

Abstract
Nucleosides with a bi(hetero)aryl nucleobase have unique potential applications as antiviral drugs and molecular probes. The need for transition metal catalysis to synthesize these nucleosides from pre‐functionalized building blocks and the use of nucleobase protection groups results in expensive and tedious syntheses. Herein we report that 5‐imidazolyl‐uracil can be obtained by scalable Van Leusen imidazole synthesis and regioselectively introduced on ribose to obtain the desired nucleoside in a 5 step synthesis (total yield 55 %). The 5‐imidazolyl moiety leads to improved fluorescence properties. The only side‐product formed was characterized by 2D‐NMR and X‐ray crystallography and could be suppressed during synthesis in favor of the desired product.
Keywords: Fleximers, Fluorescent probes, Nucleobases, Nucleosides, Green chemistry
Bi(hetero)arylic nucleosides have unique potential as antiviral drugs and molecular probes. However, their synthesis is tedious; requiring metal catalysis, pre‐functionalized building blocks, and nucleobase protection groups. Herein we report the scalable, metal‐free synthesis of 5‐imidazolyl‐uracil and its regioselective nucleoside formation. Improved fluorescence and characterization of a new fleximer side product are reported.
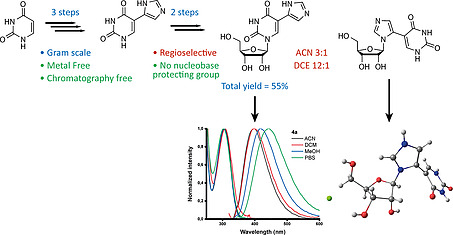
Modified nucleobases, nucleosides, and nucleotides have provided a wide range of biologically active compounds such as antibiotics, anticancer, and antiviral drugs.1 Furthermore, they provide new building blocks to construct and interfere with living systems, with potential applications in synthetic biology.2 A particular class of modified nucleosides are fleximers,3 where the nucleobase has been modified to a bi(hetero)aryl inspired by disconnecting purine nucleobases into their respective imidazole and pyrimidine ring. Both “conventional” and “reverse” fleximers have been defined[3b], the former nucleoside forms the nucleosidic bond with the imidazole ring, the latter with the pyrimidine ring. These are extremely suitable for probing helix and base pair formation.4 Additionally, both reverse and conventional fleximers, have shown promising antiviral activity, including coronaviruses.5 However, the classical way to obtain such biheteroaryl systems occurs through palladium‐catalyzed reactions such as Suzuki and Stille coupling.[3b], 4, 5, 6 Although these methods access a wide scope of compounds, they pose problems when scaling up requiring expensive reagents, pre‐functionalized starting materials (mostly halogens), and safety concerns (as is the case for stannous derivatives).
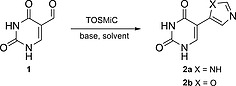
Our research group took particular interest in efficiently obtaining imidazole substituted uracil and uridine as they embody the broad applications of (reverse) fleximers[6b] and the introduction of imidazoles has led to the functionalization of DNA in order to obtain a more stable version of a ribozyme, a DNAzyme.7 Retrosynthetically, we aimed at preparing the substituted base before nucleoside formation, simultaneously avoiding the use of protection groups where possible; instead of the classical, transition metal‐catalyzed route where the heterocycle was coupled to the nucleoside. A literature study only resulting in one paper by Ressner et al.8 where the desired imidazole substituted uracil was reported. However, after the Biginelli‐condensation to obtain 5‐acetyluracil we did not obtain the α‐brominated species using several methods (Br2, NBS, HBr/AcOH) to conduct the subsequent imidazole ring formation with formamide. Therefore another route was investigated starting from commercial uracil; which was hydroxymethylated at the 5‐position and oxidized to the aldehyde 1 using potassium peroxodisulfate and a catalytic amount of silver nitrate, both using previously reported methods.9 The aldehyde was then used to investigate the Van Leusen imidazole synthesis10 using tosylmethyl isocyanide (TOSMiC) (Table 1). To the best of our knowledge, there are no examples of Van Leusen imidazole synthesis performed on a nucleobase, and mostly N‐substituted derivatives are obtained by using primary amines. NH‐analogs are either obtained by deprotection or using ammonium hydroxide as a base. In our system, using commercially available 7 m NH3 in methanol as the base in a pressurized reaction vessel results in the desired product precipitating out of the reaction mixture, which is isolated in good yield without further need for purification (Table 1, Entry 1–5). Scaling up increases the efficiency of the precipitation to yield the desired product (Table 1, Entry 1–5). Using an ammonium hydroxide solution as amine source gives a similar precipitate although less easily filtered, and much is lost during work‐up (Table 1, Entry 6–7). The amine source was exchanged for various other bases in order to obtain the oxazole analogue, however this was unsuccessful (Table 1, Entry 8–11).
Table 1.
Van Leusen reaction on 5‐formyluracil
|
| ||||||
|---|---|---|---|---|---|---|
| Entry | TOSMiC | Base | Solvent | Scale | Yielda [%] | |
| [equiv.] | (equiv.) | [g] | 2a | 2b | ||
| 1 | 3 | NH3 (6) | MeOH | 0.125 | 40 | – |
| 2 | 1.6 | NH3 (3) | MeOH | 0.125 | 48 | – |
| 3 | 1.6 | NH3 (3) | MeOH | 0.25 | 59 | – |
| 4 | 1.6 | NH3 (3) | MeOH | 0.5 | 66 | – |
| 5 | 1.6 | NH3 (3) | MeOH | 1 | 85 | – |
| 6 | 1.6 | NH4OH (3) | MeOH | 0.25 | 23 | – |
| 7 | 1.6 | NH4OH (3) | H2O | 0.25 | Degb | – |
| 8 | 1.6 | K2CO3 (3) | MeOH | 0.25 | – | m.s.c |
| 9 | 1.6 | tBuOK (3) | MeOH | 0.25 | – | m.s.c |
| 10 | 1.6 | Et3N (3) | MeOH | 0.25 | – | m.s.c |
| 11 | 1.6 | DBU (3) | MeOH | 0.25 | – | n.r.d |
Isolated yield.
Degradation of starting material.
Multiple spots, multiple products (monitored by TLC, MS).
No reaction.
Thin‐layer chromatography (TLC) showed a myriad of products forming, an intermediate before elimination of the tosyl group mediated by a base. Harsher conditions only resulted in the degradation of the uracil ring (Table 1, Entry 7–11).
Nucleoside formation with the imidazole substituted uracil was done via the Vorbrüggen method,11 by transiently protecting the nucleobase 2a with trimethylsilyl‐groups using bis(trimethylsilyl)acetamide (BSA). This was immediately reacted (one‐pot) with 1‐O‐acetyl‐2,3,5‐tri‐O‐benzoyl‐d‐ribofuranose, promoted by the addition of a Lewis acid (Scheme 1). At room temperature, no reaction products were found on TLC nor isolated (Table 2, Entry 1), gentle heating at 60 °C (Table 2, Entry 2) showed the formation of two products, a major and a minor one.
Scheme 1.
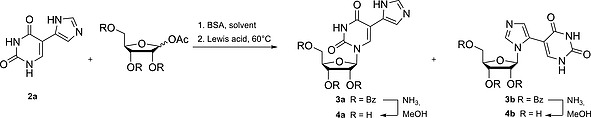
Nucleoside synthesis via Vorbrüggen reaction with 5‐imidazolyl uracil. Detailed reaction conditions for the formation of 3a/3b can be found in Table 2. BSA = bis(trimethylsilyl)acetamide.
Table 2.
Optimization of Vorbrüggen conditions
| Entry | Lewis Acid | Solvent | T | t | Yielda [%] | ||
|---|---|---|---|---|---|---|---|
| [°C] | [d] | 3a | 3b | Ratio (a/b) | |||
| 1 | TMSOTfb | ACN | r.t. | 1 | n.o.d, e | n.d.f | |
| 2 | TMSOTfb | ACN | 60 | 1 | 34 | 11 | 3:1 |
| 3 | TMSOTfb | ACN | 60 | 2 | 58 | 21 | 3:1 |
| 4 | TMSOTfb | DCE | 60 | 2 | 67 | 5 | 13:1 |
| 5 | TMSOTfc | DCE | 60 | 2 | 73 | 9 | 12:1 |
| 6 | SnCl4 b | DCE | 60 | 2 | tracese | n.d.f | |
| 7 | TMSOTfb | THF | 60 | 1 | Inseparable mixtureg | ||
| 8 | TMSOTfb | DMF | 60 | 1 | n.o.d, e | n.d.f | |
Isolated yield.
1 eq.
1.5 equiv.
Not observed.
Monitored by TLC.
Not determined.
More than 6 spots (TLC).
Longer reaction time led to increased product formation, with both products increasing similarly (Table 2, Entry 3). Changing the solvent to DCE shifted the obtained product ratio towards 3a (Table 2, Entry 4). However, it was now necessary to reflux the first silylation step in order to obtain a transparent solution, which was not necessary using ACN as a solvent. Slightly improved yields were obtained for both products when increasing the amount of TMSOTf (Table 2, Entry 5). Another Lewis acid used in nucleosidic bond formation (SnCl4), did not show any significant product formation. THF and DMF were not suitable as solvents for this reaction (Table 2, Entry 7–8). The final products 4a and 4b were obtained by overnight deprotection of the benzoyl groups using 7 m NH3 in MeOH (Scheme 1). They were analyzed by 2D‐NMR to ascertain which regioisomers were formed in the earlier mentioned Vorbrüggen reaction (Supporting Information, SI).
As the designation of the regiochemistry of 4b relied on the possibly ambiguous absence of some 2D‐NMR (HMBC) cross peaks, the structure was confirmed by X‐ray crystallography (CCDC 1996102, Figure S1). Compound 4b was crystallized as HCl salt with the imidazole ring being protonated at the N2 atom (Figure 1, Figure S1). The puckering mode of the ribose ring is C3'‐endo [pseudorotation phase angle P = 9.8(±0.4)°, puckering amplitude τm = 38.1(±0.3)°]. The torsion angle O4–C1–N1–C8 [–139.5(±0.4)°], comparable to the χ angle used to describe the orientation about the glycosyl bond, indicates an anti conformation with the bulk of the substituent pointing away from the sugar.
Figure 1.
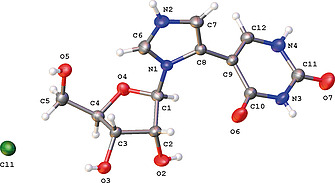
Single crystal X‐ray structure of 4b·HCl with thermal ellipsoids at 50 % probability level.
The dihedral angle between the best planes through the imidazole and uracil ring is 67.4(±0.2)°. The crystal packing is consolidated through a series of O‐H···O/Cl and N‐H···O/Cl interactions between all possible hydrogen‐bond donors and acceptor, except sugar atom O4 (Figure S1, Table S2).
Similar DNA fleximers bearing furan and oxazole moieties have practical applications as a fluorescent probe to measure the polarity at DNA abasic sites.[4a] Therefore the steady‐state spectroscopy of both products (4a and 4b) was investigated in ACN, DCM, MeOH, and phosphate‐buffered saline (PBS). The results are presented in Figure 2, S2, S3 and Table 3, S3, S4. The S0→S1 band of 4a is very similar in all four solvents, no vibrational structure is observed and the position of the maximum varies only slightly from 305 to 308 nm. The latter suggests an apolar ground state. At higher energy, the onset of a very intense transition is observed. Having the same regiochemistry as the previously mentioned fleximers, 4a exhibits similar albeit improved fluorescent properties. A broad and structureless band is observed in all solvents, shifting to longer wavelengths with increasing polarity with similar results for ACN and DCM, in agreement with literature.[4a] This suggests a slightly polar excited state. This is no surprise considering the chromophore resembles a merocyanine moiety. The quantum yield in water (PBS) is increased twofold with respect to a 5‐furan substituted deoxyuridine, from 0.03 to 0.06.[4a] In order to further evaluate its suitability as a sensor, the excited state dynamics of 4a in PBS were evaluated. In Figure S3 the fluorescence decay trace and the corresponding fit are displayed.
Figure 2.
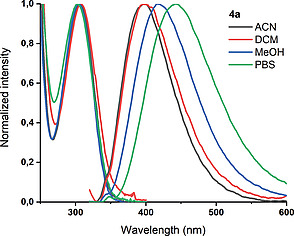
Normalized absorption and emission spectra of 4a (1 × 10–5 m) in acetonitrile (ACN), dichloromethane (DCM), methanol (MeOH), and phosphate‐buffered saline (PBS). The shoulder observed in the emission spectrum at higher energy is due to Raman scattering. The excitation wavelength for the emission spectra was 310 nm.
Table 3.
Summary of absorption and emission properties of 4a in different solvents
| Solvent | λ abs a | λ em b | ν abs – ν em c | FWHMem d | QYe |
|---|---|---|---|---|---|
| [nm] | [nm] | [cm–1] | [cm–1] | [%] | |
| ACN | 306 | 399 | 7610 ± 50 | 5200 ± 50 | 2.3 ± 0.2 |
| DCM | 305 | 400 | 7790 ± 50 | 5400 ± 50 | 3.7 ± 0.4 |
| MeOH | 307 | 417 | 8590 ± 50 | 5460 ± 50 | 5.7 ± 0.6 |
| PBS | 308 | 442 | 9840 ± 50 | 5610 ± 50 | 6.4 ± 0.6 |
Absorption maxima.
Emission maxima.
Stokes' shift.
Full width at half maximum of the emission band.
Fluorescence quantum yield. For all measurements, the concentration was 1 × 10–5 m.
The data could be fitted satisfactorily (χ 2 = 1.015) with a mono‐exponential model, resulting in a lifetime (τ) of 1.88 ns. This uncomplicated behavior is beneficial for a probe and unusual for a nucleoside.12 For 4b on the other hand, the S0→S1 transition is observed at lower wavelengths, with maxima between 270 and 276 nm. No fluorescence could be observed.
Although recent developments have introduced the use of enzymatic techniques[6b], [6c], 13 in accessing the broad scope of (reverse) fleximers, they still rely on the use of transition metal‐catalyzed reactions. Here we show that two isomers, 4a and 4b can be obtained without needing to resort to additional protecting groups and heavy metal catalysis. All this can be achieved in a simple, scalable reaction scheme of 5 steps which obtains 4a in a total yield of 55 % starting from commercially available uracil and involving only a single use of chromatographic separation. The solvent dependency of the regioselective reaction is interesting. Attempts to increase the yield of 3b/4b by means of a different Lewis acid or solvent were unfortunately unsuccessful. Further investigation was beyond the scope of this endeavor but would be interesting as 4b, belongs to the family of distal fleximers whose synthesis still remains non‐trivial.[3a], [6b], 13 The introduction of an imidazole on the 5‐position of uracil (4a), improves the spectroscopic characteristics when compared to furan, thiophene, oxazole, thiazole, and benzimidazole.[4a], 14 Furthermore, it allows the possibility of wobble base pair formation[4b] and catalytic activity.7 On the other hand, isomer 4b is a novel fleximer reminiscent of a disconnected xanthine, of which we provide the first detailed structural determination by 2D‐NMR and XRD. Testing these newly accessed privileged structures against the current COVID19 pandemic is currently underway.
Deposition Number 1996102 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Supporting information
Supporting Information
Acknowledgements
LVM thanks the Hercules Foundation for supporting the purchase of the diffractometer (project AKUL/09/ 0035). We thank Jef Rozenski for recording HRMS; supported by the Hercules Foundation of the Flemish Government (grant 20100225‐7), Luc Baudemprez for recording NMR, CA Mattelaer for graphical help and Chantal Biernaux for editorial help.
References
- 1.a) Jordheim L. P., Durantel D., Zoulim F. and Dumontet C., Nat. Rev. Drug Discovery, 2013, 12, 447; [DOI] [PubMed] [Google Scholar]; b) Serpi M., Ferrari V. and Pertusati F., J. Med. Chem, 2016, 59, 10343–10382. [DOI] [PubMed] [Google Scholar]
- 2. Benner S. A. and Sismour A. M., Nat. Rev. Genet, 2005, 6, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Seley‐Radtke K., Antiviral Chem. Chemother, 2018, 26, 1–12; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zimmermann S. C., Seley‐Radtke K. L., in: Chemical Biology of Nucleic Acids: Fundamentals and Clinical Applications (Eds.: Erdmann V. A., Markiewicz W. T. and Barciszewski J.), Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, pp. 149–165. [Google Scholar]
- 4.a) Greco N. J. and Tor Y., J. Am. Chem. Soc, 2005, 127, 10784–10785; [DOI] [PubMed] [Google Scholar]; b) Rozners E., Smicius R. and Uchiyama C., Chem. Commun, 2005, 5778–5780. [DOI] [PubMed] [Google Scholar]
- 5.a) Peters H. L., Jochmans D., de Wilde A. H., Posthuma C. C., Snijder E. J., Neyts J. and Seley‐Radtke K. L., Bioorg. Med. Chem. Lett, 2015, 25, 2923–2926; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wigerinck P., Pannecouque C., Snoeck R., Claes P., De Clercq E. and Herdewijn P., J. Med. Chem, 1991, 34, 2383–2389; [DOI] [PubMed] [Google Scholar]; c) Yates M. K., Raje M. R., Chatterjee P., Spiropoulou C. F., Bavari S., Flint M., Soloveva V. and Seley‐Radtke K. L., Bioorg. Med. Chem. Lett, 2017, 27, 2800–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Jetter M. C. and Reitz A. B., Synthesis, 1998, 829–831; [Google Scholar]; b) Vichier‐Guerre S., Dugué L., Bonhomme F. and Pochet S., Org. Biomol. Chem, 2017, 15, 8193–8203; [DOI] [PubMed] [Google Scholar]; c) Vichier‐Guerre S., Dugué L., Bonhomme F. and Pochet S., Org. Biomol. Chem, 2016, 14, 3638–3653. [DOI] [PubMed] [Google Scholar]
- 7. Silverman S. K., Acc. Chem. Res, 2015, 48, 1369–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ressner E. C., Fraher P., Edelman M. S. and Mertes M. P., J. Med. Chem, 1976, 19, 194–196. [DOI] [PubMed] [Google Scholar]
- 9. Hudson R. H. E. and Wojciechowski F., Can. J. Chem, 2005, 83, 1731–1740. [Google Scholar]
- 10. Leusen D. V., Leusen A. M. V., in: Organic Reactions, Vol. 57, John Wiley & Sons, Inc., 2004, pp. 417–666. [Google Scholar]
- 11.a) Vorbrüggen H., Krolikiewicz K. and Bennua B., Chem. Ber, 1981, 114, 1234–1255; [Google Scholar]; b) Vorbrüggen H., Ruh‐Pohlenz C., in: Organic Reactions, John Wiley & Sons, Inc., 2004. [Google Scholar]
- 12. Onidas D., Markovitsi D., Marguet S., Sharonov A. and Gustavsson T., J. Phys. Chem. B, 2002, 106, 11367–11374. [Google Scholar]
- 13. Vichier‐Guerre S., Ku T. C., Pochet S. and Seley‐Radtke K. L., ChemBioChem, 2020, 21, 1412–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo P., Xu X., Qiu X., Zhou Y., Yan S., Wang C., Lu C., Ma W., Weng X., Zhang X. and Zhou X., Org. Biomol. Chem, 2013, 11, 1610–1613. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information