

The coronavirus disease 2019 (COVID‐19) pandemic has halted many ongoing central nervous system (CNS) clinical trials, especially in Alzheimer’s disease. These long‐duration trials involve many stakeholders, especially the patients and their family members, who have demonstrated their commitment to developing new therapeutic interventions for this devastating disease. We certainly do not want to lose all the knowledge we have gained from these ongoing trials because of the pandemic.
Although some of these trials will need to restart, others can restart at different points along the trial protocol with substantial protocol amendments. However, there is an urgent need to combine useful information from the completers with those subjects undergoing complex protocols, deviations, and amendments after restart. We propose the concept of mechanistic modeling‐based virtual twin patients as a possible solution to harmonize the readouts from these complex and fragmented clinical datasets in a biologically relevant way.
BACKGROUND
A majority of ongoing clinical trials has been affected by the COVID‐19 pandemic and trials in neurodegenerative diseases are no exception. To that end, the US Food and Drug Administration (FDA) recently published guidance stating, the “FDA recognizes that protocol modifications may be required, and that there may be unavoidable protocol deviations due to COVID‐19 illness and/or COVID‐19 control measures. The necessity for, and impact of, COVID‐19 control measures on trials will vary depending on many factors, including the nature of disease under study, the trial design, and in what region(s) the study is being conducted (ref 2)” (FDA Guidance on Conduct of Clinical Trials of Medical Products during COVID‐19 Pandemic Guidance for Industry, Investigators, and Institutional Review Boards March 2020 Updated on April 2, 2020).
These events will very likely lead to unprecedented issues of “missingness” in the datasets, far beyond anything we have seen before and not yet considered in this FDA guidance. At the time of trial pause, there will be three patient categories: those who have completed the trial, those who started but had their trial interrupted, and those who enrolled but have not yet started. Some protocol deviations for the patients currently in the trial include involuntary drug holiday (especially with i.v. formulations), change in medications (anxiolytics and antidepressants) for addressing mental health issues, and missing site visits that can be partially mitigated by remote monitoring. Of particular concern are the involuntary drug holidays, because the underlying pathological mechanism that was targeted with the drug, picks up again and the patient faces a completely new pathological environment when the drug trial is restarted. In addition, when considering an 18‐month phase III study enrolling about 600 patients/year for an anticipated enrollment of 1,800 patients, up to 60% of subjects in the trial can face substantial disruptions.
There will also be subjects from the last two groups that will not return to the trial once it restarts. These factors are exacerbated for patients with neurodegenerative diseases due to their age, fragility, comorbidity, comedication, and other factors. Unfortunately, we will likely lose a sizeable cohort of trial participants due to their deaths from COVID‐19, even those who had almost completed their trial.
As we cannot afford to lose the investment by many stakeholders, not in the least the patients and their family members, it is imperative that we explore all possible avenues to generate as much knowledge as possible from these incomplete datasets. Traditional statistical methods, such as Last Observation Carried Forward, for accounting for missing data will likely be inadequate for such complex and fragmented databases. With regard to machine learning, the number of completers in a trial for a “training” set is insufficient to warrant a substantial generalization to other trial participants. In this paper, we propose a computational modeling approach that combines limited clinical data with underlying biological principles to generate predictions at the individual patient level.
“VIRTUAL TWIN” COMPUTER SIMULATION TECHNOLOGY
The virtual twin patient approach, based on mechanistic modeling, including physiologically‐based pharmacokinetics (PBPKs) and Quantitative Systems Pharmacology (QSP), is a possible novel analytical method to address this “missingness.” PBPK modeling uses virtual populations to predict drug performance across all human organs, based on in vitro and in vivo data. QSP builds on PBPK by integrating quantitative drug data with knowledge of its mechanism of action. Specifically, relevant for complex neurodegenerative diseases, such as Alzheimer’s disease (AD) and Parkinson’s disease (PD), QSP predicts how drugs modify cellular and neuronal networks in space and time and how they are impacted by both human pathophysiology and therapeutic interventions.
The virtual twin approach creates a computer‐simulated model of each patient, replicating the patient’s unique attributes that affect a drug’s fate in their body and, hence, its effects. These attributes include the patient’s age, weight, height, sex, ethnicity, and genetics of drug metabolizing enzymes and drug transporters. The virtual twin model reflects the patient’s current drug dosage, fed or fasted state, comorbid conditions, comedications, and their level of organ function that affect the activity of certain metabolic enzymes and transporters. For example, the PBPK approach accurately predicted olanzapine exposure in individual patients for model‐informed precision dosing. 1 Another application created a virtual twin to predict drug cardiotoxicity after therapeutic and supratherapeutic dosing of citalopram. 2 Encouraged by regulators, PBPK can predict drug exposure levels based on patient and drug characteristics and concomitant medications.
To model the pharmacodynamic and efficacy effects, for instance in AD, a virtual twin QSP model of trial subjects, using exactly the same comedications (many of them CNS active), genotypes, biomarkers, β‐amyloid, and tau load, as in the real trial based on key neuronal circuitry involved in cognition can be created. We can create thousands of virtual twin patients by combining the well‐documented pharmacology and target exposure of many comedications currently used in AD clinical practice with some common genotype variants derived from imaging studies, such as APOE, 5‐HTTLPR, rs23351, and COMTVal158Met. 3 , 4 Further combining the pharmacology and the pharmacokinetic profile of the investigative drug will allow to simulate the cognitive trajectory of these individual virtual patients. Introducing information from imaging and biological fluid biomarkers could further help define both levels of target engagement for the investigative drug as well as the disease state.
This mechanism‐based QSP approach has been successfully used in blindly predicting an unexpected phase I scopolamine challenge study in healthy volunteers with a drug affecting a new yet untested target. 5 Moreover, the modeling platform has generated testable hypotheses on the cognitive worsening of beta‐site cleavage enzyme inhibitors 6 and the different outcomes for the two phase III trials with aducanumab in AD, 7 whereas other examples include a Unified Parkinson Disease Rating Scale‐calibrated QSP model for motor symptoms in PD. 8
So far, many biological assumptions in these models have been extensively tested and calibrated with clinical trials, based on group average data. However, applications have included the development of a classifier to predict the complex pharmacodynamical interactions on extrapyramidal motor symptoms in a real‐world dataset in individual patients with schizophrenia on two antipsychotics 9 in the absence of a training set and with a 35% better performance than the state‐of‐the‐art calculations.
STRATEGY FOR HARMONIZING FRAGMENTED DATASETS
The model will be validated using the actual fragmented dataset of the trial. In a first step, a virtual twin population from the “completers” database can be generated with the same properties as the observed population, starting from baseline data. The QSP model will then generate a fully blinded set of expected clinical outcomes at the individual patient level based on the trial protocol, which can be compared with the real outcomes. A second level of validation would include prediction of the intermediate outcomes from patients who had reached specific time points before their trial has been interrupted. A third important level is the prediction of the clinical trajectory of those patients who have restarted with the many protocol amendments to ensure generalizability to these subjects.
Once these validation are performed satisfactorily with a predefined similarity criterion, this method can be applied to predict clinical progression of the patients that have not yet completed the trial by estimating how dropouts, protocol amendments, and deviations after the trial restart would have affected the clinical trajectory, with the ultimate goal of interpolating the functional trajectory back to the original trial design (see Figure 1 ). In principle, this would allow to “pool” these newly generated data with the data from the completers, resulting in sufficient number of subjects for “bona fide” statistical analysis and conclusions about the trial outcome.
Figure 1.
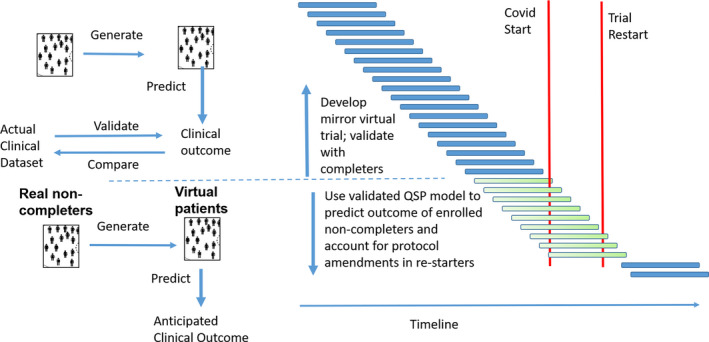
Timeline of a typical clinical trial in Alzheimer’s disease with the sudden halt caused by the coronavirus disease 2019 (COVID‐19) pandemic. A virtual twin population is created with the same characteristics as the observed population of completers (blue bars). Using those data, the Quantitative Systems Pharmacology (QSP) model generates expected clinical outcomes, which can be compared with the real outcomes of this responder population. The model takes into account pharmacokinetic and pharmacodynamic interactions among the investigative drug and comedications, genotypes, and disease states. Once this validation is performed satisfactorily, the QSP model can be applied to predict clinical progression of the noncompleter patient population (green bars) that have dropped out. The model can also “correct” for protocol amendments when the trial restarts, allowing to pool these results with the completer results. In this way, valuable information can be salvaged.
In these validation steps, modifications, such as coupling factors linking β‐amyloid levels to impact on glutamatergic or nicotinic neurotransmission or the impact of benzodiazepines on GABA receptors, can be modified within biologically relevant ranges. With over 35 CNS targets in the model, including amyloid and tau pathological processes, the virtual twin platform can cover a large range of clinical trial conditions in AD and PD.
Regulatory agencies are increasingly exposed to QSP approaches in submission. 10 However, most of the experience has been in early clinical development and QSP has played a supportive rather than a pivotal role, complementing a larger evidentiary package. Therefore, the question what would constitute a qualified (let alone validated) virtual twin QSP model from a regulatory perspective remains to be defined by key stakeholders.
The three levels of validation, as mentioned above, would generate predictions of almost the same number of subjects from the fragmented dataset as in the original design, therefore, this number will likely be sufficient to “interpolate” the clinical trajectory to the original plan.
SUMMARY
This report proposes to explore the combination of PBPK and QSP as a possible innovative way to correct the many substantial changes in the interrupted clinical trials with the purpose to “generate” the functional trajectory back to the original protocol design. We acknowledge that this has never been proposed before, but it is one of the possible approaches to salvage the information gathered so far at substantial costs.
A particularly interesting aspect of this approach is the emphasis on validation. Because the model is already calibrated on group average data both in AD and PD from publicly available sources, the relevant parameters for the additional pharmacodynamic interaction with comedications and genotypes can be reasonably constrained with even a small number of completers. For situations in which there are no or very few completers at all, data from other completed trials with similar different therapeutic interventions could be used for validation, based on the distribution of good responders, nonresponders, and subjects worsening over time. Ideally, pooling data from different sponsors might be another solution to cover a large range of targets and conditions. It has to be emphasized, that the validation at the three different levels will be performed by prospectively predicting the functional outcome solely based on the baseline characteristics but without knowledge of the outcome.
There are a number of limitations to this approach. The current version of the platform does not cover all possible therapeutic interventions in AD and PD; therefore, a number of targets currently being tested in the clinic are beyond the scope of the model. There might be other genotypes that drive the clinical outcome beyond the three implemented here, for instance, the BDNFVal66Met genotype. Although baseline biomarkers of amyloid, tau load status, and brain atrophy provide us with a fairly good estimate of the pathology, only in those cases where we have sequential biomarker readouts can we estimate the individual rate of pathology progression.
Another major limitation of this approach is the unknown central effects of the severe acute respiratory syndrome‐coronavirus 2 (SARS‐CoV‐2) virus for those patients who become infected; possibly interfering with the disease pathology or impacting the functional cognitive readout. However, in that case, lack of validation could indicate a major impact of this pathology. Accordingly, an extensive sensitivity analysis of the platform can possibly identify the impact of the SARS‐CoV‐2 virus on neuronal functioning.
In summary, virtual twin using PBPK/QSP can enable the extrapolation and “bridging” of the clinical trajectory for the many patients unable to complete the trial, helping to salvage the information gathered at substantial human and investment cost for the research participants, their family members, healthcare professionals, and sponsors. The model is individualized, so it can account for many different possibilities, including unforced drug holiday and change of medications at the individual patient level. When the trial restarts (likely with a new protocol), the approach can bridge the before and after to hopefully salvage the data and communicate continuity of information with scientists and regulators.
Funding
No funding was received for this work.
Conflict of Interest
Both authors declared no competing interests for this work.
References
- 1. Polasek, T.M. et al Prediction of olanzapine exposure in individual patients using physiologically based pharmacokinetic modelling and simulation. Br. J. Clin. Pharmacol. 84, 462–476 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Patel, N. , Wisniowska, B. , Jamei, M. & Polak, S. Real patient and its virtual twin: application of quantitative systems toxicology modelling in the cardiac safety assessment of citalopram. AAPS J. 20, 6 (2017). [DOI] [PubMed] [Google Scholar]
- 3. Slifstein, M. et al COMT genotype predicts cortical‐limbic D1 receptor availability measured with [11C]NNC112 and PET. Mol. Psychiatry 13, 821–827 (2008). [DOI] [PubMed] [Google Scholar]
- 4. Fisher, P.M. et al 5‐HTTLPR status predictive of neocortical 5‐HT4 binding assessed with [(11)C]SB207145 PET in humans. NeuroImage 62, 130–136 (2012). [DOI] [PubMed] [Google Scholar]
- 5. Timothy Nicholas, S.D. et al Systems pharmacology modeling in neuroscience: Prediction and outcome of PF‐04995274, a 5‐HT4 partial agonist, in a clinical scopolamine impairment trial. Adv. Alzheimer Dis. 2, 83–98 (2013). [Google Scholar]
- 6. Geerts, H. , Spiros, A. & Roberts, P. Impact of amyloid‐beta changes on cognitive outcomes in Alzheimer's disease: analysis of clinical trials using a quantitative systems pharmacology model. Alzheimer Res. Ther. 10, 14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Geerts, H. & Spiros, A. Learning from amyloid trials in Alzheimer's disease. A virtual patient analysis using a quantitative systems pharmacology approach. Alzheimers Dement. 10.1002/alz.12082. [e‐pub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roberts, P. , Spiros, A. & Geerts, H. A humanized clinically calibrated quantitative systems pharmacology model for hypokinetic motor symptoms in Parkinson's disease. Front. Pharmacol. 7, 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kadra, G. et al Predicting parkinsonism side‐effects of antipsychotic polypharmacy prescribed in secondary mental healthcare. J. Psychopharmacol. 32, 1191–1196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zineh, I. Quantitative systems pharmacology: a regulatory perspective on translation. CPT Pharmacometrics Syst. Pharmacol. 8, 336–339 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]