

HutC in Pseudomonas is a representative member of the GntR/HutC family of transcriptional regulators, which possess a N-terminal winged helix-turn-helix (wHTH) DNA-binding domain and a C-terminal substrate-binding domain. HutC is generally known to repress expression of histidine utilization (hut) genes through binding to the PhutU promoter with urocanate (the first intermediate of the histidine degradation pathway) as the direct inducer. Here, we first describe the detailed molecular interactions between HutC and its PhutU target site in a plant growth-promoting bacterium, P. fluorescens SBW25, and further show that HutC possesses specific DNA-binding activities with many targets in the SBW25 genome. Subsequent RNA-seq analysis and phenotypic assays revealed an unexpected global regulatory role of HutC for successful bacterial colonization in planta.
KEYWORDS: HutC, Pseudomonas, gene regulation, histidine utilization, plant-microbe interactions
ABSTRACT
HutC is known as a transcriptional repressor specific for histidine utilization (hut) genes in Gram-negative bacteria, including Pseudomonas fluorescens SBW25. However, its precise mode of protein-DNA interactions hasn’t been examined with purified HutC proteins. Here, we performed electrophoretic mobility shift assay (EMSA) and DNase I footprinting using His6-tagged HutC and biotin-labeled probe of the hut promoter (PhutU). Results revealed a complex pattern of HutC oligomerization, and the specific protein-DNA interaction is disrupted by urocanate, a histidine derivative, in a concentration-dependent manner. Next, we searched for putative HutC-binding sites in the SBW25 genome. This led to the identification of 143 candidate targets with a P value less than 10−4. HutC interaction with eight selected candidate sites was subsequently confirmed by EMSA analysis, including the type IV pilus assembly protein PilZ, phospholipase C (PlcC) for phosphatidylcholine hydrolyzation, and key regulators of cellular nitrogen metabolism (NtrBC and GlnE). Finally, an isogenic hutC deletion mutant was subjected to transcriptome sequencing (RNA-seq) analysis and phenotypic characterization. When bacteria were grown on succinate and histidine, hutC deletion caused upregulation of 794 genes and downregulation of 525 genes at a P value of <0.05 with a fold change cutoff of 2.0. The hutC mutant displayed an enhanced spreading motility and pyoverdine production in laboratory media, in addition to the previously reported growth defect on the surfaces of plants. Together, our data indicate that HutC plays global regulatory roles beyond histidine catabolism through low-affinity binding with operator sites located outside the hut locus.
IMPORTANCE HutC in Pseudomonas is a representative member of the GntR/HutC family of transcriptional regulators, which possess a N-terminal winged helix-turn-helix (wHTH) DNA-binding domain and a C-terminal substrate-binding domain. HutC is generally known to repress expression of histidine utilization (hut) genes through binding to the PhutU promoter with urocanate (the first intermediate of the histidine degradation pathway) as the direct inducer. Here, we first describe the detailed molecular interactions between HutC and its PhutU target site in a plant growth-promoting bacterium, P. fluorescens SBW25, and further show that HutC possesses specific DNA-binding activities with many targets in the SBW25 genome. Subsequent RNA-seq analysis and phenotypic assays revealed an unexpected global regulatory role of HutC for successful bacterial colonization in planta.
INTRODUCTION
Bacteria adapt to environmental changes, including nutrient availability, by altering patterns of gene expression predominately mediated by transcriptional factors (1, 2). A typical example is the HutC repressor responsible for the expression of histidine utilization (hut) genes in a concentration-dependent manner (3, 4). hutC was first identified in enteric bacteria in the 1970s, and it represents the first bacterial transcriptional factor gene whose expression is subject to self-regulation (5). However, the full nucleotide sequence of hutC was obtained in 1990 from Pseudomonas putida (6). The Pseudomonas HutC has subsequently become the representative member of the GntR/HutC family of transcriptional regulators, which possess an N-terminal winged helix-turn-helix (wHTH) DNA-binding domain and a C-terminal substrate-binding domain (7). It is known in general that HutC binds to the operator sites of hut promoters, causing repression of the hut genes, including hutC. Repression by HutC is relieved by urocanate, which interacts with the HutC repressor and presumably causes HutC to dissociate from the hut operator sites (3). However, the early protein-DNA interactions were determined using crude protein extracts instead of highly purified HutC proteins (8, 9), and thus the precise mode of HutC action is obscure in these model organisms.
HutC has recently reattracted research interest largely due to its newly discovered functions in determining bacterial virulence (10). Research on the intracellular zoonotic pathogen Brucella abortus showed that HutC directly regulates the expression of virulence genes. These include the adhesin-encoding btaE gene and virB operon encoding a type IV secretion system (11, 12). In the human opportunistic pathogen P. aeruginosa, a linkage between histidine metabolism and bacterial virulence was noted by the fact that transposon insertion into the hut locus caused a marked defect in cytotoxicity potentially through the effects on the type III secretion system (13). Furthermore, hutC was identified in a transposon screening for P. aeruginosa mutants defective in bacterial swarming motility (14). Interestingly, hutC was involved in biofilm formation for Yersinia pseudotuberculosis, a pathogenic bacterium that causes infections in humans and animals (15).
The finding that the local transcriptional regulator HutC plays a significant role in the global control of bacterial pathogenesis was initially surprising. To explain this phenomenon, we hypothesized that urocanate, accumulated in mammalian tissues such as skin, acts as a signaling molecule that elicits bacterial infection via interactions with the HutC regulator (10). Briefly, urocanate is produced by histidase (HutH) from histidine and is subsequently broken down by urocanase (HutU). The two enzymes are normally coexpressed, and urocanate is thus not usually found in natural environments. However, urocanate can potentially accumulate in certain tissues, as a result of tissue-specific inactivation of the urocanase activity. For example, urocanate accumulates in human skin and functions as a natural sunscreen (16). Therefore, HutC (and urocanate) likely represents a new mechanism of host perception, which allows pathogenic bacteria to identify suitable niches and deploy appropriate phenotypes for successful colonization and immune evasion.
The histidine catabolism of plants is currently poorly understood, and hence it remains unknown whether the above-mentioned host perception hypothesis can be extended to plant-associated bacteria such as Pseudomonas fluorescens SBW25. SBW25 is a plant growth-promoting bacterium originally isolated from the phyllosphere of field-grown sugar beet (17). It is also capable of aggressively colonizing other crops, including wheat, maize, and peas (18, 19). When growing on the plant surfaces, SBW25 activates the expression of a suite of genes for nutrient acquisition (17). These include the hut genes for histidine utilization (20, 21). Previous work has genetically established the role of HutC as a transcriptional repressor of hut genes with urocanate, not histidine, as the physiological inducer (22, 23). More specifically, deletion of hutC abolished the histidine-induced expression of all three hut promoters, resulting in constitutive expression of the hut operons. However, in the histidase-defective mutant (ΔhutH) background, histidine was no longer able to induce hut expression, suggesting that intracellular conversion of histidine to urocanate is essential for histidine-induced expression of hut (22).
Here, we report a biochemical and functional characterization of HutC in P. fluorescens SBW25. The study was prompted by an initial finding that hutC was functionally required for bacterial competitive colonization of sugar beet seedlings (22). Our work began with electrophoretic mobility shift assay (EMSA) and DNase I footprinting of the PhutU promoter using purified HutCHis6 protein. The obtained specific HutC-binding sequences were then used in a genome-wide search for candidate genes targeted by HutC. Next, eight of the predicted operator sites were subjected to experimental verification by EMSA, and their HutC-binding affinities were compared with that of the PhutU operator site. Finally, we searched for the transcriptomic and phenotypic changes associated with hutC deletion. Our data indicate that HutC plays important global regulatory roles beyond histidine catabolism and contributes to the successful colonization of P. fluorescens SBW25 in planta.
RESULTS
Determining the molecular interactions between HutC and PhutU promoter.
The DNA-binding properties of HutC were first examined by EMSA using purified HutC protein carrying a hexahistidine (His6) tag at the N terminus. The biotin-labeled probe DNA was 325 bp in length and corresponds to the −190 to +135 region of the PhutU promoter. A representative gel image is shown in Fig. 1B. The free probe produced one band in lane 1. However, with the addition of HutCHis6 at increasing concentrations in lanes 2 to 8, four significant shifted bands were observed (labeled a, b, c, and d from low to high molecular weights). More importantly, along with the increase of HutCHis6, the dominant shifted band was gradually shifted from a to b and then to c and d. Only the largest-shifted band d was present when HutCHis6 was added at the highest concentration of 2.1 μM (lane 8). Together, the EMSA data confirmed the predicted function of HutC binding to the PhutU promoter and further suggested that HutC possesses at least four oligomeric states upon binding with the PhutU promoter DNA in vitro.
FIG 1.
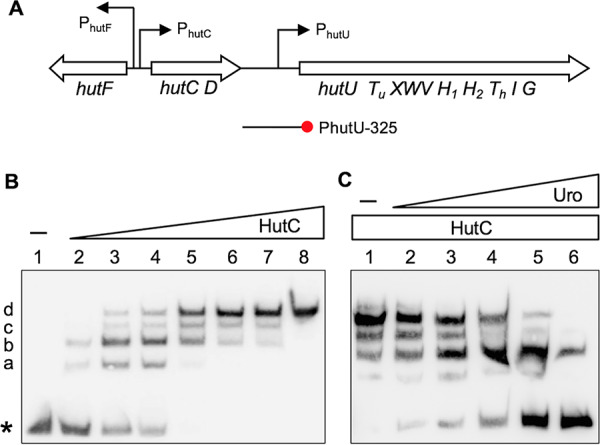
hut gene organization (A) and EMSAs showing specific binding of HutC with the PhutU promoter (B) and the effects of urocanate (C). (A) hut genes are organized in three transcriptional units: hutF, hutCD, and ten genes from hutU to hutG. The location and orientation of hut promoters are indicated by bent arrows. The red circle denotes the biotin-labeled 3′ end of the PhutU-325 probe used in the EMSAs. (B) HutCHis6 was added at increasing concentrations of 0, 35, 70, 140, 210, 315, 525, and 2,100 nM in lanes 1 to 8, respectively. The position of free probes is indicated by an asterisk. (C) EMSA was performed in reaction mixtures containing 280 nM HutCHis6 and 20 nM PhutU-325 probe with urocanate added at final concentrations of 0, 125, 250, 500, 1,000, and 1,500 μM in lanes 1 to 6, respectively.
Next, we sought to determine the effects of urocanate on HutCHis6 interactions with PhutU. EMSA was performed with the same PhutU-325 probe (20 nM), and HutCHis6 was added at a fixed concentration of 280 nM (Fig. 1C). As expected, four retarded protein-DNA complexes were observed in the absence of urocanate (lane 1). However, when urocanate was added at an increasing concentration, there was a significant decrease in the intensities of shifted bands along with an increase in band intensity of the free probe DNA (lanes 2 to 6). Of particular note is that dissociation of the HutCHis6-PhutU complex occurred at a reverse order as observed with the addition of HutCHis6 at increasing concentrations (Fig. 1C). A comparison of the DNA retardation profiles in Fig. 1B and C consistently suggests that the HutC-PhutU complexes b and d are relatively more stable than bands a and c.
Stoichiometric analysis of HutC-PhutU interactions.
The finding that HutC-PhutU can form at least four protein-DNA complexes prompted further investigation into the oligomerization of HutCHis6 in vitro. A cross-linking experiment was first performed with purified HutCHis6 using formaldehyde as the cross-linking reagent. Results shown in Fig. 2A clearly indicate that HutC is capable of forming a dimer, trimer, and tetramer in vitro. Of note, a weak band of HutC dimer was also found in the protein reaction without formaldehyde (lane 1). This was likely due to incomplete denaturation of HutC in SDS-PAGE analysis.
FIG 2.

HutC oligomerization and stoichiometry of HutC-PhutU interactions. (A) Formaldehyde cross-linking was performed with 19 μM HutCHis6 (28.6 kDa), and protein samples in lanes 1 to 4 were treated with 25 mM formaldehyde for 0, 1, 2.5, and 4 h, respectively. (B) EMSA analysis of HutCHis6 and PhutU DNA performed at the final concentration of 0.1 μM in total. The mole fractions of protein and DNA are shown below the gel image for each sample. (C) Job plots of DNA-to-protein ratios for complexes a, b, and d. The lines are the least-square fits to the rising and falling subsets of the data, and their intersection yields the binding stoichiometry of each protein/DNA complex.
Next, continuous variation analysis (or Job plot) was applied to determine the stoichiometry of specific HutCHis6 and PhutU interactions. This time EMSA was performed by keeping the total molar concentration of protein and DNA constant (100 nM) but varying in their relative compositions (Fig. 2B). Intensity of a shifted band was plotted against HutC fraction for each protein/DNA complex (Fig. 2C). The binding stoichiometry of a specific protein-DNA complex was then defined by the protein/DNA ratio when the amount of this complex reaches its maximum (24, 25). Complex a produced a stoichiometry of 1:0.96, suggesting the presence of a HutC monomer binding to the DNA probe. Complex d is most likely formed by a HutC tetramer, as it produced a stoichiometry of 1:3.64. The calculated stoichiometry for complex b was 1:1.48. The stoichiometry of complex c could not be determined due to smearing and weak bands. However, given that complex a was estimated to be a monomer and complex d a tetramer, complexes b and c are most likely the intermediate dimer and trimer, respectively. This conclusion was further supported by stoichiometric analysis using Bading’s method whereby the molecular weights of HutCHis6 proteins were calculated on the basis of their migration distance in the EMSA gel (see Table S1 in the supplemental material) (26). Taken together, the data consistently indicate that HutC possesses four oligomeric states and can form a monomer, dimer, trimer, and tetramer in a sequential manner along with the increase of HutC concentrations.
Analysis of the HutC-binding sequence in the PhutU promoter.
To determine the precise HutC-binding site, DNase I footprinting was performed with purified HutCHis6 and the PhutU-325 probe (Fig. 3A). Results show that a 49-bp region was protected by HutCHis6 from DNase I digestion, and it overlaps with the previously identified σ54-binding site (Fig. 3C). Next, a multiple-sequence alignment was performed with PhutU and PhutF promoters from 40 Pseudomonas strains representing 13 different species. A sequence logo was generated on the basis of the aligned promoter DNA (Fig. 3D). This led to the identification of a perfect inverted repeat sequence TGTA-N2-TACA, which is highly conserved in Pseudomonas and thus potentially crucial for HutC-binding activity.
FIG 3.

Sequence determination of the HutC operator site in the PhutU promoter. (A) DNase I footprinting was performed using purified HutCHis6 and a 325-bp biotin-labeled probe, PhutU-325. Lane M, G+A marker; lanes 1 and 6, no HutCHis6; lanes 2 to 5, HutCHis6 added at 0.68, 2.39, 4.08, and 5.78 μM, respectively. The HutC-protected region and Phut half sites are indicated by bars and inverted arrows, respectively. Dots denote hypersensitive residues for DNase I cleavage. (B) Effects of urocanate on DNase I analysis of HutCHis6 (2.8 μM) and the PhutU-325 probe (2.0 μM). Lane M, G+A marker; lanes 1 and 2, no HutCHis6 and no urocanate; lanes 3 to 8, urocanate added at 0, 0.2, 0.5, 1, 2, and 3 mM, respectively. The strong and weak HutC-protected regions are marked with red and blue brackets, respectively. (C) The HutCHis6-protected region is underlined, and sequences of the strong and weak protected DNA regions are shown in red and blue, respectively. β-Galactosidase activity was measured in two genetic backgrounds (wild type versus ΔhutC) for lacZ fusion to the PhutU promoter and three derived mutant alleles lacking Phut-I and/or Phut-II repeats. “(s)” denotes a significant difference between the wild-type and ΔhutC strains as revealed by the Student t test (P < 0.01). NT, not tested. (D) Sequence logo generated from the alignment of PhutU and PhutF promoters from 40 representative Pseudomonas strains. (E) EMSA was performed using 325-bp biotin-labeled probes containing the wild-type or mutant alleles (Phut-I and/or Phut-II). Lanes 1 to 7, HutCHis6 added at 0, 37, 74, 148, 222, 296, and 555 nM, respectively.
DNase I footprinting was also used to examine the effects of urocanate on HutCHis6-PhutU interaction (Fig. 3B). When urocanate was added at increasing concentrations (lanes 4 to 8), both ends of the HutC-protected DNA region gradually lost protection. The data thus indicate a strong binding of HutC to the center of the HutC-protected DNA. Importantly, the flanking weak protected regions do not contain sequence similar to the above-identified repeats for HutC binding, suggesting that they are not directly involved in the specific protein-DNA interaction. They are protected by HutC likely as a result of protein oligomerization, which occurs when HutC is present at higher concentrations and disassociates in the presence of low concentrations of urocanate.
Mutational analysis of the HutC target site in the PhutU promoter.
The data presented above identified two putative half sites (named Phut-I and Phut-II) (Fig. 3) for specific HutC binding. However, the HutCHis6-protected region is unusually long (49 bp) and potentially contains multiple Phut sites. It is also unclear if the two half sites differ in their abilities for HutC binding and how they cause repression of the promoter. To answer these questions, we constructed three PhutU variants carrying mutations of either Phut-I or Phut-II or both by site-directed mutagenesis. More specifically, the wild-type Phut-I (TGTA) and Phut-II (TACA) sites were substituted with a 4-bp randomly chosen sequence of CCGG and GGCC, respectively. The 325-bp wild-type PhutU DNA fragment and the three variants cloned into a pCR8 vector were used as templates to prepare probes for EMSA analysis. They were also subcloned into vector pUC18-mini-Tn7T-Gm-lacZ to monitor promoter activity in vivo. The lacZ reporter fusions were subsequently introduced into the genetic backgrounds of wild-type SBW25 and its derived ΔhutC mutant (Fig. 3).
Results of EMSA with purified HutCHis6 are shown in Fig. 3E. No retardation was observed for variant probe PhutU-M1&2 lacking both half sites. This excludes the possibility that the 325-bp PhutU promoter region contains any additional HutC-binding sites. One shifted band was observed for variant probes PhutU-M1 and PhutU-M2, suggesting that HutCHis6 was capable of binding to the remaining half site, likely as a monomer. The calculated Kd values for PhutU-M1 and PhutU-M2 were 472.1 nM and 478.1 nM, respectively, suggesting a reduced binding capacity with HutC compared with the wild-type promoter (probe PhutU-325, Kd of 44.6 nM).
Next, β-galactosidase activity was measured for cells grown in the absence of the hut inducer (i.e., histidine and urocanate). Data presented in Fig. 3C show that the wild-type PhutU-lacZ fusion (PhutU-325) was expressed at the basal level in the wild-type background, but expression was elevated in the ΔhutC mutant background. This was consistent with the known function of HutC as a transcriptional repressor of PhutU. However, no repression was detected for promoter activities of the two PhutU variants carrying either the Phut-I or Phut-II site mutation (Fig. 3C). The data thus indicate that simultaneous binding to both half sites (Phut-I and Phut-II) is crucial for HutC-mediated gene repression.
Taking all the data together, we are able to propose an updated model of HutC in regulating the PhutU promoter activities in response to the presence/absence of histidine (or urocanate). The model involves HutC oligomerization and specific interactions between a HutC monomer and its cognate operator site Phut-I or Phut-II. The specific protein-DNA interaction and also the protein-protein interaction can be disrupted by urocanate. As summarized in Fig. S2 in the supplemental material, on the decrease of urocanate, the apo-HutC monomer is capable of binding to either the Phut-I site or the Phut-II site in the PhutU promoter region. However, efficient repression is achieved only when HutC forms a dimer and simultaneously binds to the two half sites. As the urocanate concentration further decreases, HutC eventually forms a tetramer (Fig. 4C), which causes a stronger repression of the PhutU promoter potentially through mechanisms such as convex DNA bending (27). This dynamic process is reversed on the increase of urocanate (Fig. S2).
FIG 4.

The proposed model of HutC interaction with the PhutU promoter DNA. (A) A homology model generated in the SWISS-MODEL server showing the side view of the HutC homodimer. Helices are represented in violet and strands in green. (B) Structural prediction of HutC protein and operator DNA complex. Only the dimeric wHTH DNA-binding domains are shown for clarity, and the conserved Phut-I and Phut-II sites in the Watson strand are highlighted in cyan. (C) Schematic representation of the HutC tetramer bound with the PhutU promoter. This involves direct interactions between the HutC dimer and PhutU with the formation of a HutC tetramer via protein-protein interactions.
The Phut-I and Phut-II elements are located in two adjacent major grooves at opposite sides of the DNA molecule (Fig. 4). To gain further insights into the HutC actions, homology modeling was performed in the SWISS-MODEL server using the crystal structure of DasR from Streptomyces coelicolor (PDB ID 4ZS8) as a template. As shown in Fig. 4B, the recognition helix of each wHTH domain is oriented toward the major groove, while the β-sheet forms the wing domain and inserts in the adjacent minor groove.
In silico identification of putative HutC-binding sites at a genomic scale.
Armed with the knowledge of HutC-mediated regulation of the PhutU promoter, we proceeded to identify potential HutC targets beyond the hut locus in the genome of P. fluorescens SBW25. First, the motif discovery tool MEME was used to generate a position-dependent letter-probability matrix based on the above-mentioned sequence alignment of the PhutU and PhutF promoters. The resulting matrix was inputted into the FIMO program (version 4.11.2) to search for genome-wide occurrence of this binding motif. This analysis led to the identification of 143 motif occurrences with a P value of less than 10−4 (see Table S2 in the supplemental material). Sixty-three out of the 143 sites (44.1%) are located within 200 bp upstream of an open reading frame, which represents 17.6% of the genome, which thus suggests a functional selection for HutC target sites in promoter regions.
A cursory examination of the candidate sites shows that a wide range of biological activities can potentially be influenced by HutC-mediated gene regulation (Table S2). Of particular note are ten genes involved in amino acid metabolism, e.g., hisB and argA for histidine and arginine biosynthesis and nadB and speA for the breakdown of aspartate and arginine, respectively. Five candidate genes have important functions in cellular carbon and energy metabolism: aceE and glcB for pyruvate oxidation; gcl, kdgD, and garP required for glucarate utilization; and treP and treR involved in bacterial growth on trehalose. Three genes (pilZ, pilV, and pflu4885) have predicted roles in bacterial motility, specifically the assembly of type IV pilus. Some candidate genes are involved in the enzymatic breakdown of cell envelope components. These include plcC, encoding an extracellular phospholipase C for phosphatidylcholine (PC) hydrolyzation, and amiA, encoding a peptidoglycan amidase. Intriguingly, the in silico analysis has also identified two key regulators of cellular nitrogen metabolism: glnE and ntrBC. The adenylyltransferase GlnE is involved in modifying the glutamine synthetase (GlnA) activity, whereas NtrBC is a well-studied two-component system coordinating the expression of many genes involved in nitrogen assimilation.
Experimental verification of the candidate HutC target sites.
EMSA analysis was performed with a panel of eight Phut candidates, representing different biological functions of our interest (Table 1). Location of a candidate site in the predicted promoter structure of the related gene(s) was additionally considered (Table S2). In addition to the above-mentioned six loci (ntrBC, glnE, pilZ, hisB, plcC, and amiA), the pflu2467 and tnpS genes were included to represent an AraC-type transcriptional regulator and a transposable element, respectively. First, EMSA and DNase I footprinting were performed with a biotin-labeled probe containing the predicted Phut site in the ntrBC locus (Fig. 5). Consistent with our expectation, a shifted band was observed, and its intensity increased, coupled with decreased free probe DNA, when HutCHis6 was added at increasing concentrations (Fig. 5C). More importantly, DNA retardation was abolished with probe DNA carrying mutations in the putative Phut site (PntrBC-mut2) (Fig. 5C). The DNase I profiles clearly indicate that a 38-bp region was protected by HutCHis6 (Fig. 5A), and the protection was interrupted with the addition of urocanate (Fig. 5B).
TABLE 1.
HutC target sites whose activity was confirmed in vitro in this work
| Site sequenceb | Locationa | Locus tag | Gene product and function |
|---|---|---|---|
| TGTATATACA | ← • → | Pflu0848 | PlcC, phosphatidylcholine-hydrolyzing phospholipase C |
| Pflu0849 | 5-Dehydro-4-deoxyglucarate dehydratase | ||
| TGTATAAACA | ← • → | Pflu2466 | Hypothetical protein |
| Pflu2467 | AraC family transcriptional regulator | ||
| CGTATATACA | ← • → | Pflu0460 | AceE, pyruvate dehydrogenase subunit E1 |
| Pflu0461 | GlnE, bifunctional glutamine synthetase adenylyltransferase/deadenyltransferase | ||
| TGTCCGAACA | ← •← | Pflu0344 | NtrB, sensor kinase for nitrogen metabolism |
| Pflu0345 | Putative lipoprotein | ||
| TGTATGCACA | → • → | Pflu0325 | Transporter in major facilitator family |
| Pflu0327 | HisB, imidazoleglycerol-phosphate dehydratase in histidine biosynthesis | ||
| TGTATGCACA | ←• → | Pflu6087 | AmiA, N-acetylmuramoyl-l-alanine |
| Pflu6088 | Putative GTP cyclohydrolase | ||
| CGTTTGTACA | ← • ← | Pflu4947 | PilZ, type IV pilus assembly protein |
| Pflu4948 | Putative lipoprotein | ||
| TGTATATACG | ← • → | Pflu2987 | TnpT, putative cointegrase resolution protein |
| Pflu2988 | TnpS, cointegrase |
HutC target site (circle) is shown together with genes located immediately upstream and downstream in the genome of P. fluorescens SBW25.
Putative Phut half sites are underlined.
FIG 5.

Characterization of the HutC-binding site in the ntrBC promoter region. (A) DNase I footprinting was performed using the PntrBC-154 probe labeled at the 3′ end. Lane M, G+A marker; lane 6, no HutCHis6; lanes 1 to 5, HutCHis6 added at 0.26, 0.77, 1.8, 3.34 and 5.14 μM, respectively. (B) DNase I analysis showing the effect of urocanate on HutCHis6 binding. Lane 1, no HutCHis6 and no urocanate; lanes 2 to 5, HutCHis6 (5.14 μM) with urocanate added at 0, 0.2, 0.5, and 2.0 mM, respectively. (C) EMSA of HutCHis6 using 3′-end biotin-labeled probe PntrBC-154 or mutant probe PntrBC-mut2 carrying mutations in the Phut motif. Lanes 1 to 4, HutCHis6 added at 0, 0.64, 1.28, and 1.92 μM, respectively. The position of free probe is marked by an asterisk. (D) Sequence of the HutC-protected region as revealed by DNase I footprinting. The two HutC-binding half sites are shown by underlined letters, and the corresponding sequence was mutated in the PntrBC-mut2 probe DNA.
To test the binding affinity with the plcC promoter, a 387-bp biotin-labeled probe was used for EMSA analysis with HutCHis6 added at seven different concentrations (Fig. 6B). Multiple shifted bands were observed, and the calculated Kd was 673.2 ± 38.9 nM, suggested a relatively weaker interaction compared with the PhutU promoter (Kd = 44.6 ± 5.1 nM). Finally, probes for the six other candidate sites were amplified by PCR and then end-labeled by digoxigenin (DIG). Results of subsequent EMSA confirmed the binding of HutCHis6 with target sites at the promoter regions of pflu2467, glnE, hisB, amiA, pilZ, and tnpS (Fig. 6A).
FIG 6.
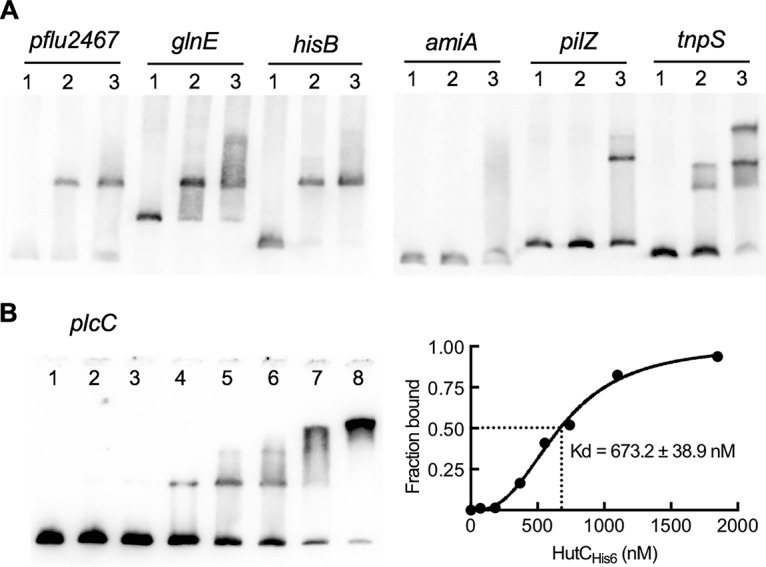
Interactions between HutCHis6 and seven candidate target DNAs. (A) EMSA was performed with DIG-labeled probes containing the Phut site in the promoter region of the gene shown above the gel image. Lanes 1 to 3, HutCHis6 added at 0, 0.73, and 2.2 μM, respectively. (B) EMSA of HutCHis6 with a biotin-labeled probe for the plcC promoter. HutCHis6 was added at 0, 0.074, 0.185, 0.370, 0.555, 0.74, 1.10, and 1.85 μM in lanes 1 to 8, respectively. The strength of binding is calculated as the equilibrium dissociation shown at the right.
hutC deletion analysis revealed regulatory roles beyond histidine catabolism.
To further determine the global regulatory roles of hutC, we performed a transcriptome sequencing (RNA-seq) analysis and searched for phenotypic differences between the wild-type SBW25 and an isogenic ΔhutC mutant. Total RNA was prepared from three biological replicates of bacterial cells exponentially grown on succinate (20 mM) and histidine (10 mM), a laboratory nutrient condition that requires hutC (see below [Fig. 7]). The mRNA fraction was then subjected to RNA-seq analysis. Expression was detected for ∼97% of predicted genes in the SBW25 genome (Fig. 7A). Of the 1,319 genes differentially expressed, 794 genes were expressed at higher levels and 525 at lower levels in the ΔhutC mutant (P < 0.05; fold change cutoff, 2.0).
FIG 7.

Comparative RNA-seq and phenotypic analysis between wild-type SBW25 and the isogenic mutant devoid of hutC. (A) Volcano plot displaying the differentially expressed genes. Green dots represent upregulated genes, whereas red dots indicate downregulated genes in the ΔhutC mutant. (B) Promoter activities of ′lacZ fusions to PntrBC and its variant PntrBC-mut2 in the wild-type background. β-Galactosidase assays were performed for cells grown in the defined medium at 6 h after inoculation. Values are means and standard errors from four replicate cultures. (C) Expression of PplcC-lacZ fusion in wild-type and ΔhutC mutant backgrounds. (D) Agar plates showing the spreading of the wild-type (WT) and ΔhutC strains in LB and minimal salt medium supplemented with succinate (Suc) (20 mM) and histidine (His) (10 mM) or urocanate (Uro) (10 mM). Photos were taken 16 and 36 h after inoculation in 0.3% and 0.5% agar, respectively. (E) Diameters were measured for ten replicate plates in the succinate-plus-histidine medium. (F) Growth dynamics of wild-type SBW25 and the ΔhutC mutant in minimal broth of succinate plus histidine. (G to I) Production of pyoverdine, expressed as relative fluorescence units (RFU), was measured in ten media at 12 (G), 24 (H), and 48 (I) hours after inoculation. Data are means and standard errors from four replicate cultures. Asterisks indicate significant differences (P < 0.05).
The detailed results of RNA-seq are available in Data Set S1 in the supplemental material. For the eight Phut sites described above (Table 1), significant upregulation was detected for plcC, aceE, pflu6087/6088, and pilZ, suggesting a repressor role of HutC in their expression. Of particular note is significant downregulation of genes in the wss operon for cellulose production and the WspD regulator. In contrast, genes involved in the production of flagella, type IV pili, and the viscosin biosurfactant were mostly upregulated. These data thus suggest a potential role of HutC in coordinating bacterial motility and biofilm formation. Forty-six genes with a predicted role in nitrogen metabolism were differentially expressed, including hisI encoding phosphoribosyl-AMP cyclohydrolase for histidine biosynthesis. Four genes encoding the type III secretion system were downregulated, whereas nine genes for the type VI secretion system were upregulated in the ΔhutC mutant background. Notably, the most affected functional group is iron metabolism. As a result of hutC deletion, 24 genes involved in pyoverdine synthesis, uptake, and regulation were upregulated from 5.7- to 262.1-fold. Interestingly, two putative genes encoding the bacterioferritin for iron storage was expressed at lower levels in the ΔhutC mutant.
To catch a glimpse of the HutC global function in vivo, we first compared expression levels of the above-identified plcC gene in the wild-type versus ΔhutC mutant backgrounds. Consistent with our expectation, the plcC-lacZ fusion was expressed at a significantly higher level in the ΔhutC mutant than in wild-type SBW25 (Fig. 7C) when bacteria were grown on succinate and histidine. However, no significant difference was observed in LB and minimal medium with succinate and ammonium. In these two media, the PhutU promoter was expressed at very low or basal levels and subjected to strong repression by HutC (22). Next, we compared the expression of two PntrBC-lacZ fusions carrying wild-type and Phut mutant alleles separately in the same genetic background of wild-type SBW25. The results revealed a slightly increased but significant (P < 0.05) expression of the mutant PntrBC promoter for bacteria grown on succinate and histidine (Fig. 7B). The data thus suggest that HutC may repress PntrBC activities by directly targeting the Phut site in the ntrBC promoter region.
In a previous work, we showed that the ΔhutC mutant displayed a severe defect in competitive bacterial growth on the surfaces of sugar beets. To search for more phenotypes on the basis of the new evidence presented above, we first subjected the ΔhutC mutant to a standard bacterial motility assay in three nutrient agar plates: LB and minimal salt medium supplemented with succinate plus histidine or urocanate (Fig. 7D). No significant difference was observed in LB and the succinate-plus-urocanate medium. However, when grown on succinate plus histidine, the ΔhutC mutant showed an enhanced ability to spread in both 0.3% and 0.5% agar plates (Fig. 7D and E). The ΔhutC mutant grew similarly to the wild type in succinate-plus-histidine broth (Fig. 7F), and the observed effects are thus attributable to bacterial motilities. Next, we compared biofilm formation for bacteria grown in LB and minimal media containing succinate with histidine, urocanate, or ammonium as the sole nitrogen source. Biofilm was quantitively measured in both microtiter plates and glass tubes. The result (summarized in Fig. S4 in the supplemental material) showed that biofilms were pronouncedly produced in succinate-plus-histidine medium, but the differences between the wild type and the ΔhutC mutant were not significant (P < 0.05). Finally, we measured pyoverdine production for bacteria grown in KB broth and nine minimal salt media supplemented with various combinations of succinate, histidine, urocanate, and ammonium. Data from 12, 24, and 48 h after inoculation are shown in Fig. 7. In general, pyoverdine was more pronouncedly produced in histidine-containing media than in those containing urocanate. Significant differences were found in KB and succinate-ammonium media at day 2 and at 12 h for the succinate-histidine media with and without the addition of ammonium (Fig. 7G). Additionally, an enhanced production of pyoverdine, a yellowish pigment, was visible for the ΔhutC mutant in the 0.5% swarming succinate-histidine agar plates (Fig. 7D). Given the fact that the two strains (wild type and ΔhutC) grew similarly in these media (22, 23), the data thus consistently indicate that hutC deletion caused a significant increase of pyoverdine production under certain nutrient conditions.
DISCUSSION
Despite the fact that HutC is a representative member of the GntR/HutC protein family, the molecular interaction between HutC and its target DNAs hasn’t been examined using purified HutC proteins in Pseudomonas. In the present study, we performed EMSA and DNase I analysis of the PhutU promoter DNA using His6-tagged HutC from P. fluorescens SBW25. The data allowed us to propose an updated model of HutC function, which involves complex oligomerization of the HutC repressor when bound with PhutU. Transition of oligomeric status from monomer to tetramer (and vice versa) was determined by varying intracellular concentrations of urocanate. Notably, EMSA produced four DNA retardation bands, and HutC protected an unusually large DNA region (49 bp) from DNase I digestion. These findings were initially against our expectation, as previous work with B. abortus HutC revealed only one shifted band in EMSA and a much shorter protected DNA region (20 bp) in DNase I footprinting (12). The HutC-binding sequences are highly similar between hut promoters in Pseudomonas and Brucella. It is thus interesting to further understand the biological significance and also the underlying mechanisms of the observed HutC oligomerization.
Oligomerization of DNA-binding proteins can occur through mechanisms such as intrinsic protein aggregation and nonspecific binding to DNA surrounding the preceding protein (28). The latter is likely true for the HutC-PhutU interaction in Pseudomonas. The HutC target site contains a highly conserved dyad symmetric sequence, suggesting that HutC can potentially bind to PhutU as a dimer, without involving a higher order of oligomerization. This is further supported by crystal structural analysis that the effector-binding domain (UTRA domain, PF07702) of proteins in the GntR/HutC family can serve as a dimerization domain (29, 30). Significantly, no HutC-binding activity was detected for mutant probe with the elimination of both Phut-I and Phut-II half sites (Fig. 3E). This result strongly suggests that any possible protein/DNA interactions in the flanking weakly protected DNA regions are not sequence specific. Oligomerizations likely occur after two HutC monomers are first bound to their target sites at the center of the HutC-protected region. This also explains why only one shifted band was observed for variant probes lacking either half site (Fig. 3E), most likely because of insufficient HutC proteins in the local PhutU promoter area. A third probable mechanism of repressor oligomerization is the presence of multiple binding sites in the promoter DNA. Clearly, this does not apply to the PhutU promoter, as we showed by mutation analysis that the two Phut-I and Phut-II half sites are crucial for HutC function as a dimer.
In terms of biological significance, oligomerization is often involved in a tighter repression of gene expression. First, oligomerization causes repression of a large DNA region that may contain multiple promoter activities, with DnaA being a well-studied example. Expression of dnaA is autoregulated by targeting an operator site (DnaA box) located between two active promoters. Oligomerization enables DnaA to occlude/block the access of RNA polymerase to both promoters through binding to a single operator site (28). Second, oligomerization is crucial for repressors like LacI in Escherichia coli to achieve a maximal repression. Tetramer assembly by self-association of LacI causes the formation of a looped DNA structure, which can prevent access of the transcriptional activators, thus enhancing repression of the lac operon (31). Third, multiple repressor proteins can lengthen the occupancy of operator DNA, leading to an increased local concentration of the repressor (32, 33). Here, we found that the HutC-protected region overlaps the σ54-binding sequence as a result of HutC oligomerization. This strongly suggests that an enhanced repression of hut genes is necessary for Pseudomonas strains to thrive in their natural habitats, including the plant environments.
The global regulatory role of hutC was first evidenced in an in vivo competitive colonization assay between wild-type SBW25 and an isogenic ΔhutC mutant (22). When growing on the surfaces of sugar beet seedlings, the ΔhutC mutant was defective in competitive colonization, with a measured fitness of −2.28 and −1.9 relative to the wild-type strain in the shoot and rhizosphere, respectively (of note, the relative fitness would be zero in terms of selection rate constant if the two strains were equally fit) (34). This result was initially surprising as plant surfaces do contain histidine, albeit at a low concentration of ∼4 μM (20). Even in the absence of histidine the cost of fitness for constitutive hut expression was very low, and only a slight decrease of fitness was observed for the ΔhutC mutant (−0.71). A large fitness reduction can thus be explained only by the as-yet-unknown functions of HutC beyond a local hut repressor.
Data presented here provide further support that HutC plays a global regulatory role in determining the ecological success of P. fluorescens SBW25 in the plant environments. RNA-seq analysis identified 1,319 differentially expressed genes in the ΔhutC mutant. Interestingly, these include 37 plant-inducible genes revealed in previous work using a promoter-trapping technique known as in vivo expression technology (IVET) (17, 21). These genes are located immediately downstream of the detected plant-inducible promoters, and their expression was mostly downregulated in the ΔhutC mutant. Furthermore, our phenotypic analysis essentially confirmed two functional traits with established roles in bacterial colonization in planta, i.e., bacterial motility and the production of iron-chelating siderophores.
Finally, Phut sites in the ntrBC and plcC operons have been selected for further functional analysis in vivo. plcC (pflu0848) encodes a putative phospholipase for releasing choline from phosphatidylcholine (PC) (35). The reverse reaction is catalyzed by phosphatidylcholine synthase encoded by pcs (pflu2780). It is interesting to note that plcC and pcs are copresent in the genome of P. fluorescens SBW25. PCs are major components of cellular membranes of plant (and animal) cells. plcC can thus confer on Pseudomonas the ability to utilize plant-derived PCs as a valuable source of nutrients. However, it is puzzling to understand why pcs is required, as PCs are not a structural component of bacterial cells. Nevertheless, both plcC and pcs are commonly found in Pseudomonas strains that form intimate associations with eukaryotic hosts, and thus they likely play important roles in bacterium-host interaction. Indeed, a previous mutational analysis indicated that plcC is functionally involved in biofilm formation (36). In this work, we show that HutC was capable of binding to the plcC promoter, and the binding affinity was 15.1 times lower than that of the PhutU promoter. Deletion of hutC caused a significant increase of the plcC-lacZ expression. Taken together, our data consistently indicate that the histidine-responsive repressor HutC plays an important global regulatory role in fine-tuning the expression of genes such as ntrBC and plcC and contributes to the successful colonization of P. fluorescens on plant surfaces.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
Bacterial strains and plasmids used in this work are summarized in Table 2. P. fluorescens and E. coli strains were routinely grown in lysogeny broth (LB) medium at 28°C and 37°C, respectively. When grown in the M9 minimal salt medium (MSM), carbon and nitrogen substrates were supplemented at the specified concentrations (37). E. coli DH5αλpir was used for general gene cloning and triparental conjugation into P. fluorescens SBW25 and its derived mutants. When required, antibiotics were used at the following concentrations (μg/ml): ampicillin (Ap), 100; tetracycline (Tc), 15; spectinomycin (Sp), 100; kanamycin (Km), 50; gentamicin (Gm), 25; and nitrofurantoin (NF), 100.
TABLE 2.
Bacterial strains and plasmids used in this study
| P. fluorescens strain or plasmid | Genotype or characteristics | Source or reference |
|---|---|---|
| Strains | ||
| SBW25 | Wild-type strain isolated from phyllosphere of sugar beet | 47 |
| MU60-1 | ΔhutC, SBW25 devoid of pflu0359 | This work |
| MU60-2 | SBW25 carrying mini-Tn7T-Gm-lacZ::PhutU | This work |
| MU60-3 | SBW25ΔhutC carrying mini-Tn7T-Gm-lacZ::PhutU | This work |
| MU60-4 | SBW25 carrying mini-Tn7T-Gm-lacZ::PhutU-M1 | This work |
| MU60-5 | SBW25ΔhutC carrying mini-Tn7T-Gm-lacZ::PhutU-M1 | This work |
| MU60-6 | SBW25 carrying mini-Tn7T-Gm-lacZ::PhutU-M2 | This work |
| MU60-7 | SBW25ΔhutC carrying mini-Tn7T-Gm-lacZ::PhutU-M2 | This work |
| MU60-11 | SBW25 carrying mini-Tn7T-Gm-lacZ::PntrBC | This work |
| MU60-12 | SBW25 carrying mini-Tn7T-Gm-lacZ::PntrBC-mut2 | This work |
| MU60-8 | SBW25 carrying mini-Tn7T-Gm-lacZ::PplcC | This work |
| MU60-9 | SBW25ΔhutC carrying mini-Tn7T-Gm-lacZ::PplcC | This work |
| MU60-10 | SBW25 hutC overexpression strain carrying pME6010-hutC | This work |
| Plasmids | ||
| pRK2013 | Helper plasmid, Tra+ Kmr | 48 |
| pUIC3 | Integration vector with promoterless ′lacZ, Mob+ Tcr | 21 |
| pCR2.1-TOPO | Cloning vector, Kmr Apr | Invitrogen |
| pCR8/GW/TOPO | Cloning vector, Spr | Invitrogen |
| pCR8-PhutU-M1 | Recombinant plasmid containing PhutU-M1 promoter variant | This work |
| pCR8-PhutU-M2 | Recombinant plasmid containing PhutU-M2 promoter variant | This work |
| pCR2.1-PhutU-MW | Recombinant plasmid containing PhutU-M1&2 promoter variant | This work |
| pTrc99A | Protein expression vector, Ptac promoter, Apr | 40 |
| pTrc99A-hutC | pTrc99A carrying HutCHis6 from SBW25, Apr | This work |
| pUX-BF13 | Helper plasmid for transposition of mini-Tn7 element, Apr | 49 |
| pUC18-mini-Tn7T-Gm-lacZ | Mini-Tn7 vector for transcriptional fusion to promoterless lacZ, Apr Gmr | 38 |
| pUC18-Tn7T-lacZ-PhutU | pUC18-mini-Tn7T-Gm-lacZ containing lacZ fusion to PhutU promoter | This work |
| pUC18-Tn7T-lacZ-PhutU-M1 | pUC18-mini-Tn7T-Gm-lacZ containing lacZ fusion to PhutU-M1 promoter variant | This work |
| pUC18-Tn7T-lacZ-PhutU-M2 | pUC18-mini-Tn7T-Gm-lacZ containing lacZ fusion to PhutU-M2 promoter variant | This work |
| pCR2.1-PntrB-Mut2 | Recombinant plasmid containing PntrBC-mut2 promoter variant | This work |
| pUC18-Tn7T-lacZ-PntrBC | pUC18-mini-Tn7T-Gm-lacZ containing lacZ fusion to PntrBC promoter | This work |
| pUC18-Tn7T-lacZ-PntrBC-mut2 | pUC18-mini-Tn7T-Gm-lacZ containing lacZ fusion to PntrBC-mut2 promoter variant | This work |
| pUC18-Tn7T-lacZ-PplcC | pUC18-mini-Tn7T-Gm-lacZ containing lacZ fusion to PplcC promoter | This work |
| pME6010 | Broad-host-range vector, Tcr | 50 |
| pME6010-hutC | Recombinant plasmid for hutC overexpression | This work |
Growth kinetics of P. fluorescens strains were examined by first growing bacteria stored in a –80°C freezer in LB broth. To ensure that strains being compared were physiologically equal, bacterial cells in the overnight culture were washed twice and then subjected to carbon and nitrogen starvation at 28°C for 2 h using the MSM salt solution. Next, 2 μl of the starved culture was inoculated into 200 μl of the tested medium per well in a 96-well microplate. Turbidity was measured at a wavelength of 450 nm using a Synergy 2 plate reader installed with Gen5 software (Bio-Tek). Data were collected at 5-min intervals over a period of 24 h.
Strain construction.
Standard protocols were used for plasmid DNA isolation, restriction endonuclease digestion, ligation, and PCR (37). PCRs were performed using Taq DNA polymerase purchased from Invitrogen Ltd. (Auckland, New Zealand) and oligonucleotide primers synthesized by Integrated DNA Technologies Inc. (Singapore) (see Table S1 in the supplemental material). Deletion of the hutC gene in vivo was achieved by a previously established procedure of splicing by overlapping extension PCR (SOE-PCR) in conjunction with a two-step allelic-exchange strategy using the suicide-integration vector pUIC3 (22). The detailed procedures are outlined in Fig. S5 in the supplemental material. SOE-PCR was also used for site-directed mutagenesis of the PhutU promoter.
To construct a PhutU-lacZ fusion in vivo, an error-free DNA fragment was subcloned into the suicide vector pUC18-mini-Tn7T-Gm-lacZ (38). The obtained recombinant plasmid was then electroporated into P. fluorescens together with the helper plasmid pUX-BF13. The mini-Tn7 element carrying the lacZ reporter was integrated into a unique Tn7 site located downstream of glmS (39). β-Galactosidase activity was assayed for bacterial cells grown in tested media at ∼6 h after inoculation with a starting absorbance (A600) of ∼0.1. The assay was performed using 4-methylumbelliferyl-β-d-galactoside (4MUG) as the enzymatic substrate (20). The fluorescent product, 7-hydroxy-4-methylcoumarin (4MU), was detected at 460 nm with an excitation wavelength of 365 nm using a Synergy 2 plate reader (Bio-Tek). The enzyme activity was expressed as the amount of 4MU (μM) produced per minute per cell (A600).
EMSAs.
The HutC-coding region was amplified by PCR from the genomic DNA of P. fluorescens SBW25 with the integration of His6 at the N terminus (Table S3). The PCR product was directly cloned into the protein expression vector pTrc99A at the NcoI and HindIII sites (40). The resulting recombinant plasmid pTrc99A-hutC was then transformed into E. coli BL21(DE3) (41). IPTG (isopropyl-β-d-thiogalactopyranoside) was added at 1 mM to induce the expression of HutCHis6, which was subsequently purified using the Talon metal affinity resin (Clontech Laboratories Inc.). Probe DNAs were either biotin labeled at the 5′ ends through PCR with a biotinylated primer or digoxigenin (DIG) labeled using the DIG oligonucleotide 3′-end labeling kit (Roche) (see Fig. S3 in the supplemental material). Protein-DNA interaction was examined in a 20-μl reaction mixture containing 1 μg salmon sperm DNA, 10 mM HEPES, 50 mM KCl, 5 mM MgCl2, and 1 mM dithiothreitol (DTT) (pH 7.5). After 30 min of incubation at room temperature, the samples were subjected to 6% polyacrylamide gel electrophoresis in 0.5× Tris-borate-EDTA (TBE) buffer at low temperatures (∼4°C). Next, DNAs in the gel were transferred by electroblotting to a positively charged Whatman Nytran SuPerCharge nylon membrane (Sigma-Aldrich) and were heat immobilized at 80°C for 30 min. The biotin- or DIG-labeled probes were detected using the LightShift chemiluminescent electrophoretic mobility shift assay (EMSA) kit (Thermo Fisher Scientific) or the DIG nucleic acid detection kit (Roche), respectively. The image was visualized with a LAS-4000 luminescent imager equipped with ImageQuant LAS 4000 software (Fujifilm). Relative abundance of the free probe and protein-bound probe was determined using the ImageJ program. The data were then used to calculate Kd, referring to the HutCHis6 concentration when 50% of the DNA is protein bound.
DNase I footprinting and formaldehyde cross-linking.
DNase I footprinting was performed at room temperature in a 50-μl reaction under conditions similar to those described above for EMSA. After 30 min of coincubation, the reaction mixture was mixed with 50 μl of cofactor solution (5 mM CaCl2 and 10 mM MgCl2), followed by a 5-min treatment with 0.02 unit of DNase I (Invitrogen). The enzymatic reaction was terminated by adding 100 μl DNase I stop solution (200 mM NaCl, 20 mM EDTA, and 1% SDS). Next, the DNAs were extracted with an equal volume of 1:1 phenol-chloroform and precipitated with the addition of 1 μl glycogen (20 mg/ml), 1/10 volume of 3 M sodium acetate (pH 5.2), and three volumes of ethanol. After precipitation at –20°C for at least 1 h, DNAs were collected by centrifugation and then dissolved in 8 μl loading buffer (95% formamide, 0.05% bromophenol blue, and 20 mM EDTA). After heat denaturation (95°C, 10 min), DNAs were separated in a 6% urea-polyacrylamide gel (21 by 40 cm) in 1× TBE buffer using the Sequi-Gen GT electrophoresis system (Bio-Rad Laboratories Pty). The DNAs were then transferred from the gel to a positively charged nylon membrane by contact blotting (42) and subjected to detection using the LightShift chemiluminescent EMSA kit as described above for EMSA. A G+A chemical sequencing reaction was included to determine the sequence of the probe DNA (43).
HutC cross-linking was performed in 20-μl reaction mixtures containing 19 μM HutC protein, 20 mM HEPES (pH 8.2), 100 mM NaCl, 0.1 mM EDTA (pH 8.0), and 25 mM formaldehyde (44). The reaction was terminated after 1, 2.5, and 4 h of incubation at 25°C by adding an equal volume of SDS buffer (20% glycerol, 5% β-mercaptoethanol, 4% SDS, 0.003% Coomassie blue, and 0.125 M Tris-HCl, pH 6.8). The cross-linked proteins were separated in a standard 12% SDS-polyacrylamide gel.
Phenotypic assays.
Bacterial motility was examined on 0.3% (swimming) and 0.5% (swarming) nutrient agars using the standard method of stab inoculation, and the inoculants were prepared by growing the strains in comparison on 0.3% LB agar plates overnight. Pyoverdine was estimated by measuring fluorescence of the supernatant at 460 nm with an excitation wavelength of 365 nm in a Synergy 2 multimode microplate reader (Bio-Tek Instruments). The data were expressed as relative fluorescence units (RFU). The ability to form biofilm on the inner surfaces of microtiter plates and glass tubes was quantitatively assessed using the standard method of crystal violet staining as previously described (45, 46).
RNA-seq and in silico analysis.
Wild-type and ΔhutC mutant cells were grown to mid-log phase in M9 minimal salt medium supplemented with succinate (20 mM) and histidine (10 mM) as carbon and nitrogen sources. Total RNA was then prepared using the SV total RNA isolation system (Promega, Madison, WI). The RNA concentration was spectrophotometrically quantified (NanoDrop Technologies, Inc.), and its integrity was determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.). Sequencing was then performed on an Illumina HiSeq 4000 platform using the services provided by Novogene Technology Co. Ltd. (Beijing, China). The obtained data of 150-bp paired-end reads were processed in the Geneious 9.0.5 program (Geneious, Auckland, New Zealand). Reads were mapped to the reference genome of P. fluorescens SBW25. Reads per kilobase per million (RPKM) and transcripts per million (TPM) were subsequently calculated for each assembly. Differentially expressed genes were determined based on transcript comparison normalized using the Median of Gene Expression Ratios method implemented in Geneious 9.0.5.
The deduced amino acid sequence of P. fluorescens HutC was used to generate the homology model in the SWISS_MODEL server. Template searching identified five regulatory proteins in the GntR family whose crystal structures are currently available, and the highest homologue, DasR from Streptomyces coelicolor (PDB ID 4ZS8), was then used as a template to predict the HutC structure, and its interactions with the PhutU promoter DNA were predicted with the w3DNA web server (http://web.x3dna.org/). The protein-DNA interactions were displayed using the PyMOL 2.3.2 program.
Data availability.
The RNA-seq data have been deposited in NCBI’s Gene Expression Omnibus with accession number GSE146488.
Supplementary Material
ACKNOWLEDGMENTS
We thank Paul Rainey for his advice and continued support and Kiran Jayan for helpful discussions.
N.N. was a recipient of a Massey University Doctoral Scholarship. X.-X.Z. acknowledges support from the Massey University Research Foundation (MURF).
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Balleza E, López-Bojorquez LN, Martínez-Antonio A, Resendis-Antonio O, Lozada-Chávez I, Balderas-Martínez YI, Encarnación S, Collado-Vides J. 2009. Regulation by transcription factors in bacteria: beyond description. FEMS Microbiol Rev 33:133–151. doi: 10.1111/j.1574-6976.2008.00145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bervoets I, Charlier D. 2019. Diversity, versatility and complexity of bacterial gene regulation mechanisms: opportunities and drawbacks for applications in synthetic biology. FEMS Microbiol Rev 43:304–339. doi: 10.1093/femsre/fuz001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bender RA. 2012. Regulation of the histidine utilization (hut) system in bacteria. Microbiol Mol Biol Rev 76:565–584. doi: 10.1128/MMBR.00014-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Itoh Y, Nishijyo T, Nakada Y. 2007. Histidine catabolism and catabolite regulation, p 371–395. In Ramos H-L, Fillous A (ed), Pseudomonas. Springer, Berlin, Germany. [Google Scholar]
- 5.Smith GR, Magasanik B. 1971. Nature and self-regulated synthesis of the repressor of the hut operons in Salmonella typhimurium. Proc Natl Acad Sci U S A 68:1493–1497. doi: 10.1073/pnas.68.7.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allison SL, Phillips AT. 1990. Nucleotide sequence of the gene encoding the repressor for the histidine utilization genes of Pseudomonas putida. J Bacteriol 172:5470–5476. doi: 10.1128/jb.172.9.5470-5476.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorelik M, Lunin VV, Skarina T, Savchenko A. 2006. Structural characterization of GntR/HutC family signaling domain. Protein Sci 15:1506–1511. doi: 10.1110/ps.062146906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hagen DC, Magasanik B. 1973. Isolation of the self-regulated repressor protein of the Hut operons of Salmonella typhimurium. Proc Natl Acad Sci U S A 70:808–812. doi: 10.1073/pnas.70.3.808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu L, Allison SL, Phillips AT. 1989. Identification of multiple repressor recognition sites in the hut system of Pseudomonas putida. J Bacteriol 171:4189–4195. doi: 10.1128/jb.171.8.4189-4195.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang XX, Ritchie SR, Rainey PB. 2014. Urocanate as a potential signaling molecule for bacterial recognition of eukaryotic hosts. Cell Mol Life Sci 71:541–547. doi: 10.1007/s00018-013-1527-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sieira R, Bialer MG, Roset MS, Ruiz-Ranwez V, Langer T, Arocena GM, Mancini E, Zorreguieta A. 2017. Combinatorial control of adhesion of Brucella abortus 2308 to host cells by transcriptional rewiring of the trimeric autotransporter btaE gene. Mol Microbiol 103:553–565. doi: 10.1111/mmi.13576. [DOI] [PubMed] [Google Scholar]
- 12.Sieira R, Arocena GM, Bukata L, Comerci DJ, Ugalde RA. 2010. Metabolic control of virulence genes in Brucella abortus: HutC coordinates virB expression and the histidine utilization pathway by direct binding to both promoters. J Bacteriol 192:217–224. doi: 10.1128/JB.01124-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rietsch A, Wolfgang MC, Mekalanos JJ. 2004. Effect of metabolic imbalance on expression of type III secretion genes in Pseudomonas aeruginosa. Infect Immun 72:1383–1390. doi: 10.1128/iai.72.3.1383-1390.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeung AT, Torfs EC, Jamshidi F, Bains M, Wiegand I, Hancock RE, Overhage J. 2009. Swarming of Pseudomonas aeruginosa is controlled by a broad spectrum of transcriptional regulators, including MetR. J Bacteriol 191:5592–5602. doi: 10.1128/JB.00157-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joshua GW, Atkinson S, Goldstone RJ, Patrick HL, Stabler RA, Purves J, Camara M, Williams P, Wren BW. 2015. Genome-wide evaluation of the interplay between Caenorhabditis elegans and Yersinia pseudotuberculosis during in vivo biofilm formation. Infect Immun 83:17–27. doi: 10.1128/IAI.00110-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gibbs NK, Norval M. 2011. Urocanic acid in the skin: a mixed blessing? J Invest Dermatol 131:14–17. doi: 10.1038/jid.2010.276. [DOI] [PubMed] [Google Scholar]
- 17.Silby MW, Cerdeno-Tarraga AM, Vernikos GS, Giddens SR, Jackson RW, Preston GM, Zhang XX, Moon CD, Gehrig SM, Godfrey SA, Knight CG, Malone JG, Robinson Z, Spiers AJ, Harris S, Challis GL, Yaxley AM, Harris D, Seeger K, Murphy L, Rutter S, Squares R, Quail MA, Saunders E, Mavromatis K, Brettin TS, Bentley SD, Hothersall J, Stephens E, Thomas CM, Parkhill J, Levy SB, Rainey PB, Thomson NR. 2009. Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens. Genome Biol 10:R51. doi: 10.1186/gb-2009-10-5-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaderlund L, Hellman M, Sundh I, Bailey MJ, Jansson JK. 2008. Use of a novel nonantibiotic triple marker gene cassette to monitor high survival of Pseudomonas fluorescens SBW25 on winter wheat in the field. FEMS Microbiol Ecol 63:156–168. doi: 10.1111/j.1574-6941.2007.00420.x. [DOI] [PubMed] [Google Scholar]
- 19.Humphris SN, Bengough AG, Griffiths BS, Kilham K, Rodger S, Stubbs V, Valentine TA, Young IM. 2005. Root cap influences root colonisation by Pseudomonas fluorescens SBW25 on maize. FEMS Microbiol Ecol 54:123–130. doi: 10.1016/j.femsec.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 20.Zhang XX, George A, Bailey MJ, Rainey PB. 2006. The histidine utilization (hut) genes of Pseudomonas fluorescens SBW25 are active on plant surfaces, but are not required for competitive colonization of sugar beet seedlings. Microbiology 152:1867–1875. doi: 10.1099/mic.0.28731-0. [DOI] [PubMed] [Google Scholar]
- 21.Rainey PB. 1999. Adaptation of Pseudomonas fluorescens to the plant rhizosphere. Environ Microbiol 1:243–257. doi: 10.1046/j.1462-2920.1999.00040.x. [DOI] [PubMed] [Google Scholar]
- 22.Zhang XX, Rainey PB. 2007. Genetic analysis of the histidine utilization (hut) genes in Pseudomonas fluorescens SBW25. Genetics 176:2165–2176. doi: 10.1534/genetics.107.075713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang XX, Rainey PB. 2008. Dual involvement of CbrAB and NtrBC in the regulation of histidine utilization in Pseudomonas fluorescens SBW25. Genetics 178:185–195. doi: 10.1534/genetics.107.081984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang CY. 1982. Determination of binding stoichiometry by the continuous variation method: the Job plot. Methods Enzymol 87:509–525. doi: 10.1016/s0076-6879(82)87029-8. [DOI] [PubMed] [Google Scholar]
- 25.Beno I, Rosenthal K, Levitine M, Shaulov L, Haran TE. 2011. Sequence-dependent cooperative binding of p53 to DNA targets and its relationship to the structural properties of the DNA targets. Nucleic Acids Res 39:1919–1932. doi: 10.1093/nar/gkq1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bading H. 1988. Determination of the molecular weight of DNA-bound protein(s) responsible for gel electrophoretic mobility shift of linear DNA fragments examplified with purified viral myb protein. Nucleic Acids Res 16:5241–5248. doi: 10.1093/nar/16.12.5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guazzaroni ME, Krell T, del Arroyo PG, Velez M, Jimenez M, Rivas G, Ramos JL. 2007. The transcriptional repressor TtgV recognizes a complex operator as a tetramer and induces convex DNA bending. J Mol Biol 369:927–939. doi: 10.1016/j.jmb.2007.04.022. [DOI] [PubMed] [Google Scholar]
- 28.Lee YS, Hwang DS. 1997. Occlusion of RNA polymerase by oligomerization of DnaA protein over the dnaA promoter of Escherichia coli. J Biol Chem 272:83–88. doi: 10.1074/jbc.272.1.83. [DOI] [PubMed] [Google Scholar]
- 29.Fillenberg SB, Friess MD, Korner S, Bockmann RA, Muller YA. 2016. Crystal structures of the global regulator DasR from Streptomyces coelicolor: implications for the allosteric regulation of GntR/HutC repressors. PLoS One 11:e0157691. doi: 10.1371/journal.pone.0157691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Resch M, Schiltz E, Titgemeyer F, Muller YA. 2010. Insight into the induction mechanism of the GntR/HutC bacterial transcription regulator YvoA. Nucleic Acids Res 38:2485–2497. doi: 10.1093/nar/gkp1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chakerian AE, Matthews KS. 1992. Effect of lac repressor oligomerization on regulatory outcome. Mol Microbiol 6:963–968. doi: 10.1111/j.1365-2958.1992.tb02162.x. [DOI] [PubMed] [Google Scholar]
- 32.Rojo F. 1999. Repression of transcription initiation in bacteria. J Bacteriol 181:2987–2991. doi: 10.1128/JB.181.10.2987-2991.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lloyd G, Landini P, Busby S. 2001. Activation and repression of transcription initiation in bacteria. Essays Biochem 37:17–31. doi: 10.1042/bse0370017. [DOI] [PubMed] [Google Scholar]
- 34.Lenski RE. 1991. Quantifying fitness and gene stability in microorganisms. Bio/Technology 15:173–192. doi: 10.1016/b978-0-409-90199-3.50015-2. [DOI] [PubMed] [Google Scholar]
- 35.Wilderman PJ, Vasil AI, Martin WE, Murphy RC, Vasil ML. 2002. Pseudomonas aeruginosa synthesizes phosphatidylcholine by use of the phosphatidylcholine synthase pathway. J Bacteriol 184:4792–4799. doi: 10.1128/jb.184.17.4792-4799.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rossignol G, Merieau A, Guerillon J, Veron W, Lesouhaitier O, Feuilloley MG, Orange N. 2008. Involvement of a phospholipase C in the hemolytic activity of a clinical strain of Pseudomonas fluorescens. BMC Microbiol 8:189. doi: 10.1186/1471-2180-8-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory Mannual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 38.Choi KH, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods 2:443–448. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- 39.Zhang XX, Rainey PB. 2007. Construction and validation of a neutrally-marked strain of Pseudomonas fluorescens SBW25. J Microbiol Methods 71:78–81. doi: 10.1016/j.mimet.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 40.Amann E, Ochs B, Abel KJ. 1988. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene 69:301–315. doi: 10.1016/0378-1119(88)90440-4. [DOI] [PubMed] [Google Scholar]
- 41.Studier FW, Moffatt BA. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 42.Petersen I, Reichel MB, Dietel M. 1996. Use of non-radioactive detection in SSCP, direct DNA sequencing and LOH analysis. Clin Mol Pathol 49:M118–21. doi: 10.1136/mp.49.2.m118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maxam AM, Gilbert W. 1980. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol 65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 44.Nadeau OW, Carlson GM. 2007. Protein interactions captured by chemical cross-linking: one-step cross-linking with formaldehyde. CSH Protoc 2007:pdb.prot4634. doi: 10.1101/pdb.prot4634. [DOI] [PubMed] [Google Scholar]
- 45.O’Toole GA. 2011. Microtiter dish biofilm formation assay. J Vis Exp doi: 10.3791/2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu S, Zhang XX. 2016. Small colony variants are more susceptible to copper-mediated contact killing for Pseudomonas aeruginosa and Staphylococcus aureus. J Med Microbiol 65:1143–1151. doi: 10.1099/jmm.0.000348. [DOI] [PubMed] [Google Scholar]
- 47.Bailey MJ, Lilley AK, Thompson IP, Rainey PB, Ellis RJ. 1995. Site directed chromosomal marking of a fluorescent pseudomonad isolated from the phytosphere of sugar beet; stability and potential for marker gene transfer. Mol Ecol 4:755–763. doi: 10.1111/j.1365-294x.1995.tb00276.x. [DOI] [PubMed] [Google Scholar]
- 48.Ditta G, Stanfield S, Corbin D, Helinski DR. 1980. Broad host range DNA cloning system for gram-negative bacteria: construction of a gene bank of Rhizobium meliloti. Proc Natl Acad Sci U S A 77:7347–7351. doi: 10.1073/pnas.77.12.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bao Y, Lies DP, Fu H, Roberts GP. 1991. An improved Tn7-based system for the single-copy insertion of cloned genes into chromosomes of gram-negative bacteria. Gene 109:167–168. doi: 10.1016/0378-1119(91)90604-a. [DOI] [PubMed] [Google Scholar]
- 50.Heeb S, Itoh Y, Nishijyo T, Schnider U, Keel C, Wade J, Walsh U, O'Gara F, Haas D. 2000. Small, stable shuttle vectors based on the minimal pVS1 replicon for use in gram-negative, plant-associated bacteria. Mol Plant Microbe Interact 13:232–237. doi: 10.1094/MPMI.2000.13.2.232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data have been deposited in NCBI’s Gene Expression Omnibus with accession number GSE146488.