochondrial calcium uniporter incompletely inhibits calcium uptake and induction of the permeability transition pore in brain mitochondria. J Biol Chem. 2018;293:15652–63. doi: 10.1074/jbc.RA118.002926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qiu J, Tan YW, Hagenston AM, Martel MA, Kneisel N, Skehel PA, et al. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun. 2013;4:1–12. doi: 10.1038/ncomms3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Medvedeva YV, Lin B, Shuttleworth CW, Weiss JH. Intracellular Zn2+ accumulation contributes to synaptic failure, mitochondrial depolarization, and cell death in an acute slice oxygen-glucose deprivation model of ischemia. J Neurosci. 2009;29:1105–14. doi: 10.1523/JNEUROSCI.4604-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yin HZ, Wang HL, Ji SG, Medvedeva YV, Tian G, Bazrafkan AK, et al. Rapid Intramitochondrial Zn2+ Accumulation in CA1 Hippocampal Pyramidal Neurons After Transient Global Ischemia: A Possible Contributor to Mitochondrial Disruption and Cell Death. J Neuropathol Exp Neurol. 2019;78:655–64. doi: 10.1093/jnen/nlz042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giulian D, Li J, Leara B, Keenen C. Phagocytic microglia release cytokines and cytotoxins that regulate the survival of astrocytes and neurons in culture. Neurochem Int. 1994;25:227–33. doi: 10.1016/0197-0186(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 67.Barone FC, Hillegass LM, Price WJ, White RF, Lee EV, Feuerstein GZ, et al. Polymorphonuclear leukocyte infiltration into cerebral focal ischemic tissue: Myeloperoxidase activity assay and histologic verification. J Neurosci Res. 1991;29:336–45. doi: 10.1002/jnr.490290309. [DOI] [PubMed] [Google Scholar]
- 68.Zhang C, Brandon NR, Koper K, Tang P, Xu Y, Dou H. Invasion of peripheral immune cells into brain parenchyma after cardiac arrest and resuscitation. Aging Dis. 2018;9:412–25. doi: 10.14336/AD.2017.0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qin C, Zhou LQ, Ma XT, Hu ZW, Yang S, Chen M, et al. Dual Functions of Microglia in Ischemic Stroke. Neurosci Bull 2019. 10.1007/s12264-019-00388-3. [DOI] [PMC free article] [PubMed]
- 70.Kim JY, Kim N, Yenari MA. Mechanisms and Potential Therapeutic Applications of Microglial Activation after Brain Injury. CNS Neurosci Ther. 2015;21:309–19. doi: 10.1111/cns.12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Walter L, Neumann H. Role of microglia in neuronal degeneration and regeneration. Semin Immunopathol. 2009;31:513–25. doi: 10.1007/s00281-009-0180-5. [DOI] [PubMed] [Google Scholar]
- 72.Dabrowska S, Andrzejewska A, Lukomska B, Janowski M. Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles. J Neuroinflammation 2019;16. 10.1186/s12974-019-1571-8. [DOI] [PMC free article] [PubMed]
- 73.Pluta R, Lossinsky AS, Wiśniewski HM, Mossakowski MJ. Early blood-brain barrier changes in the rat following transient complete cerebral ischemia induced by cardiac arrest. Brain Res. 1994;633:41–52. doi: 10.1016/0006-8993(94)91520-2. [DOI] [PubMed] [Google Scholar]
- 74.Egashira Y, Suzuki Y, Azuma Y, Takagi T, Mishiro K, Sugitani S, et al. The growth factor progranulin attenuates neuronal injury induced by cerebral ischemia-reperfusion through the suppression of neutrophil recruitment. J Neuroinflammation 2013;10. 10.1186/1742-2094-10-105. [DOI] [PMC free article] [PubMed]
- 75.Jackman K, Kahles T, Lane D, Garcia-Bonilla L, Abe T, Capone C, et al. Progranulin deficiency promotes post-ischemic blood-brain barrier disruption. J Neurosci. 2013;33:19579–89. doi: 10.1523/JNEUROSCI.4318-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kanazawa M, Kawamura K, Takahashi T, Miura M, Tanaka Y, Koyama M, et al. Multiple therapeutic effects of progranulin on experimental acute ischaemic stroke. Brain. 2015;138:1932–48. doi: 10.1093/brain/awv079. [DOI] [PubMed] [Google Scholar]
- 77.Mizuma A, Yenari MA. Anti-inflammatory targets for the treatment of reperfusion injury in stroke. Front Neurol 2017;8. 10.3389/fneur.2017.00467. [DOI] [PMC free article] [PubMed]
- 78.Xiao F, Arnold TC, Zhang S, Brown C, Steven Alexander J, Carden DL, et al. Cerebral cortical aquaporin-4 expression in brain edema following cardiac arrest in rats. Acad Emerg Med. 2004;11:1001–7. doi: 10.1197/j.aem.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 79.Wallisch JS, Janesko-Feldman K, Alexander H, Jha RM, Farr GW, McGuirk PR, et al. The aquaporin-4 inhibitor AER-271 blocks acute cerebral edema and improves early outcome in a pediatric model of asphyxial cardiac arrest. Pediatr Res. 2019;85:511–7. doi: 10.1038/s41390-018-0215-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hayman EG, Patel AP, Kimberly WT, Sheth KN, Simard JM. Cerebral Edema After Cardiopulmonary Resuscitation: A Therapeutic Target Following Cardiac Arrest? Neurocrit Care. 2018;28:276–87. doi: 10.1007/s12028-017-0474-8. [DOI] [PubMed] [Google Scholar]
- 81.Panickar KS, Noremberg MD. Astrocytes in cerebral ischemic injury: Morphological and general considerations. Glia. 2005;50:287–98. doi: 10.1002/glia.20181. [DOI] [PubMed] [Google Scholar]
- 82.Deng G, Carter J, Traystman RJ, Wagner DH, Herson PS. Pro-inflammatory T-lymphocytes rapidly infiltrate into the brain and contribute to neuronal injury following cardiac arrest and cardiopulmonary resuscitation. J Neuroimmunol. 2014;274:132–40. doi: 10.1016/j.jneuroim.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huang K, Wang Z, Gu Y, Hu Y, Ji Z, Wang S, et al. Glibenclamide Is Comparable to Target Temperature Management in Improving Survival and Neurological Outcome After Asphyxial Cardiac Arrest in Rats. J Am Heart Assoc. 2016;5:1–11. doi: 10.1161/JAHA.116.003465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nakayama S, Taguchi N, Isaka Y, Nakamura T, Tanaka M. Glibenclamide and Therapeutic Hypothermia Have Comparable Effect on Attenuating Global Cerebral Edema Following Experimental Cardiac Arrest. Neurocrit Care. 2018;29:119–27. doi: 10.1007/s12028-017-0479-3. [DOI] [PubMed] [Google Scholar]
- 85.Walter EJ, Carraretto M. The neurological and cognitive consequences of hyperthermia. Crit Care 2016;20. 10.1186/s13054-016-1376-4. [DOI] [PMC free article] [PubMed]
- 86.Lee BH, Inui D, Suh GY, Kim JY, Kwon JY, Park J, et al. Association of body temperature and antipyretic treatments with mortality of critically ill patients with and without sepsis: Multi-centered prospective observational study. Crit Care 2012;16. 10.1186/cc11211. [DOI] [PMC free article] [PubMed]
- 87.de Jonge JC, Wallet J, van der Worp HB. Fever worsens outcomes in animal models of ischaemic stroke: A systematic review and meta-analysis. Eur Stroke J. 2019;4:29–38. doi: 10.1177/2396987318776421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kiyatkin EA, Sharma HS. Permeability of the blood-brain barrier depends on brain temperature. Neuroscience. 2009;161:926–39. doi: 10.1016/j.neuroscience.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zlotnik A, Gurevich B, Artru AA, Gruenbaum SE, Dubilet M, Leibowitz A, et al. The effect of hyperthermia on blood glutamate levels. Anesth Analg. 2010;111:1497–504. doi: 10.1213/ANE.0b013e3181fc0112. [DOI] [PubMed] [Google Scholar]
- 90.Campos F, Pérez-Mato M, Agulla J, Blanco M, Barral D, Almeida Á, et al. Glutamate Excitoxicity Is the Key Molecular Mechanism Which Is Influenced by Body Temperature during the Acute Phase of Brain Stroke. PLoS One 2012;7. 10.1371/journal.pone.0044191. [DOI] [PMC free article] [PubMed]
- 91.Jackson TC, Kochanek PM. A New Vision for Therapeutic Hypothermia in the Era of Targeted Temperature Management: A Speculative Synthesis. Ther Hypothermia Temp Manag. 2019;9:13–47. doi: 10.1089/ther.2019.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Anderson CA, Arciniegas DB. Cognitive sequelae of hypoxic-ischemic brain injury: A review. NeuroRehabilitation. 2010;26:47–63. doi: 10.3233/NRE-2010-0535. [DOI] [PubMed] [Google Scholar]
- 93.Lybeck A, Friberg H, Aneman A, Hassager C, Horn J, Kjærgaard J, et al. Prognostic significance of clinical seizures after cardiac arrest and target temperature management. Resuscitation. 2017;114:146–51. doi: 10.1016/j.resuscitation.2017.01.017. [DOI] [PubMed] [Google Scholar]
- 94.Walker AC, Johnson NJ. Targeted Temperature Management and Postcardiac arrest Care. Emerg Med Clin North Am. 2019;37:381–93. doi: 10.1016/j.emc.2019.03.002. [DOI] [PubMed] [Google Scholar]
- 95.Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med 2009;37. 10.1097/CCM.0b013e3181aa5241. [DOI] [PubMed]
- 96.Wang Q, Miao P, Modi HR, Garikapati S, Koehler RC, Thakor NV. Therapeutic hypothermia promotes cerebral blood flow recovery and brain homeostasis after resuscitation from cardiac arrest in a rat model. J Cereb Blood Flow Metab. 2019;39:1961–73. doi: 10.1177/0271678X18773702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gunn AJ, Laptook AR, Robertson NJ, Barks JD, Thoresen M, Wassink G, et al. Therapeutic hypothermia translates from ancient history in to practice. Pediatr Res. 2017;81:202–9. doi: 10.1038/pr.2016.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tooley JR, Satas S, Porter H, Silver IA, Thoresen M. Head cooling with mild systemic hypothermia in anesthetized piglets is neuroprotective. Ann Neurol. 2003;53:65–72. doi: 10.1002/ana.10402. [DOI] [PubMed] [Google Scholar]
- 99.Wassink G, Gunn ER, Drury PP, Bennet L, Gunn AJ. The mechanisms and treatment of asphyxial encephalopathy. Front Neurosci. 2014;8:1–11. doi: 10.3389/fnins.2014.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sun YJ, Zhang ZY, Fan B, Li GY. Neuroprotection by therapeutic hypothermia. Front Neurosci. 2019;13:1–11. doi: 10.3389/fnins.2019.00586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Laptook AR, Corbett RJ, Sterett R, Garcia D, Tollefsbol G. Quantitative relationship between brain temperature and energy utilization rate measured in vivo using 31P and 1H magnetic resonance spectroscopy. Pediatr Res. 1995;38:919–25. doi: 10.1203/00006450-199512000-00015. [DOI] [PubMed] [Google Scholar]
- 102.Rosomoff HL, Holaday DA. Cerebral blood flow and cerebral oxygen consumption during hypothermia. Am J Physiol. 1954;179:85–8. doi: 10.1152/ajplegacy.1954.179.1.85. [DOI] [PubMed] [Google Scholar]
- 103.Khalilov RA, Dzhafarova AM, Khizrieva SI. Effect of Hypothermia on Kinetic Characteristics of Lactate Dehydrogenase in Rat Brain under Conditions of Global Ischemia and Reperfusion. Bull Exp Biol Med. 2017;163:334–7. doi: 10.1007/s10517-017-3797-8. [DOI] [PubMed] [Google Scholar]
- 104.Busto R, Globus MY, Dietrich WD, Martinez E, Valdés I, Ginsberg MD. Effect of mild hypothermia on ischemia-induced release of neurotransmitters and free fatty acids in rat brain. Stroke. 1989;20:904–10. doi: 10.1161/01.STR.20.7.904. [DOI] [PubMed] [Google Scholar]
- 105.Nakashima K, Todd MM. Effects of hypothermia on the rate of excitatory amino acid release after ischemic depolarization. Stroke. 1996;27:913–8. doi: 10.1161/01.STR.27.5.913. [DOI] [PubMed] [Google Scholar]
- 106.Ooboshi H, Ibayashi S, Takano K, Sadoshima S, Kondo A, Uchimura H, et al. Hypothermia inhibits ischemia-induced efflux of amino acids and neuronal damage in the hippocampus of aged rats. Brain Res. 2000;884(1–2):23–30. 10.1016/S0006-8993(00)02861-4. [DOI] [PubMed]
- 107.Bart RD, Takaoka S, Pearlstein RD, Dexter F, Warner DS. Interactions between hypothermia and the latency to ischemic depolarization: implications for neuroprotection. Anesthesiology. 1998;88:1266–73. doi: 10.1097/00000542-199805000-00018. [DOI] [PubMed] [Google Scholar]
- 108.McManus T, Sadgrove M, Pringle AK, Chad JE, Sundstrom LE. Intraischaemic hypothermia reduces free radical production and protects against ischaemic insults in cultured hippocampal slices. J Neurochem. 2004;91:327–36. doi: 10.1111/j.1471-4159.2004.02711.x. [DOI] [PubMed] [Google Scholar]
- 109.Lei B, Adachi N, Arai T. The effect of hypothermia on H2O2 production during ischemia and reperfusion: a microdialysis study in the gerbil hippocampus. Neurosci Lett. 1997;222:91–4. doi: 10.1016/S0304-3940(97)13349-3. [DOI] [PubMed] [Google Scholar]
- 110.Baumann E, Preston E, Slinn J, Stanimirovic D. Post-ischemic hypothermia attenuates loss of the vascular basement membrane proteins, agrin and SPARC, and the blood-brain barrier disruption after global cerebral ischemia. Brain Res. 2009;1269:185–97. doi: 10.1016/j.brainres.2009.02.062. [DOI] [PubMed] [Google Scholar]
- 111.Akbari Y, Mulder M, Razmara A, Geocadin R. Cool Down the Inflammation: Hypothermia as a Therapeutic Strategy for Acute Brain Injuries. Immunol. Mech. Ther. Brain Inj. Stroke. Chen J, Hu X, Stenzel-Poore M, Zhang JH, editors. New York, NY: Springer New York; 2014. Chapter 19 p.349–375. 10.1007/978-1-4614-8915-3.
- 112.Webster CM, Kelly S, Koike MA, Chock VY, Giffard RG, Yenari MA. Inflammation and NFκB activation is decreased by hypothermia following global cerebral ischemia. Neurobiol Dis. 2009;33:301–12. doi: 10.1016/j.nbd.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Eberspächer E, Werner C, Engelhard K, Pape M, Laacke L, Winner D, et al. Long-term effects of hypothermia on neuronal cell death and the concentration of apoptotic proteins after incomplete cerebral ischemia and reperfusion in rats. Acta Anaesthesiol Scand. 2005;49:477–87. doi: 10.1111/j.1399-6576.2005.00649.x. [DOI] [PubMed] [Google Scholar]
- 114.Vitkovic L, Maeda S, Sternberg E. Anti-inflammatory cytokines: Expression and action in the brain. Neuroimmunomodulation. 2001;9:295–312. doi: 10.1159/000059387. [DOI] [PubMed] [Google Scholar]
- 115.Stahel PP, Yatsiv I, Morganti-Kossmann MC, Ferez D, Novick D, Rubinstein M, et al. Elevated intracranial il-18 and evidence of neuroprotective effects of IL-18 binding protein (IL-18bp) after experimental closed head injury. Langenbeck’s Arch Surg. 2001;386:474–5. [Google Scholar]
- 116.Hofstetter C, Boost KA, Flondor M, Basagan-Mogol E, Betz C, Homann M, et al. Anti-inflammatory effects of sevoflurane and mild hypothermia in endotoxemic rats. Acta Anaesthesiol Scand. 2007;51:893–9. doi: 10.1111/j.1399-6576.2007.01353.x. [DOI] [PubMed] [Google Scholar]
- 117.Huet O, Kinirons B, Dupic L, Lajeunie E, Mazoit JX, Benhamou D, et al. Induced mild hypothermia reduces mortality during acute inflammation in rats. Acta Anaesthesiol Scand. 2007;51:1211–6. doi: 10.1111/j.1399-6576.2007.01419.x. [DOI] [PubMed] [Google Scholar]
- 118.Polderman KH, Herold I. Therapeutic hypothermia and controlled normothermia in the intensive care unit: Practical considerations, side effects, and cooling methods. Crit Care Med. 2009;37:1101–20. doi: 10.1097/CCM.0b013e3181962ad5. [DOI] [PubMed] [Google Scholar]
- 119.Frank SM, Satitpunwaycha P, Bruce SR, Herscovitch P, Goldstein DS. Increased myocardial perfusion and sympathoadrenal activation during mild core hypothermia in awake humans. Clin Sci 2003;104(5):503–8. 10.1042/CS20020256. [DOI] [PubMed]
- 120.Nabel EG, Ganz P, Gordon JB, Alexander RW, Selwyn AP. Dilation of normal and constriction of atherosclerotic coronary arteries caused by the cold pressor test. Circulation. 1988;77:43–52. doi: 10.1161/01.cir.77.1.43. [DOI] [PubMed] [Google Scholar]
- 121.Zhou SE, Maciel CB, Ormseth CH, Beekman R, Gilmore EJ, Greer DM. Distinct predictive values of current neuroprognostic guidelines in post-cardiac arrest patients. Resuscitation. 2019;139:343–50. doi: 10.1016/j.resuscitation.2019.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kavi T, Desai M, Yilmaz FM, Kakadia B, Burakgazi-Dalkilic E, Shrestha GS. Inter-predictability of Neuroprognostic Modalities After Cardiac Arrest. Cureus 2019;11. 10.7759/cureus.4489. [DOI] [PMC free article] [PubMed]
- 123.Greer DM, Rosenthal ES, Wu O. Neuroprognostication of hypoxic–ischaemic coma in the therapeutic hypothermia era. Nat Rev Neurol. 2014;10:190–203. doi: 10.1038/nrneurol.2014.36. [DOI] [PubMed] [Google Scholar]
- 124.Sandroni C, D’Arrigo S. Neurologic prognostication: Neurologic examination and current guidelines. Semin Neurol. 2017;37:40–7. doi: 10.1055/s-0036-1593857. [DOI] [PubMed] [Google Scholar]
- 125.Eertmans W, Tran TMP, Genbrugge C, Peene L, Mesotten D, Dens J, et al. A prediction model for good neurological outcome in successfully resuscitated out-of-hospital cardiac arrest patients. Scand J Trauma Resusc Emerg Med 2018;26. 10.1186/s13049-018-0558-2. [DOI] [PMC free article] [PubMed]
- 126.Willoughby JO, Leach BG. Relation of Neurological Findings after Cardiac Arrest to Outcome. BMJ. 1974;3:437–9. doi: 10.1136/bmj.3.5928.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Snyder BD, Ramirez-Lassepas M, Lippert DM. Article abstract Neurologic status and prognosis after cardiopulmonary arrest: 1. A retrospective study. Neurology 1977;27(9):807–807. 10.1212/WNL.27.9.807. [DOI] [PubMed]
- 128.Levy DE, Caronna JJ, Singer BH, Lapinski RH, Frydman H, Plum F. Predicting Outcome From Hypoxic-lschemic Coma. JAMA J Am Med Assoc. 1985;253:1420. doi: 10.1001/jama.1985.03350340072020. [DOI] [PubMed] [Google Scholar]
- 129.Perkins GD, Jacobs IG, Nadkarni VM, Berg RA, Bhanji F, Biarent D, et al. Cardiac arrest and cardiopulmonary resuscitation outcome reports: Update of the Utstein resuscitation registry templates for out-of-hospital cardiac arrest: A statement for healthcare professionals from a task force of the international liaison committee. Circulation. 2015;132:1286–300. doi: 10.1161/CIR.0000000000000144. [DOI] [PubMed] [Google Scholar]
- 130.Sandroni C, D’Arrigo S, Nolan JP. Prognostication after cardiac arrest. Crit Care. 2018;22:1–9. doi: 10.1186/s13054-018-2060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Riker RR, Sawyer ME, Fischman VG, May T, Lord C, Eldridge A, et al. Neurological Pupil Index and Pupillary Light Reflex by Pupillometry Predict Outcome Early After Cardiac Arrest. Neurocrit Care 2019. 10.1007/s12028-019-00717-4. [DOI] [PubMed]
- 132.Oddo M, Friberg H. Neuroprognostication after cardiac arrest in the light of targeted temperature management. Curr Opin Crit Care. 2017;23:244–50. doi: 10.1097/MCC.0000000000000406. [DOI] [PubMed] [Google Scholar]
- 133.Torbey MT, Selim M, Knorr J, Bigelow C, Recht L. Quantitative analysis of the loss of distinction between gray and white matter in comatose patients after cardiac arrest. Stroke. 2000;31:2163–7. doi: 10.1161/01.STR.31.9.2163. [DOI] [PubMed] [Google Scholar]
- 134.Yanagawa Y, Un-No Y, Sakamoto T, Okada Y. Cerebral density on CT immediately after a successful resuscitation of cardiopulmonary arrest correlates with outcome. Resuscitation. 2005;64:97–101. doi: 10.1016/j.resuscitation.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 135.Choi SP, Park HK, Park KN, Kim YM, Ahn KJ, Choi KH, et al. The density ratio of grey to white matter on computed tomography as an early predictor of vegetative state or death after cardiac arrest. Emerg Med J. 2008;25:666–9. doi: 10.1136/emj.2007.053306. [DOI] [PubMed] [Google Scholar]
- 136.Metter RB, Rittenberger JC, Guyette FX, Callaway CW. Association between a quantitative CT scan measure of brain edema and outcome after cardiac arrest. Resuscitation. 2011;82:1180–5. doi: 10.1016/j.resuscitation.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wu O, Batista LM, Lima FO, Vangel MG, Furie KL, Greer DM. Predicting clinical outcome in comatose cardiac arrest patients using early noncontrast computed tomography. Stroke. 2011;42:985–92. doi: 10.1161/STROKEAHA.110.594879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hun D. Combining brain computed tomography and serum neuron specific enolase improves the prognostic performance compared to either alone in comatose cardiac arrest survivors treated with therapeutic hypothermia. Resuscitation. 2013;84:1387–92. doi: 10.1016/j.resuscitation.2013.05.026. [DOI] [PubMed] [Google Scholar]
- 139.Silva S, Peran P, Kerhuel L, Malagurski B, Chauveau N, Bataille B, et al. Brain gray matter MRI morphometry for neuroprognostication after cardiac arrest. Crit Care Med. 2017;45:e763–71. doi: 10.1097/CCM.0000000000002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Greer D, Scripko P, Bartscher J, Sims J, Camargo E, Singhal A, et al. Clinical MRI interpretation for outcome prediction in cardiac arrest. Neurocrit Care. 2012;17:240–4. doi: 10.1007/s12028-012-9716-y. [DOI] [PubMed] [Google Scholar]
- 141.Hirsch KG, Mlynash M, Eyngorn I, Pirsaheli R, Okada A, Komshian S, et al. Multi-Center Study of Diffusion-Weighted Imaging in Coma After Cardiac Arrest. Neurocrit Care. 2016;24:82–9. doi: 10.1007/s12028-015-0179-9. [DOI] [PubMed] [Google Scholar]
- 142.Hirsch KG, Mlynash M, Jansen S, Persoon S, Eyngorn I, Krasnokutsky MV, et al. Prognostic Value of A Qualitative Brain MRI Scoring System After Cardiac Arrest. J Neuroimaging. 2015;25:430–7. doi: 10.1111/jon.12143. [DOI] [PubMed] [Google Scholar]
- 143.Oren NC, Chang E, Yang CWY, Lee SK. Brain Diffusion Imaging Findings May Predict Clinical Outcome after Cardiac Arrest. J Neuroimaging. 2019;29:540–7. doi: 10.1111/jon.12626. [DOI] [PubMed] [Google Scholar]
- 144.Jang J, Oh SH, Nam Y, Lee K, Choi HS, Jung SL, et al. Prognostic value of phase information of 2D T2*-weighted gradient echo brain imaging in cardiac arrest survivors: A preliminary study. Resuscitation. 2019;140:142–9. doi: 10.1016/j.resuscitation.2019.05.026. [DOI] [PubMed] [Google Scholar]
- 145.Parra-Morales AM, Rudas J, Vargas JA, Gómez F, Enciso-Olivera CO, Trujillo-Rodriguez D, et al. Structural and functional connectivity of ascending reticular activating system in a patient with impaired consciousness after a cardiac arrest: A case report. Medicine (Baltimore) 2019;98:e15620. doi: 10.1097/MD.0000000000015620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jeon CH, Park JS, Lee JH, Kim H, Kim SMSC, Park KH, et al. Comparison of brain computed tomography and diffusion-weighted magnetic resonance imaging to predict early neurologic outcome before target temperature management comatose cardiac arrest survivors. Resuscitation. 2017;118:21–6. doi: 10.1016/j.resuscitation.2017.06.021. [DOI] [PubMed] [Google Scholar]
- 147.Mörtberg E, Cumming P, Wiklund L, Rubertsson S. Cerebral metabolic rate of oxygen (CMRO2) in pig brain determined by PET after resuscitation from cardiac arrest. Resuscitation. 2009;80:701–6. doi: 10.1016/j.resuscitation.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 148.Synek VM. Value of a Revised EEG Coma Scale for Prognosis after Cerebral Anoxia and Diffuse Head Injury. Clin EEG Neurosci. 1990;21:25–30. doi: 10.1177/155005949002100111. [DOI] [PubMed] [Google Scholar]
- 149.Backman S, Cronberg T, Friberg H, Ullén S, Horn J, Kjaergaard J, et al. Highly malignant routine EEG predicts poor prognosis after cardiac arrest in the Target Temperature Management trial. Resuscitation. 2018;131:24–8. doi: 10.1016/j.resuscitation.2018.07.024. [DOI] [PubMed] [Google Scholar]
- 150.Rossetti AO, Oddo M, Logroscino G, Kaplan PW. Prognostication after cardiac arrest and hypothermia: A prospective study. Ann Neurol. 2010;67:301–7. doi: 10.1002/ana.21984. [DOI] [PubMed] [Google Scholar]
- 151.Burjek NE, Wagner CE, Hollenbeck RD, Wang L, Yu C, McPherson JA, et al. Early Bispectral Index and Sedation Requirements During Therapeutic Hypothermia Predict Neurologic Recovery Following Cardiac Arrest*. Crit Care Med. 2014;42:1204–12. doi: 10.1097/CCM.0000000000000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Park JH, Oh JH, Choi SP, Wee JH. Neurologic outcome after out-of-hospital cardiac arrest could be predicted with the help of bispectral-index during early targeted temperature management. Scand J Trauma Resusc Emerg Med 2018;26. 10.1186/s13049-018-0529-7. [DOI] [PMC free article] [PubMed]
- 153.Hoedemaekers CWE, Abdo WF. Bispectral index for prognostication after cardiac arrest: The holy grail at last? Crit Care Med. 2014;42:1312–3. doi: 10.1097/CCM.0000000000000187. [DOI] [PubMed] [Google Scholar]
- 154.Gul SS, Huesgen KW, Wang KK, Mark K, Tyndall JA. Prognostic utility of neuroinjury biomarkers in post out-of-hospital cardiac arrest (OHCA) patient management. Med Hypotheses. 2017;105:34–47. doi: 10.1016/j.mehy.2017.06.016. [DOI] [PubMed] [Google Scholar]
- 155.Stammet P, Collignon O, Hassager C, Wise MP, Hovdenes J, Åneman A, et al. Neuron-specific enolase as a predictor of death or poor neurological outcome after out-of-hospital cardiac arrest and targeted temperature management at 33°C and 36°C. J Am Coll Cardiol. 2015;65:2104–14. doi: 10.1016/j.jacc.2015.03.538. [DOI] [PubMed] [Google Scholar]
- 156.Stammet P, Dankiewicz J, Nielsen N, Fays F, Collignon O, Hassager C, et al. Protein S100 as outcome predictor after out-of-hospital cardiac arrest and targeted temperature management at 33 °C and 36 °C. Crit Care 2017;21. 10.1186/s13054-017-1729-7. [DOI] [PMC free article] [PubMed]
- 157.Duez CHV, Grejs AM, Jeppesen AN, Schrøder AD, Søreide E, Nielsen JF, et al. Neuron-specific enolase and S-100b in prolonged targeted temperature management after cardiac arrest: A randomised study. Resuscitation. 2018;122:79–86. doi: 10.1016/j.resuscitation.2017.11.052. [DOI] [PubMed] [Google Scholar]
- 158.Wiberg S, Hassager C, Stammet P, Winther-Jensen M, Thomsen JH, Erlinge D, et al. Single versus serial measurements of neuron-specific enolase and prediction of poor neurological outcome in persistently unconscious patients after out-of-hospital cardiac arrest - A TTM-trial substudy. PLoS One 2017;12. 10.1371/journal.pone.0168894. [DOI] [PMC free article] [PubMed]
- 159.Jang JH, Bin PW, Lim YS, Choi JY, Cho JS, Woo JH, et al. Combination of S100B and procalcitonin improves prognostic performance compared to either alone in patients with cardiac arrest: A prospective observational study. Medicine (Baltimore) 2019;98:e14496. doi: 10.1097/MD.0000000000014496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sandroni C, Cariou A, Cavallaro F, Cronberg T, Friberg H, Hoedemaekers C, et al. Prognostication in comatose survivors of cardiac arrest: An advisory statement from the European Resuscitation Council and the European Society of Intensive Care Medicine. Intensive Care Med. 2014;40:1816–31. doi: 10.1007/s00134-014-3470-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Boileau A, Somoza AS, Dankiewicz J, Stammet P, Gilje P, Erlinge D, et al. Circulating Levels of miR-574-5p Are Associated with Neurological Outcome after Cardiac Arrest in Women: A Target Temperature Management (TTM) Trial Substudy. Dis Markers. 2019;2019:1–10. doi: 10.1155/2019/1802879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.O’Sullivan TD, Cerussi AE, Cuccia DJ, Tromberg BJ. Diffuse optical imaging using spatially and temporally modulated light. J Biomed Opt. 2012;17:0713111. doi: 10.1117/1.jbo.17.7.071311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Stankovic MR, Maulik D, Rosenfeld W, Stubblefield PG, Kofinas AD, Drexler S, et al. Real-time optical imaging of experimental brain ischemia and hemorrhage in neonatal piglets. J Perinat Med. 1999;27:279–86. doi: 10.1515/JPM.1999.039. [DOI] [PubMed] [Google Scholar]
- 164.Chang YS, Park WS, Lee M, Kim KS, Shin SM, Choi JH. Near infrared spectroscopic monitoring of secondary cerebral energy failure after transient global hypoxia-ischemia in the newborn piglet. Neurol Res. 1999;21:216–24. doi: 10.1080/01616412.1999.11740921. [DOI] [PubMed] [Google Scholar]
- 165.Sinha N, Parnia S. Monitoring the Brain After Cardiac Arrest: a New Era. Curr Neurol Neurosci Rep 2017;17. 10.1007/s11910-017-0770-x. [DOI] [PubMed]
- 166.Storm C, Leithner C, Krannich A, Wutzler A, Ploner CJ, Trenkmann L, et al. Regional cerebral oxygen saturation after cardiac arrest in 60 patients-A prospective outcome study. Resuscitation. 2014;85:1037–41. doi: 10.1016/j.resuscitation.2014.04.021. [DOI] [PubMed] [Google Scholar]
- 167.Genbrugge C, Dens J, Meex I, Boer W, Eertmans W, Sabbe M, et al. Regional Cerebral Oximetry during Cardiopulmonary Resuscitation: Useful or Useless? J Emerg Med. 2016;50:198–207. doi: 10.1016/j.jemermed.2015.03.043. [DOI] [PubMed] [Google Scholar]
- 168.Cournoyer A, Iseppon M, Chauny JM, Denault A, Cossette S, Notebaert É. Near-infrared Spectroscopy Monitoring During Cardiac Arrest: A Systematic Review and Meta-analysis. Acad Emerg Med. 2016;23:851–62. doi: 10.1111/acem.12980. [DOI] [PubMed] [Google Scholar]
- 169.Sanfilippo F, Serena G, Corredor C, Benedetto U, Maybauer MO, Al-Subaie N, et al. Cerebral oximetry and return of spontaneous circulation after cardiac arrest: A systematic review and meta-analysis. Resuscitation. 2015;94:67–72. doi: 10.1016/j.resuscitation.2015.06.023. [DOI] [PubMed] [Google Scholar]
- 170.Wang Q, Miao P, Modi HR, Garikapati S, Koehler RC, Thakor N V. Therapeutic hypothermia promotes cerebral blood flow recovery and brain homeostasis after resuscitation from cardiac arrest in a rat model. J Cereb Blood Flow Metab 2018. 10.1177/0271678X18773702. [DOI] [PMC free article] [PubMed]
- 171.Lee JK, Brady KM, Mytar JO, Kibler KK, Carter EL, Hirsch KG, et al. Cerebral blood flow and cerebrovascular autoregulation in a swine model of pediatric cardiac arrest and hypothermia*. Crit Care Med. 2011;39:2337–45. doi: 10.1097/CCM.0b013e318223b910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Elwell C, Leung T. Oxygen Transport to Tissue XXXVII. Elwell CE, Leung TS, Harrison DK, editors. Advances in Experimental Medicine and Biology. New York, NY: Springer New York. V–Vi. (Advances in Experimental Medicine and Biology; vol. 876). Chapter 19: Detection of ROSC in Patients with Cardiac Arrest During Chest Compression Using NIRS: A Pilot Study 2016;p.151-157. 10.1007/978-1-4939-3023-4. [DOI] [PubMed]
- 173.Peters J, Van Wageningen B, Hoogerwerf N, Tan E. Near-Infrared Spectroscopy: A Promising Prehospital Tool for Management of Traumatic Brain Injury. Prehosp Disaster Med. 2017;32:414–8. doi: 10.1017/S1049023X17006367. [DOI] [PubMed] [Google Scholar]
- 174.Allen BS, Ko Y, Buckberg GD, Sakhai S, Tan Z. Studies of isolated global brain ischaemia: I. A new large animal model of global brain ischaemia and its baseline perfusion studies. Eur J Cardio-Thoracic Surg. 2012;41:1138–46. doi: 10.1093/ejcts/ezr316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ferradal SL, Yuki K, Vyas R, Ha CG, Yi F, Stopp C, et al. Non-invasive assessment of cerebral blood flow and oxygen metabolism in neonates during hypothermic cardiopulmonary bypass: Feasibility and clinical implications. Sci Rep. 2017;7:1–9. doi: 10.1038/srep44117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dehaes M, Aggarwal A, Lin PY, Rosa Fortuno C, Fenoglio A, Roche-Labarbe N, et al. Cerebral oxygen metabolism in neonatal hypoxic ischemic encephalopathy during and after therapeutic hypothermia. J Cereb Blood Flow Metab. 2014;34:87–94. doi: 10.1038/jcbfm.2013.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Pham P, Bindra J, Chuan A, Jaeger M, Aneman A. Are changes in cerebrovascular autoregulation following cardiac arrest associated with neurological outcome? Results of a pilot study. Resuscitation. 2015;96:192–8. doi: 10.1016/j.resuscitation.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 178.Crouzet C, Wilson RH, Lee D, Bazrafkan A, Tromberg BJ, Akbari Y, et al. Dissociation of Cerebral Blood Flow and Femoral Artery Blood Pressure Pulsatility After Cardiac Arrest and Resuscitation in a Rodent Model: Implications for Neurological Recovery. J Am Heart Assoc. 2020;9:e012691. doi: 10.1161/JAHA.119.012691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Shang Y, Chen L, Toborek M, Yu G. Diffuse optical monitoring of repeated cerebral ischemia in mice. Opt Express. 2011;19:20301. doi: 10.1364/oe.19.020301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Farkas E, Bari F, Obrenovitch TP. Multi-modal imaging of anoxic depolarization and hemodynmic changes induced by cardiac arrest in the rat cerebral cortex. Neuroimage. 2010;51:734–42. doi: 10.1016/j.neuroimage.2010.02.055. [DOI] [PubMed] [Google Scholar]
- 181.Bere Z, Obrenovitch TP, Bari F, Farkas E. Ischemia-induced depolarizations and associated hemodynamic responses in incomplete global forebrain ischemia in rats. Neuroscience. 2014;260:217–26. doi: 10.1016/j.neuroscience.2013.12.032. [DOI] [PubMed] [Google Scholar]
- 182.Chalak LF, Zhang R. New Wavelet Neurovascular Bundle for Bedside Evaluation of Cerebral Autoregulation and Neurovascular Coupling in Newborns with Hypoxic-Ischemic Encephalopathy. Dev Neurosci. 2017;39:89–96. doi: 10.1159/000457833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sussman CB, Rossignol C, Zhang Q, Jiang H, Zheng T, Steindler D, et al. Photoacoustic tomography can detect cerebral hemodynamic alterations in a neonatal rodent model of hypoxia-ischemia. Acta Neurobiol Exp (Wars) 2012;72:253–63. doi: 10.55782/ane-2012-1898. [DOI] [PubMed] [Google Scholar]
- 184.Konecky SD, Wilson RH, Hagen N, Mazhar A, Tkaczyk TS, Frostig RD, et al. Hyperspectral optical tomography of intrinsic signals in the rat cortex. Neurophotonics. 2015;2:045003. doi: 10.1117/1.nph.2.4.045003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Rummel C, Basciani R, Nirkko A, Schroth G, Stucki M, Reineke D, et al. Spatially extended versus frontal cerebral near-infrared spectroscopy during cardiac surgery: a case series identifying potential advantages. J Biomed Opt. 2018;23:1. doi: 10.1117/1.jbo.23.1.016012. [DOI] [PubMed] [Google Scholar]
- 186.Kawauchi S, Sato S, Ooigawa H, Nawashiro H, Ishihara M, Kikuchi M. Light scattering change precedes loss of cerebral adenosine triphosphate in a rat global ischemic brain model. Neurosci Lett. 2009;459:152–6. doi: 10.1016/j.neulet.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 187.Wilson RH, Crouzet C, Torabzadeh M, Bazrafkan A. High-speed spatial frequency domain imaging of rat cortex detects dynamic optical and physiological properties following cardiac arrest and resuscitation. Neurophotonics. 2017;4:1. doi: 10.1117/1.nph.4.4.045008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wilson RH, Crouzet C, Lee DE, Donga DP, Patel AH, Bazrafkan A, et al. Spreading depolarization and repolarization during cardiac arrest as an ultra-early marker of neurological recovery in a preclinical model. BioRxiv [Preprint] 2019. 10.1101/786210.
- 189.Crouzet C, Wilson RH, Bazrafkan A, Farahabadi MH, Lee D, Alcocer J, et al. Cerebral blood flow is decoupled from blood pressure and linked to EEG bursting after resuscitation from cardiac arrest. Biomed Opt Express. 2016;7:4660. doi: 10.1364/boe.7.004660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wilson RH, Crouzet C, Torabzadeh M, Bazrafkan AK, Maki N, Alocer J, et al. Cerebral perfusion and metabolism coupling during a critical time window provides rapid assessment of cardiac arrest severity and prognosis in a preclinical model. BioRxiv [Preprint] 2019. 10.1101/785972.
- 191.Wilson RH, Crouzet C, Torabzadeh M, Bazrafkan A, Maki N, Tromberg BJ, et al. High-speed quantitative optical imaging of absolute metabolism in the rat cortex. BioRxiv [Preprint] 2019. 10.1101/786244. [DOI] [PMC free article] [PubMed]
- 192.Leao AAP. SPREADING DEPRESSION OF ACTIVITY IN THE CEREBRAL CORTEX. J Neurophysiol. 1944;7:359–90. doi: 10.1152/jn.1944.7.6.359. [DOI] [PubMed] [Google Scholar]
- 193.Leão AAP. The slow voltage variation of cortical spreading depression of activity. Electroencephalogr Clin Neurophysiol. 1951;3:315–21. doi: 10.1016/0013-4694(51)90079-X. [DOI] [PubMed] [Google Scholar]
- 194.Kramer DR, Fujii T, Ohiorhenuan I, Liu CY. Cortical spreading depolarization: Pathophysiology, implications, and future directions. J Clin Neurosci. 2016;24:22–7. doi: 10.1016/j.jocn.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 195.Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med. 2011;17:439–47. doi: 10.1038/nm.2333. [DOI] [PubMed] [Google Scholar]
- 196.van Harreveld A, Ochs S. Cerebral impedance changes after circulatory arrest. Am J Physiol. 1956;187:180–92. doi: 10.1152/ajplegacy.1956.187.1.180. [DOI] [PubMed] [Google Scholar]
- 197.Hartings JA, Shuttleworth CW, Kirov SA, Ayata C, Hinzman JM, Foreman B, et al. The continuum of spreading depolarizations in acute cortical lesion development: Examining Leão’s legacy. J Cereb Blood Flow Metab. 2017;37:1571–94. doi: 10.1177/0271678X16654495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Dreier JP, Lemale CL, Kola V, Friedman A, Schoknecht K. Spreading depolarization is not an epiphenomenon but the principal mechanism of the cytotoxic edema in various gray matter structures of the brain during stroke. Neuropharmacology. 2018;134:189–207. doi: 10.1016/j.neuropharm.2017.09.027. [DOI] [PubMed] [Google Scholar]
- 199.Chung DY, Oka F, Ayata C. Spreading depolarizations: A therapeutic target against delayed cerebral ischemia after subarachnoid hemorrhage. J Clin Neurophysiol. 2016;33:196–202. doi: 10.1097/WNP.0000000000000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Shuttleworth CW, Andrew RD, Akbari Y, Ayata C, Balu R, Brennan KC, et al. Which Spreading Depolarizations Are Deleterious To Brain Tissue? Neurocrit Care 2019. 10.1007/s12028-019-00776-7. [DOI] [PMC free article] [PubMed]
- 201.Bernard SASA, Gray TWTW, Buist MDMD, Jones BM, Silvester W, Gutteridge G, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;11:82–3. doi: 10.1016/s1062-1458(02)00737-7. [DOI] [PubMed] [Google Scholar]
- 202.Peberdy MA, Callaway CW, Neumar RW, Geocadin RG, Zimmerman JL, Donnino M, et al. Part 9: Post-cardiac arrest care: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122:768–86. doi: 10.1161/CIRCULATIONAHA.110.971002. [DOI] [PubMed] [Google Scholar]
- 203.Hypothermia after Cardiac Arrest Study Group Mild Therapeutic Hypothermia to Improve the Neurologic Outcome after Cardac Arrest. N Engl J Med. 2002;346:549–56. doi: 10.1056/NEJMoa012689. [DOI] [PubMed] [Google Scholar]
- 204.Zeiner A, Holzer M, Sterz F, Schörkhuber W, Eisenburger P, Havel C, et al. Hyperthermia after cardiac arrest is associated with an unfavorable neurologic outcome. Arch Intern Med. 2001;161:2007–12. doi: 10.1001/archinte.161.16.2007. [DOI] [PubMed] [Google Scholar]
- 205.Bro-Jeppesen J, Hassager C, Wanscher M, Søholm H, Thomsen JH, Lippert FK, et al. Post-hypothermia fever is associated with increased mortality after out-of-hospital cardiac arrest. Resuscitation. 2013;84:1734–40. doi: 10.1016/j.resuscitation.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 206.Leary M, Grossestreuer AV, Iannacone S, Gonzalez M, Shofer FS, Povey C, et al. Pyrexia and neurologic outcomes after therapeutic hypothermia for cardiac arrest. Resuscitation. 2013;84:1056–61. doi: 10.1016/j.resuscitation.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 207.Nielsen N, Wetterslev J, Cronberg T, Erlinge D, Gasche Y, Hassager C, et al. Targeted temperature management at 33°c versus 36°c after cardiac arrest. N Engl J Med. 2013;369:2197–206. doi: 10.1056/NEJMoa1310519. [DOI] [PubMed] [Google Scholar]
- 208.Nuzzo A, Peron N, Voicu S, Mégarbane B, Deye N. Targeted temperature management for non-shockable cardiac arrests: The debate must go on. J Thorac Dis. 2018;10:1304–7. doi: 10.21037/jtd.2018.03.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Sung G, Bosson N, Kaji AH, Eckstein M, Shavelle D, French WJ, et al. Therapeutic Hypothermia After Resuscitation From a Non-Shockable Rhythm Improves Outcomes in a Regionalized System of Cardiac Arrest Care. Neurocrit Care. 2016;24:90–6. doi: 10.1007/s12028-015-0184-z. [DOI] [PubMed] [Google Scholar]
- 210.Lascarrou JB, Merdji H, Le Gouge A, Colin G, Grillet G, Girardie P, et al. Targeted temperature management for cardiac arrest with nonshockable rhythm. N Engl J Med. 2019;381:2327–37. doi: 10.1056/NEJMoa1906661. [DOI] [PubMed] [Google Scholar]
- 211.Frydland M, Kjaergaard J, Erlinge D, Wanscher M, Nielsen N, Pellis T, et al. Target temperature management of 33°C and 36°C in patients with out-of-hospital cardiac arrest with initial non-shockable rhythm - A TTM sub-study. Resuscitation. 2015;89:142–8. doi: 10.1016/j.resuscitation.2014.12.033. [DOI] [PubMed] [Google Scholar]
- 212.Thomsen JH, Nielsen N, Hassager C, Wanscher M, Pehrson S, Køber L, et al. Bradycardia during Targeted Temperature Management: An Early Marker of Lower Mortality and Favorable Neurologic Outcome in Comatose Out-of-Hospital Cardiac Arrest Patients∗. Crit Care Med. 2016;44:308–18. doi: 10.1097/CCM.0000000000001390. [DOI] [PubMed] [Google Scholar]
- 213.Kirkegaard H, Taccone FS, Skrifvars MB, Søreide E. Prolonged targeted temperature management in patients suffering from out-of-hospital cardiac arrest. J Thorac Dis. 2018;10:E752–4. doi: 10.21037/jtd.2018.09.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kirkegaard H, Søreide E, De Haas I, Pettilä V, Taccone FS, Arus U, et al. Targeted temperature management for 48 vs 24 hours and neurologic outcome after out-of-hospital cardiac arrest: A randomized clinical trial. JAMA - J Am Med Assoc. 2017;318:341–50. doi: 10.1001/jama.2017.8978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Cho E, Lee SE, Park E, Kim HH, Lee JS, Choi S, et al. Pilot study on a rewarming rate of 0.15°C/hr versus 0.25°C/hr and outcomes in post cardiac arrest patients. Clin Exp Emerg Med. 2019;6:25–30. doi: 10.15441/ceem.17.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Cocchi MN, Boone MD, Giberson B, Giberson T, Farrell E, Salciccioli JD, et al. NIH Public Access. 2014;29:365–9. doi: 10.1177/0885066613491932.Fever. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Calabró L, Bougouin W, Cariou A, De Fazio C, Skrifvars M, Soreide E, et al. Effect of different methods of cooling for targeted temperature management on outcome after cardiac arrest: a systematic review and meta-analysis. Crit Care 2019;23. 10.1186/s13054-019-2567-6. [DOI] [PMC free article] [PubMed]
- 218.De Fazio C, Skrifvars MB, Søreide E, Creteur J, Grejs AM, Kjærgaard J, et al. Intravascular versus surface cooling for targeted temperature management after out-of-hospital cardiac arrest: An analysis of the TTH48 trial. Crit Care. 2019;23:1–9. doi: 10.1186/s13054-019-2335-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Goury A, Poirson F, Chaput U, Voicu S, Garçon P, Beeken T, et al. Targeted temperature management using the “Esophageal Cooling Device” after cardiac arrest (the COOL study): A feasibility and safety study. Resuscitation. 2017;121:54–61. doi: 10.1016/j.resuscitation.2017.09.021. [DOI] [PubMed] [Google Scholar]
- 220.Nordberg P, Taccone FS, Truhlar A, Forsberg S, Hollenberg J, Jonsson M, et al. Effect of Trans-Nasal Evaporative Intra-arrest Cooling on Functional Neurologic Outcome in Out-of-Hospital Cardiac Arrest: The PRINCESS Randomized Clinical Trial. Jama. 2019;321:1677–85. doi: 10.1001/jama.2019.4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Cronberg T, Lilja G, Horn J, Kjaergaard J, Wise MP, Pellis T, et al. Neurologic function and health-related quality of life in patients following targeted temperature management at 33°C vs 36°C after out-of-hospital cardiac arrest a randomized clinical trial. JAMA Neurol. 2015;72:634–41. doi: 10.1001/jamaneurol.2015.0169. [DOI] [PubMed] [Google Scholar]
- 222.Lilja G, Nielsen N, Friberg H, Horn J, Kjaergaard J, Nilsson F, et al. Cognitive Function in survivors of out-of-hospital cardiac arrest after target temperature management at 33°C Versus 36°C. Circulation. 2015;131:1340–9. doi: 10.1161/CIRCULATIONAHA.114.014414. [DOI] [PubMed] [Google Scholar]
- 223.Hu HJ, Song M. Disrupted Ionic Homeostasis in Ischemic Stroke and New Therapeutic Targets. J Stroke Cerebrovasc Dis. 2017;26:2706–19. doi: 10.1016/j.jstrokecerebrovasdis.2017.09.011. [DOI] [PubMed] [Google Scholar]
- 224.King ZA, Sheth KN, Kimberly WT, Simard JM. Profile of intravenous glyburide for the prevention of cerebral edema following large hemispheric infarction: Evidence to date. Drug Des Devel Ther. 2018;12:2539–52. doi: 10.2147/DDDT.S150043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sheth KN, Petersen NH, Cheung K, Elm JJ, Hinson HE, Molyneaux BJ, et al. Long-term outcomes in patients aged ≥70 years with intravenous glyburide from the Phase II GAMES-RP study of large hemispheric infarction an exploratory analysis. Stroke. 2018;49:1457–63. doi: 10.1161/STROKEAHA.117.020365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Kang Y-J, Tian G, Bazrafkan A, Farahabadi MH, Azadian M, Abbasi H, et al. Recovery from coma post-cardiac arrest is dependent on the orexin pathway. J Neurotrauma 2017:neu.2016.4852. 10.1089/neu.2016.4852. [DOI] [PMC free article] [PubMed]
- 227.Modi HR, Wang Q, Sahithi GD, Sherman D, Greenwald E, Savonenko A V., et al. Intranasal post-cardiac arrest treatment with orexin-A facilitates arousal from coma and ameliorates neuroinflammation. PLoS One 2017;12. 10.1371/journal.pone.0182707. [DOI] [PMC free article] [PubMed]
- 228.Shaik JSB, Poloyac SM, Kochanek PM, Alexander H, Tudorascu DL, Clark RSB, et al. 20-Hydroxyeicosatetraenoic Acid Inhibition by HET0016 Offers Neuroprotection, Decreases Edema, and Increases Cortical Cerebral Blood Flow in a Pediatric Asphyxial Cardiac Arrest Model in Rats. J Cereb Blood Flow Metab. 2015;35:1757–63. doi: 10.1038/jcbfm.2015.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Quaegebeur A, Segura I, Schmieder R, Verdegem D, Decimo I, Bifari F, et al. Deletion or inhibition of the oxygen sensor PHD1 protects against ischemic stroke via reprogramming of neuronal metabolism. Cell Metab. 2016;23:280–91. doi: 10.1016/j.cmet.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Vrselja Z, Daniele SG, Silbereis J, Talpo F, Morozov YM, Sousa AMM, et al. Restoration of brain circulation and cellular functions hours post-mortem. Nature. 2019;568:336–43. doi: 10.1038/s41586-019-1099-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Reardon S. Pig brains kept alive outside body for hours after death. Nature. 2019;568:283–4. doi: 10.1038/d41586-019-01216-4. [DOI] [PubMed] [Google Scholar]
- 232.Li W, Yang S. Targeting oxidative stress for the treatment of ischemic stroke: Upstream and downstream therapeutic strategies. Brain Circ. 2016;2:153. doi: 10.4103/2394-8108.195279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. 2018;122:877–902. doi: 10.1161/CIRCRESAHA.117.311401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Rabadi MH, Kristal BS. Effect of vitamin C supplementation on stroke recovery: a case-control study. Clin Interv Aging. 2007;2:147–51. doi: 10.2147/ciia.2007.2.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Firuzi O, Miri R, Tavakkoli M, Saso L. Antioxidant Therapy: Current Status and Future Prospects. Curr Med Chem. 2012;18:3871–88. doi: 10.2174/092986711803414368. [DOI] [PubMed] [Google Scholar]
- 236.Nomani F, Kamran KA. Citicoline in the treatment of acute ischaemic stroke: An international, randomized, multicentre, placebo-controlled study (ICTUS trial)Is the use of Citicoline is beneficial for acute ischaemic stroke? J Pak Med Assoc. 2013;63:1445. [PubMed] [Google Scholar]
- 237.Diener HC, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM, et al. NXY-059 for the treatment of acute stroke: Pooled analysis of the SAINT I and II trials. Stroke. 2008;39:1751–8. doi: 10.1161/STROKEAHA.107.503334. [DOI] [PubMed] [Google Scholar]
- 238.Teixeira G, Szyndralewiez C, Molango S, Carnesecchi S, Heitz F, Wiesel P, et al. Therapeutic potential of NADPH oxidase 1/4 inhibitors. Br J Pharmacol. 2017;174:1647–69. doi: 10.1111/bph.13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Vereczki V, Martin E, Rosenthal RE, Hof PR, Hoffman GE, Fiskum G. Normoxic resuscitation after cardiac arrest protects against hippocampal oxidative stress, metabolic dysfunction, and neuronal death. J Cereb Blood Flow Metab. 2006;26:821–35. doi: 10.1038/sj.jcbfm.9600234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Allen BS, Ko Y, Buckberg GD, Tan Z. Studies of isolated global brain ischaemia: III. Influence of pulsatile flow during cerebral perfusion and its link to consistent full neurological recovery with controlled reperfusion following 30 min of global brain ischaemia. Eur J Cardio-Thoracic Surg. 2012;41:1147–54. doi: 10.1093/ejcts/ezr317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Dell’Anna AM, Lamanna I, Vincent JL, Taccone FS. How much oxygen in adult cardiac arrest? Crit Care 2014;18. 10.1186/s13054-014-0555-4. [DOI] [PMC free article] [PubMed]
- 242.Jakkula P, Reinikainen M, Hästbacka J, Pettilä V, Loisa P, Karlsson S, et al. Targeting low- or high-normal Carbon dioxide, Oxygen, and Mean arterial pressure After Cardiac Arrest and REsuscitation: Study protocol for a randomized pilot trial. Trials. 2017;18:1–9. doi: 10.1186/s13063-017-2257-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Sekhon MS, Griesdale DE. Individualized perfusion targets in hypoxic ischemic brain injury after cardiac arrest. Crit Care. 2017;21:1–5. doi: 10.1186/s13054-017-1832-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Mickel HS, Vaishnav YN, Kempski O, Lubitz D Von, Weiss JF, Feuerstein G. Breathing 100% oxygen after global brain ischemia in mongolian gerbils results hi increased lipid peroxidation and increased mortality. Stroke 1987;18:426–30. 10.1161/01.STR.18.2.426. [DOI] [PubMed]
- 245.Liu Y, Rosenthal RE, Haywood Y, Miljkovic-Lolic M, Vanderhoek JY, Fiskum G. Normoxic ventilation after cardiac arrest reduces oxidation of brain lipids and improves neurological outcome. Stroke. 1998;29:1679–86. doi: 10.1161/01.STR.29.8.1679. [DOI] [PubMed] [Google Scholar]
- 246.Jakkula P, Reinikainen M, Hästbacka J, Loisa P, Tiainen M, Pettilä V, et al. Targeting two different levels of both arterial carbon dioxide and arterial oxygen after cardiac arrest and resuscitation: a randomised pilot trial. Intensive Care Med. 2018;44:2112–21. doi: 10.1007/s00134-018-5453-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Kim B, Park I, Lee JH, Kim S, Lee MJ, Jo YH. Effect of Electrical Vagus Nerve Stimulation on Cerebral Blood Flow and Neurological Outcome in Asphyxial Cardiac Arrest Model of Rats. Neurocrit Care. 2019;30:572–80. doi: 10.1007/s12028-018-0640-7. [DOI] [PubMed] [Google Scholar]
- 248.Allen BS, Ko Y, Buckberg GD, Tan Z. Studies of isolated global brain ischaemia: II. Controlled reperfusion provides complete neurologic recovery following 30 min of warm ischaemia - the importance of perfusion pressure. Eur J Cardiothorac Surg. 2012;41:1147–54. doi: 10.1093/ejcts/ezr317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Gao C-J, Niu L, Ren P-C, Wang W, Zhu C, Li Y-Q, et al. Hypoxic preconditioning attenuates global cerebral ischemic injury following asphyxial cardiac arrest through regulation of delta opioid receptor system. Neuroscience. 2012;202:352–62. doi: 10.1016/j.neuroscience.2011.11.060. [DOI] [PubMed] [Google Scholar]
- 250.Jensen HA, Loukogeorgakis S, Yannopoulos F, Rimpiläinen E, Petzold A, Tuominen H, et al. Remote Ischemic Preconditioning Protects the Brain Against Injury After Hypothermic Circulatory Arrest. Circulation. 2011;123:714–21. doi: 10.1161/CIRCULATIONAHA.110.986497. [DOI] [PubMed] [Google Scholar]
- 251.Pérez-Pinzón MA, Xu GP, Dietrich WD, Rosenthal M, Sick TJ. Rapid preconditioning protects rats against ischemic neuronal damage after 3 but not 7 days of reperfusion following global cerebral ischemia. J Cereb Blood Flow Metab. 1997;17:175–82. doi: 10.1097/00004647-199702000-00007. [DOI] [PubMed] [Google Scholar]
- 252.Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7:437–48. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- 253.Dharap A, Vemuganti R. Ischemic pre-conditioning alters cerebral microRNAs that are upstream to neuroprotective signaling pathways. J Neurochem. 2010;113:1685–91. doi: 10.1111/j.1471-4159.2010.06735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Correia SC, Carvalho C, Cardoso S, Santos RX, Santos MS, Oliveira CR, et al. Mitochondrial preconditioning: A potential neuroprotective strategy. Front Aging Neurosci. 2010;2:1–13. doi: 10.3389/fnagi.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Constantino LC, Tasca CI, Boeck CR. The role of NMDA receptors in the development of brain resistance through pre- and postconditioning. Aging Dis. 2014;5:430–42. doi: 10.14336/AD.2014.0500430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Zhao H, Sapolsky RM, Steinberg GK. Interrupting reperfusion as a stroke therapy: Ischemic postconditioning reduces infarct size after focal ischemia in rats. J Cereb Blood Flow Metab. 2006;26:1114–21. doi: 10.1038/sj.jcbfm.9600348. [DOI] [PubMed] [Google Scholar]
- 257.Ren C, Yan Z, Wei D, Gao X, Chen X, Zhao H. Limb remote ischemic postconditioning protects against focal ischemia in rats. Brain Res. 2009;1288:88–94. doi: 10.1016/j.brainres.2009.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Meng R, Asmaro K, Meng L, Liu Y, Ma C, Xi C, et al. Upper limb ischemic preconditioning prevents recurrent stroke in intracranial arterial stenosis. Neurology. 2012;79:1853–61. doi: 10.1212/WNL.0b013e318271f76a. [DOI] [PubMed] [Google Scholar]
- 259.Mitchell HM, White DM, Domowicz MS, Kraig RP. Cold pre-conditioning neuroprotection depends on TNF-α and is enhanced by blockade of interleukin-11. J Neurochem. 2011;117:187–96. doi: 10.1111/j.1471-4159.2010.07103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Yang Q, Guo M, Wang X, Zhao Y, Zhao Q, Ding H, et al. Ischemic preconditioning with a ketogenic diet improves brain ischemic tolerance through increased extracellular adenosine levels and hypoxia-inducible factors. Brain Res. 1667;2017:11–8. doi: 10.1016/j.brainres.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 261.Zhang J, Zhang W, Gao X, Zhao Y, Chen D, Xu N, et al. Preconditioning with partial caloric restriction confers long-term protection against grey and white matter injury after transient focal ischemia. J Cereb Blood Flow Metab. 2019;39:1394–409. doi: 10.1177/0271678X18785480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Azadian M, Tian G, Bazrafkan A, Maki N, Rafi M, Desai M, et al. Overnight caloric restriction prior to cardiac arrest and resuscitation leads to improved survival and neurological outcome in a rodent model. BioRxiv 2019. 10.1101/786871. [DOI] [PMC free article] [PubMed]
- 263.Busl KM, Greer DM. Hypoxic-ischemic brain injury: Pathophysiology, neuropathology and mechanisms. NeuroRehabilitation. 2010;26:5–13. doi: 10.3233/NRE-2010-0531. [DOI] [PubMed] [Google Scholar]
- 264.Rossetti AO, Rabinstein AA, Oddo M. Neurological prognostication of outcome in patients in coma after cardiac arrest. Lancet Neurol. 2016;15:597–609. doi: 10.1016/S1474-4422(16)00015-6. [DOI] [PubMed] [Google Scholar]
- 265.Böttiger BW, Schneider A, Popp E. Number needed to treat = six: Therapeutic hypothermia following cardiac arrest - An effective and cheap approach to save lives. Crit Care. 2007;11:4–5. doi: 10.1186/cc6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 173 kb)