

Abstract
For the past 150 years, platelets have been recognized as the major blood component that mediates hemostasis and thrombosis. In more recent years, however, we have come to appreciate that platelets also perform profound immune functions during infection with various pathogens. We now recognize that platelets can also mediate a response to various RNA viruses such as influenza and that many viral infections, including severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), can affect platelet count. Thrombocytopenia and increased coagulation have been independently associated with increased mortality. This article provides a perspective on the potential roles of platelets during coronavirus disease 2019.
Keywords: COVID‐19, immunity, platelets, SARS‐CoV‐2, thrombosis
Coronavirus disease 2019 (COVID‐19) has already caused severe health and economic burdens worldwide and, as of May 15, 2020, has claimed 60 300 total deaths in the United States. 1 COVID‐19 presents with various symptoms which include increased inflammatory cytokines, dysregulated coagulation parameters, increased D‐dimer, increased microthrombi and reduced platelet count. 2 In COVID‐19 patients, disseminated intravascular coagulation and reduced platelet count are associated with poor prognosis and increased risk of mortality. 2 , 3 , 4 These clinical observations suggest that platelets may serve important functions during COVID‐19 progression. To understand how the incidence of microthrombosis and reduced platelet count may occur during severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) infection, it is necessary to look at the potential active versus reactive roles of platelets during progression of SARS‐CoV‐2 infection.
The first stage of infection involves viral replication in epithelial cells of the respiratory tract. SARS‐CoV‐2 is a single‐stranded RNA virus classified as β‐coronavirus. It is an upper respiratory virus that, similarly to influenza, becomes problematic when it reaches the lower lung epithelial cells. These cells comprise the lining of the alveolar sacs and are located in very close proximity to the capillary endothelial cells. This proximity established by both cell types is instrumental to gas exchange. In the capillary, platelets are essential in mediating endothelial integrity by performing their hemostatic function. 5 During infection with respiratory viruses, however, the integrity of infected epithelial and endothelial cells becomes compromised, leading to increased permeability that may allow virus to crossover into the circulation. Limited presence of SARS‐CoV2 RNA in blood has been reported in some patients. 6 Of note, viral RNA tested by quantitative polymerase chain reaction (qPCR) does not indicate presence of infectious virus in blood. qPCR can amplify fragmented RNA while plaque assays are appropriate for determination of infectious virus.
Due to their abundance, platelets may be the first blood component to internalize viral particles and induce a response once the pathogen reaches circulation. For example, although viremia (infectious virus in blood) is considered rare, platelets from influenza‐infected patients have been found to contain influenza particles. 7 Influenza, similarly to SARS‐CoV‐2, is a single‐stranded RNA respiratory virus that can infect the alveolar epithelial cells. In human platelets, the initial response to single‐stranded viral RNA (vRNA) is mediated predominantly by Toll‐like receptor 7 (TLR7). 7 , 8 TLR7 is located in the endolysosomes of platelets and requires both internalization of the viral particle and the acidic pH of the endolysosome for proper activation and signaling. 7 , 8 During influenza infection, 1 platelet can internalize many viral particles, which colocalize with TLR7 in the lysosomes. 7 Activation of TLR7 leads to α‐granule release in an AKT‐ and p38‐dependent manner and consequently leads to interaction of platelets with neutrophils via P‐selectin and CD40L. 8 In addition, platelet TLR7 leads to complement C3 release, which pushes neutrophils to release their DNA in the process of netosis. 7 C3‐mediated netosis does not require the attachment of neutrophils to the vascular bed, and netting neutrophils can circulate in blood. 7 Neutrophil extracellular traps (NETs) capture and protect from viral challenge 9 but are also highly prothrombotic and, when dysregulated, may induce intravascular coagulation. Thrombin generated from coagulation in turn can activate C3 and, consequently, the entire proinflammatory complement cascade. 10 In support of intravascular DNA release from neutrophils and potential applicability to this mechanism, sera from patients with COVID‐19 contain highly specific markers for netosis. 11
Netosis at the site of infection is necessary for protection against viral challenge. 9 Conversely, uncontrolled netosis can cause severe cell and organ damage suggesting that limiting the levels of netosis is crucial for vascular maintenance. Interestingly, in addition to C3, influenza‐exposed platelets also secrete granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) in a neutrophil‐dependent manner. 7 GM‐CSF in turn controls the levels of DNA released from neutrophils. 7 Thus, platelets exhibit 2 major functions when single‐stranded vRNA is recognized by TLR7: They initiate netosis, which is necessary to remove virus from the circulation, and they modulate the level of DNA release (Figure 1). This process likely occurs in some form at the start of infection before the host becomes symptomatic. As infection progresses, this C3–NET–GM‐CSF response can become dysregulated and amplified due to increased levels of inflammatory cytokines and tissue damage, and from direct neutrophil activation by the virus. Increased C3 and GM‐CSF have been reported in patients with COVID‐19 12 , 13 ; but the contribution of platelets is yet to be established.
FIGURE 1.
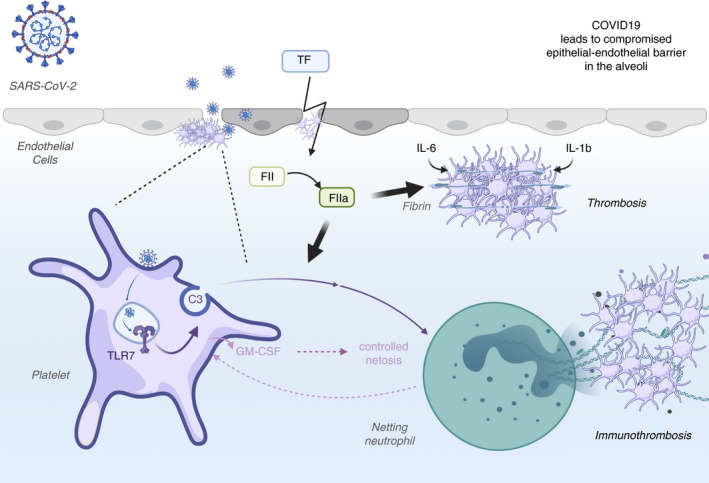
Potential roles of platelets during COVID‐19. As infection progresses and the virus infects the epithelial cells of the alveoli, some of the viral particles have the potential to cross over into the circulation. Once in the circulation, platelets can internalize the virus and the viral single‐stranded RNA can activate TLR7. Platelet TLR7 activation leads to release of complement C3 from their alpha granules. C3 leads to netosis and the released neutrophilic DNA captures any virus that may have been missed by platelets. Platelets may also control the magnitude of netosis by releasing GM‐CSF as a result of a (unknown) signal that comes from neutrophils. As infection progresses, other cells also contribute to the C3 pool and further increase netosis. At this stage, platelets may no longer be able to regulate the level of netosis by secreting GM‐CSF due to their exhaustion or reduction in number. Additionally, infection‐damaged tissue will also lead to tissue factor (TF) release, which will generate thrombin (activated factor II [FIIa]) through the intrinsic coagulation cascade. Thrombin, in turn, can increase fibrin generation (from fibrinogen), direct platelet aggregation, and C3 activation. Inflammatory cytokines such as IL‐1b or IL‐6 can further contribute to platelet aggregation. All of these mechanisms would be of particular relevance in the small vessels where flow velocity is reduced and the protective role of the endothelial lining is compromised due to the direct infection. COVID‐19, coronavirus disease 2019; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; IL, interleukin; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; TLR7, Toll‐like receptor 7
The plausibility of the TLR7‐C3‐netosis mechanism is dependent on internalization of SARS‐CoV‐2. The surface receptor that recognizes SARS‐CoV‐2 is angiotensin‐converting enzyme 2 (ACE2), and binding of the 2 initiates internalization by receptor‐mediated endocytosis. It is unclear if platelets express the ACE2 protein on their surface and whether this expression changes during infection. ACE2 mRNA, however, is not detected by sequencing of human platelets. 14 , 15 Single‐stranded RNA viruses such as influenza can also be internalized by other mechanisms that are independent of receptor recognition. Processes such as micropinocytosis, leading to uptake of large‐sized cargo through endocytic vesicles, and phagocytosis of fragments from apoptotic influenza‐infected cells have been described as a nonspecific point of viral entry into other cell types. 16 , 17 Other mechanisms of internalization specific to platelets may also be involved. It is well accepted that platelets can actively acquire RNA from endothelial cells and internalize circulating vesicles, debris, mitochondria, pollen particles, and so on. 18 The exact mechanisms of internalization by platelets are not clear, but due to their abundance, lack of a nucleus and open canalicular system, the likelihood of platelets sticking to SARS‐CoV‐2 particles is very high. A better understanding of viral entry into platelets could be instrumental in targeting platelet function during COVID‐19.
As previously mentioned, platelets are important in maintaining vascular homeostasis and endothelial integrity in the alveolar capillaries. As infection progresses, shedding of epithelial cells is also observed. In addition to the profound cytokine storm that occurs, some SARS‐CoV‐2 viral particles can reach and infect the endothelial cells that are highly ACE2 positive. 19 Infected endothelium, particularly in the small vessels, where blood flow is much slower than in the big arteries, can lead to thrombosis. 20 Thus, multiple mechanisms may be contributing to thrombosis at this stage of COVID‐19. Direct infection of endothelial cells may lead to damage of the endothelial lining by apoptosis or pyroptosis. As a result, exposed subendothelial matrix could trigger platelets to enter their hemostatic mode, aggregate, and attempt to repair the damage. 18 Even if infection of the endothelium does not occur, the vast tissue inflammation and propensity for netosis described in patients with COVID‐19 11 , 21 can lead to compromised endothelial function and increased expression of adhesion molecules. 20 As more endothelial cells become infected, more platelets will aggregate and form microthrombi in the vessels. The size of thrombus formation and, consequently, thrombus dissolution is limited and controlled by healthy endothelium that would generally release prostaglandins and nitric oxide and increase levels of adenosine. 20 In that sense, if the endothelium is compromised due to direct infection and/or inflammation, it will be unable to limit thrombus formation. Additionally, the damage caused by inflammation and netosis during infection can also lead to tissue factor release. Tissue factor would further amplify the thrombotic response of platelets by generating thrombin and fibrin fibers. Lack of functional endothelium and tissue factor can further contribute to the increased propensity for thrombosis in small vessels presenting as pulmonary embolism and deep vein thrombosis despite anticoagulation treatment. 22
Inflammatory cytokines underlying the etiology of COVID‐19 can directly affect platelet function and further contribute to their thrombotic propensity. The relevance of acute inflammation is even greater when considered in addition to the chronic inflammation underlying obesity and aging (Figure 1). Interleukin (IL)‐6 is increased with obesity and tumor necrosis factor‐α (TNF‐α) is increased with aging in humans. IL‐6, IL‐1b and TNF‐α are also elevated in the plasma of patients with COVID‐19. 12 , 23 IL‐6 or IL‐1b can lead to platelet hyperactivation and spreading, 24 and both of these cytokines can augment agonist‐induced platelet aggregation. 25 , 26 Exposure of whole blood to IL‐6 and IL‐1b may lead to increased hypercoagulability and pathological changes that affect not only platelets but also erythrocytes. 24 TNF‐α, on the other hand, promotes the development of age‐related platelet hyperactivity and consequently increases thrombosis risk. 27 Overall, these inflammatory cytokines can amplify the thrombotic response of platelets and lead to further activation and platelet exhaustion.
As infection progresses and the adaptive immune response begins, there is a possibility that platelets can become further activated by antibodies to the virus similarly to influenza. 28 In this case, platelet‐FcγRIIa recognizes immunoglobulin G antibodies that have scaffolded viral particles and subsequently leads to platelet activation through thrombin generation. 28
In summary, over the course of SARS‐CoV‐2 infection, platelets may perform various functions in concert with neutrophils, erythrocytes, and endothelial cells that can increase the cumulative incidence of thrombosis. At the beginning of infection, platelets may actively induce netosis, which can become dysregulated as infection progresses. Additionally, platelets can react to inflamed and infected endothelium, inflammatory cytokines, and antibody level changes, which can all contribute to amplification of their thrombotic function. Keeping in mind that thrombocytopenia and microthrombosis are difficult to resolve simultaneously, perhaps targeting inflammatory cytokine signaling and netosis during COVID‐19 may be a better approach than providing antiplatelet drugs. Further investigations are necessary to examine the factual role of platelets in response to SARS‐CoV‐2.
RELATIONSHIP DISCLOSURE
The authors declare nothing to report.
AUTHOR CONTRIBUTIONS
MK wrote the manuscript and created the figure using biorender.com.
Koupenova M. Potential role of platelets in COVID‐19: Implications for thrombosis. Res Pract Thromb Haemost. 2020;4:737–740. 10.1002/rth2.12397
Handling Editor: Alisa Wolberg
REFERENCES
- 1. CDC . Daily Updates of Totals by Week and State. Provisional Death Counts for Coronavirus Disease (COVID‐19). Center for Disease Control and Prevention; 2020. [Accessed 2020 May 15] Available from https://www.cdc.gov/nchs/nvss/vsrr/covid19/index.htm
- 2. Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18(4):844–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang X, Yang Q, Wang Y, Wu Y, Xu J, Yu Y, et al. Thrombocytopenia and its association with mortality in patients with COVID‐19. J Thromb Haemost. 2020;18(6):1469–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lippi G, Plebani M, Henry BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID‐19) infections: a meta‐analysis. Clin Chim Acta. 2020;13(506):145–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weyrich AS, Zimmerman GA. Platelets in lung biology. Annu Rev Physiol. 2013;75:569–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang W, Xu Y, Gao R, Lu R, Han K, Wu G, et al. Detection of SARS‐CoV‐2 in different types of clinical specimens. JAMA. 2020;323(18):1843–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun. 2019;10(1):1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, et al. Platelet‐TLR7 mediates host survival and platelet count during viral infection in the absence of platelet‐dependent thrombosis. Blood. 2014;124(5):791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jenne CN, Wong CH, Zemp FJ, McDonald B, Rahman MM, Forsyth PA, et al. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe. 2013;13(2):169–80. [DOI] [PubMed] [Google Scholar]
- 10. Amara U, Rittirsch D, Flierl M, Bruckner U, Klos A, Gebhard F, et al. Interaction between the coagulation and complement system. Adv Exp Med Biol. 2008;632:71–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil extracellular traps in COVID‐19. JCI Insight. 2020;5(11):138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID‐19 infection: a report of five cases. Transl Res. 2020. 10.1016/j.trsl.2020.04.007. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clancy L, Beaulieu LM, Tanriverdi K, Freedman JE. The role of RNA uptake in platelet heterogeneity. Thromb Haemost. 2017;117(5):948–61. [DOI] [PubMed] [Google Scholar]
- 15. Rowley JW, Oler AJ, Tolley ND, Hunter BN, Low EN, Nix DA, et al. Genome‐wide RNA‐seq analysis of human and mouse platelet transcriptomes. Blood. 2011;118(14):e101–e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Edinger TO, Pohl MO, Stertz S. Entry of influenza A virus: host factors and antiviral targets. J Gen Virol. 2014;95(Pt 2):263–77. [DOI] [PubMed] [Google Scholar]
- 17. Hashimoto Y, Moki T, Takizawa T, Shiratsuchi A, Nakanishi Y. Evidence for phagocytosis of influenza virus‐infected, apoptotic cells by neutrophils and macrophages in mice. J Immunol. 2007;178(4):2448–57. [DOI] [PubMed] [Google Scholar]
- 18. Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res. 2018;122(2):337–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID‐19. Lancet. 2020;395(10234):1417–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koupenova M, Kehrel BE, Corkrey HA, Freedman JE. Thrombosis and platelets: an update. Eur Heart J. 2017;38(11):785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barnes BJ, Adrover JM, Baxter‐Stoltzfus A, Borczuk A, Cools‐Lartigue J, Crawford JM, et al. Targeting potential drivers of COVID‐19: neutrophil extracellular traps. J Exp Med. 2020;217(6):e20200652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Helms J, Tacquard C, Severac F, Leonard‐Lorant I, Ohana M, Delabranche X, et al. High risk of thrombosis in patients in severe SARS‐CoV‐2 infection: a multicenter prospective cohort study. Intensive Care Med. 2020. 10.1007/s00134-020-06062-x. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu J, Li S, Liu J, Liang B, Wang X, Wang H, et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS‐CoV‐2 infected patients. EBioMedicine. 2020;55:102763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bester J, Pretorius E. Effects of IL‐1beta, IL‐6 and IL‐8 on erythrocytes, platelets and clot viscoelasticity. Sci Rep. 2016;26(6):32188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oleksowicz L, Mrowiec Z, Zuckerman D, Isaacs R, Dutcher J, Puszkin E. Platelet activation induced by interleukin‐6: evidence for a mechanism involving arachidonic acid metabolism. Thromb Haemost. 1994;72(2):302–8. [PubMed] [Google Scholar]
- 26. Beaulieu LM, Lin E, Mick E, Koupenova M, Weinberg EO, Kramer CD, et al. Interleukin 1 receptor 1 and interleukin 1beta regulate megakaryocyte maturation, platelet activation, and transcript profile during inflammation in mice and humans. Arterioscler Thromb Vasc Biol. 2014;34(3):552–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Davizon‐Castillo P, McMahon B, Aguila S, Bark D, Ashworth K, Allawzi A, et al. TNF‐alpha‐driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood. 2019;134(9):727–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boilard E, Pare G, Rousseau M, Cloutier N, Dubuc I, Levesque T, et al. Influenza virus H1N1 activates platelets through FcgammaRIIA signaling and thrombin generation. Blood. 2014;123(18):2854–63. [DOI] [PubMed] [Google Scholar]