

Abstract
The novel coronavirus (CoV), severe acute respiratory syndrome (SARS)‐CoV‐2 is an international public health emergency. Until now, the intermediate host and mechanisms of the interspecies jump of this virus are unknown. Phylogenetic analysis of all available bat CoV complete genomes was performed to analyze the relationships between bat CoV and SARS‐CoV‐2. To suggest a possible intermediate host, another phylogenetic reconstruction of CoV genomes obtained from animals that were hypothetically commercialized in the Chinese markets was also carried out. Moreover, mutation analysis was executed to suggest genomic regions that may have permitted the adaptation of SARS‐CoV‐2 to the human host. The phylogenetic analysis demonstrated that SARS‐CoV‐2 formed a cluster with the bat CoV isolate RaTG13. Possible CoV interspecies jumps among bat isolates were also observed. The phylogenetic tree reconstructed from CoV strains belonging to different animals demonstrated that SARS‐CoV‐2, bat RaTG13, and pangolin CoV genomes formed a monophyletic cluster, demonstrating that pangolins may be suggested as SARS‐CoV‐2 intermediate hosts. Three AA substitutions localized in the S1 portion of the S gene were observed, some of which have been correlated to structural modifications of the S protein which may facilitate SARS‐CoV‐2 tropism to human cells. Our analysis shows the tight relationship between SARS‐CoV‐2 and bat SARS‐like strains. It also hypothesizes that pangolins might have been possible intermediate hosts of the infection. Some of the observed AA substitutions in the S‐binding protein may serve as possible adaptation mutations in humans but more studies are needed to elucidate their function.
Keywords: bat coronaviruses, CoV, intermediate host, phylogeny, SARS‐CoV‐2, zoonotic infection
Highlights
The intermediate host of SARS‐CoV‐2 is unknown.
Zoonotic genomic surveillance is important to understant SARS‐CoV‐2 evolutionary history.
Bat coronavirus internspecies jumps are important for the evolution of coronaviruses.
Specific aminoacid substitutions might have helped for the adaptation of SARS‐CoV‐2 in the human population.
1. INTRODUCTION
Coronaviruses (CoVs) are comprised of a large family of single‐stranded RNA viruses with a large genome (~30 kb), which cause (referring coronaviruses) respiratory and intestinal infections in animals and humans. Their role as emerging infections was first recognized during the severe acute respiratory syndrome (SARS) outbreak in China in 2002 caused by the SARS‐CoV. The SARS epidemic affected more than 8000 people and caused 916 deaths in 29 countries. 2 Palm civets (Paguma larvata) were involved in the SARS‐CoV jump to the human population as intermediate hosts. Ten years later, a second CoV outbreak caused by the Middle East respiratory syndrome (MERS)‐CoV occurred. It was characterized by 2040 confirmed cases and 712 deaths between 2012 and 2017. 3 The majority of the MERS‐CoV cases (80%) were reported in Saudi Arabia 4 and dromedary camels were suggested as MERS‐CoV intermediate hosts.
Currently, the world is facing the largest CoV pandemic caused by SARS‐CoV‐2 with millions of cases throughout the world. The disease caused by SARS‐CoV‐2, coronavirus disease 2019, ranges from asymptomatic infection to severe respiratory failure. 5 In the early infection phases, anti‐SARS‐CoV immunoglobulin G (IgG) and IgM are produced within a period of approximately 13 days after symptom onset. 6 The infection can be controlled by the timely detection of SARS‐CoV‐2 RNA in patients. Similar to SARS‐CoV, SARS‐CoV‐2 can be treated by population isolation and antiviral and symptomatic treatments, all of them with varying levels of success. 7 Many efforts for more effective SARS‐CoV‐2 treatment are in progress now, including compounds targeting viral products that have an essential function, especially the viral main protease. 8 , 9
One of the most intriguing questions is related to the origin of SARS‐CoV‐2 and its interspecies jump to the human population. The first cases of unknown causes of pneumonia originated from the Huanan seafood and animal market in the city of Wuhan, Hubei province, China. 7 So far, the mechanism of the interspecies jump of SARS‐CoV‐2 has not been fully understood although Malayan pangolins (Manis javanica) have been suggested as possible intermediate hosts due to the high similarity of pangolin and SARS‐CoV‐2 genomes 7 , 10 while bats are regarded as the original source of the infection. 11
The aim of this study is to analyze the available data of complete zoonotic CoV genomes in regard to SARS‐CoV‐2 to examine possible relationships, interspecies evolution, and suggest a possible intermediate host of SARS‐CoV‐2. Furthermore, knowing the importance of viral proteins for the viral cycle and host adaptation, we have investigated the S gene which is responsible for viral adsorption and probably participates in the host shifts.
2. MATERIALS AND METHODS
2.1. Dataset of bat CoV genomes
The complete genomes of 145 CoV strains were used, 144 originating from different bat genera (Asiatic bats, 115 genomes; European bats, 8 genome; African bats, 19 genomes; and American bats, 2 genomes) and 1 SARS‐CoV‐2 genome obtained in China. All complete CoV genomes were obtained from the GenBank (http://www.ncbi.nlm.nih.gov/genbank/). The Asiatic bat CoV genomes originated from China (92 genomes), Hong Kong (13 genomes), Singapore (1 genome), Vietnam (10 genomes), Japan (1 genome), and South Korea (1 genome); the European bat CoV were obtained from Bulgaria (1 genome), Finland (2 genomes), and Italy (5 genomes); the African bat CoV were obtained from Kenya (7 genomes), South Africa (2 genomes), Uganda (1 genome), Ghana (4 genomes), and Cameroon (4 genomes); and the American bat CoV were obtained from USA (1 genome) and Canada (1 genome). In the study, we included CoV genomes obtained from different bat hosts including the genera Rhinolophus, Scotophilus, Chaerephon, Rousettus, Cynopterus, Tylonycteris, Pipistrellus, Hypsugo, Vespertilio, Hipposideros, Myotis, Miniopterus, Nyctalus, Triaenops, and Eidolon (see Table S1).
We also obtained information of the geographic origin of the host, year of virus detection/isolation, and the type of the infected host (species name or genus). All of this information was represented on the tree branches. We used the following keyword combinations for the search of complete bat CoV genomes in the GenBank: (a) bat + coronavirus + complete + genome; (b) bat + CoV + complete + genome; (c) bat + SARS + complete + genome; (d) bat + SARS‐CoV‐2 + complete + genome; and (e) bat + SARS‐CoV + complete + genome.
2.2. Dataset of CoV complete genomes obtained from different animal sources
We also performed a phylogenetic analysis of CoV strains isolated from possible animals which could be commercialized in Chinese markets. From this approach, we would like to suggest a potential intermediate host for SARS‐CoV‐2 which may have served as a source of viral dissemination in the human population. For this analysis, we used complete animal CoV genomes obtained from the GenBank (excluding bats). Although we observed that there is a wide variety of wild and domestic animals which could be commercialized in the Chinese markets, we obtained and analyzed from the GenBank 77 complete animal CoV genomes, distributed as follows: murine (20 CoV genomes), camelids (9 CoV genomes), deer (5 CoV genomes), equine (4 CoV genomes), civets (14 CoV genomes), pangolin (5 CoV genomes), canine (12 CoV genomes), and swine (8 complete CoV genomes).
For the construction of the dataset, the first applied criterion was if the genome was complete, but we also added other characteristics including the geographic origin of the host, year of virus detection/isolation, and the type of the infected host (species name or genus). All of this information was represented on the tree branches. The sequences of animal CoV were not directly obtained from Chinese markets but from countries including the USA (murine, deer, horse, and canine), Nigeria, Morocco, Ethiopia, Saudi Arabia, United Arab Emirates (camelid CoV), South Korea (deer CoV), Japan (equine CoV), China (civet, pangolin, canine, and swine) and Italy, Germany, Taiwan (canine CoVs), (see Table S2).
We used the following keyword combinations for the search of complete animal CoV genomes in the GenBank: (a) murine + coronavirus + complete + genome; (b) camel + coronavirus + complete + genome; (c) deer + coronavirus + complete + genome; (d) equine + coronavirus + complete + genome; (e) civets + coronavirus + complete + genome; (f) pangolin + coronavirus + complete + genome; (g) canine + coronavirus + complete + genome; and (h) swine + coronavirus + complete + genome.
2.3. Phylogenetic analysis
The sequence dataset was aligned using MAFTT v.7.450 software 12 and manually edited by Bioedit program v. 7.0.5. To check the phylogenetic signal, we used the TREE‐PUZZLE v. 5.3. software. 13 Maximum likelihood trees were reconstructed using generalized time‐reversible (GTR) plus empirical codon frequencies (F) and invariant (I) sites plus Gamma distribution (G4), (GTR + F + I + G4), chosen according to Bayesian information criterion using IQ‐TREE v. 1.6.8. 14 Sequences identified as duplicates in the phylogenetic reconstruction were excluded. For visualization of the tree and for its editing, we used the FigTree v. 1.4. software. 15
We opted for the division of the datasets into two groups and consequently into two different phylogenetic trees due to the following reasons: (a) the phylogenetic tree which included CoV complete genomes isolated from bats aimed at investigating bat CoV diversity in relation to the bat genera as well as the evolutionary behavior including interspecies jumps which can increase the spread of certain CoV isolates; and (b) the phylogenetic tree which included CoV obtained from animal candidates which could hypothetically be commercialized in the Chinese markets was reconstructed to suggest a possible intermediate host for SARS‐CoV‐2 which could have served as a source for viral introduction into the human population.
2.4. Analysis of evolutionary mutations
Initially, to evaluate the impact of the evolutionary pressure for virus‐host adaptation, we performed similarity analysis using the BLAST ring image generator (BRIG) software v. 0.95 16 comparing the bat CoV isolate RaTg13 obtained from Rhinolophus affinis (MN996532) and the first sequenced human strain of SARS‐CoV‐2, Wuhan‐Hu‐1 (MN908947). In this analysis, we obtain a circular graph that verifies the similarity between the human and bat strains and attempts to outline areas with significant mutation coverage.
Consequently, the exact localization of the mutations in high resolution was shown using the Pipmaker program. 17 For a more comprehensive view of the possible mutations among the human SARS‐CoV‐2 strains, we used the available complete genomes in the Genbank (44 strains until 28 February 2020). We limited the mutation search to the S gene region, to decrease the computational time and focus on an area of major importance for the host shift. The S regions were aligned using ClustalW 18 and subsequently edited and translated into protein sequences using BioEdit software 19 v. 7.0.5. The identified mutations were analyzed using protein variation effect analyzer software 20 to verify whether the amino acid substitutions or indels have an impact on the biological function of the S protein. Finally, we used the PredictProtein software 21 to predict aspects of protein structure and function.
3. RESULTS
All available in the GenBank bat CoV full‐length genomes and the human SARS‐CoV‐2 Wuhan strain were compared, enabling us to combine information regarding the evolution of SARS‐CoV‐2. The phylogenetic analysis performed allowed us to assess also the evolutionary position of SARS‐CoV‐2 in comparison to zoonotic CoV strains to trace a possible intermediate host of this infection before its jump to the human population.
The generated phylogenetic tree of the bat CoV demonstrated the expected separation with high bootstrap support on almost all branches (100%), showing high reliability. The CoV clustering was strongly dependent on the type of bat host and can be observed in Figure 1. The basal evolutionary position of the SARS‐CoV‐2 clustered with RaTG13 bat strain isolated from Rhinolophus bats demonstrates that probably RaTg13 has been circulating as a zoonotic infection between Rhinolophus bats long before the introduction of SARS‐CoV‐2 into the human population. This phylogenetic tree aimed to examine SARS‐CoV‐2 relationships in regard to other bat CoV isolates and therefore we used only one reference strain for SARS‐CoV‐2 isolated in Wuhan.
Figure 1.
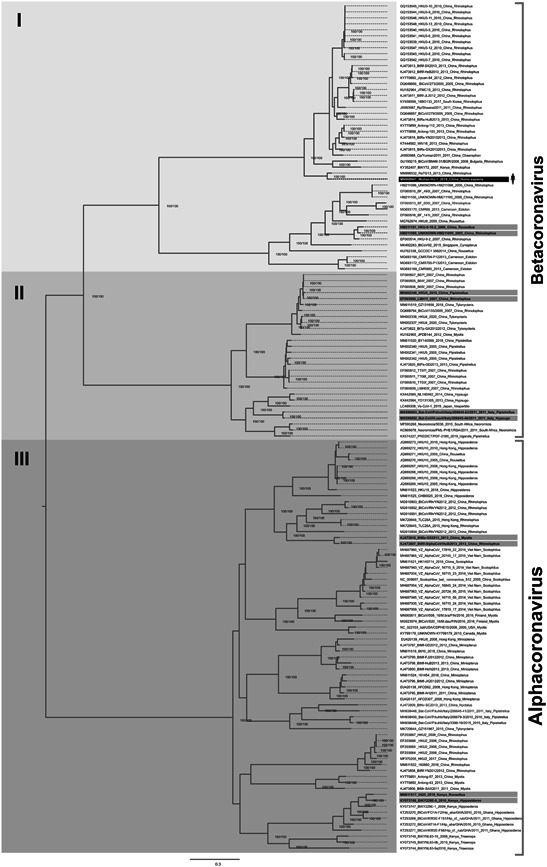
Phylogenetic analyses of the bat and other mammalian coronaviruses in regard to the SARS‐CoV‐2. Phylogenetic analysis of bat CoV strains obtained from different geographical localizations. Mostly, the dataset was composed from Asiatic strains including bat SARS‐like viral agents. The human SARS‐CoV‐2 Wuhan‐Hu‐1 strain was used to show the genetic distance between the zoonotic and human strains. In the tree, it is highlighted the possible interspecies jump between different bat CoVs. To explain better the cluster formation, the tree branches were grouped into three groups designated with roman numerals I‐III. Only complete genomes for tracing the phylogenetic history of bat CoV were used. The nucleotide substitution model used was GTR + F + I + G4 for tree reconstruction, which was chosen by the Bayesian information criterion statistic model, utilizing 10 000 ultrafast bootstrap replicates for statistical significance. Only values of above 99% were demonstrated on important tree branches. The phylogenetic tree was constructed using the IQ‐TREE software v.16.12, applying the maximum likelihood approach. F, frequency; G4, gamma distribution; GTR, generalized time‐reversible; I, invariant; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
The cluster of SARS‐CoV‐2 (Figure 1, division I) was basally located compared to the SARS‐like isolates obtained from Rhinolophus bats, which formed a monophyletic cluster. This demonstrates that SARS‐CoV‐2 is genetically distant from bat SARS‐like viruses. The SARS‐CoV‐2 strain was clustered with the highest bootstrap support with the RaTG13 strain isolated in 2013 in China, and this demonstrates the lack of genomic CoV surveillance between the date of isolation if the bat RaTG13 strain and its closest relative SARS‐CoV‐2 at the end of 2019. Given the information from the obtained phylogenetic tree, we were able to identify a possible interspecies jump: Rousettus bat CoV in the group of SARS‐like CoV obtained from Rhinolophus bats (Figure 1, division I), presence of Pipistrellus CoV isolates among Rhinolophus isolates, and Pipistrellus CoV isolates with high genetic similarity to Hypsugo CoV isolates in cluster II (Figure 1, division II). Interspecies jumps were also detected in isolates which were basally located, that is, Myotis isolates with high genetic similarity to Rhinolophus CoVs and Rousettus CoV located in the cluster of Hipposideros CoV isolates (Figure 1, division III).
Interspecies jumps were suspected in almost all of the clusters of the phylogenetic tree. However, these events probably occurred in a different manner. We observed that some cases, such as CoV jumps from Hipposideros to Rousettus, occurred in a clade where there were only CoV genomes obtained from Hipposideros. The same event occurred for Pipistrellus CoV isolates located among Rhinolophus and Rousettus CoV isolates (see the gray lines in Figure 1). However, we can consider as interspecies jumps the presence of CoV strains with high genetic similarity isolated from evolutionary divergent bat genera, as in the cases of Pipistrellus CoV isolates with high genetic similarity to CoV obtained from Hypsugo bats and Myotis CoV isolates with high genetic similarity to CoV strains isolated from Rhinolophus bats (Figure 1).
On the other hand, we observed that the clusters I‐III (Figure 1) were characterized by the presence of predominantly Asiatic CoV bat genomes with lower presence of CoV sampled in Europe, Africa. and America. While in the betacoronavirus cluster, there were almost exclusively Asiatic CoV strains, the highest diversity regarding place of origin was observed in the cluster of alphacoronaviruses (Figure 1, division III). This probably shows that the highest diversity of betacoronaviruses is concentrated in Asia, while alphacoronaviruses encompass CoV strains originating from all continents. However, the majority of bat CoV in the GenBank belonged to Asiatic species and as a consequence, they represented a greater majority in the final phylogenetic tree. Therefore, there is probably a gap regarding availability of complete genomes of bat CoV obtained from Europe, Africa, and America, and thus genomic surveillance with the characterization of more complete bat CoV genomes from these locations is necessary to fulfill the lack in their phylogeny.
To suggest a possible intermediate SARS‐CoV‐2 host, we also performed a separate phylogenetic analysis of CoV strains obtained from wild and domestic animals which were hypothetically commercialized in the Chinese markets (camels, civets, dogs, donkeys, horses, swine, rats, deer, and pangolins). The obtained clustering demonstrated the division of the animal CoV into three well‐supported clusters, that is, alpha‐, beta‐, and deltacoronaviruses. The largest cluster which comprised almost all the analyzed genomes belonged to betacoronaviruses, which is directly linked to their higher diversity. The alphacoronaviruses encompassed canine sequences, and the deltacoronaviruses were represented by swine CoV. Human SARS‐CoV‐2 strains isolated in China, Italy, and Brazil formed a monophyletic cluster and were grouped into a large cluster with pangolin and bat RaTG13 CoV strains. The pangolin sequences obtained in 2017 were located in a basal position compared to pangolin sequences from 2019, RaTG13 bat strain and SARS‐CoV‐2, which means that these sequences are evolutionarily much older but can accumulate changes throughout the years, which suggests that pangolin may be suggested as a possible intermediate host of SARS‐CoV‐2. In this phylogenetic tree, the civet CoV genomes were distantly located compared to SARS‐CoV‐2 precluding their involvement as a hypothetic intermediate host of SARS‐CoV‐2. The phylogenetic tree obtained from the analysis of SARS‐CoV‐2 and zoonotic CoV strains obtained from different animals is shown in Figure 2.
Figure 2.

Phylogenetic analysis of zoonotic CoV strain obtained from different geographical localizations and outbreaks. In this analysis, different kinds of animals were included that were probably commercialized in the Chinese markets (camels, civets, dogs, donkeys, horses, swine, rats, deer, and pangolins). The human isolate Wuhan‐Hu‐1 strain was used to show the genetic distance between the zoonotic and human strains. We used only complete genomes for tracing the phylogenetic history of zoonotic CoVs. The nucleotide substitution model used was GTR + F + I + G4 for tree reconstruction, which was chosen by the Bayesian information criterion statistic model, utilizing 10 000 ultrafast bootstrap replicates for statistical significance. Only values of above 99% were demonstrated on important tree branches. The phylogenetic tree was constructed using the IQ‐TREE software v.16.12, applying the maximum likelihood approach. CoV, coronavirus; F, frequency; G4, gamma distribution; GTR, generalized time‐reversible; I, invariant
The analysis of the possible mutations of SARS‐CoV‐2 (Wuhan‐Hu‐1 strain) in comparison to the zoonotic bat sequence (RaTg13) revealed the existence of 1141 single‐nucleotide polymorphisms with 96% identity between the two sequences. As was suspected, most of the identified mutations were located in the S (spike) gene, which encompasses positions between 21 563 and 25 384 base pair (GenBank accession number: MN908947). Therefore, we focused our analyses on the spike (S)‐protein portion (with both its subregions S1/S2 and the junction region), where we found 30 residue mutations in different positions, including insertion in position 680. Consequently, we checked for possible deleterious action of these AA changes on the protein residues. All possible mutations found in the S protein were considered neutral, that is, evolutionarily beneficial for the virus, without any possible harmful effect on virus survival processes. The functional prediction of protein demonstrated the presence of three amino acid mutations (see Table 1). They were characterized by two AA substitutions in positions 32 (S→F), 218 (P→Q), and one insertion in position 680 (PRRA), localized in the junction region of the S1/S1 subunits composing the S protein.
Table 1.
Detected mutations and their implications for binding sites in the S portion of the SARS‐CoV‐2
| AA a position in the S gene | Bat‐CoV b | SARS‐CoV‐2 c | Mutation involved in binding sites |
|---|---|---|---|
| 32 | S | F | X |
| 50 | L | S | |
| 76 | I | T | |
| 218 | P | Q | X |
| 324 | D | E | |
| 346 | T | R | |
| 372 | T | A | |
| 403 | T | R | |
| 439 | K | N | |
| 440 | H | N | |
| 441 | I | L | |
| 443 | A | S | |
| 445 | E | V | |
| 449 | F | Y | |
| 459 | A | S | |
| 478 | K | T | |
| 483 | Q | V | |
| 484 | T | E | |
| 486 | L | F | |
| 490 | Y | F | |
| 493 | Y | Q | |
| 494 | R | S | |
| 498 | Y | Q | |
| 501 | D | N | |
| 505 | H | Y | |
| 519 | N | H | |
| 604 | A | T | |
| 680 | S | SPRRA d | X |
| 1121 | S | N | |
| 1224 | I | V |
Abbreviation: SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2.
AA, amino acid position.
AA in the S portion of the genome of the bat coronavirus strain RaTg13.
AA change in the SARS‐CoV‐2 genome.
SPRRA insertion in the SARS‐CoV‐2 genome.
After carrying out the analyzes using BRIG software, we found a similarity rate of 96% between RaTG13 and SARS‐CoV‐2 (Wuhan isolate). However, the mutations which were present in the human strain were randomly distributed along the S gene and not concentrated in a specific location, which did not permit their graphical representation.
4. DISCUSSION
The performed study reveals the phylogenetic relationships between SARS‐CoV‐2 and CoV genomes obtained from bats from different parts of the world. Moreover, we performed an additional phylogenetic reconstruction which suggests the possibility of a specific intermediate host for SARS‐CoV mediating its jump into the human population.
The reconstructed phylogenetic tree of bat CoV genomes (Figure 1) demonstrated, as expected, that SARS‐CoV‐2 formed a cluster with the zoonotic strain RaTG13 obtained from a fecal swab from Rhinolophus affinis bats in 2013. The basal position of the SARS‐CoV‐2 compared to the other bat SARS‐like viruses demonstrates their genetic divergence and probably shows that a SARS‐CoV‐2‐like virus has been intensively circulating among bat populations before its emergence as a human pathogen. Although SARS‐CoV‐2 was positioned in one cluster with SARS‐like bat CoV sequences, they were distantly located in a monophyletic cluster and were also related to Rhinolophus bats. This probably demonstrates that Rhinolophus bats may participate in CoV host shifts with a higher frequency compared to other bat species and the close contact between bats, humans, and intermediate mammalian hosts may have helped in the establishment of the SARS‐CoV‐2 outbreak. 22 In addition, SARS‐like viruses with high‐sequence homology to SARS‐CoV were isolated from Rhinolophus sinicus, which supports the hypothesis that these bats can be definitive hosts with the extensive circulation of SARS‐like viruses. 7 Moreover, characteristics of the bat CoV are very high mutation rates, 23 recombination capacity, and presence of multiple open‐reading frames in the large CoV genome, 24 which allows for rapid adaptation to new hosts and expanded tissue tropism. This was also suggested by our study, where we identified frequent bat host shifts among the bat CoV. Moreover, the receptors responsible for productive SARS‐CoV infection (ACE2) are present in different bat species, 25 which can be important for the interspecies jumps of the SARS‐like viruses. The ability of the CoV to undergo frequent host shifts supports the hypothesis that these viruses are one of the most important groups of pathogenic agents responsible for the appearance of emerging diseases, as discussed by other authors, and are probably the most important hosts of emerging viruses. 22
We analyzed CoV genomes obtained from animals which could be probable intermediate hosts for SARS‐CoV‐2 due to their frequent commercialization in the Chinese markets. Although none of the analyzed CoV sequences originated directly from animals sold in the Chinese markets, we suggest that intermediate SARS‐CoV‐2 exists based on the genomic surveillance of closely related animal CoV. Moreover, the virus needs initially to be established into an intermediate host with high population density and presence of specific receptors which will further enable the interspecies jump to the human population as occurred in SARS‐CoV, MERS‐CoV, and probably SARS‐CoV‐2 26 (our hypothesis for SARS‐CoV‐2 is presented in Figure 3). In this respect, we have to examine animal CoV strains with older sampling dates. Although in the performed phylogenetic analysis, the SARS‐CoV‐2 sequences obtained from China, Italy, and Brazil were more closely related to the bat RaTG13 strain similar to the bat CoV phylogenetic tree, they also formed a cluster with CoV strains obtained from Malayan pangolin (Manis javanica) obtained in 2017 and 2019. Such clustering may be suggestive of a zoonotic transmission chain where pangolins served as intermediate hosts of SARS‐CoV‐2. In addition, pangolin CoV exhibits strong sequence similarity with the RBD region of SARS‐CoV‐2, which strongly suggests that the spike protein of SARS‐CoV‐2 which binds to the ACE2 receptor results from natural selection with the help of an intermediate host. 26 Therefore, obtaining more sequence information, especially from animal sources, and performing zoonotic surveillance of the CoV is the most categorical way to examine the virus origin and its dissemination in the human population.
Figure 3.
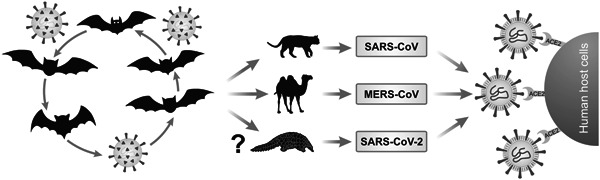
Possible routes of SARS‐CoV, MERS‐CoV, and SARS‐CoV‐2 interspecies jumps and transmission to the human population due to genomic adaptations that help the virus in the invasion of subsequent species. The image accentuates the zoonotic circulation of the viral agents in bat populations with subsequent jumps to intermediate hosts and finally, due to anthropogenic factors, the viruses can be disseminated in the human population. CoV, coronavirus; MERS, Middle East respiratory syndrome; SARS, severe acute respiratory syndrome
The performed mutation analysis demonstrated several AA changes which were localized in the S region of the SARS‐CoV‐2 genome compared to RaTG13 bat strain. The AA insertion at position 680 (PRRA) is already described in the literature and is located at the S1‐S2 junction of the S protein and is a polybasic cleavage site leading to the generation of the binding (S1) and fusion (S2) subunits. 26 The generation of the furin subunit can be an important adaptation factor for SARS‐CoV‐2 replication in humans. Moreover, more R in the S1‐S2 cleavage site can enhance the cleavage of S1 and S2 subunits with the removal of the structural constraints and as a consequence, the insert peptide S2 is exposed and can be inserted into the host cell membrane, which suggests an increase in the efficiency of the viral entry into the human cell and fusion of the membranes. 27 The other two identified AA changes in the S protein, P32F and P218Q, are still not described in the literature. However, molecular changes of SARS‐CoV during the early transition and expansion into middle‐phase epidemics include A3047V and A3072V in the replicase gene and D778Y and E1163K in the S gene, and, in general, during the SARS‐CoV epidemic, the S gene has been submitted to strong positive selection. 24 Therefore, similar to SARS‐CoV, the observed AA substitutions in SARS‐CoV‐2 may help us in expanding the epidemic in human hosts or represent adaptive changes in the S gene. Nevertheless, a broader investigation and validations are needed to know how the AA substitutions in the S gene affect, or not, the mechanisms of the virus to produce infection in a new host.
The SARS‐CoV‐2 is a major world health emergency that has led to breakdowns in the health systems of the affected countries. Therefore, detailed studies of the origins of SARS‐CoV‐2 and the possible mechanisms of viral interspecies jump to the human population are fundamental to elaborate strategies for restraining further emergence of zoonotic pandemic viruses. The high diversity of bat CoV and the unique characteristics of the CoV‐like high mutation rates and recombination are prognostic markers that increase the possibility of further outbreaks. There is a significant amount of evidence that SARS‐CoV‐2 used an intermediate host for viral adaptation before its jump into the human population but there is an urgent need for genomic surveillance of CoV genomes obtained from different animal sources which will show in a direct way the evolutionary relationships of SARS‐CoV‐2 and will help to create hypotheses for the emergence and spread of SARS‐CoV‐2 in the human population.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
The authors are grateful to Sandra N. Bresciani for the figures. This study was supported by the São Paulo Research Foundation (FAPESP) with the following Grant Numbers: 2017/23205‐8, 2018/15826‐5, 2018/22009‐3, 2019/07861‐8, and 2019/08528‐0.
dos Santos Bezerra R, Valença IN, de Cassia Ruy P, et al. The novel coronavirus SARS‐CoV‐2: From a zoonotic infection to coronavirus disease 2019. J Med Virol. 2020;92:2607–2615. 10.1002/jmv.26072
REFERENCES
- 1. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17(3):181‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Drosten C, Günther S, Preiser W, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1967‐1976. [DOI] [PubMed] [Google Scholar]
- 3. Chafekar A, Fielding BC. MERS‐CoV: understanding the latest human coronavirus threat. Viruses. 2018;10(2):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yin Y, Wunderink RG. MERS, SARS and other coronaviruses as causes of pneumonia. Respirology. 2018;23(2):130‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pascarella G, Strumia A, Piliego C, et al. COVID‐19 diagnosis and management: a comprehensive review [published online ahead of print April 29, 2020]. J Intern Med. 10.1111/joim.13091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Long QX, Deng HJ, Chen J, et al. Antibody responses to SARS‐CoV‐2 in COVID‐19 patients: the perspective application of serological tests in clinical practice. medRxiv. 2020. 10.1101/2020.03.18.20038018 [DOI] [Google Scholar]
- 7. Xu J, Zhao S, Teng T, et al. Systematic comparison of two animal‐to‐human transmitted human coronaviruses: SARS‐CoV‐2 and SARS‐CoV. Viruses. 2020;12(2):244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang L, Lin D, Sun X, et al. Crystal structure of SARS‐CoV‐2 main protease provides a basis for design of improved α‐ketoamide inhibitors. Science. 2020;368(6489):409‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Joshi RS, Jagdale SS, Bansode SB, et al. Discovery of potential multi‐target‐directed ligands by targeting host‐specific SARS‐CoV‐2 structurally conserved main protease. J Biomol Struct Dyn. 2020;24:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu P, Jiang JZ, Wan XF, et al. Are pangolins the intermediate host of the 2019 novel coronavirus (2019‐nCoV)? bioRxiv. 2020. 10.1101/2020.02.18.954628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Katoh K, Misawa K, Kuma KI, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059‐3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE‐PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18(3):502‐504. [DOI] [PubMed] [Google Scholar]
- 14. Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Mol Biol Evol. 2015;32(1):268‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rambaut A. FigTree v1. 4. Molecular Evolution, Phylogenetics and Epidemiology. Edinburgh, UK: Institute of Evolutionary Biology, University of Edinburgh; 2012. [Google Scholar]
- 16. Alikhan NF, Petty NK, Ben ZNL, Beatson SA. BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12(1):402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schwartz S, Zhang Z, Frazer KA, et al. PipMaker: a web server for aligning two genomic DNA sequences. Genome Res. 2000;10(4):577‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thompson JD, Toby JG, Des GH. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics. 2003;1:2‐3. [DOI] [PubMed] [Google Scholar]
- 19. Hall T, Biosciences I, Carlsbad C. BioEdit: an important software for molecular biology. GERF Bull Biosci. 2011;2(1):60‐61. [Google Scholar]
- 20. Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7(10):e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rost B, Yachdav G, Liu J. The predictprotein server. Nucleic Acids Res. 2004;32(suppl 2):W321‐W326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cui J, Han N, Streicker D, et al. Evolutionary relationships between bat coronaviruses and their hosts. Emerg Infect Dis. 2007;13(10):1526‐1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eckerle LD, Becker MM, Halpin RA, et al. Infidelity of SARS‐CoV Nsp14‐exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010;6(5):e1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bolles M, Donaldson E, Baric R. SARS‐CoV and emergent coronaviruses: viral determinants of interspecies transmission. Curr Opin Virol. 2011;1(6):624‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hou Y, Peng C, Yu M, et al. Angiotensin‐converting enzyme 2 (ACE2) proteins of different bat species confer variable susceptibility to SARS‐CoV entry. Arch Virol. 2010;155(10):1563‐1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS‐CoV‐2. Nature Med. 2020;26(4):450‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Meng T, Cao H, Zhang H, et al. The insert sequence in SARS‐CoV‐2 enhances spike protein cleavage by TMPRSS. bioRxiv. 2020. 10.1101/2020.02.08.926006 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information