

Abstract
Renin–angiotensin system (RAS) blockers are extensively used worldwide to treat many cardiovascular disorders, where they are effective in reducing both mortality and morbidity. These drugs are known to induce an increased expression of angiotensin‐converting enzyme 2 (ACE2). ACE2 acts as receptor for the novel SARS coronavirus‐2 (SARS‐CoV‐2) which raising the important issue of possible detrimental effects that RAS blockers could exert on the natural history and pathogenesis of the coronavirus disease‐19 (COVID‐19) and associated excessive inflammation, myocarditis and cardiac arrhythmias. We review the current knowledge on the interaction between SARS‐CoV‐2 infection and RAS blockers and suggest a scientific rationale for continuing RAS blockers therapy in patients with COVID‐19 infection.
Keywords: cardiovascular system, COVID‐19, SARS coronavirus (CoV)‐2, pandemic, angiotensin‐converting enzyme 2 (ACE2), ACEIs/ARBs, RAAS

Introduction
The past 2 decades have witnessed three lethal zoonotic diseases of humans caused by novel coronaviruses: the severe acute respiratory syndrome (SARS) in 2002, the Middle East respiratory syndrome (MERS) in April 2012, and currently, coronavirus disease‐19 (COVID‐19) caused by SARS‐CoV, MERS‐CoV and SARS‐CoV‐2, respectively. All three coronaviruses are listed in the WHO Blueprint list for priority pathogens [1]. Coronaviruses are members of the Coronaviridae family, a heterogeneous family of RNA viruses that cause respiratory tract infections in humans. HCoV‐229E, HCoV‐NL63, HCoV‐OC43 and HCoV‐HKU1 cause mild infections whilst MERS‐CoV, like SARS‐CoV and SARS‐CoV‐2, can cause a spectrum of clinical manifestations from mild to severe disease and death [2].
The ongoing global pandemic of SARS coronavirus‐2 (SARS‐CoV‐2) has affected almost 3 millions of people in all continents [3]. The high death rate has been attributed to comorbidities of diabetes, hypertension, coronary artery disease amongst others [4]. Renin–angiotensin system (RAS) blockers are extensively used worldwide to treat many cardiovascular disorders, where they are effective in reducing both mortality and morbidity. These drugs are known to induce an increased expression of angiotensin‐converting enzyme 2 (ACE2).
SARS‐CoV‐2 binds to human cells via angiotensin‐converting enzyme 2 (ACE2), which is expressed on epithelial cells of the lung, kidney, intestinal tract and blood vessels [5]. ACE2 expression is increased in patients with diabetes. Hypertension, treated with ACE inhibitors and ARBs, results in an upregulation of ACE2, which theoretically could enhance SARS‐CoV‐2 infection [6]. Therefore, since SARS‐CoV‐2 strongly interacts with ACE2, patients undergoing chronic RAS blocker treatment may be more prone to SARS‐CoV2 infection or develop worse forms of COVID‐19. In this review, we will discuss some aspects of the viral–cell interaction, pathophysiology and the ACEIs/ARBs action mechanism. Lastly, we suggest a scientific rationale for continuing RAS blockers therapy in patients with COVID‐19 infection, thus providing both those clinicians working in the emergency setting and facing with acute cardiovascular diseases, and those following outpatients receiving ACEI/ARBs during the COVID‐19 pandemic, a basis for their therapeutic decision‐making.
Renin–angiotensin–aldosterone system blockade in cardiovascular diseases
As widely described in literature [7], the complex renin–angiotensin system (RAS) plays a crucial role in the pathophysiology of several morbidities, such as hypertension, diabetes mellitus, myocarditis [8], heart failure and myocardial infarction, where the use of RAS blockers is essential for management. ACE inhibitors (ACEIs) and/or angiotensin II receptor blockers (ARBs) delay or prevent the onset of heart failure in asymptomatic patients with left ventricular systolic dysfunction and, importantly, reduce both mortality and morbidity of patients with heart failure and reduced ejection fraction. In these patients, ACEIs/ARBs are titrated to the upper tolerated dose to achieve adequate RAS inhibition [9, 10]. The most recent ESC guidelines recommend employing ACEIs/ARBs as antihypertensive treatment to prevent cardiovascular events and heart failure in diabetic patients, particularly in those with cardiovascular complications [11]. Also, ESC guidelines on hypertension give IA recommendation for employment of ACEIs and ARBs as antihypertensive drugs with demonstrated efficacy in blood pressure and cardiovascular events reduction [12]. Further, hemodynamic changes after an acute myocardial infarction cause a strong activation of both the circulating and the local RAS [13]. This adaptive response [14] could be dangerous and contributes to the development of cardiac structural and electrophysiological remodelling [15, 16]. The effects of RAS inhibition are beneficial [17] in all patients after a myocardial infarction and are more prominent in more severe patients [18]. Further, in one systematic overview including 100,000 patients, it was observed that the most significant short‐term survival benefit occurs if treatment with ACEIs is started within the first 7 days (40% in days 0 to 1, 45% in days 2 to 7, 15% subsequently) [19]. In line with this, guidelines provide class IA/IB recommendation to commence ACEIs/ARBs treatment within the first 24h after the acute event in all ST‐segment elevation myocardial infarction (STEMI) patients with left ventricular systolic dysfunction, heart failure, anterior STEMI or diabetes [18, 20]. Treatment with ACEIs/ARBs is also recommended (class IA/IB) in non‐ST‐segment elevation myocardial infarction (NSTEMI) patients with left ventricular systolic dysfunction, heart failure, hypertension or diabetes mellitus, in order to reduce mortality and risk of recurrent ischaemic event or hospitalization for heart failure [21, 22].
Angiotensin‐Converting enzyme 2 (ACE2)
In the context of SARS‐CoV‐2 infection, it is important to note that ACEIs/ARBs increase the local expression and activity of angiotensin‐converting enzyme 2 (ACE2) [23], a zinc metalloprotease that exists both as a membrane‐associated form and as a secreted form. ACE2 is ubiquitously expressed, with the highest levels of transcripts being found in the lung, the heart and the gastrointestinal and urinary systems. ACE2 is mostly expressed in lung alveolar type II (AT2) cells, myocardial cells, vascular endothelia, buccal epithelial cells, T and B lymphocytes, on the proximal tubule epithelial cells in the kidney, and in testes [24, 25]. All cardiac cell components (i.e. endothelial cells, smooth muscle cells, cardiac myocytes, fibroblasts and macrophages) express ACE2, although the highest levels were observed in vascular cells, especially pericytes [26, 27]. Upregulation of ACE2 expression has been documented in patients affected by myocardial infarction or heart failure. However, in animal models of ageing, pressure overload and chronic ischaemia, ACE2 downregulation has been associated with cardiac hypertrophy, adverse remodelling and fibrosis [28, 29, 30]. Conversely, ACE2 expression is reduced in hypertension, advanced diabetes mellitus and atherosclerotic plaques and, experimentally, by both angiotensin II (Ang II) and endothelin [28, 29, 30, 31]. Importantly, it has been estimated that heritability accounts for 67% of the variability in ACE2 circulating levels. Moreover, ACE2 genetic variants have been associated with both essential hypertension and coronary heart disease (CHD), whilst they act as disease modifiers of both hypertrophic and dilated cardiomyopathies [31]. Concerning development, it was shown that ACE2 protein levels and activity are high during gestation and decline postnatally [32].
ACE2 acts as a negative regulator of RAS, by degrading AngII to the heptapeptide angiotensin 1‐7 (Ang1‐7) [33], that exerts its biological activity via the Mas receptor, modulating the release of nitric oxide (NO), antagonizing the intracellular effect of AngII stimulation (thus tempering the production of reactive oxygen species), at least in part by activating intracellular phosphatases [34]. As a result, Ang1‐7 exerts many positive effects on the cardiovascular system (e.g. increased endothelial function, reduced fibrosis, anti‐proliferative effects on smooth muscle cells, anti‐cardiac hypertrophy), as well as on other organs, such as the lungs, where it exerts anti‐fibrotic, anti‐inflammatory and anti‐apoptotic effects [35, 36, 37]. Moreover, ACE2 cleaves other peptides and plays a central role in inactivating des‐arginine(9)‐bradykinin (des‐Arg(9)‐BK), a potent metabolite of the kinin–kallikrein system that increases vascular permeability, thus promoting angioedema, acting on B1 type receptors, which are, in turn, upregulated by inflammatory cytokines [38, 39]. According to some authors, the leakage of plasma in the subendothelial compartment in conjunction with the inflammatory response triggered by the host–virus interaction may have a central pathogenetic role in the endotheliitis and disseminated intravascular coagulation that have been documented to occur in COVID‐19 patients [39, 40, 41]. ACE2 plays an important role in heart failure, in diabetic microvascular or macrovascular disease [42] and in inflammatory lung disease [43], and ACEIs/ARBs‐mediated increase in ACE2 exerts a protective role in many conditions such as atherosclerosis, myocardial infarction, myocardial dysfunction, heart failure, diabetes mellitus and acute lung injury (ALI).
SARS‐COV‐2 Infection and host injury
Although the details have not been completely elucidated yet, SARS‐CoV‐2 was originated either as a result of natural selection that took place in a host before zoonotic transfer or by natural selection in humans after the zoonotic transfer had occurred [44, 45]. Importantly, during the selection process, the virus acquires a furin cleavage site and three predicted O‐linked glycans around the site that are very likely associated with the acquisition of a highly pathogenic phenotype [44].
Virus – entry and spread
Coronaviruses’ (SARS‐CoV, SARS‐CoV‐2) Spike (S) protein, binds with high affinity to human ACE2 [45] (Figure 1). Before SARS‐CoV‐2 and SARS‐CoV cell entry, their S protein is subjected to a priming process via serine protease TMPRSS2 in order permit the attachment of viral particles to ACE2 and thus on cell surface [46]. This entry mechanism is confirmed by the fact that TMPRSS2 inhibition or TMPRSS2‐KO mice show both decreased, though not abolished, S protein priming, and reduced viral entry, spread, and inflammatory chemokine and cytokine release [46, 47]. SARS‐CoV‐2 is then internalized mainly by endocytosis. In this process, phosphatidylinositol 3‐phosphate 5‐kinase (PIKfyve), two pore channel subtype 2 (TPC2) and cathepsin L are critical for virus entry [48]. SARS‐CoV binding also induces the release/shedding of catalytically active ACE2 ectodomains via ADAM‐17⁄TACE activity [49, 50, 51, 52]. In line, inhibition of ADAM17/TACE blocks ACE2 shedding and limits viral entry in both in vitro and in vivo [53]. Intriguingly, recent data show that soluble ACE2 (sACE2) could inhibit S‐mediated infection in vitro. Although sACE2 was not able to completely abrogate viral entry, ACE2 shedding may help in contrasting viral diffusion to different organs [54]. Conversely, it has been demonstrated that ACE2 downregulation following infection has a detrimental effect in the pathogenesis of ALI [55], although this effect could be limited, judging by the amount of ACE2 expressed by the AT2 cells of the lungs, or the ratio between ACE2 and circulating sACE2 [24, 56, 57]. Altogether, these results suggest that ACE2 might not be the only receptor for SARS‐CoV‐2. In fact, recent structural and predictive studies proposed other receptor‐mediated mechanisms for SARS‐CoV‐2 cell entry, indicating three other aminopeptidases, alanyl aminopeptidase (ANPEP), glutamyl aminopeptidase (ENPEP) and dipeptidyl peptidase 4 (DPP4), as the most probable additional receptors [58, 59].
Figure 1.
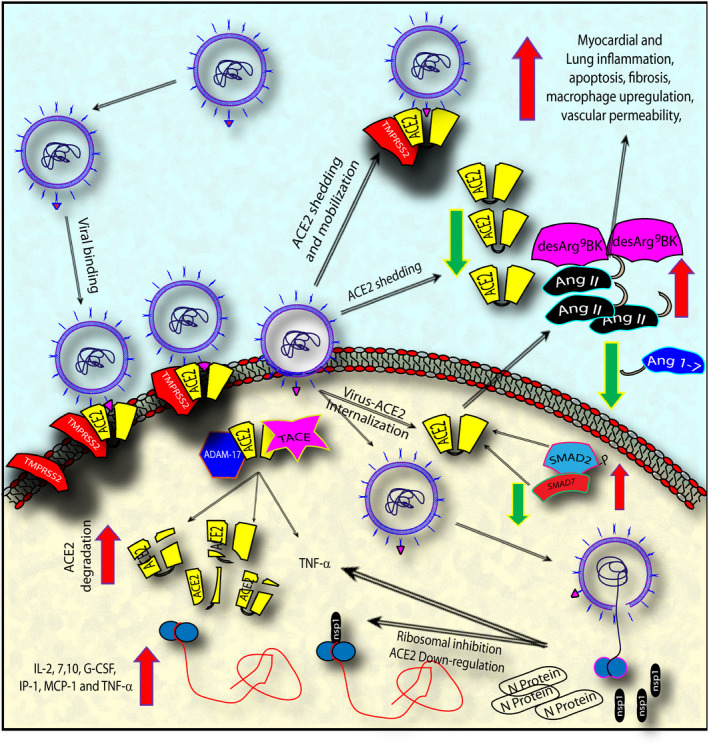
The figure summarizes SARS‐CoV, and presumably SARS‐CoV‐2, confirmed and hypothesized mechanism of viral binding, internalization and shedding via ACE2 interaction, and the successive downregulative effect on ACE2 and the resulting pathogenic effect on lung and cardiac tissues. Abbreviations: angiotensin‐converting enzyme 2 (ACE2); angiotensin (Ang); angiotensin 1‐7 (Ang 1‐7); transmembrane protease, serine 2 (TMPRSS2); ADAM metallopeptidase domain 17 (ADAM‐17); tumour necrosis factor‐α converting enzyme (TACE); SMAD family member2, 7 (SMAD2, SMAD7); nonstructural protein 1 (nsp‐1); SARS‐CoV nucleocapsid (N) protein (N protein); interleukin‐2, 7, 10 (IL‐2, 7,10); granulocyte colony‐stimulating factor (G‐CSF); transforming growth factor‐α (TNF‐α); monocyte chemoattractant protein 1 (MCP‐1).
Virus‐direct negative effects
It is known that the virus survival strategy is to elude and suppress host innate immune defences through gene deactivation or inhibition [57, 60, 61]. In line, Huang et al. demonstrated that SARS‐CoV nonstructural protein (nsp1) binding to ribosomes inhibits host gene translation [62]. More precisely, a marked downregulation of ACE2 mRNA and protein expression was associated with the presence of SARS‐CoV and (HCoV)‐NL63 (another human coronavirus, causing mild to severe respiratory tract infections, excluding SARS) in target tissues [63]. However, whilst SARS‐CoV induced ACE2 shedding from the cell surface, (HCoV)‐NL63 did not, suggesting that ACE2 shedding is directly related with prognostic severity [30, 52, 53]. Additionally, a viral titre‐ and infection time‐related SARS‐CoV effect on ACE2 mRNA has been shown, where lower viral titre is associated with upregulation instead, higher dosage and time after infection are correlated to reduced ACE2 expression [64]. Last, since interleukin (IL)‐2, IL‐7, IL‐10, G‐CSF, IP‐1 MCP‐1 MIP‐1a and TNF‐α are highly increased in patients with severe SARS‐CoV infection, with respect to patients with mild symptoms, it was hypothesized that ACE2 suppression could result from a direct cell driven effect, rather than virus mediated. In line, in vitro data could support a suppressive role for IL7 and IL2 on Ace2 expression [64].
Acute Lung Injury – ALI
Part of the conundrum related to the role of ACE2 in SARS‐CoV‐2 infection (designated coronavirus disease‐19 or COVID‐19) is related to the role exerted by this enzyme in ALI. Indeed, ALI that can be caused by a number of aetiological agents (e.g. influenza and coronaviruses, bacterial pneumonia, sepsis [65], cigarette smoking [66], particulate matter [67] and aspiration pneumonia [68]) has been associated with the activation of the local RAS [69]. Ang II, by acting on AngII type 1 receptor (AT1R), activates several signal transduction pathways, including mitogen‐activated protein kinase (MAPK) and Janus‐activated kinase (JAK)/ signal transducer and activator of transcription 3 (STAT3), phosphatidylinositol 3 kinase (PI3K), promoting vascular permeability, vasoconstriction, myofibroblast, smooth muscle cell and macrophage activation, fibrosis, and the expression of inflammatory cytokines [65, 70, 71]. The latter effect has been mainly attributed to the ability of AngII to activate the transcription factor NFκB via AT1R [72]. AngII may also bind to other receptors, the most studied of which is type 2 receptor (AT2R). However, the net effect of AT2R stimulation is less clear since both pro‐ and anti‐inflammatory effects have been described [57]. To corroborate the crucial role exerted by pulmonary RAS on ALI, it was shown that AngII levels are increased in animal models of ALI [73]. Importantly, recent reports on SARS‐CoV‐2‐infected patients showed a significant increase in Ang II plasma level that was inversely correlated with viral load [74]. In line, survivors of acute respiratory distress syndrome (ARDS) have been associated with lower plasmatic ACE levels, in a preliminary report [75]. Moreover, by evaluating the levels of angiotensinogen metabolites, it was shown that ARDS outcome was associated with higher Ang1‐9/Ang1‐10 and Ang1‐7/Ang1‐10 ratios, suggesting higher activity of both ACE and ACE2 in survivors [76]. Conversely, AT1R blockage resulted in the protection of animal models of lung injury [77]. Further, an accumulating body of literature is showing the protective role exerted by ACE2 on ALI. Indeed, ACE2 knockout animals develop more severe forms of ALI in response to a series of injurious stimuli [67, 73, 75]. Conversely, the results of clinical trials that experimented the administration of soluble ACE2 to treat patients suffering from ARDS, although in early phase and not designed to test efficacy, did not show very promising results [78]. Moreover, as anticipated, ACE2 downregulation causes also accumulation of des‐Arg(9)‐BK, which interacts with type B1 bradykinin receptor, possibly leading to the observed development of angioedema, and coagulation cascade triggering [39]. Last, the protective effect of ACE2 may be also postulated considering both that ACE2 levels decrease with age and that, although children are vulnerable to the infection, these patients are usually less severe [79].
Myocardial injury
The relevance of myocardial involvement in SARS‐CoV‐2 infection is related to the high frequency of patients showing evidence of acute cardiac injury, especially amongst those that necessitated ICU care [80]. This finding is similar to that observed in patients that succumbed to the SARS crisis in Toronto, where the duration of disease was almost 10 times shorter in patients that showed cardiac involvement vs. those that had no evidence of cardiac infection [30]. In line, a recent report suggests that the SARS‐CoV‐2 infection can be complicated by acute fulminant myocarditis [81]. In this scenario, left ventricular function completely recovered early after immunomodulation therapy [82] suggesting that organ damage in these patients is more the consequence of a 'cytokine storm', than of an uncontrolled viral replication [80, 82, 83]. To complete the picture, the presence of viral particles infecting myocardial interstitial cells, but neither endothelial cells nor myocytes, was recently described to occur in one patient that died of COVID‐19, with a clinical scenario characterized by respiratory distress, hypotension and cardiogenic shock [84].
Consistently, SARS‐CoV‐2‐infected patients were characterized by high amounts of plasmatic IL1β, IFNγ, IP10 and MCP1 levels. Additionally, more severe patients had higher concentrations of G‐CSF, IP10, MCP1, MIP1A and TNF‐α, strongly supporting the relevance of cytokines in dictating prognosis [80]. This observation has stimulated early trials experimenting IL6 axis inhibition in COVID‐19 patients [82]. Cytokine‐mediated systemic response to infections can be associated with transient cardiac dilatation and dysfunction [85]. However, local viral replication could depress cardiac function too. Indeed, SARS‐CoV can activate the NLRP3 inflammasome [86], thus promoting the release of IL1β and possibly IL18, a mediator known to cause systolic impairment [87]. Notably, extrapulmonary, multiorgan SARS‐CoV dissemination at the time of death was described in patients who died of SARS during the Toronto outbreak [88] and SARS‐CoV viral RNA was detected in 35‐40% of cardiac autopsy samples [30, 88]. Histology studies conducted on these hearts showed cardiomyocyte hypertrophy, fibrosis and an inflammatory infiltrate, mainly constituted by CD68+ macrophages [30]. However, necropsy of 39‐year‐old woman who died from SARS‐CoV, with severe left ventricular dysfunction at echocardiographic evaluation, has shown slight left ventricular enlargement without evidence of interstitial lymphocytic infiltrate or cardiomyocyte necrosis [89]. Importantly, in one postmortem cardiac specimen of a 50‐year‐old patient who died from SARS‐CoV‐2 infection, only limited interstitial mononuclear cell infiltrates without significant injury were described [90]. Similar findings, characterized by CD68+ macrophage infiltrates showing evidence of a cytopathic effect, were recently reported in a case report of a 69‐year‐old patient that died from COVID‐19 [84]. The paucity of lymphocyte infiltrate observed in these reports may be explained by the severe lymphopenia that accompanies COVID‐19 [80].
The mechanism of negative effect on myocardial tissue of SARS‐CoV infection was hypothesized to be related to both ACE2 downregulation and cytokine storm [63, 82]. As outlined above, ACE2 expression may be reduced both by the binding of SARS‐CoV to ACE2, leading to the endocytosis of the latter [91], and as a result of the activation of ADAM‐17/TACE by SARS spike protein, known to cleave, internalize and promote ACE2 shedding in endothelial cells [49]. Downregulation of ACE2 results in an unbalance between the detrimental action of AngII and the cardioprotective effects of Ang1‐7. Further, more chronic negative consequences on the cardiovascular system might be related to ACE2 downregulation via des‐Arg(9)‐BK accumulation and its consequential increase in vascular permeability, inflammation that leads to an increased atherosclerotic plaques formation [92]. Further, ADAM‐17⁄TACE activation, induced by the spike protein, promotes TNF‐α release, thus increasing macrophage mobilization and activation [49, 52]. Moreover, SARS‐CoV nucleocapsid (N) protein potentiates TGF‐β‐mediated fibrosis [93].
ACEIs/ARBs treatment during the COVID‐19 pandemic
On the basis of the increase of ACE2, associated with ACEIs/ARBs treatment, it has been speculated that these drugs might rise the chances of severe COVID‐19 [94]. According to the last report (29th April 2020) from the Italian Istituto Superiore di Sanità, 25.452 patients positive to SARS‐CoV‐2 died. The observed mortality was higher amongst males in all age groups and in patients with comorbidities. In 9.23% of the total sample, data regarding comorbidities were available; 81.7% of the deceased patients had 2 or more comorbidities, such as arterial hypertension (69.2%), diabetes mellitus (31.8%) and CHD (28.2%). Similar data regarding comorbidities and gender differences in mortality were observed during China COVID‐19 emergency [95]. Prior to hospitalization, 24% of COVID‐19‐positive deceased Italian patients were on treatment with ACEIs and 16% were receiving ARBs.
The gender difference in mortality could be related to the smoking prevalence and sex‐based immunological differences. Moreover, a recent study suggests that ACE2 expression seems to be higher in women than men and to decrease with age [64, 96, 97]. The more severe prognosis of patients of older age may also been related to distinct mechanisms, such as the age‐associated decline in the molecular machinery involved in removing cytosolic DNA [98]. Also, patients with hypertension or diabetes show high rate of ACE2 genetic polymorphism that could influence ACE2 or Ang1–7 levels in the human body [64, 99, 100, 101]. Therefore, a negative correlation between ACE2 expression and mortality has been suggested [64].
Should indications for ACEIs/ARBs treatment change for patients with COVID‐19?
An enormous body of literature shows that ACEIs/ARBs should be given as early as possible to all patients with acute myocardial infarction, in asymptomatic patients with left ventricular systolic dysfunction, in patients with heart failure and reduced ejection fraction, as well in hypertensive or diabetic patients, for their important benefits for patient survival. Conversely, an abrupt discontinuation of ACEIs, in survivors of myocardial infarction on long‐term treatment [102, 103] or in patients with heart failure [104], can be potentially harmful. Further, at the time of this pandemic COVID‐19, there are no experimental or strong clinical data to contraindicate this therapy whilst, on the contrary, some are supporting the opposite [105, 106].
Calcium channel blockers were suggested as alternative treatment for ACEIs and ARBs [107]. However, since the development and progression of the remodelling process after an myocardial infarction, in setting of heart failure, hypertension or diabetes mellitus depends on neurohormonal activation, only medications, which block the effect of neurohormones [108], have demonstrated to reduce morbidity and mortality of patients. For calcium channel blockers, in spite of their beneficial hemodynamic activities, there is no evidence supporting their ability to exert favourable effect early after an acute myocardial infarction on preventing heart failure in diabetics with hypertension and on general mortality reduction [18, 109, 110]. Lastly, calcium channel blockers have a strong interaction with antiviral drugs (lopinavir/ritonavir) used to treat COVID‐19 patients.
In a recent retrospective observational study from Wuhan, including 188 hypertensive subjects affected by COVID‐19, treatment ACEIs/ARBs was associated with lower all‐cause mortality [105]. Furthermore, a large population‐based case–control study performed in Lombardy during the ongoing COVID‐19 epidemic spread on more than 6000 COVID‐19 patients did not show evidence that the use of either ARBs, or ACEIs, affects the risk of COVID‐19 infection [111]. Italian Regulatory Agency (AIFA) recommended on 17 March 2020 do not to modify the therapy in progress with anti‐hypertensives (whatever the therapeutic class) in well‐controlled hypertensive patients, as they expose frail patients to potential new side effects or an increase in risk of adverse cardiovascular events does not appear justified. For the same reasons, compared to the hypothesis of using ACE inhibitors and sartans also in healthy people for prophylactic purposes, remembered that these drugs should be used exclusively for the treatment of pathologies for which there are approved [112].
Conclusions
As depicted in Figure 1, the interaction between SARS‐CoV‐2 and the host cell promotes a series of complex alterations leading to both the downregulation of ACE2 expression and the increased production and release of inflammatory cytokines. The net loss of the ACE2/Ang1‐7 anti‐inflammatory properties, along with a dysregulated vascular permeability ACE2/des‐Arg(9)‐BK mediated and the pro‐inflammatory properties of secreted cytokines, may be therefore responsible for a large part of the detrimental effects caused by COVID‐19. Taking into account, the evidences presented in this review, and at present, there are no data demonstrating adverse outcomes amongst COVID‐19 patients in therapy with ACE/ARBs; on the contrary, it seems that the treatment discontinuation could have plausible negative effects. Thus, according with the recommendations of the main cardiovascular international [113, 114] and Italian scientific societies [115, 116], patients chronically taking ACEI/ARBs who contract COVID‐19 infection should not discontinue the treatment, neither temporarily (Figure 2). Further researches are needed to indagate some uncleared and controversial aspects of the pandemics.
Figure 2.
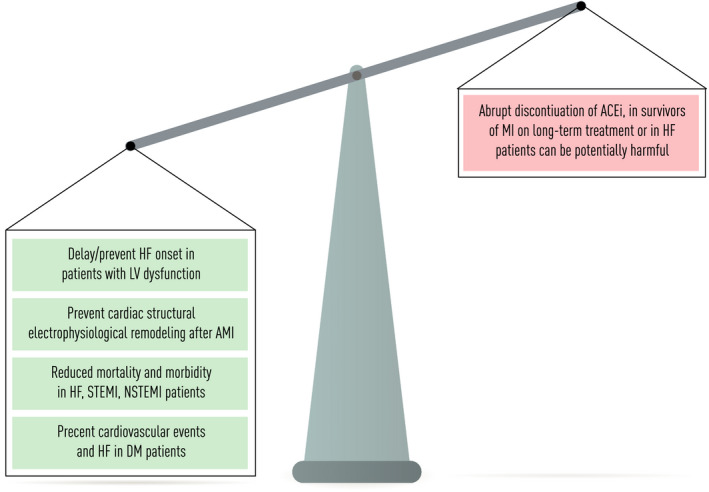
ACEIs/ARBs effects in cardiovascular disease. Abbreviations: heart failure (HF); left ventricular systolic disfunction (LVSD); acute myocardial infarction (AMI); ST‐segment elevation myocardial infarction (STEMI); non‐ST‐segment elevation myocardial infarction (NSTEMI); diabetes mellitus (DM); ACE inhibitors (ACEIs), myocardial infarction (MI).
Author contribution
Aneta ALEKSOVA: Conceptualization (lead); Writing‐original draft (lead); Writing‐review & editing (lead). Federico Ferro: Writing‐original draft (supporting); Writing‐review & editing (supporting). Giulia Gagno: Writing‐original draft (supporting). Chaira Cappelletto: Writing‐original draft (supporting). Daniela Santon: Writing‐original draft (supporting). Maddalena Rossi: Writing‐original draft (supporting). Giuseppe Ippolito: Supervision (supporting); Writing‐original draft (supporting); Writing‐review & editing (equal). Alimuddin I Zumla: Supervision (equal); Writing‐original draft (supporting); Writing‐review & editing (equal). Antonio Paolo Beltrami: Conceptualization (supporting); Supervision (equal); Writing‐original draft (supporting); Writing‐review & editing (equal). Gianfranco Sinagra: Supervision (equal); Validation (equal).
Conflict of interest statement
All authors declare no conflicts of interest.
Acknowledgements
AZ and GI are members of the Pan‐African Network on Emerging and Re‐Emerging Infections (PANDORA‐ID‐NET – https://www.pandora‐id.net/) funded by the European and Developing Countries Clinical Trials Partnership the EU Horizon 2020 Framework Programme for Research and Innovation. Sir Zumla is in receipt of a National Institutes of Health Research senior investigator award. Professor Ippolito is supported by the Italian Ministry of Health (Ricerca Corrente Linea 1).
Aleksova A, Ferro F, Gagno G, Cappelletto C, Santon D, Rossi M, Ippolito G, Zumla A, Beltrami AP, Sinagra G (University of Trieste, Trieste; National Institute for Infectious Diseases Lazzaro Spallanzani ‐ IRCCS, Rome, Italy; University College London, London; University College London Hospitals NHS Foundation Trust, London, UK; and University of Udine, Udine, Italy). COVID‐19 and renin‐angiotensin system inhibition: role of angiotensin converting enzyme 2 (ACE2) ‐ Is there any scientific evidence for controversy? (Review). J Intern Med 2020; 288: 410–421. 10.1111/joim.13101
References
- 1. WHO . A research and development Blueprint for action to prevent epidemics. 2020. https://www.who.int/blueprint/en/.
- 2. Memish ZA, Perlman S, Van Kerkhove MD, Zumla A. Middle East respiratory syndrome. Lancet 2020; 395: 1063–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. WHO . Coronavirus. 2020. https://www.who.int/health‐topics/coronavirus#tab=tab_1.
- 4. Zhou F, Yu T, Du R et al. Clinical course and risk factors for mortality of adult inpatients with COVID‐19 in Wuhan, China: a retrospective cohort study. Lancet 2020; 395: 1054–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade‐long structural studies of SARS coronavirus. J Virol 2020; 947:e00127‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guo T, Fan Y, Chen M et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID‐19). JAMA Cardiol 2020.e201017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Patel S, Rauf A, Khan H, Abu‐Izneid T. Renin‐angiotensin‐aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed Pharmacother 2017; 94: 317–25. [DOI] [PubMed] [Google Scholar]
- 8. Watanabe K, Arumugam S, Thandavarayan RA. Experimental autoimmune myocarditis: role of renin angiotensin system. Myocarditis 2011; 309–20. [Google Scholar]
- 9. Ponikowski P, Voors AA, Anker SD et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016; 37: 2129–200. [DOI] [PubMed] [Google Scholar]
- 10. Yancy CW, Jessup M, Bozkurt B et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J Card Fail 2017; 23: 628–51. [DOI] [PubMed] [Google Scholar]
- 11. Cosentino F, Grant PJ, Aboyans V et al. 2019 ESC Guidelines on diabetes, pre‐diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur Heart J 2020; 41: 255–323. [DOI] [PubMed] [Google Scholar]
- 12. Williams B, Mancia G, Spiering W et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur Heart J 2018; 39: 3021–104. [DOI] [PubMed] [Google Scholar]
- 13. Dargie HJ, Byrne J. Pathophysiological aspects of the renin‐angiotensin‐aldosterone system in acute myocardial infarction. J Cardiovasc Risk 1995; 2: 389–95. [DOI] [PubMed] [Google Scholar]
- 14. Greenberg B. An ACE in the hole alternative pathways of the renin angiotensin system and their potential role in cardiac remodeling. J Am Coll Cardiol 2008; 52: 755–7. [DOI] [PubMed] [Google Scholar]
- 15. De Mello WC. Novel aspects of angiotensin II action in the heart. Implications to myocardial ischemia and heart failure. Regul Pept 2011; 166: 9–14. [DOI] [PubMed] [Google Scholar]
- 16. Padoan L, Beltrami AP, Stenner E et al. Left ventricular adverse remodeling after myocardial infarction and its association with vitamin D levels. Int J Cardiol 2019; 277: 159–65. [DOI] [PubMed] [Google Scholar]
- 17. Jalowy A, Schulz R, Dorge H, Behrends M, Heusch G. Infarct size reduction by AT1‐receptor blockade through a signal cascade of AT2‐receptor activation, bradykinin and prostaglandins in pigs. J Am Coll Cardiol 1998; 32: 1787–96. [DOI] [PubMed] [Google Scholar]
- 18. Ibanez B, James S, Agewall S et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST‐segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST‐segment elevation of the European Society of Cardiology (ESC). Eur Heart J 2018; 39: 119–77. [DOI] [PubMed] [Google Scholar]
- 19. Indications for ACE inhibitors in the early treatment of acute myocardial infarction: systematic overview of individual data from 100,000 patients in randomized trials. ACE Inhibitor Myocardial Infarction Collaborative Group. Circulation 1998; 97: 2202–12. [DOI] [PubMed] [Google Scholar]
- 20. O'Gara PT, Kushner FG, Ascheim DD et al. 2013 ACCF/AHA guideline for the management of ST‐elevation myocardial infarction: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013; 127: 529–55. [DOI] [PubMed] [Google Scholar]
- 21. Roffi M, Patrono C, Collet JP et al. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST‐segment elevation: Task Force for the Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST‐Segment Elevation of the European Society of Cardiology (ESC). Eur Heart J 2016; 37: 267–315. [DOI] [PubMed] [Google Scholar]
- 22. Amsterdam EA, Wenger NK, Brindis RG et al. 2014 AHA/ACC guideline for the management of patients with non‐ST‐elevation acute coronary syndromes: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 130: e344–426. [DOI] [PubMed] [Google Scholar]
- 23. Ferrario CM, Jessup J, Chappell MC et al. Effect of angiotensin‐converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin‐converting enzyme 2. Circulation 2005; 111: 2605–10. [DOI] [PubMed] [Google Scholar]
- 24. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single‐cell RNA‐seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019‐nCoV infection. Front Med 2020; 14: 185–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu H, Zhong L, Deng J et al. High expression of ACE2 receptor of 2019‐nCoV on the epithelial cells of oral mucosa. Int J Oral Sci 2020; 12: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Burrell LM, Risvanis J, Kubota E et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur Heart J 2005; 26(4): 369–75; discussion 22–4. [DOI] [PubMed] [Google Scholar]
- 27. Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS‐CoV‐2. Cardiovasc Res 2020; 116: 1097–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gallagher PE, Ferrario CM, Tallant EA. Regulation of ACE2 in cardiac myocytes and fibroblasts. Am J Physiol Heart Circ Physiol 2008; 295: H2373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tikellis C, Thomas MC. Angiotensin‐Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int J Pept 2012; 2012: 256294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oudit GY, Kassiri Z, Jiang C et al. SARS‐coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Invest 2009; 39: 618–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fan Z, Wu G, Yue M et al. Hypertension and hypertensive left ventricular hypertrophy are associated with ACE2 genetic polymorphism. Life Sci 2019; 225: 39–45. [DOI] [PubMed] [Google Scholar]
- 32. Song R, Preston G, Yosypiv IV. Ontogeny of angiotensin‐converting enzyme 2. Pediatr Res 2012; 71: 13–9. [DOI] [PubMed] [Google Scholar]
- 33. Ferrario CM, Chappell MC, Tallant EA, Brosnihan KB, Diz DI. Counterregulatory actions of angiotensin‐(1–7). Hypertension 1997; 30: 535–41. [DOI] [PubMed] [Google Scholar]
- 34. Chappell MC, Marshall AC, Alzayadneh EM, Shaltout HA, Diz DI. Update on the Angiotensin converting enzyme 2‐Angiotensin (1–7)‐MAS receptor axis: fetal programing, sex differences, and intracellular pathways. Front Endocrinol (Lausanne) 2014; 4: 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Santos RA. Angiotensin‐(1–7). Hypertension 2014; 63: 1138–47. [DOI] [PubMed] [Google Scholar]
- 36. Meng Y, Li T, Zhou GS et al. The angiotensin‐converting enzyme 2/angiotensin (1–7)/Mas axis protects against lung fibroblast migration and lung fibrosis by inhibiting the NOX4‐derived ROS‐mediated RhoA/Rho kinase pathway. Antioxid Redox Signal 2015; 22: 241–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang W, Bodiga S, Das SK, Lo J, Patel V, Oudit GY. Role of ACE2 in diastolic and systolic heart failure. Heart Fail Rev 2012; 17: 683–91. [DOI] [PubMed] [Google Scholar]
- 38. Vickers C, Hales P, Kaushik V et al. Hydrolysis of biological peptides by human angiotensin‐converting enzyme‐related carboxypeptidase. J Biol Chem 2002; 277: 14838–43. [DOI] [PubMed] [Google Scholar]
- 39. van de Veerdonk F, Netea MG, van Deuren M et al. Kinins and Cytokines in COVID‐19: A Comprehensive Pathophysiological Approach. Preprints 2020.2020040023 [Google Scholar]
- 40. Varga Z, Flammer AJ, Steiger P et al. Endothelial cell infection and endotheliitis in COVID‐19. Lancet 2020; 395: 1417–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Salvagno GL, Lippi G, Gelati M, Poli G, Favaloro EJ. Analytical performance of the new D‐dimer and antithrombin assay on Roche cobas t 711 analyzer. Int J Lab Hematol 2019; 41: e54–e6. [DOI] [PubMed] [Google Scholar]
- 42. Soro‐Paavonen A, Gordin D, Forsblom C et al. Circulating ACE2 activity is increased in patients with type 1 diabetes and vascular complications. J Hypertens 2012; 30: 375–83. [DOI] [PubMed] [Google Scholar]
- 43. Zhang X, Zheng J, Yan Y et al. Angiotensin‐converting enzyme 2 regulates autophagy in acute lung injury through AMPK/mTOR signaling. Arch Biochem Biophys 2019; 672: 108061. [DOI] [PubMed] [Google Scholar]
- 44. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS‐CoV‐2. Nat Med 2020; 26: 450–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhou P, Yang XL, Wang XG et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020; 579: 270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hoffmann M, Kleine‐Weber H, Schroeder S et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020; 181: 271–280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Iwata‐Yoshikawa N, Okamura T, Shimizu Y, Hasegawa H, Takeda M, Nagata N. TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J Virol 2019; 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang H, Yang P, Liu K et al. SARS coronavirus entry into host cells through a novel clathrin‐ and caveolae‐independent endocytic pathway. Cell Res 2008; 18: 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Haga S, Yamamoto N, Nakai‐Murakami C et al. Modulation of TNF‐alpha‐converting enzyme by the spike protein of SARS‐CoV and ACE2 induces TNF‐alpha production and facilitates viral entry. Proc Natl Acad Sci U S A 2008; 105: 7809–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jia HP, Look DC, Tan P et al. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am J Physiol Lung Cell Mol Physiol 2009; 297: L84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lambert DW, Hooper NM, Turner AJ. Angiotensin‐converting enzyme 2 and new insights into the renin‐angiotensin system. Biochem Pharmacol 2008; 75: 781–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Glowacka I, Bertram S, Herzog P et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J Virol 2010; 84: 1198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Haga S, Nagata N, Okamura T et al. TACE antagonists blocking ACE2 shedding caused by the spike protein of SARS‐CoV are candidate antiviral compounds. Antiviral Res 2010; 85: 551–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hofmann H, Geier M, Marzi A et al. Susceptibility to SARS coronavirus S protein‐driven infection correlates with expression of angiotensin converting enzyme 2 and infection can be blocked by soluble receptor. Biochem Biophys Res Commun 2004; 319: 1216–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kuba K, Imai Y, Rao S et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus‐induced lung injury. Nat Med 2005; 11: 875–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Danser AHJ, Epstein M, Batlle D. Renin‐angiotensin system blockers and the COVID‐19 pandemic. Hypertension 2020; 75: 1382–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 2004; 203: 631–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vankadari N, Wilce JA. Emerging WuHan (COVID‐19) coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes Infect 2020; 9: 601–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Qi F, Qian S, Zhang S, Zhang Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem Biophys Res Commun 2020;526( 1):135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li W, Moore MJ, Vasilieva N et al. Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003; 426: 450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ni G, Ma Z, Damania B. cGAS and STING: At the intersection of DNA and RNA virus‐sensing networks. PLoS Pathog 2018; 14: e1007148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Huang C, Lokugamage KG, Rozovics JM, Narayanan K, Semler BL, Makino S. SARS coronavirus nsp1 protein induces template‐dependent endonucleolytic cleavage of mRNAs: viral mRNAs are resistant to nsp1‐induced RNA cleavage. PLoS Pathog 2011; 7: e1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dijkman R, Jebbink MF, Deijs M et al. Replication‐dependent downregulation of cellular angiotensin‐converting enzyme 2 protein expression by human coronavirus NL63. J Gen Virol 2012; 93: 1924–9. [DOI] [PubMed] [Google Scholar]
- 64. Chen J, Jiang Q, Xia X et al. Individual variation of the SARS‐CoV2 receptor ACE2 gene expression and Regulation. 2020. [DOI] [PMC free article] [PubMed]
- 65. Jia H. Pulmonary angiotensin‐converting enzyme 2 (ACE2) and inflammatory lung disease. Shock 2016; 46: 239–48. [DOI] [PubMed] [Google Scholar]
- 66. Hung YH, Hsieh WY, Hsieh JS et al. Alternative Roles of STAT3 and MAPK Signaling Pathways in the MMPs Activation and Progression of Lung Injury Induced by Cigarette Smoke Exposure in ACE2 Knockout Mice. Int J Biol Sci 2016; 12: 454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lin CI, Tsai CH, Sun YL et al. Instillation of particulate matter 2.5 induced acute lung injury and attenuated the injury recovery in ACE2 knockout mice. Int J Biol Sci 2018; 14: 253–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mandell LA, Niederman MS. Aspiration Pneumonia. N Engl J Med 2019; 380: 651–63. [DOI] [PubMed] [Google Scholar]
- 69. Marshall RP. The pulmonary renin‐angiotensin system. Curr Pharm Des 2003; 9: 715–22. [DOI] [PubMed] [Google Scholar]
- 70. Husain K, Hernandez W, Ansari RA, Ferder L. Inflammation, oxidative stress and renin angiotensin system in atherosclerosis. World J Biol Chem 2015; 6: 209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jeong J, Lee J, Lim J et al. Soluble RAGE attenuates AngII‐induced endothelial hyperpermeability by disrupting HMGB1‐mediated crosstalk between AT1R and RAGE. Exp Mol Med 2019; 51: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Xianwei W, Magomed K, Ding Z et al. Cross‐talk between inflammation and angiotensin II: studies based on direct transfection of cardiomyocytes with AT1R and AT2R cDNA. Exp Biol Med 2012; 237: 1394–401. [DOI] [PubMed] [Google Scholar]
- 73. Imai Y, Kuba K, Rao S et al. Angiotensin‐converting enzyme 2 protects from severe acute lung failure. Nature 2005; 436: 112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Liu Y, Yang Y, Zhang C et al. Clinical and biochemical indexes from 2019‐nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci 2020; 63: 364–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Annoni F, Cortés DO, Irazabal M et al. Angiotensin converting enzymes in patients with acute respiratory distress syndrome. Intens Care Med Exp 2015; 3: A91. [DOI] [PubMed] [Google Scholar]
- 76. Reddy R, Asante I, Liu S et al. Circulating angiotensin peptides levels in Acute Respiratory Distress Syndrome correlate with clinical outcomes: A pilot study. PLoS One 2019; 14: e0213096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nahmod KA, Nahmod VE, Szvalb AD. Potential mechanisms of AT1 receptor blockers on reducing pneumonia‐related mortality. Clin Infect Dis 2013; 56: 1193–4. [DOI] [PubMed] [Google Scholar]
- 78. Vrigkou E, Tsangaris I, Bonovas S, Tsantes A, Kopterides P. The evolving role of the renin‐angiotensin system in ARDS. Crit Care 2017; 21: 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Dong Y, Mo X, Hu Y et al. Epidemiological characteristics of 2143 pediatric patients with 2019 coronavirus disease in China. Pediatrics 2020. [Google Scholar]
- 80. Huang C, Wang Y, Li X et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020; 395: 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hu H, Ma F, Wei X, Fang Y. Coronavirus fulminant myocarditis saved with glucocorticoid and human immunoglobulin. Eur Heart J 2020; ehaa190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. Hlh Across Speciality Collaboration UK. COVID‐ 19: consider cytokine storm syndromes and immunosuppression. Lancet 2020; 395: 1033–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Peiris JS, Chu CM, Cheng VC et al. Clinical progression and viral load in a community outbreak of coronavirus‐associated SARS pneumonia: a prospective study. Lancet 2003; 361: 1767–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tavazzi G, Pellegrini C, Maurelli M et al. Myocardial localization of coronavirus in COVID‐19 cardiogenic shock. Eur J Heart Fail 2020.1: 10.1002/ejhf.1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Parrillo JE. Pathogenetic mechanisms of septic shock. N Engl J Med 1993; 328: 1471–7. [DOI] [PubMed] [Google Scholar]
- 86. Shi CS, Nabar NR, Huang NN, Kehrl JH. SARS‐Coronavirus Open Reading Frame‐8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov 2019; 5: 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Toldo S, Mezzaroma E, O'Brien L et al. Interleukin‐18 mediates interleukin‐1‐induced cardiac dysfunction. Am J Physiol Heart Circ Physiol 2014; 306: H1025–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Farcas GA, Poutanen SM, Mazzulli T et al. Fatal severe acute respiratory syndrome is associated with multiorgan involvement by coronavirus. J Infect Dis 2005; 191: 193–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Li SS, Cheng CW, Fu CL et al. Left ventricular performance in patients with severe acute respiratory syndrome: a 30‐day echocardiographic follow‐up study. Circulation 2003; 108: 1798–803. [DOI] [PubMed] [Google Scholar]
- 90. Xu Z, Shi L, Wang Y et al. Pathological findings of COVID‐19 associated with acute respiratory distress syndrome. Lancet Respirat Med 2020; 8: 420–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wang S, Guo F, Liu K et al. Endocytosis of the receptor‐binding domain of SARS‐CoV spike protein together with virus receptor ACE2. Virus Res 2008; 136: 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Liu T, Liu H, Feng L, Xiao B. Kinin B1 receptor as a novel, prognostic progression biomarker for carotid atherosclerotic plaques. Mol Med Rep 2017; 16: 8930–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhao X, Nicholls JM, Chen YG. Severe acute respiratory syndrome‐associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor‐beta signaling. J Biol Chem 2008; 283: 3272–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zheng YY, Ma YT, Zhang JY, Xie X. COVID‐19 and the cardiovascular system. Nat Rev Cardiol 2020; 17: 259–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Guan WJ, Ni ZY, Hu Y et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 2020; 382: 1708–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Liu J, Ji H, Zheng W et al. Sex differences in renal angiotensin converting enzyme 2 (ACE2) activity are 17beta‐oestradiol‐dependent and sex chromosome‐independent. Biol Sex Differ 2010; 1: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Xie X, Chen J, Wang X, Zhang F, Liu Y. Age‐ and gender‐related difference of ACE2 expression in rat lung. Life Sci 2006; 78: 2166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Takahashi A, Loo TM, Okada R et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun 2018; 9: 1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Luo Y, Liu C, Guan T et al. Correction: Association of ACE2 genetic polymorphisms with hypertension‐related target organ damages in south Xinjiang. Hypertens Res 2019; 42: 744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Xu HY, Liu MM, Wang X, He XY. Association of angiotensin‐converting enzyme insertion/deletion polymorphism with type 1 diabetic nephropathy: a meta‐analysis. Ren Fail 2016; 38: 1320–7. [DOI] [PubMed] [Google Scholar]
- 101. Liu D, Chen Y, Zhang P et al. Association between circulating levels of ACE2‐Ang‐(1–7)‐MAS axis and ACE2 gene polymorphisms in hypertensive patients. Medicine (Baltimore) 2016; 95: e3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Westendorp B, Schoemaker RG, Buikema H, Boomsma F, van Veldhuisen DJ, van Gilst WH. Progressive left ventricular hypertrophy after withdrawal of long‐term ACE inhibition following experimental myocardial infarction. Eur J Heart Fail 2006; 8: 122–30. [DOI] [PubMed] [Google Scholar]
- 103. Westendorp B, Schoemaker RG, van Gilst WH, Buikema H. Improvement of EDHF by chronic ACE inhibition declines rapidly after withdrawal in rats with myocardial infarction. J Cardiovasc Pharmacol 2005; 46: 766–72. [DOI] [PubMed] [Google Scholar]
- 104. Halliday BP, Wassall R, Lota AS et al. Withdrawal of pharmacological treatment for heart failure in patients with recovered dilated cardiomyopathy (TRED‐HF): an open‐label, pilot, randomised trial. Lancet 2019; 393: 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zhang P, Zhu L, Cai J et al. Association of inpatient use of angiotensin converting enzyme inhibitors and angiotensin II receptor blockers with mortality among patients with hypertension hospitalized with COVID‐19. Circ Res 2020; 10.1161/CIRCRESAHA.120.317134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Battistoni A, Volpe M. Might renin‐angiotensin system blockers play a role in the COVID‐19 pandemic? Eur Heart J Cardiovasc Pharmacother 2020; 10.1093/ehjcvp/pvaa030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Fang L, Karakiulakis G, Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID‐19 infection? Lancet Respirat Med 2020; 8: e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Sorbets E, Steg PG, Young R et al. beta‐blockers, calcium antagonists, and mortality in stable coronary artery disease: an international cohort study. Eur Heart J 2019; 40: 1399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Grossman E, Messerli FH. Are calcium antagonists beneficial in diabetic patients with hypertension? Am J Med 2004; 116: 44–9. [DOI] [PubMed] [Google Scholar]
- 110. Cohn JN, Ziesche S, Smith R et al. Effect of the calcium antagonist felodipine as supplementary vasodilator therapy in patients with chronic heart failure treated with enalapril: V‐HeFT III. Vasodilator‐Heart Failure Trial (V‐HeFT) Study Group. Circulation 1997; 96: 856–63. [DOI] [PubMed] [Google Scholar]
- 111. Mancia G, Rea F, Ludergnani M, Apolone G, Corrao G. Renin–angiotensin–aldosterone system blockers and the risk of covid‐19. N Engl J Med 2020; 10.1056/NEJMoa2006923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. AIFA . Precisazioni AIFA su Malattia da coronavirus Covid‐19 ed utilizzo di ACE‐Inibitori e Sartani. 2020. https://www.aifa.gov.it/‐/precisazioni‐aifa‐su‐malattia‐da‐coronavirus‐covid‐19‐ed‐utilizzo‐di‐ace‐inibitori‐e‐sartani.
- 113. ESC . Position Statement of the ESC Council on Hypertension on ACE‐Inhibitors and Angiotensin Receptor Blockers. 2020. https://www.escardio.org/Councils/Council‐on‐Hypertension‐(CHT)/News/position‐statement‐of‐the‐esc‐council‐on‐hypertension‐on‐ace‐inhibitors‐and‐ang.
- 114. HFSA/ACC/AHA . Statement Addresses Concerns Re: Using RAAS Antagonists in COVID‐19. 2020. https://www.acc.org/latest‐in‐cardiology/articles/2020/03/17/08/59/hfsa‐acc‐aha‐statement‐addresses‐concerns‐re‐using‐raas‐antagonists‐in‐covid‐19. [DOI] [PMC free article] [PubMed]
- 115. (SIIA) SIdIA . Terapia farmacologica Antipertensiva e COVID‐19: la posizione della SIIA ribadita da Società Scientifiche Europee ed Internazionali. 2020. https://siia.it/wp‐content/uploads/2020/03/Statetment‐SIIA.pdf.
- 116. (SIC) SIdC . GUIDA CLINICA COVID‐19 PER CARDIOLOGI. 2020. https://www.sicardiologia.it/public/Documento‐SIC‐COVID‐19.pdf.