

Abstract
The recent pandemic of severe acute respiratory syndrome–coronavirus2 (SARS-CoV-2) infection (COVID-19) has put the world on serious alert. The main protease of SARS-CoV-2 (SARS-CoV-2-MPro) cleaves the long polyprotein chains to release functional proteins required for replication of the virus and thus is a potential drug target to design new chemical entities in order to inhibit the viral replication in human cells. The current study employs state of art computational methods to design novel molecules by linking molecular fragments which specifically bind to different constituent sub-pockets of the SARS-CoV-2-MPro binding site. A huge library of 191678 fragments was screened against the binding cavity of SARS-CoV-2-MPro and high affinity fragments binding to adjacent sub-pockets were tailored to generate new molecules. These newly formed molecules were further subjected to molecular docking, ADMET filters and MM-GBSA binding energy calculations to select 17 best molecules (named as MP-In1 to MP-In17), which showed comparable binding affinities and interactions with the key binding site residues as the reference ligand. The complexes of these 17 molecules and the reference molecule with SARS-CoV-2-MPro, were subjected to molecular dynamics simulations, which assessed the stabilities of their binding with SARS-CoV-2-MPro. Fifteen molecules were found to form stable complexes with SARS-CoV-2-MPro. These novel chemical entities designed specifically according to the pharmacophoric requirements of SARS-CoV-2-MPro binding pockets showed good synthetic feasibility and returned no exact match when searched against chemical databases. Considering their interactions, binding efficiencies and novel chemotypes, they can be further evaluated as potential starting points for SARS-CoV-2 drug discovery. 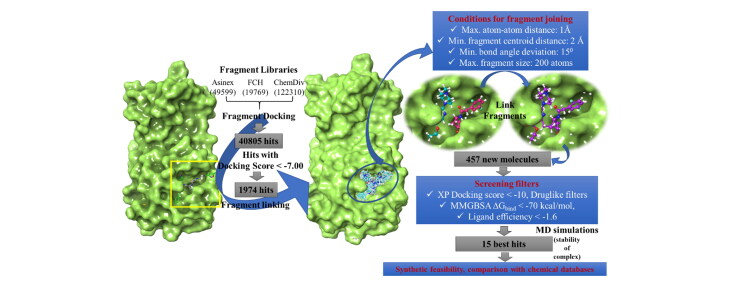
Communicated by Ramaswamy H. Sarma
Keywords: Covid-19, main protease, fragment-based drug discovery, docking, molecular dynamics
Introduction
The recent pandemic of novel corona virus infection (COVID19) has put the world on alert. This is caused by a positive sense RNA virus of coronaviridae family and nidovirales order which is known to cause respiratory tract infections in mammals including humans. A recent form of the virus, the novel coronavirus has emerged in china and has been named as SARS-CoV-2 because of the acute respiratory distress syndrome developing with these cases in infections which become severe with course of time. This is a zoonotic corona virus mediated disease which is third incidence after SARS and MERS (Gu et al., 2020) The source has later been shown to have sequence homology as high as 96% with SARS-CoV of bats (Xu et al., 2020). According to the latest WHO reports (https://www.who.int/emergencies/diseases/novel-coronavirus-2019/events-as-they-happen), 4234821 confirmed cases has been reported with 285913 deaths around a total of 166 countries which includes 70768 confirmed cases and 2294 deaths in India. This pandemic is spreading exponentially and has become an issue of serious concern for the whole world. In the absence of any specific drugs and treatment measures, WHO is emphasizing on hand washing, personal protection, use of hand sanitizers and social distancing and isolation for prevention of spread of disease and contamination. This has proven effective in some countries to curtail the spread and they are still in phase 1 and 2 of the epidemic spread. However, in certain parts of the world it has become an issue of major and immediate concern due to advanced phases of epidemic. Possible treatment strategies and methods have become urgent needs for the world. Various possible drug treatments have been used with some success and some negative studies. Repurposing the existing drugs as low hanging fruits is being used as the first strategy in search of a possible treatment for the disease. The various drugs tried so far are oseltamivir (Li et al., 2020), systemic steroids in severe respiratory involvement which is still inconclusive (Khot & Nadkar, 2020), Lopinavir/Ritonavir with both positive (Bhatnagar et al., 2020; Cao et al., 2020; Muralidharan et al., 2020), chloroquine (Sahraei et al., 2020) phosphate/Hydroxychloroquine (Zhou et al., 2020). Also, some results have been seen with Ramdesivir (Al-Tawfiq et al., 2020; Ko et al., 2020), Tocilizumab, RNA polymerase inhibitors like Favipiravir (Al-Tawfiq et al., 2020) and JAK-STAT inhibitors like Baricitinib, Fedratinib and ruxolitinib (Dong et al., 2020). The 32 kb long RNA genome of SARS-CoV-2 (Lu et al., 2020) codes for its structural proteins such as spike glycoprotein which facilitates the entry of the virus into the host cells through interaction with the host enzyme ACE2 (Hasan et al., 2020), the nucleocapsid (Lu et al., 2020), envelope (Gupta et al., 2020) and other membrane proteins and the non-structural proteins such as the chymotrypsin like main protease (Jin et al., 2020) which cleaves the long polyprotein chains to release functional proteins required for replication. Thus, these proteins can be exploited as potential drug targets and hunting for new chemical entities (NCEs) with fewer side effects is the need of the hour to combat COVID19 (Boopathi et al., 2020). However, successful discovery of NCEs will hugely depend on proper understanding of the structure, interactions and dynamics of validated targets and the unexplored potential of their binding sites to bind new chemotypes (Boopathi et al., 2020). Computational methods have become indispensable for infectious disease drug discovery in last few decades (Njogu et al., 2016) not only to understand the drug-target interactions (Choudhury et al., 2014; Njogu et al., 2016; Schuler et al., 2017) and delineate the structure activity relationship of small druglike molecules (Gahtori et al., 2019; Srivastava et al., 2012), but also for screening huge chemical libraries providing a fast and less expensive alternative to the traditional high throughput screening (Choudhury et al., 2015, 2016; Murgueitio et al., 2012). The recent literature reports several interesting computational approaches including computational drug repurposing on the TMPRSS2 (Elmezayen et al., 2020), reverse vaccinology (Hasan et al., 2019), in silico screening of novel guanosine derivatives against MERS CoV polymerase ((Elfiky & Azzam 2020, Elfiky, 2020a), ayurvedic anti-tussive medicines, anti-viral phytochemicals and synthetic anti-virals against SARS-CoV-2 MPro, ACE-2 and RNA dependent RNA polymerase (RdRp) (Joshi et al., 2020; Elfiky, 2020b), in silico investigation of natural product compounds against the substrate-binding domain β of cell-surface heat shock protein A5, which reported to be the recognition site for the SARS-CoV-2 spike (Elfiky, 2020), in silico study of binding potency of different Saikosaponins with targets NSP15 and fusion spike glycoprotein (Sinha et al., 2020) and computational evaluation of stilbene based compounds such as resveratrol, as anti-COVID-19 drug candidates acting through disruption of the spike proteins (Wahedi et al., 2020). Among a plethora of computational drug design strategies, fragment based de novo design of molecules has gained immense popularity (Coutard et al., 2014; Hoffer et al., 2011; Kumar et al., 2019; Loving et al., 2010). Usually, fragment hits show very high binding affinity with the receptors pertaining to their size. As starting points, they provide profound opportunity for subsequent optimization leading to chemical entities with improved pharmacokinetic properties compared to molecules obtained as hits from high throughput screenings (Hoffer et al., 2011; Loving et al., 2010). Another major plus point of fragments is the low molecular complexity as compared to that of drug-like molecules, and thus reducing the search space to be explored (Hoffer et al., 2011). Further, possibility of an exponentially huge number of combinatorial molecules by linking high affinity fragments ensures novel drug like chemotypes. So, with experimental high throughput screenings, simultaneous development of new computational fragment screening strategies would surely prove useful to significantly reduce the number of molecules to be tested experimentally (Kanakaveti et al., 2020). In this study, we have considered the main protease of the SARS-CoV-2 (SARS-CoV-2- MPro) as our target of interest for fragment-based design of new inhibitors (Jin et al., 2020). As the protease binding pocket is a large cavity with three to four prominent sub pockets, it provides an interesting scope for screening fragments for these sub pockets and then linking them to design new molecules with optimal binding with the protease. With the arrival of the very first structure (6LU7) of this protein in PDB (Jin et al., 2020), several groups have come up with interesting strategies such as artificial intelligence based de novo design (Bung et al., 2020), repurposing existing drugs that can bind this protein (Khan et al., 2020) or virtually screening large chemical databases to identify peptide like small molecules (Pant et al., 2020), natural products such as Moroccan medicinal plants products (Aanouz et al., 2020), against this protein (Islam et al., 2020; Khan et al., 2020; Sarma et al., 2020), identification of Andrographolide as a potential inhibitor of SARS-CoV-2 main protease through in silico screening (Enmozhi et al., 2020) using molecular docking, molecular dynamics simulations and PCA based quantitative structure-activity relationship (QSAR) for pattern recognition of the best ligands (Islam et al., 2020), to mention a few. Till date RCSB protein data bank (PDB) reports more than 80 structures of the SARS-Cov-2 MP binding more than 80 different fragment like molecules (https://doi.org/10.2210/pdb5R7Y/pdb). These fragments bind to a particular sub-pocket of the large binding pocket of SARS-Cov-2 main protease. These recent additions in PDB have further strengthened our idea of designing inhibitors through fragment linking.
Materials and methods
Preparation of datasets
Crystal structure of SARS-CoV-2-MPro
More than 80 different crystal structures of the SARS-CoV-2- MPro bound to diverse ligands have been deposited in PDB till date. For this study, we have considered one 6LU7, the first one to be deposited in PDB which binds a potential peptide-based inhibitor N3 (Jin et al., 2020). The Protein Preparation Wizard (PPW) module (Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, 2019) of Schrödinger software package, version 2019-2 was used to pre-process the macromolecular structure downloaded from PDB. Missing hydrogens were added, water molecules beyond 5 Å of the active site were removed and appropriate bond orders were assigned to the structure. The residues/side chains unresolved in some of the crystal structures were repaired with prime (Prime, Schrödinger, LLC, New York, NY, 2019) module in the PPW the protonation states of the polar residues were optimized with the protassign module of PPW, which uses PROPKA to predict pKa values (pH 7.0 ± 2.0) and side chain functional group orientations. The structure was then subjected to restrained minimization (cutoff RMSD 0.3 Å) with impref to avoid steric clashes. The prepared structure was further used for preparation of grids, molecular docking and molecular dynamics (MD) simulations.
Fragment libraries
Fragment structures were downloaded in the form of .sdf files from 4 different publicly available libraries, viz., Asinex fragments and building blocks (http://www.asinex.com/fragments/), FCH group’s (http://fchgroup.net/fragment-libraries.php) ‘all purpose’ fragment library, fluorine fragment library, fragment like acids, fragment like amines, fragment like amino acids, high fsp3 fragment library, spiro fragments and FCH special selection of fragment library and ChemDiv (https://www.chemdiv.com/) fragment library. A consolidated set of 191678 unique fragments were verified for the ‘rule of 3′ (Najjar et al., 2019) agreement and considered for fragment-based design. All the fragments were subjected to preparation in LigPrep (LigPrep, Schrödinger, LLC, New York, NY, 2019), generating their ionization states at pH 7.0 (± 2.0) using Epik ionizer.
Fragment screening
The prepared structure of 6LU7.pdb was directly used for receptor grid generation. ‘Receptor Grid Generation’ module of Schrödinger was utilized to define interaction grids for molecular docking keeping the centroid of the peptide like cocrystal-ligand as grid the centre. The size of the interaction grid was fixed to 14 Å for inner box and 20 Å as outer box. Docking enrichment analysis was performed with Glide module of Schrödinger software (Glide, Schrödinger, LLC, New York, NY, 2019.) package. 76 unique fragment structures, bound to different crystal structures of SARS-CoV-2-MPro were used as the actives and the respective decoy set of 1362 molecules with similar size downloaded from the protease subset of DUD.E (Mysinger et al., 2012) database. These 1438 (1362 decoy + 76 active) molecules were subjected to Glide SP docking with the active site grid of SARS-CoV-2-MPro and the resultant docked poses were used for enrichment calculations. Upon observing a satisfactory ROC value, the fragment library with 191678 fragments was subjected to docking calculations. Molecular docking calculations were performed using the Glide module of Schrödinger software package with standard precision (SP) mode. 3 best poses were generated for each fragment. OPLS_2005 force field (Shivakumar et al., 2010) was used for docking with all default parameters.
Fragment joining and de novo compound design
Top fragment hits with docking score < −7.00 were retained from each SP fragment-docking calculations for further design. The ‘combine fragments’ panel from the library design module was used for direct joining of the fragments prepositioned at different regions of the SARS-CoV-2- MPro binding site to design new compounds. The panel joins the fragments by identifying feasible bonds that can be formed between the fragments. The angle between the bond directions were set to be 15°. The maximum distances between the two bonding atoms from different fragments were set to be 1 Å, while the minimum distance between the centroids two fragments was set to be 2 Å. All bonds attached to hydrogen or halogens in a fragment were chosen for breaking and re-joining to another fragment. All atoms of the newly built molecule were subjected to minimization. 3 rounds of such fragment joining were performed where, in the first-round pairs of fragments were joined and in the next round the resultants of the first round were used as inputs to combine up to 4 fragments and so on.
Molecular docking and binding energy estimation
All the newly formed molecules were docked to the binding site of SARS-CoV-2- MPro using Glide module of Schrodinger Suite. The same grid that was used for fragment screening was used for this docking too. Docking was performed in two sub-steps i.e. the SP docking and an extra precision (XP) docking (Friesner et al., 2006). 5 best docked poses were generated for each newly designed molecule and OPLS_2005 force field was used for docking with all default parameters. The resultant complexes of the molecules with SARS-CoV-2-MPro were further submitted for binding energy estimation, where Molecular Mechanics-Generalized Born Surface Area (MM/GBSA) based binding free energy (ΔGbind) were computed for the complexes using Prime module.
Evaluation of physicochemical and pharmacokinetic properties and synthetic accessibility
QuickProp module (QikProp, Schrödinger, LLC, New York, NY, 2019) of Schrodinger were employed to calculate the drug like properties and predict the physicochemical and pharmacokinetic (absorption, distribution, metabolism, excretion and toxicity) properties of all the new molecule hits selected in the previous section. A synthetic accessibility score was also predicted for each molecule from SwissADME server (Daina et al., 2017). All the molecules which violate no drug likeliness rules were identified and then, 17 best molecules were selected based on their ΔGbind and ligand efficiency.
Molecular dynamics (MD) simulations
MD simulations were carried out on the complexes of SARS-CoV-2-MPro with 17 selected molecules and the crystal structure 6LU7 binding the reference molecule N3, using the Desmond MD simulation package (release 2018) of Schrodinger (Desmond Research, 2018). The OPLS_2005 force field was employed for the protein-ligand complexes. Using the system builder tool of Desmond, the complexes were solvated in a cubical water box (TIP3P water model) keeping 12 Å buffer space in x, y and z dimensions. Each system was neutralized by adding appropriate counter ions and an ionic concentration of 0.15 M was maintained by adding Na+ and Cl− ions. The systems were minimized with 10000 steepest descent steps followed by gradual heating from 0 to 300 K, under NVT ensemble. The systems were thermally relaxed before the production run using Nose-Hoover Chain thermostat method for 5 ns and 5 ns of pressure relaxation with Martyna-Tobias-Klein barostat method.
Finally, 50 ns production run under NPT ensemble was carried out for each system using a cutoff distance of 12 Å for non-bonded interactions. Coordinates were saved at each 10 ps to generate trajectories of 5000 frames each. Simulation interaction diagrams were used for trajectory analyses. Figure 1 shows the overall workflow of the study.
Figure 1.

Overall workflow of the study.
Results and discussion
Binding site of SARS-CoV-2-MPro and the experimental structures
Crystal structure of SARS-CoV-2- MPro reveals that the overall structure of SARS-CoV-2-MPro is a combination of three domains (Jin et al., 2020). The first and second domains (DI and DII) have an antiparallel β-barrel structure, where residue 8-100 comprise the DI and residues 102 to 184 form the DII. Residues 201 to 303 form the third domain (DIII) which is a combination of 5 α-helices and the connecting link between DII and DIII is a long loop (L1) formed by residues 185 to 200. The substrate binding site of SARS-CoV-2-MPro is situated at the junction between DI and DII, which also extents up to L1. Figure S1 shows the domains and the overall secondary structure of the protein. Figure 2 shows the binding site sub-pockets and interactions of N3 with SARS-CoV-2-MPro.
Figure 2.

Positioning of N3 in the binding site sub-pockets and molecular interactions of N3 with SARS-CoV-2-MPro, from the structure 6LU7.
As of now 88 different crystal structures of SARS-CoV-2-MPro have been reported to PDB, most of which are from the PanDDA analysis group depositions [36], where each of the structures binds a unique ligand at different cavities all over the protein structure. Some of these small fragment-like ligands binding to the substrate binding site, mostly occupy one or two sub-pockets of the huge substrate binding cavity. A quick look at the positioning of the peptide-based ligand N3 in 6LU7.pdb reveals that, different constituent fragments of N3 comfortably occupy almost all sub-pockets of the huge binding site exploiting the potential of the binding cavity to bind bigger molecules. N3 makes H-bonds with G143, H164, E166 and Q189. The initial MMGBSA binding energy of N3 was calculated to be −77.36 kcal/mol. Our study is inspired by these crystal structures as we attempt to computationally screen a huge fragment library against the crystal structure 6LU7 and then combine the best fragment hits from different sub-pockets to design new molecules.
Fragment screening and linking
To validate the potential of the docking program to screen active compounds, a docking enrichment analysis was carried out taking 76 fragments bound to different SARS-CoV-2-MPro structures reported in PDB (excluding the 6LU7 used for our screening) and a decoy set of 1362 molecules with similar size as the actives. The docking program was able to screen 74 active compounds. The AUC of the ROC curve, which is considered to be a reliable metric to evaluate the performance of the program is shown in (Fig. S2). Results of this analysis achieved good value of 0.82 AUC and 0.84 ROC, indicating the power of the docking score ranking over random distribution. A huge fragment library consisting of 191678 fragments was constructed from the publicly available Asinex, FCH and ChemDiv fragment libraries. These fragments were then screened against the energy grid created by keeping the N3 molecule as the grid centre and the dimensions of the inner and outer grid boxes were kept as large as 14 Å and 20 Å to cover all the sub-pockets inside and adjacent to the main binding pocket. Glide SP protocol was applied initially for fast screening of the fragments, which returned 40805 fragment hits with docking scores ranging from −9.79 to −5.55. 1974 top hits with SP docking score < −7 were chosen for further study. The nearest atoms of two fragments binding to different sub-pockets and are that are pre-positioned with respect to each other were joined to form a new molecule. The potential bonds that can be formed between two fragments placed in the adjacent sub-pockets were identified. These potential new bonds were identified by based on i) the distance between the atom that remains in one fragment and the atom that leaves in the other fragment must be less than 1 Å after adjustment of the bond lengths in each fragment to the ideal bond length for the new bond, ensuring the right alignment of fragments, ii) the angle between the bond directions should be less than 15° for a right rotational alignment of the fragments and iii) the distance between the fragment centroids should be below 2 Å to make sure that the fragments do not occupy the same location in the receptor cavities (Figure 1). However, the fragments can have some overlapping regions and both internal and peripheral (H and halogen) bonds were considered for breaking to form a bond with another fragment. Once such potential new bonds are identified, the fragments were linked in three rounds. In the first round, pairs of fragments were joined and in the next rounds of joining, the resultant molecules from the first round are considered for joining based on the above criteria and so on. The fragments, having no atoms as close as 1 Å, but are lying in adjacent binding pockets are still considered for linking by introducing methylene groups to each fragment, and then if the bond formation criteria are satisfied, the two fragments were joined by maximum two methylene linkers. The minimum and maximum fragments to be joined were set to be 2 and 4 respectively. The fragments were randomly sampled in several non-redundant trials in order to manage the huge number of combinations. The number of such trials were set to be 20 for this study. Once the fragments are joined, the resultant structures are subjected to energy minimization restraining all heavy atoms in the fragments except for the linker atoms with a restraint of 100 kcal/mol. The fragment linking process taking 1974 selected high scoring fragments pre-positioned in different subpackets generated 487 novel molecules which were further screened using several levels of screening filters.
Screening of newly designed molecules
The 487 newly formed molecules were subjected to further screening using four different levels of filters. XP docking scores were used as the first level of screening. XP docking of calculations were performed using the same huge interaction grid generated for fragment docking in order to provide enough space for several conformers of the newly formed molecules to access all the sub-pockets and place themselves in binding site in a conformation that is energetically most favourable. The XP docking scores ranged from −14.13 to −6.997 for these molecules. The top scoring 172 molecules with Glide XP docking score <−10 were then subjected to the second level of filter i.e. calculation of ADMET properties with QuickProp module of Schrodinger which predicts many significant and pharmacologically relevant properties to estimate the drug likeliness of a given molecules. One can compare certain properties of a particular molecule with the given ranges of those of 95% of known drugs. Also, QuickProp can identify the presence of 30 types of reactive functional groups that may cause false positives during virtual screening studies. The important properties that are calculated and can be compared with the ranges of known drugs are MW, dipole, IP, EA, SASA, FOSA, FISA, PISA, WPSA, PSA, volume, #rotor, donorHB, accptHB, glob, QPpolrz, QPlogPC16, QPlogPoct, QPlogPw, QPlogPo/w, logS, QPLogKhsa, QPlogBB, #metabol, etc. The descriptions of all these properties are listed in List S1. We have prioritized our screened compounds based on the number of descriptor values that fall outside the 95% range of similar values for 95% of known drugs (#stars) calculated by QuickProp. Hence a smaller #stars suggests that a molecule is more drug-like than molecules with more #stars. We screened all the compounds that have passed our previous filters which have #star as 0. Thus, we obtained a total of 83 molecules which violate no drug likeness rules. Table 1 lists the important ADMET properties of the selected.
Table 1.
ADMET properties* of the 17 selected molecules.
| Name | #stars | QPlogPC16 | QPlogPoct | QPlogPw | QPlogKhsa | QPlogHERG | QPlogPo/w | QPlogKp | QPlogBB | #metab | QPlogS | CNS | FOSA | FISA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MP-In1 | 0 | 15.27 | 25.32 | 12.57 | 1.10 | −5.16 | 5.31 | −4.35 | −0.57 | 5 | −6.33 | 1 | 440.80 | 119.48 |
| MP-In2 | 0 | 16.82 | 28.47 | 15.97 | 0.43 | −5.70 | 4.06 | −4.64 | −0.87 | 4 | −6.27 | 0 | 562.96 | 131.24 |
| MP-In3 | 0 | 15.25 | 27.87 | 15.26 | 0.84 | −6.97 | 4.03 | −7.18 | −0.20 | 5 | −4.92 | 0 | 315.15 | 150.09 |
| MP-In4 | 0 | 15.04 | 27.54 | 19.16 | −0.09 | −6.35 | 1.51 | −5.81 | −1.81 | 6 | −3.22 | −2 | 332.10 | 211.19 |
| MP-In5 | 0 | 15.70 | 26.50 | 14.47 | 0.79 | −6.87 | 4.76 | −4.29 | −0.54 | 8 | −5.79 | 0 | 151.48 | 140.40 |
| MP-In6 | 0 | 14.79 | 28.35 | 19.53 | 0.08 | −6.52 | 1.29 | −7.70 | −0.93 | 6 | −1.89 | −1 | 317.85 | 190.63 |
| MP-In7 | 0 | 17.92 | 31.04 | 18.74 | 0.10 | −3.80 | 1.76 | −5.21 | −1.85 | 8 | −5.09 | −2 | 219.90 | 216.91 |
| MP-In8 | 0 | 17.12 | 30.53 | 17.10 | 0.78 | −6.64 | 3.23 | −8.80 | −1.21 | 4 | −4.23 | −2 | 288.03 | 238.17 |
| MP-In9 | 0 | 16.65 | 29.50 | 21.06 | 0.03 | −6.20 | 1.85 | −7.47 | −1.16 | 5 | −2.22 | −2 | 340.75 | 195.09 |
| MP-In10 | 0 | 15.16 | 22.74 | 12.72 | 0.78 | −7.40 | 4.43 | −3.70 | −0.78 | 6 | −5.76 | −1 | 437.09 | 104.38 |
| MP-In11 | 0 | 13.42 | 25.89 | 17.92 | −0.23 | −4.55 | 2.02 | −4.58 | −0.69 | 5 | −3.38 | 0 | 358.08 | 130.79 |
| MP-In12 | 0 | 15.24 | 26.47 | 16.37 | 0.22 | −3.09 | 2.85 | −5.22 | −1.03 | 6 | −4.07 | −2 | 604.43 | 146.03 |
| MP-In13 | 0 | 16.10 | 31.73 | 22.05 | 0.00 | −5.34 | 1.83 | −7.50 | −0.78 | 4 | −2.09 | −1 | 361.04 | 183.14 |
| MP-In14 | 0 | 16.72 | 34.43 | 20.69 | 0.42 | −6.68 | 2.12 | −9.43 | −1.61 | 5 | −4.13 | −2 | 393.44 | 264.88 |
| MP-In15 | 0 | 16.75 | 31.15 | 17.16 | 0.75 | −6.54 | 2.99 | −8.12 | −1.10 | 6 | −4.09 | −2 | 585.43 | 192.52 |
| MP-In16 | 0 | 13.19 | 25.58 | 20.28 | −0.30 | −4.21 | 0.79 | −6.43 | −1.52 | 7 | −1.87 | −2 | 366.70 | 209.63 |
| MP-In17 | 0 | 14.24 | 26.21 | 12.71 | 0.99 | −6.05 | 4.26 | −6.25 | −1.18 | 4 | −6.38 | −2 | 396.43 | 195.27 |
*Description of all the fields are listed in List S1 (QikProp, Schrödinger, LLC, New York, NY, 2019).
The next level filter used for our screening was ΔGbind-Ligand efficiency. Mathematically, ligand efficiency is the ratio of Gibbs free energy (ΔG) to the number of non-hydrogen atoms of the compound. As the binding energy and docking scores of the ligands are biased towards the size of the ligands, ligand efficiency is a more appropriate parameter to normalize and compare the binding affinities of ligands of different sizes (Abad-Zapatero & Metz, 2005).
Ligand efficiency measures the binding energy per atom of a ligand to its receptor and is popularly used in drug discovery projects to narrow down the focus to lead molecules along with optimal combinations of ADMET and physicochemical properties. The initial ΔGbind of N3 with SARS-CoV-2-MPro was calculated to be −77. 36 kcal/mol and the ligand efficiency was calculated to be −1.58. Hence, we set a cut-off of −70.00 for the ΔGbind and −1.6 for the ΔGbind - Ligand efficiency as the 4th level filter. 17 molecules with MM-GBSA ΔGbind ranging from −70.00 to −80.97 and ligand efficiencies ranging from −1.65 to −2.52 were obtained after applying 4th level filter, which were named as MP-In1 to MP-In17. The various components of the XP docking score, MM-GBSA ΔGbind and Ligand efficiencies of the 17 molecules have been given in Figure 3 and various components of these values are given in Table S1.
Figure 3.

XP docking score and MM-GBSA binding energies (kcal/mol) and Prime ligand efficiencies of the 17 selected molecules with MMGBSA binding energy <−70 kcal/mol.
In order to evaluate the novelty of these molecules, they were searched against the 10’639’400 drug like molecules of the most popular public chemical database ZINC and 177’000 bioactive compounds (activity <10µM) of ChEMBL using SwissSimilarity server (Zoete et al., 2016). Table 2 lists the ZINC and ChEMBL IDs of the most similar (measured by Tanimoto coefficient) molecules returned for MP-In (1-17). We did not get any exact matches from both the databases ensuring the 17 molecules represent novel chemotypes. Interestingly, none of the molecules were found to have close similarities (Tanimoto coefficient) with the ChEMBL compounds. However, MP-In1, MP-In6, MP-In10, MP-In12, MP-In14, MP-In15 and MP-In17 showed close similarities with some of the ZINC compounds. The synthetic accessibilities of these compounds as predicted with SwissADME ranged between 3.9 to 5.6 (Table 2) indicating that these molecules are reasonably synthesizable.
Table 2.
Synthetic feasibility and similarity of the 17 selected molecules with ZINC and ChEMBL compounds.
| Name | Synthetic feasibility¥ | ChEMBL ID | Similarity Score (Tanimoto) | ZINC ID | Similarity Score (Tanimoto) |
|---|---|---|---|---|---|
| MP-In1 | 4.98 | CHEMBL1744037 | 0.530 | ZINC19582044 | 0.907 |
| MP-In2 | 5.58 | CHEMBL2179016 | 0.171 | ZINC72342120 | 0.304 |
| MP-In3 | 4.3 | CHEMBL1760664 | 0.337 | ZINC72340395 | 0.793 |
| MP-In4 | 4.6 | CHEMBL3128188 | 0.248 | ZINC19656462 | 0.272 |
| MP-In5 | 4.31 | CHEMBL463225 | 0.539 | ZINC72337341 | 0.405 |
| MP-In6 | 4.85 | CHEMBL472125 | 0.127 | ZINC72356404 | 0.911 |
| MP-In7 | 4.82 | CHEMBL126780 | 0.137 | ZINC05097750 | 0.399 |
| MP-In8 | 3.9 | CHEMBL2403868 | 0.219 | ZINC76893596 | 0.501 |
| MP-In9 | 4.6 | CHEMBL3128188 | 0.248 | ZINC19656462 | 0.272 |
| MP-In10 | 4.85 | CHEMBL1092573 | 0.255 | ZINC91486444 | 0.957 |
| MP-In11 | 5.1 | CHEMBL113436 | 0.374 | ZINC20118875 | 0.748 |
| MP-In12 | 4.6 | CHEMBL115600 | 0.531 | ZINC00571472 | 0.859 |
| MP-In13 | 5.59 | none | none | ZINC72410748 | 0.250 |
| MP-In14 | 5 | CHEMBL2029718 | 0.628 | ZINC67967404 | 0.956 |
| MP-In15 | 4.98 | CHEMBL1744037 | 0.530 | ZINC19582044 | 0.907 |
| MP-In16 | 5.35 | CHEMBL327990 | 0.120 | ZINC79021439 | 0.215 |
| MP-In17 | 4.42 | CHEMBL2387076 | 0.305 | ZINC33126995 | 0.835 |
¥Value ranges from 1 to 10, a value of 1 indicates easily synthesizable, while 10 indicates very difficult to synthesize.
SARS-CoV-2-MPro residues which participated in H-bonds or salt bridge interactions with the 17 selected ligands are mostly F140, L141, G143, S144, C145, H163, E166, Q189, T190.
Interestingly, all these residues make salt bridge interactions with E166 and only MP-In7 makes a π-π stacking with H41. Apart from making H-bond and salt bridge interactions with residues of one or two sub-pockets, these molecules occupy the other sub-pockets by shape complementarity and hydrophobic contacts. We also observed a fused tricyclic fragment (SMILES: Cn1c2CCCCc2c2ccccc12) which occurs in four of the top 17 screened molecules, which makes factorable contacts with the sub-pocket present at the interface between DI, DII and L1, surrounded by residues C44 to M49 of D1, P168 to H172 of DII and F185 to Q192 of L1. Figure 4 shows the molecular interactions and binding pocket occupancy of the top 16 molecules with SARS-CoV-2-MPro. The complexes of MP-In1 to MP-In17 were further subjected to molecular dynamics simulations in order to assess their structural and enthalpic stabilities and to analyse the nature of their interactions with SARS-CoV-2-MPro binding pockets.
Figure 4.

Molecular interactions and binding pocket occupancy of 16 out of 17 selected molecules with comparable MM-GBSA binding energy and ligand efficiency with SARS-CoV-2-MPro as compared to N3.
Understanding the stabilities of MP-in (1-17) and SARS-CoV-2-MPro complexes
MD simulation is a technique of apt choice in order to estimate the stability of the identified MP-In (1-17) and SARS-CoV-2-MPro interactions under dynamical conditions. It also significantly enhances strong binding of ligands with the target (Guterres & Im, 2020). The generated 17 complexes were submitted to MD simulations for 50 ns in an aqueous environment to study the evolution of these systems with respect to time. The PDB structure 6LU7 binding the peptide inhibitor N3 was also subjected to MD simulations as the reference system. Various analyses were carried out on the MD trajectories to evaluate the stabilities of the complexes. The Root Mean Square Deviation (RMSD) was used to measure the average change in displacement of the whole protein-ligand complexes and the ligands solely for all 5000 frames in the trajectory with respect to the first frame. Figures 5a and 5b show the RMSDs of the protein (All heavy atoms) and protein (Cα atoms). RMSD of the protein in all systems indicated that the simulations have equilibrated, the fluctuations towards the end of the simulation are around some thermal average structures. Changes in RMSD values of the protein considering all heavy atoms and the Cα atoms in all protein-ligand complexes were of the order of 1-3 Å, which is normally acceptable for small, globular proteins. We performed Principal component analysis (PCA) to understand conformational distribution during the simulation time and investigate large-scale collective motions of the protein in protein-ligand complexes on the trajectories generated by our simulations. Essential dynamics (ED) analysis script of the Desmond program (trj_essential_dynamics.py) (Amadei et al., 1993) was used through command line for predicting the dynamic behaviours of a protein. This script which calculates the principal components of the protein C-alpha atoms. The resulting mode vectors were stored as atom level properties in the output structure, and the cross-correlation plots and per-frame conformational deviations projected onto the modes were also be generated (Figure S3 and S4). Projection of the motion for the protein in phase space along the PC1 and PC2 for all these complexes showed uniform distribution of the conformations throughout the simulations, while, the cross-correlation plots showed no significant difference in the inter domain motions in SARS-CoV-2-MPro structure in the 18 complexes (including the reference structure). No highly anti-correlated movements were noticed among the binding site residues. In order to further characterize local changes along the protein chain, The Root Mean Square Fluctuation (RMSF) of each residue in each complex throughout the simulations were comparatively analysed (Figure 5c). RMSF of most of the residues (except the N and C-terminal ones) in all the systems were found to be well below 2.5 Å. The residues D47-P52, Y154, relatively high fluctuations, which are not involved in ligand binding. However, Q189-N193, which are involved in ligand binding showed slightly high flexibilities in the complexes with MP-In10, MP-In11, MP-In17 and the reference system, as they are part of a long loop L1, joining D2 and D3. These analyses, led to the conclusion that the simulations produced stable trajectories of the receptor structures, thus providing a suitable basis for further investigation with the structural evolutions of the ligands within the binding sites. First, RMSDs of the ligands with respect to the receptors were analysed for the 18 protein-ligand complexes throughout the simulations (Figure 5d). The ligand RMSD values were calculated when the protein-ligand complex is first aligned on the protein backbone of the reference and then the RMSD of the ligand heavy atoms were measured. As the RMSD of the ligands with respect to the protein were observed to be maintained below 4 Å for 14 complexes (MP-In1 to MP-In5, MP-In-7 to MP-In10, MP-In12 to MP-In14 and MP-In16 to MP-In17), indicating that these ligands bind stably inside the binding pockets. For systems binding MP-In6, MP-In11 and MP-In15 the ligands underwent higher structural changes with respect to their receptors, which showed some probability of these ligands to diffuse out of the binding site.
Figure 5.

Analyses of structural and enthalpic stabilities of the 17 complexes from their MD trajectories a) RMSD of the protein (All heavy atoms) b) RMSD of the ligands with respect to the protein c) MM-GBSA binding energy and d) MM-GBSA binding energy normalized by the number of heavy atoms (MM-GBSA binding energy: ligand efficiency).
However, when the trajectories were played (Movie clip S1a, S1b and S1c) and the positioning of these three ligands inside their respective binding pockets were closely observed, we found that MP-In6, MP-In11 do not show tendencies to diffuse out of the binding pockets, while MP-In15 shows such tendency. In order to draw further observations on the stabilities of the ligands inside the binding pocket, the radius of gyration (RGyr), which measures the ‘extendedness/compactness’ of a ligand, and the solvent accessible surface area (SASA), corresponding to the surface area of the ligands accessible by the water molecules were calculated and analysed (Figure 5e and 5f). Both RGyr and SASA values for the reference ligand N3 were pretty high as compared to the screened molecules owing to its size and peptide nature. MP-In11 shows an increased Rgyr after 20 ns, which eventually gets stabilized, due to torsional rearrangements leading to a higher RMSD. However, its SASA remains relatively lower as compared to others ligands indicating it remains embedded in the binding pocket. At the same time MP-In15 shows a stable Rgyr profile, but the SASA shows a sudden increase, correlating with its RMSD profile (Figure 5d) indicates that, the molecule diffuses out of the binding pocket. The RGyr and SASA of rest of the ligands showed stable profiles suggesting stable binding. The stabilities of the complexes were further examined in terms of MM-GBSA ΔGbind and ligand efficiencies, which were calculated for snapshots of the complexes taken at every 5 ns (10 snapshots per system were collected). Figure 5g and 5h show the MM-GBSA ΔGbind and ligand efficiencies of the 18 complexes including the reference system throughout the simulations. As depicted from Figures 5c and 5d, the systems with MP-In4 and MP-In16 showed very good binding energies, while MP-In15 and MP-In17 show the weakest binding energy and ligand efficiency profiles, which were maintained much lower than that of the reference ligand. Hence, it may not be a good idea to consider MP-In15 and MP-In17 for further studies. Various protein-ligand interactions of the ligands monitored throughout the simulation have been given in Figure 6.
Figure 6.
Protein-Ligand Contacts of the top 16 molecules and the reference molecule monitored throughout the simulation. The stacked bar charts are normalized over the course of the trajectory; e.g. a value of 0.7 suggests that 70% of the simulation time the specific interaction is maintained. Values over 1.0 are possible when a protein residue make multiple contacts of same subtype with the ligand.


Interactions that occur more than 30% of the simulation time in each trajectory through 0 to 50 are is shown in Figure S5. These interactions were categorized into 4 types: Hydrogen Bonds, Hydrophobic, Ionic and Water Bridges. As shown in Figure 6 and Figure S3, the residues G143, S144, C145, E166, Q189, T190, Q192 mostly make stable H-bonds with the ligands. Hydrogen-bonding properties of the molecules in drug design is considered important because of their strong influence on drug specificity, metabolization and adsorption. The hydrophobic contacts shown in the figure include π-Cation; π-π; and Other, non-specific interactions which generally hydrophobic amino acids and aromatic or aliphatic groups on the ligands. The residues H41, M49, M165, L167 and P168 mostly formed hydrophobic contacts with the hydrophobic fragments of MP-In (1-17). H41 was also shown to form π-π stacking with the aromatic rings of MP-In7 and MP-In8 (Figure S5).
Ionic or polar interactions, between two oppositely charged atoms were mostly shown by E166 while T26, N142 formed stable water bridges i.e. hydrogen-bonded protein-ligand interactions mediated by a water molecule in complexes with MP-In-8 MP-In9, MP-In-12 and MP-In15. The novel molecules make interactions with almost all key binding pocket residues closely resembling the interactions of the reference ligand N3. Hence, 15 out of 17 selected molecules, excluding MP-In15 and MP-In17 might be considered as potential inhibitors of SARS-CoV-2- MPro based on their stable molecular interactions, good binding energy comparable to the reference ligand and good shape complementarity and ligand efficiencies.
Summary
The current study attempts to design new molecules by tailoring fragments that bind to various sub-pockets of the binding sites of SARS-CoV-2- MPro. A huge library of publicly available molecular fragments was screened against SARS-CoV-2- MPro binding site in order to obtain sub-pocket specific fragments. These fragments prepositioned in adjacent binding sub-pockets were linked to form new molecules. these new molecules were further screened against SARS-CoV-2- MPro using extra precision docking, ADMET and druglike filters and MMGBSA free energy of binding and ligand efficiency to find 17 potential molecules named as MP-In (1-17), with better ligand efficiencies as compared to the reference inhibitor N3. MD simulations were run on the complexes of these 17 molecules with SARS-CoV-2- MPro and also the reference PDB structure 6LU7 to ensure the stabilities of their binding and interactions. 15 of them showed stable binding through various stable molecular interactions such as H-bonding, salt bridges, hydrophobic contacts and water bridged H-bonds. These novel chemical entities designed specifically according to the pharmacophoric requirements of SARS-CoV-2- MPro binding pockets showed good synthetic feasibilities and returned no exact match when searched against chemical databases. Considering their interactions, binding efficiencies and novel chemotypes, we propose these fifteen molecules as potential starting points for medicinal chemists working on SARS-CoV-2- MPro inhibitor design.
Supplementary Material
Funding Statement
CC thanks Department of Science and Technology for financial assistance in the form of DST-INSPIRE Faculty award, Dr. Anuradha Chakravarti, Head, Department of Experimental Medicine and Biotechnology, PGIMER, Chandigarh for providing required infrastructure and fruitful discussions and Schrodinger for providing short term licences for some of the modules.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Aanouz I., Belhassan A., El Khatabi K., Lakhlifi T., El Idrissi M., & Bouachrine M. (2020). Moroccan Medicinal plants as inhibitors of COVID-19: Computational investigations. Journal of Biomolecular Structure and Dynamics, 38(8), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abad-Zapatero C., & Metz J. T. (2005). Ligand efficiency indices as guideposts for drug discovery. Drug Discovery Today, 10(7), 464–469. 10.1016/S1359-6446(05)03386-6 [DOI] [PubMed] [Google Scholar]
- Al-Tawfiq J. A., Al-Homoud A. H., & Memish Z. A. (2020). Remdesivir as a possible therapeutic option for the COVID-19. Travel Medicine and Infectious Disease, 34, 101615 10.1016/j.tmaid.2020.101615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadei A., Linssen A. B., & Berendsen H. J. (1993). Essential dynamics of proteins. Proteins: Structure, Function, and Genetics, 17(4), 412–425. 10.1002/prot.340170408 [DOI] [PubMed] [Google Scholar]
- Bhatnagar T., Murhekar M. V., Soneja M., Gupta N., Giri S., Wig N., & Gangakhedkar R. (2020). Lopinavir/ritonavir combination therapy amongst symptomatic coronavirus disease 2019 patients in India: Protocol for restricted public health emergency use. Indian Journal of Medical Research, 151(2), 184 10.4103/ijmr.IJMR_502_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boopathi S., Poma A. B., & Kolandaivel P. (2020). Novel 2019 Coronavirus Structure, Mechanism of Action, Antiviral drug promises and rule out against its treatment. Journal of Biomolecular Structure and Dynamics, 38, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bung N., Krishnan S. R., Bulusu G., & Roy A. (2020). De novo design of new chemical entities (NCEs) for SARS-CoV-2 using artificial intelligence. ChemRxiv Preprint. 10.26434/chemrxiv.11998347.v2 [DOI] [PMC free article] [PubMed]
- Cao B., Wang Y., Wen D., Liu W., Wang J., Fan G., Ruan L., Song B., Cai Y., Wei M., Li X., Xia J., Chen N., Xiang J., Yu T., Bai T., Xie X., Zhang L., Li C., … Wang C. (2020). A trial of lopinavir–ritonavir in adults hospitalized with severe Covid-19. The New England Journal of Medicine, 382(19), 1787–1799. 10.1056/NEJMoa2001282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury C., Priyakumar U. D., & Sastry G. N. (2014). Molecular dynamics investigation of the active site dynamics of mycobacterial cyclopropane synthase during various stages of the cyclopropanation process. Journal of Structural Biology, 187(1), 38–48. 10.1016/j.jsb.2014.04.007 [DOI] [PubMed] [Google Scholar]
- Choudhury C., Priyakumar U. D., & Sastry G. N. (2015). Dynamics based pharmacophore models for screening potential inhibitors of mycobacterial cyclopropane synthase. Journal of Chemical Information and Modeling, 55(4), 848–860. 10.1021/ci500737b [DOI] [PubMed] [Google Scholar]
- Choudhury C., Priyakumar U. D., & Sastry G. N. (2016). Dynamic ligand-based pharmacophore modeling and virtual screening to identify mycobacterial cyclopropane synthase inhibitors. Journal of Chemical Sciences, 128(5), 719–732. 10.1007/s12039-016-1069-1 [DOI] [PubMed] [Google Scholar]
- Coutard B., Decroly E., Li C., Sharff A., Lescar J., Bricogne G., & Barral K. (2014). Assessment of Dengue virus helicase and methyltransferase as targets for fragment-based drug discovery. Antiviral Research, 106, 61–70. 10.1016/j.antiviral.2014.03.013 [DOI] [PubMed] [Google Scholar]
- Daina A., Michielin O., & Zoete V. (2017). SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports, 7, 42717 10.1038/srep42717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY. (2018). Maestro-desmond interoperability tools. Schrödinger. [Google Scholar]
- Dong L., Hu S., & Gao J. (2020). Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discoveries & Therapeutics, 14(1), 58–60. 10.5582/ddt.2020.01012 [DOI] [PubMed] [Google Scholar]
- Elfiky A. A. (2020. a). Natural products may interfere with SARS-CoV-2 attachment to the host cell. Journal of Biomolecular Structure and Dynamics, 38, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A. A. (2020. b). SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. Journal of Biomolecular Structure and Dynamics, 38, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A. A., & Azzam E. B. (2020). Novel Guanosine Derivatives against MERS CoV polymerase: An in silico perspective. Journal of Biomolecular Structure and Dynamics, 38, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmezayen A. D., Al-Obaidi A., Şahin A. T., & Yelekçi K. (2020). Drug repurposing for coronavirus (COVID-19): In silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. Journal of Biomolecular Structure and Dynamics, 38, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enmozhi S. K., Raja K., Sebastine I., & Joseph J. (2020). Andrographolide As a Potential Inhibitor of SARS-CoV-2 Main Protease: An In Silico Approach. Journal of Biomolecular Structure and Dynamics, 38, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A., Murphy R. B., Repasky M. P., Frye L. L., Greenwood J. R., Halgren T. A., Sanschagrin P. C., & Mainz D. T. (2006). Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein − ligand complexes. Journal of Medicinal Chemistry, 49(21), 6177–6196. 10.1021/jm051256o [DOI] [PubMed] [Google Scholar]
- Gahtori J., Pant S., & Srivastava H. K. (2019). Modeling antimalarial and antihuman African trypanosomiasis compounds: A ligand-and structure-based approaches. Molecular Diversity, 23, 1–18. 10.1007/s11030-019-10015-y [DOI] [PubMed] [Google Scholar]
- Glide, Schrödinger, LLC, New York, NY, (2019). [Google Scholar]
- Gu X., Cao B., & Wang J. (2020). Full spectrum of COVID-19 severity still being depicted–Authors’ reply. The Lancet, 395(10228), 948–949. 10.1016/S0140-6736(20)30371-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M. K., Vemula S., Donde R., Gouda G., Behera L., & Vadde R. (2020). In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. Journal of Biomolecular Structure and Dynamics, 38 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guterres H., & Im W. (2020). Improving protein-ligand docking results with high-throughput molecular dynamics simulations. Journal of Chemical Information and Modeling, 60(4), 2189–2198. 10.1021/acs.jcim.0c00057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan M., Ghosh P. P., Azim K. F., Mukta S., Abir R. A., Nahar J., & Khan M. M. H. (2019). Reverse vaccinology approach to design a novel multi-epitope subunit vaccine against avian influenza A (H7N9) virus. Microbial Pathogenesis, 130, 19–37. 10.1016/j.micpath.2019.02.023 [DOI] [PubMed] [Google Scholar]
- Hasan A., Paray B. A., Hussain A., Qadir F. A., Attar F., Aziz F. M., … Shahpasand K. (2020). A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. Journal of Biomolecular Structure and Dynamics, 38, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffer L., Renaud J. P., & Horvath D. (2011). Fragment-based drug design: Computational and experimental state of the art. Combinatorial Chemistry & High Throughput Screening, 14(6), 500–520. 10.2174/138620711795767884 [DOI] [PubMed] [Google Scholar]
- Islam R., Parves R., Paul A. S., Uddin N., Rahman M. S., Mamun A. A., … Halim M. A. (2020). A Molecular Modeling Approach to Identify Effective Antiviral Phytochemicals against the Main Protease of SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 38, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., … Duan Y. (2020). Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature, 580. 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- Joshi R. S., Jagdale S. S., Bansode S. B., Shankar S. S., Tellis M. B., Pandya V. K., … Kulkarni M. J. (2020). Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease$. Journal of Biomolecular Structure and Dynamics,38, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanakaveti V., Shanmugam A., Ramakrishnan C., Anoosha P., Sakthivel R., Rayala S. K., & Gromiha M. M. (2020). Computational approaches for identifying potential inhibitors on targeting protein interactions in drug discovery. Advances in Protein Chemistry and Structural Biology, 121, 25–47. 10.1016/bs.apcsb.2019.11.013 [DOI] [PubMed] [Google Scholar]
- Khan R. J., Jha R. K., Amera G., Jain M., Singh E., Pathak A., … Singh A. K. (2020). Targeting SARS-Cov-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like Proteinase and 2’-O-RiboseMethyltransferase. Journal of Biomolecular Structure and Dynamics, 38,1–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S. A., Zia K., Ashraf S., Uddin R., & Ul-Haq Z. (2020). Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. Journal of Biomolecular Structure and Dynamics, 38, 1–13. [DOI] [PubMed] [Google Scholar]
- Khot W. Y., & Nadkar M. Y. (2020). The 2019 novel coronavirus outbreak-A global threat. The Journal of the Association of Physicians of India, 68(3), 67–71. [PubMed] [Google Scholar]
- Ko W. C., Rolain J. M., Lee N. Y., Chen P. L., Huang C. T., Lee P. I., & Hsueh P. R. (2020). Arguments in favour of remdesivir for treating SARS-CoV-2 infections. International Journal of Antimicrobial Agents, 55(4), 105933 10.1016/j.ijantimicag.2020.105933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Rathi E., & Kini S. G. (2019, November). Fragment-based design of novel inhibitors of HPV 16 E6 oncoprotein: Molecular docking, molecular dynamics simulation and in silico ADME analysis [Paper presentation]. Conference on Drug Design and Discovery Technologies. (Vol. 355, p. 25). Royal Society of Chemistry. [Google Scholar]
- Li H., Wang Y. M., Xu J. Y., & Cao B. (2020). [Potential antiviral therapeutics for 2019 Novel Coronavirus]. Zhonghua Jie he he hu xi za Zhi = Zhonghua Jiehe he Huxi Zazhi = Chinese Journal of Tuberculosis and Respiratory Diseases, 43(0), E002–E002. 10.3760/cma.j.issn.1001-0939.2020.0002 [DOI] [PubMed] [Google Scholar]
- LigPrep, Schrödinger, LLC, New York, NY, (2019). [Google Scholar]
- Loving K., Alberts I., & Sherman W. (2010). Computational approaches for fragment-based and de novo design. Current Topics in Medicinal Chemistry, 10(1), 14–32. 10.2174/156802610790232305 [DOI] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., Bi Y., Ma X., Zhan F., Wang L., Hu T., Zhou H., Hu Z., Zhou W., Zhao L., … Tan W. (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. The Lancet, 395(10224), 565–574. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan N., Sakthivel R., Velmurugan D., & Gromiha M. M. (2020). Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 Protease against COVID-19. Journal of Biomolecular Structure and Dynamics, 38, 1–7. [DOI] [PubMed] [Google Scholar]
- Murgueitio M. S., Bermudez M., Mortier J., & Wolber G. (2012). In silico virtual screening approaches for anti-viral drug discovery. Drug Discovery Today: Technologies, 9(3), e219–e225. 10.1016/j.ddtec.2012.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger M. M., Carchia M., Irwin J. J., & Shoichet B. K. (2012). Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. Journal of Medicinal Chemistry, 55(14), 6582–6594. 10.1021/jm300687e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najjar A., Olğaç A., Ntie-Kang F., & Sippl W. (2019). Fragment-based drug design of nature-inspired compounds. Physical Sciences Reviews, 4(9), 1-13. 10.1515/psr-2018-0110 [DOI] [Google Scholar]
- Njogu P. M., Guantai E. M., Pavadai E., & Chibale K. (2016). Computer-aided drug discovery approaches against the tropical infectious diseases malaria, tuberculosis, trypanosomiasis, and leishmaniasis. ACS Infectious Diseases, 2(1), 8–31. 10.1021/acsinfecdis.5b00093 [DOI] [PubMed] [Google Scholar]
- Pant S., Singh M., Ravichandiran V., Murty U. S. N., & Srivastava H. K. (2020). Peptide-like and small-molecule inhibitors against Covid-19. Journal of Biomolecular Structure and Dynamics, 38, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prime, Schrödinger, LLC, New York, NY, (2019). [Google Scholar]
- Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, (2019). [Google Scholar]
- QikProp, Schrödinger, LLC, New York, NY, (2019). [Google Scholar]
- Sahraei Z., Shabani M., Shokouhi S., & Saffaei A. (2020). Aminoquinolines against coronavirus disease 2019 (COVID-19): Chloroquine or hydroxychloroquine. International Journal of Antimicrobial Agents, 55(4), 105945 10.1016/j.ijantimicag.2020.105945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma P., Sekhar N., Prajapat M., Avti P., Kaur H., Kumar S., … Medhi B. (2020). In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). Journal of Biomolecular Structure and Dynamics, 38, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler J., Hudson M. L., Schwartz D., & Samudrala R. (2017). A systematic review of computational drug discovery, development, and repurposing for Ebola virus disease treatment. Molecules, 22(10), 1777 10.3390/molecules22101777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivakumar D., Williams J., Wu Y., Damm W., Shelley J., & Sherman W. (2010). Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. Journal of Chemical Theory and Computation, 6(5), 1509–1519. 10.1021/ct900587b [DOI] [PubMed] [Google Scholar]
- Sinha S. K., Shakya A., Prasad S. K., Singh S., Gurav N. S., Prasad R. S., & Gurav S. S. (2020). An in-silico evaluation of different Saikosaponins for their potency against SARS-CoV-2 using NSP15 and fusion spike glycoprotein as targets. Journal of Biomolecular Structure and Dynamics, 38, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava H. K., Choudhury C., & Narahari Sastry G. (2012). The efficacy of conceptual DFT descriptors and docking scores on the QSAR models of HIV protease inhibitors. Medicinal Chemistry, 8(5), 811–825. 10.2174/157340612802084351 [DOI] [PubMed] [Google Scholar]
- Wahedi H. M., Ahmad S., & Abbasi S. W. (2020). Stilbene-based Natural Compounds as Promising Drug Candidates against COVID-19. Journal of Biomolecular Structure and Dynamics, 38, 1–16. [DOI] [PubMed] [Google Scholar]
- www.asinex.com/fragments/
- www.chemdiv.com/
- www.fchgroup.net/fragment-libraries.php
- www.who.int/emergencies/diseases/novel-coronavirus-2019/events-as-they-happen
- Xu Z., Li S., Tian S., Li H., & Kong L. Q. (2020). Full spectrum of COVID-19 severity still being depicted. The Lancet, 395(10228), 947–948. 10.1016/S0140-6736(20)30308-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D., Dai S. M., & Tong Q. (2020). COVID-19: A recommendation to examine the effect of hydroxychloroquine in preventing infection and progression. Journal of Antimicrobial Chemotherapy, dkaa114, 1–4. 10.1093/jac/dkaa114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoete V., Daina A., Bovigny C., & Michielin O. (2016). SwissSimilarity: A web tool for low to ultra high throughput ligand-based virtual screening, 56(8), 1399–1404e) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.