

Abstract
Endurance exercise training promotes numerous biochemical adaptations within skeletal muscle fibers culminating into a phenotype that is safeguarded against numerous perils including doxorubicin-induced myopathy and inactivity-induced muscle atrophy. This exercise-induced protection of skeletal muscle fibers is commonly termed “exercise preconditioning”. This review will discuss the biochemical mechanisms responsible for exercise-induced protection of skeletal muscle fibers against these harmful events. The first segment of this report highlights the evidence that endurance exercise training provides cytoprotection to skeletal muscle fibers against several potentially damaging insults. The second and third sections of the review will discuss the cellular adaptations responsible for exercise-induced protection of skeletal muscle fibers against doxorubicin-provoked damage and inactivity-induced fiber atrophy, respectively. Importantly, we also identify gaps in our understanding of exercise preconditioning in hopes of stimulating future research.
Keywords: Doxorubicin, Endurance exercise, Diaphragm, Mechanical ventilation, Muscle atrophy, Heart, Muscle wasting
Graphical abstract

1. Introduction
It is established that endurance exercise training promotes numerous biochemical adaptations in skeletal muscle fibers. In fact, the first reports describing the biochemical adaptations of skeletal myocytes in response to exercise were published over 60 years ago. The earliest investigations were descriptive and chronicled the exercise-induced increases in the abundance of key bioenergetic enzymes in both cardiac and skeletal muscles [18,21,22]. In the decades to follow, investigations explored exercise-induced changes in a variety of skeletal muscle proteins including antioxidant enzymes and heat shock proteins. The observation that exercise training increases the abundance of endogenous antioxidants and heat shock proteins in muscle fibers inspired the hypothesis that exercise-induced biochemical adaptations in skeletal muscles have the potential to protect fibers against damaging events.
During the past decade, numerous studies confirm that endurance exercise training results in “exercise preconditioning” of skeletal muscle fibers. Specifically, the term exercise preconditioning is used to describe exercise-induced adaptations in cardiac and skeletal muscle fibers that are associated with protection against a variety of harmful threats to muscle health. For example, endurance exercise training protects skeletal muscle fibers against doxorubicin-induced wasting and the muscle fiber atrophy associated with prolonged muscle inactivity (reviewed in Refs. [47,55]). Although studies investigating exercise-induced biochemical changes in skeletal muscles have spanned several decades, investigations into the mechanisms responsible for exercise preconditioning of skeletal muscle is a relatively new endeavor. Nonetheless, significant progress has been made toward delineating the mechanisms responsible for exercise-induced preconditioning of skeletal muscle fibers.
This report provides a systematic review of the cellular mechanism(s) responsible for exercise-induced preconditioning of skeletal muscle fibers. We begin with an overview of the evidence that endurance exercise training results in a protective phenotype in skeletal muscle fibers. We then discuss the biochemical changes in muscle fibers that are postulated to contribute to exercise preconditioning. In particular, we provide a critical analysis of the evidence delineating the mechanisms responsible for exercise-induced protection against both doxorubicin-induced muscle wasting and inactivity-induced muscle atrophy.
2. Exercise-induced preconditioning in skeletal muscle fibers
The term “exercise preconditioning” evolved from the parent term “preconditioning” that is commonly used in biology to describe a process whereby a person or animal is exposed to a sub-lethal stress that confers subsequent protection against a potentially harmful stressor. Over the last three decades, the term exercise preconditioning has been commonly used to describe the protective phenotype that occurs in both cardiac and skeletal muscle fibers following endurance exercise training.
In regard to exercise preconditioning of skeletal muscle, endurance exercise training protects skeletal muscle fibers against a variety of stresses including muscle damage associated with sepsis, contraction-induced muscle injury, doxorubicin-mediated muscle wasting and inactivity-induced muscle atrophy [25,47,55,70]. To illustrate two specific examples of the magnitude of exercise preconditioning-induced protection of skeletal muscle fibers, the next segment will highlight the evidence that endurance exercise results in a skeletal muscle phenotype that is protected against both doxorubicin-induced muscle wasting and inactivity-induced muscle atrophy.
2.1. Exercise preconditioning protects against doxorubicin-induced muscle wasting
Prior to discussing the evidence that exercise training protects against doxorubicin-induced muscle wasting, a brief introduction to doxorubicin-induced muscle atrophy is warranted. Doxorubicin (DOX), a quinone-containing anthracycline antibiotic, is a highly effective antitumor agent that is widely used in the treatment of several cancers (i.e., solid tumors and hematologic malignancies) [63]. Unfortunately, prolonged use of DOX in cancer therapy is not possible because of the deleterious side effects that occur in off-target tissues including heart and skeletal muscles.
DOX-induced skeletal muscle wasting occurs due to increases in mitochondrial production of reactive oxygen species (ROS) resulting in damage to both cellular membranes and proteins [37]. Specifically, DOX promotes increases in mitochondrial ROS production via interactions with mitochondrial NADH that result in a redox cycling event [61]. Explicitly, ROS producing enzymes in the mitochondria (i.e., NADPH oxidase) transform DOX into a semiquinone through one-electron reduction of the quinone moiety in ring C; this semiquinone can then react with oxygen to form superoxide radicals [61]. This DOX-mediated oxidant production oxidizes mitochondrial proteins and other macromolecules in the fiber leading to activation of all major proteolytic systems (i.e., ubiquitin proteasome system, calpain, caspase-3, and autophagy) in skeletal muscle fibers resulting in accelerated proteolysis and muscle atrophy [37,56,57]. To illustrate the high toxicity of DOX on skeletal muscle fibers, a single dose of DOX promotes myonuclear apoptosis and a loss of myonuclei in rodent skeletal muscle fibers [37].
Numerous preclinical studies confirm that endurance exercise results in protection against DOX-induced damage to skeletal muscle. Indeed, as few as 10 days of moderate intensity (e.g., 60 min/day at ~60–70% VO2 max) endurance exercise training protects rodent skeletal muscle against DOX-induced muscle oxidative damage and accelerated proteolysis [56,57]. For example, exercise results in a fiber phenotype that is protected against DOX-induced oxidative modification of skeletal muscle proteins as evidenced by a reduction in the levels of both carbonyl derivatives within myofibrillar proteins and the conjugation of 4-hydroxy-2-nonenal to muscle proteins [57]. Further, exercise preconditioning protects skeletal muscles against DOX-induced activation of major proteolytic systems including the activation of the proteolytic enzymes calpain and caspase-3 [57]. In addition, exercise-induced protection against the DOX-mediated activation of proteolysis in skeletal muscles is associated with a reduced expression of several autophagy genes, the forkhead-box O (FoxO) transcription of the E3 ubiquitin ligase, muscle RING finger-1 (MuRF-1), and the pro-apoptotic protein, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) [30,56] (Fig. 1).
Fig. 1.

Endurance exercise training performed prior to treatment with doxorubicin (DOX) protects skeletal muscles against DOX-induced damage to mitochondria, oxidative damage to macromolecules, accelerated proteolysis, myonuclear apoptosis and muscle fiber atrophy.
2.2. Exercise preconditioning protects against inactivity-induced muscle atrophy
Prior to a discussion of the evidence that exercise training protects against inactivity-induced muscle atrophy, a brief overview of the events leading to disuse muscle atrophy is provided. Skeletal muscle fiber size is regulated by the balance between the rates of protein synthesis versus degradation and prolonged skeletal muscle inactivity results in fiber atrophy due to both accelerated proteolysis and decreased protein synthesis [51]. In reference to the mechanism(s) responsible for inactivity-induced muscle atrophy, evidence exists that oxidative stress plays a key role in regulation of the signaling pathways that regulate both proteolysis and protein synthesis in skeletal muscles during prolonged inactivity [52]. Indeed, studies confirm that treatment of animals with select antioxidants can protect skeletal muscle against inactivity-induced oxidative stress, accelerated proteolysis, and muscle atrophy [4,35,38,48,64,73].
The sources of ROS production in inactive skeletal muscles have been investigated for over a decade and results reveal that superoxide production occurs at numerous cellular locations in the muscle fiber; sites of ROS production include NAD(P)H oxidase, xanthine oxidase, and mitochondria [13,36,37,48,72]. Of these primary sites of superoxide production within inactive muscle, several lines of evidence indicate that mitochondrial ROS emission plays a dominant role. For example, it is established that mitochondrial ROS emission is greater in the “resting” state of respiration where the ADP supply to the mitochondria is limited (i.e., state 4 respiration) compared to the active ADP stimulated condition (i.e., state 3 respiration) [2,19,48]. This is relevant because conditions in inactive skeletal muscle correspond to state 4 respiration whereas conditions in actively contracting muscle corresponds to state 3 [31]. Further, mitochondria isolated from diaphragm muscle of rats exposed to prolonged mechanical ventilation (MV) (i.e., resulting in diaphragm inactivity) release more ROS than mitochondria from the diaphragm of animals that are spontaneously breathing [48]. While these experiments reveal that mitochondrial ROS emission is increased in inactive skeletal muscle, direct evidence that increased mitochondrial ROS emission plays a key role in promoting inactivity-induced muscle atrophy is provided by experiments demonstrating that treatment of animals with a mitochondrial-specific antioxidant (i.e., SS-31) that protects locomotor muscles against immobilization induced oxidative stress, accelerated proteolysis, and fiber atrophy [38,64]. Similarly, treatment with SS-31 protects diaphragm muscle against inactivity-induced oxidative stress and fiber atrophy [48]. Lastly, experimental evidence also links the inactivity-induced increase in mitochondria ROS emission to depressed protein synthesis and accelerated proteolysis in skeletal muscle fibers [24,38,64]. Together, these findings support the concept that mitochondrial ROS emission is a primary source of oxidants in skeletal muscle fibers during prolonged inactivity.
As discussed earlier, endurance exercise training has been shown to improve the antioxidant properties of skeletal muscle fibers and therefore, it is predicted that exercise training can potentially protect against inactivity-induced muscle atrophy. Indeed, several studies corroborate that endurance exercise training protects against disuse-induced oxidative stress, proteolysis, and muscle atrophy. Specifically, exercise preconditioning protects limb muscles against disuse muscle atrophy [16,43,66,74]. Further, exercise also protects inspiratory muscles (e.g., diaphragm) against the inactivity-induced muscle atrophy that occurs during prolonged MV. This exercise training-induced protection against diaphragmatic atrophy is accompanied by protection against inactivity-induced mitochondrial uncoupling, oxidative stress, and activation of key proteases [58]. In the next segments, we discuss the mechanisms responsible for exercise-induced protection against both DOX-induced muscle wasting and inactivity-induced muscle atrophy.
3. Mechanisms responsible for exercise-induced protection against DOX-induced muscle wasting
Currently, a complete understanding of the mechanism(s) responsible for exercise-induced protection against DOX-induced muscle wasting does not exist and this topic remains an active area of investigation. To date, three primary mechanisms have been proposed to explain exercise-induced cytoprotection against DOX-mediated damage to skeletal muscle fibers (Fig. 3).
Fig. 3.

Endurance exercise training results in numerous biochemical changes within skeletal muscle fibers. Three hypotheses have been proposed to explain the mechanism behind exercise training-induced protection against doxorubicin-induced muscle damage; these include increased expression of: 1) endogenous antioxidant enzymes; 2) heat shock protein 72; and 3) ATP-binding cassette proteins (ABC transporters).
3.1. Exercise training-induced increases in cellular antioxidants
As introduced earlier, DOX-stimulated wasting of skeletal muscles is due, at least in part, to increased mitochondrial production of ROS resulting in oxidative damage to cellular components and accelerated proteolysis [29,30,56]. It follows that exercise-induced increases in endogenous antioxidants have the potential to provide protection against DOX-induced muscle wasting.
In reference to endogenous antioxidants, muscle fibers and other cells are protected against ROS by a complex network of endogenous antioxidants. Specifically, superoxide dismutase (SOD), glutathione reductase (GPX), and catalase (CAT) are three major antioxidant enzymes within cells (reviewed in Ref. [50]). SOD is the first line of defense against superoxide radicals and dismutates superoxide to form hydrogen peroxide (H2O2) and oxygen. Two isoforms of SOD (SOD1, SOD2) exist within cells. SOD1 resides primarily in the cytosol whereas SOD2 is located within the mitochondrial matrix. The relative abundance of SOD1 and SOD2 varies across skeletal muscle fiber types with the highest activities of both isoforms being found in type I (slow) fibers [50].
Five glutathione peroxidase isoforms exist in mammals (GPX1-GPX5) and each of these GPX isoforms function to reduce H2O2 and/or organic hydroperoxides to form water and alcohol, respectively [6]. Three GPX isoforms exist within muscle fibers that are strategically compartmentalized to protect against ROS-mediated oxidation at the site of H2O2 production.
Finally, the antioxidant enzyme CAT is responsible for the breakdown of H2O2 into water and oxygen. CAT is primarily located in the cytosol of the cell [50]. Interestingly, CAT has one of the highest turnover rates of all enzymes as turnover number for the decomposition of hydrogen peroxide is 2.5 × 106 to 5 × 106 mol/min [34].
The impact of endurance exercise training on the abundance of antioxidant enzymes in skeletal muscles has been extensively studied for over three decades. Although a few studies report that endurance exercise training does not increase the abundance of SOD1 and SOD2 in muscle fibers, a large body of research demonstrates that endurance exercise training increases the abundance of both SOD1 and SOD2 in skeletal muscles [49,50]. The magnitude of exercise-induced increases in muscle SOD activity (i.e., 10–40%) conforms to a training dose-response curve as high intensity and/or longer durations (i.e., up to 60 min) of exercise training results in higher increases in SOD activity (Fig. 2) [45,46].
Fig. 2.
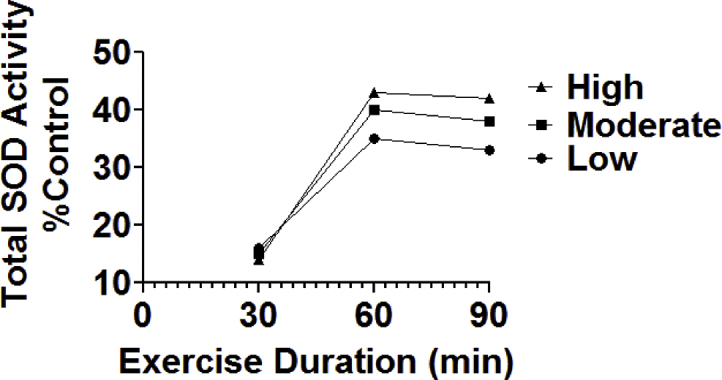
Total SOD activity in the rat soleus muscle following 10 weeks (4 days/week) of endurance exercise training (treadmill) at varying durations and intensities of exercise. Exercise intensities varied from low (~55% VO2 max), moderate (~65% VO2 max), and high intensity (~75% VO2 max). Values are means and data are from Ref. [45].
Identical to SOD, GPX is also inducible in skeletal muscle fibers and GPX activity rapidly increases in muscle fibers within the first five days of exercise training [49,50]. The magnitude of the exercise-induced increase in muscle GPX activity following 10 weeks of exercise training ranges from 20 to 177%; this exercise-induced increase in GPX abundance increases as a function of the both exercise intensity and duration (reviewed in Refs. [49,50]). Further, training-induced increases in GPX within muscle fibers occurs in both the cytosolic and mitochondrial compartments.
Finally, it remains unclear if exercise training increases the activity or abundance of CAT in skeletal muscle fibers; indeed, studies report both increases and decreases of CAT abundance following exercise training (reviewed in Ref. [49]). This lack of consistency across investigations likely results from experimental problems associated with the measurement of CAT activity in vitro; see Powers and Jackson for more details [49].
Research confirms that increased mitochondrial emission of ROS and activation of proteases are required for DOX-induced skeletal muscle atrophy [37,39]. Specifically, administration of a mitochondrial-targeted antioxidant (i.e., SS-31) protects against both DOX-induced increases in mitochondrial ROS emission and skeletal muscle atrophy [37,39]. Hence, it is feasible that exercise-induced increases in mitochondrial antioxidants (e.g., GPX1 and SOD2) are potential mechanisms to explain why exercise training protects skeletal muscle against DOX-induced wasting [56]. Nonetheless, to date, experiments that provide definitive cause and effect of this supposition do not currently exist and therefore, future work will be required to resolve this issue. The role that SOD2 plays in exercise preconditioning against inactivity-induced muscle atrophy will be addressed later in this report.
3.2. Exercise training increases the expression of HSP72 in muscle fibers
Several members of the heat shock protein family are expressed in skeletal muscles. Of particular interest is the 72 kDa member (HSP72) of the heat shock protein family. HSP72 is inducible by exercise training; importantly, increased abundance of HSP72 can protect cells against a variety of stresses. Studies confirm that 30–60 min of endurance exercise training increases the expression of HSP72 in active skeletal muscles of both rodents and humans [14,33,42,53]. Specifically, endurance exercise training can increase the abundance of HSP72 in rodent skeletal muscles by 22–243% depending upon the duration of the exercise bout and the fiber type composition of the muscle [42].
Exercise-induced increased expression of HSP72 can protect against DOX-induced muscle atrophy in several ways. First, HSP72 can provide cytoprotection by preventing protein unfolding [65]. For example, HSP72 is capable of binding to sarcoplasmic reticulum Ca2+ ATPase1a and stabilizing it's nucleotide binding domain to prevent oxidative stress-induced inactivation [69]. In addition, HSP72 is hypothesized to protect mitochondria against oxidative damage through its chaperone activity [3,17]. Although SOD2 functions as an antioxidant within the mitochondrial matrix, SOD2 is first synthesized in the cytosol and then translocated into the mitochondria via HSP72 acting as a chaperone [3,17]. Evidence to support the importance of HSP72 as a chaperone for SOD2 comes from the observation that SOD2 levels in the mitochondria are increased in response to HSP72 overexpression [10].
Overexpression of HSP72 in skeletal muscles can also inhibit the transcriptional activity of both forkhead box O 3a (FOXO3a) and nuclear factor kappa β (NF-κβ) in muscle fibers during prolonged periods of muscle inactivity [11,54]. This is important because activation of both FOXO3a and NF-κβ are linked with the increased expression of the muscle-specific E3 ubiquitin ligases, atrogin-1/muscle atrophy F-box (MAFbx), and MuRF1 which are key players in the ubiquitin proteasome pathway of skeletal muscle protein degradation. Hence, in theory, increases in HSP72 has the potential to protect muscle fibers against DOX-induced muscle atrophy in several ways.
Because of the protective properties of HSP72, it has been hypothesized that increases in HSP72 levels in muscle fibers can protect against DOX-provoked skeletal muscle wasting [56]. In this regard, transgenic overexpression of HSP72 in skeletal muscles (+500% above control) is sufficient to protect skeletal muscles against inactivity-induced muscle atrophy in both young and senescent animals [11,54]. Nonetheless, studies investigating the mechanism(s) responsible for exercise-induced protection against DOX-induced mitochondrial damage in the heart have concluded that exercise-induced increases in HSP72 are not required to achieve protection against DOX-induced damage to the cardiac myocyte [29]. Unfortunately, similar experiments in skeletal muscle have not been performed and this remains an interesting area for future work.
3.3. Exercise training increases the abundance of multidrug resistant proteins in skeletal muscle
Growing evidence suggests that endurance exercise training increases the expression of several members of the ATP-binding cassette (ABC) transporters in both cardiac and skeletal muscle fibers. These ABC transporters belong to a protein superfamily that transport targeted substrates across cell membranes; transported substrates include cellular proteins, lipids, and xenobiotic drugs [75]. Because of their role in drug resistance, ABC transporters are commonly labeled as multidrug resistant proteins [9].
Emerging evidence suggests that ABC transporters can play important roles in prevention of DOX accumulation in cells. Indeed, transgene overexpression of the ABC transporter ABCB8 in the heart is sufficient to protect cardiac myocytes against DOX accumulation [27]. Further, genetic depletion of the ABCB8 gene results in rapid cardiomyopathy in response to treatment with DOX [26,27]. Together, these experiments suggest that overexpression of specific ABC transporters in cardiac and skeletal muscle fibers can protect against DOX accumulation in cells and subsequently, DOX-induced cellular damage.
In reference to exercise and expression of ABC transporters, recent studies reveal that endurance exercise training increases the expression of several cell membrane ABC transporters (e.g., MRP-1, MRP-2, ABCB6, ABCB7, ABCB8, and ABCB10) in both heart and skeletal myocytes [40,44]. Importantly, this exercise-induced increase in ABC transporters is associated with a decrease in the accumulation of DOX in both tissues [20,40,44]. Notably, it was recently discovered that exercise training promotes the expression of several mitochondrial-localized ABC transporters in both heart and skeletal muscle; importantly, this increase in mitochondrial ABC transporters is associated with lower DOX accumulation within the mitochondria [40]. Collectively, these results are consistent with the concept that exercise-induced increases in ABC transporters could contribute to exercise-induced preconditioning against DOX-induced skeletal muscle wasting. However, direct experimental evidence that an increase in ABC transporters is essential for exercise-induced protection against DOX-provoked skeletal muscle atrophy does not currently exist.
3.4. Exercise-induced protection against DOX-induced myopathy: summary and future directions
Abundant evidence demonstrates that performing endurance exercise training prior to DOX treatment protects skeletal muscle against DOX-induced atrophy. This protection is achieved by the prevention of mitochondrial damage, oxidative stress, and activation of skeletal muscle proteolytic systems. Unfortunately, the mechanism(s) responsible for exercise-provoked protection against DOX-induced muscle wasting remain unknown. To date, three primary hypotheses have been proposed to explain why exercise training provides cytoprotection against DOX-mediated damage to skeletal muscles; these include exercise-induced increases in: 1) cytosolic and mitochondrial antioxidants; 2) HSP72 levels; and/or 3) the expression of both sarcolemmal and mitochondrial-specific ABC transporters (Fig. 3). Clearly, each of these claims are testable hypotheses and warrant future research. Rigorous studies to test each of these postulates will likely include both loss of function and gain of function experiments. For example, studies that selectively prevent exercise-mediated increases in individual proteins would provide key information about the role that each protein plays in exercise-induced cytoprotection against DOX-induced damage. Moreover, studies that utilize molecular tools to overexpress specific target proteins, independent of exercise, can provide key information about the role that each protein plays in protection of skeletal muscle fibers against DOX-induced wasting. This topic remains important for future research.
4. Mechanism(s) responsible for exercise-induced protection against inactivity-induced muscle atrophy
Significant progress has recently been made toward delineating the mechanism(s) responsible for exercise-induced protection against inactivity-induced muscle atrophy. The foundation for these studies arose from proteomic experiments investigating the global changes in the abundance of both mitochondrial and soluble proteins in skeletal muscles following endurance exercise training [1,8,60]. These studies identified exercise-induced increases in numerous muscle proteins that possess cytoprotective properties. Hence, each of these protective proteins become candidates to elucidate exercise-induced protection against inactivity-induced muscle atrophy. In response to these proteomic studies, four major hypotheses have evolved to explain the mechanism(s) responsible for exercise-mediated protection against inactivity-induced skeletal muscle atrophy (Fig. 4). A discussion of the evidence to support these postulates follows.
Fig. 4.

Endurance exercise training promotes many biochemical changes in skeletal muscle fibers. Four primary hypotheses have been proposed to explain the exercise training-induced protection against inactivity-induced muscle atrophy; these include: 1) endogenous antioxidant enzymes; 2) Changes in HDAC4/Gadd45 axis; 3) Increased TFAM expression; and 4) heat shock protein 72.
4.1. Exercise training alters the HDAC4/Gadd45 axis
Growth arrest and DNA damage-inducible 45α (Gadd45α) is a soluble, primarily nuclear protein that promotes skeletal muscle atrophy [7]. Specifically, expression of Gadd45α increases in skeletal muscles during prolonged periods of muscle inactivity in both young and old animals [5,62,74]. Importantly, increased expression of Gadd45α appears to be required for the muscle atrophy that occurs during some conditions (e.g., starvation and prolonged disuse) [12]. Indeed, forced expression of Gadd45α in skeletal muscles is sufficient to promote fiber atrophy independent of muscle activity levels [12]. At present, the specific mechanisms connecting Gadd45α to muscle atrophy remain unknown but recent evidence reveals that increased muscle levels of Gadd45α forms a complex with the mitogen-activated protein kinase kinase kinase (MEKK4) to promote atrophy [7]. The mechanisms by which the Gadd45α-MEKK4 complex promotes muscle wasting is unspecified but it is predicted to promote muscle atrophy by phosphorylating one or more downstream muscle proteins involved in muscle wasting [7].
Recently, Yoshihara et al. proposed that exercise preconditioning attenuates inactivity-induced muscle atrophy in old animals via decreased expression of Gadd45α [74]. This work reveals that a single bout of endurance exercise protects old rats against disuse muscle atrophy and this protection is accompanied with decreased levels of both Gadd45α and the class II histone deacetylase 4 (HDAC4) [74]. This decrease in HDAC4 is potentially important because elevated expression of HDAC4 is sufficient to increase Gadd45α mRNA resulting in increased Gadd45α expression in muscle fibers [5]. While these results are consistent with the thesis that exercise training protects against inactivity-induced muscle atrophy by decreasing the expression of Gadd45α, these findings do not demonstrate cause and effect. Therefore, future studies are required to determine if decreased expression of Gadd45α plays a key role in exercise-induced protection against inactivity-induced muscle atrophy.
4.2. TFAM overexpression
Mitochondrial transcription factor A (TFAM) is a mitochondrial DNA binding protein that plays several roles in cells including mitochondrial transcription and protection against oxidative stress [68]. For example, TFAM binds to mitochondrial DNA forming histone-like nucleoid structures to protect DNA from ROS-mediated damage [32]. Further, overexpression of TFAM increases the abundance of both SOD1 and SOD2 in skeletal muscle [68]. Moreover, a recent study reveals that transgenic animals that overexpress TFAM (i.e., five-fold increase) have increased SOD1 and SOD2 abundance and are resistant to skeletal muscle atrophy during hindlimb suspension [67]. This observation provides proof of concept that overexpression of TFAM is associated with increased muscle antioxidants and a rescue from disuse muscle atrophy [67].
In regard to TFAM and exercise, it is established that exercise training increases the abundance of TFAM in the skeletal muscles (reviewed in Ref. [23]). However, it is unclear if this exercise-induced increase in TFAM is sufficient to protect against inactivity-induced muscle atrophy. For example, the five-fold increase in TFAM overexpression in the aforementioned transgenic animals is several times greater than the exercise training-induced increase in TFAM in skeletal muscle. Therefore, it remains unclear if exercise-induced increases in TFAM expression is sufficient to provide protection against disuse muscle atrophy.
4.3. Exercise training-induced increases cellular antioxidants
As introduced previously, decades of research confirms that endurance exercise training increases the abundance of the mitochondrial antioxidant SOD2 in both locomotor and diaphragm muscle [28,41,45,58,60,71]. Improved mitochondrial antioxidant capacity is an attractive hypothesis to explain the exercise training-induced protection against inactivity-induced muscle wasting because increased mitochondrial ROS emission is a requirement for muscle atrophy during prolonged inactivity [48]. This hypothesis was recently tested in experiments using a gene-silencing approach (i.e., antisense oligonucleotide targeted against SOD2) to prevent the exercise-induced increase in SOD2 in the diaphragm. Prevention of exercise-mediated increases in diaphragmatic SOD2 diminished the protection against MV-induced diaphragmatic atrophy, suggesting that increased SOD2 is required to achieve the full benefit of exercise-induced protection (Fig. 5) [41]. This study also revealed that, independent of exercise, transgenic overexpression of SOD2 provided only partial protection against MV-induced diaphragm atrophy [41]. Together, these experiments suggest that while increased levels of SOD2 contributes to the exercise-mediated protection of the diaphragm, the elevation in SOD2 acts in concert with other exercise-mediated changes to protect against inactivity-induced muscle atrophy.
Fig. 5.

A) Prevention of endurance exercise-induced increases in SOD2 in the diaphragm attenuates exercise-provoked protection against MV-induced diaphragmatic atrophy. B) Transgenic overexpression of SOD2 in the diaphragm does not protect against ventilator-induced diaphragmatic atrophy. Data are from Morton et al. [41]. Key: A) SB = sedentary animals, spontaneously breathing; MV = animals exposed to prolonged mechanical ventilation; MVEX = exercise trained animals exposed to prolonged mechanical ventilation; MVEX-SOD2AO = exercise trained animals treated with an antisense oligonucleotide against SOD2 and exposed to prolonged mechanical ventilation. Key: B) SB = sedentary animals, spontaneously breathing; MV = animals exposed to prolonged mechanical ventilation; SOD2OExp = spontaneously breathing animals overexpression an SOD2 transgene; MVSOD2OExp = animals overexpression an SOD2 transgene and exposed to prolonged mechanical ventilation; * = different from SB at P < 0.05.
4.4. Exercise-induced increases in HSP72
Endurance exercise training increases the abundance of HSP72 in skeletal muscles and elevated levels of HSP72 have the potential to protect muscle fibers in several ways (e.g., increased mitochondrial biogenesis/turnover, protection against oxidative damage to mitochondria, refolding of damaged proteins, and prevention of proteolysis). Indeed, transgenic overexpression of HSP72 in skeletal muscles provides protection against inactivity-induced atrophy in limb muscles [11,54]. Importantly, recent research confirms that increases in HSP72 in diaphragm muscle are required to achieve exercise-mediated protection against ventilator-induced diaphragm atrophy [59]. Specifically, when exercise-induced increases in diaphragmatic HSP72 are prevented by an antisense oligonucleotide directed toward HSP72, the exercise-induced protection against MV-mediated diaphragmatic atrophy is lost (Fig. 6). Indeed, prevention of the increases in HSP72 in the diaphragm eliminates the exercise-induced protection against protease activation, oxidative stress, and the mitochondrial uncoupling that occurs in diaphragm fibers during prolonged MV [59]. Furthermore, evidence indicates that both genetic and pharmacological overexpression of HSP72 are sufficient to protect the diaphragm against ventilator-induced fiber atrophy, protease activation, and mitochondrial uncoupling [59]. Collectively, these findings make a strong case for the important role that exercise-induced increases in HSP72 plays in exercise preconditioning of diaphragm muscle.
Fig. 6.

A) Prevention of endurance exercise-stimulated increases in HSP72 in the diaphragm results in the loss of exercise-mediated protection against mechanical ventilation-induced diaphragmatic atrophy. B) Independent of exercise, transgenic overexpression of HSP72 in the diaphragm protects against ventilator-induced diaphragmatic atrophy. Data are from Smuder et al. [59]. Key A): SED = sedentary animals; EX = exercise trained animals; EX-HSP72AO = exercise trained animals treated with an antisense oligonucleotide against HSP72. SB = spontaneously breathing animals; MV = animals exposed to prolonged mechanical ventilation. Key B): SB = spontaneously breathing animals; HSP72 = animals overexpressing HSP72 transgene; MV = animals exposed to prolonged mechanical ventilation; * = different from SB at P < 0.05.
4.5. Exercise-induced protection against inactivity-induced muscle atrophy: summary and future directions
Abundant evidence demonstrates that endurance exercise training protects skeletal muscles against inactivity-induced muscle atrophy. This exercise-induced defense is achieved by preventing the key hallmarks of muscle atrophy: oxidative stress, mitochondrial damage/dysfunction, and the activation of muscle proteases. The mechanism(s) responsible for exercise-induced protection against disuse muscle atrophy have been explored in numerous studies and four principal hypotheses have emerged; these include exercise-induced: 1) increases in cellular antioxidants; 2) changes in the HDAC4/Gadd45 axis; 3) increases in TFAM expression; and 4) elevation in HSP72 levels (Fig. 4). In theory, each of these exercise-mediated biochemical changes in muscle fibers has the potential to protect against inactivity-induced muscle wasting. Nonetheless, to date, only two of these hypotheses have been rigorously tested. Specifically, robust evidence exists that exercise-induced increases in the expression of both SOD2 and HSP72 play important roles in protection against disuse muscle atrophy.
In particular, preventing exercise-induced increase in SOD2 in muscle results in diminished protection against inactivity-induced muscle atrophy. Nonetheless, transgenic overexpression of SOD2 in muscle fibers provides limited protection against disuse muscle atrophy. Therefore, while increased expression of SOD2 contributes to the exercise-induced protection against inactivity-induced muscle atrophy, this increase in SOD2 requires interaction with other exercise-induced cytoprotective molecules to provide the full exercise-induced protection.
New evidence reveals that increased HSP72 expression is essential to achieve exercise-induced protection against ventilator-induced diaphragmatic atrophy. Indeed, prevention of exercise-induced increases in HSP72 results in a complete loss of exercise-mediated protection. Moreover, independent of exercise, transgenic overexpression of HSP72 protects the diaphragm against MV-induced atrophy. Therefore, it is feasible that exercise-induced increases HSP72 in skeletal muscles works in concert with SOD2 to protect against inactivity-induced muscle atrophy.
Several unanswered questions remain regarding the mechanisms responsible for exercise-induced protection against disuse muscle atrophy. As discussed, only two of the four aforementioned hypotheses have been rigorously tested and additional research is required to determine the relative contributions of both TFAM and Gadd45α to exercise-induced protection against disuse muscle atrophy. Moreover, if future studies reveal that exercise-induced changes in these proteins are essential for exercise protection against inactivity-induced atrophy, additional studies will be important to delineate the downstream mechanism(s) by which they provide cytoprotective effects.
Lastly, future translational studies are required to determine the specific details of dose and timing of drug delivery to prevent inactivity-induced muscle atrophy in specific patient populations. For instance, evidence exists that pharmacological overexpression of HSP72 protects against muscle atrophy; however, the efficacy of these pharmacological agents in overexpressing HSP72 must be determined in clinical populations. For example, some patients would likely be treated with pharmacological-inducers of HSP72 at the beginning of hospitalization rather than treatment several days prior to the initiation of prolonged muscle inactivity. Therefore, future studies are essential to determine the time course of effective pharmacological treatment to protect patients against inactivity-induced muscle wasting.
Acknowledgements
This work was supported by grants from the National Institutes of Health (NIH R01AR064189 and R21AR073956) to SKP.
References
- 1.Alves R.M., Vitorino R., Figueiredo P., Duarte J.A., Ferreira R., Amado F. Lifelong physical activity modulation of the skeletal muscle mitochondrial proteome in mice. J. Gerontol. A Biol. Sci. Med. Sci. 2010;65:832–842. doi: 10.1093/gerona/glq081. [DOI] [PubMed] [Google Scholar]
- 2.Anderson E.J., Neufer P.D. Type II skeletal myofibers possess unique properties that potentiate mitochondrial H(2)O(2) generation. Am. J. Physiol. Cell Physiol. 2006;290:C844–C851. doi: 10.1152/ajpcell.00402.2005. [DOI] [PubMed] [Google Scholar]
- 3.Beddoe T., Lithgow T. Delivery of nascent polypeptides to the mitochondrial surface. Biochim. Biophys. Acta. 2002;1592:35–39. doi: 10.1016/s0167-4889(02)00262-8. [DOI] [PubMed] [Google Scholar]
- 4.Betters J.L., Criswell D.S., Shanely R.A., Van Gammeren D., Falk D., Deruisseau K.C., Deering M., Yimlamai T., Powers S.K. Trolox attenuates mechanical ventilation-induced diaphragmatic dysfunction and proteolysis. Am. J. Respir. Crit. Care Med. 2004;170:1179–1184. doi: 10.1164/rccm.200407-939OC. [DOI] [PubMed] [Google Scholar]
- 5.Bongers K.S., Fox D.K., Ebert S.M., Kunkel S.D., Dyle M.C., Bullard S.A., Dierdorff J.M., Adams C.M. Skeletal muscle denervation causes skeletal muscle atrophy through a pathway that involves both Gadd45a and HDAC4. Am. J. Physiol. Endocrinol. Metab. 2013;305:E907–E915. doi: 10.1152/ajpendo.00380.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brigelius-Flohe R. Glutathione peroxidases and redox-regulated transcription factors. Biol. Chem. 2006;387:1329–1335. doi: 10.1515/BC.2006.166. [DOI] [PubMed] [Google Scholar]
- 7.Bullard S.A., Seo S., Schilling B., Dyle M.C., Dierdorff J.M., Ebert S.M., DeLau A.D., Gibson B.W., Adams C.M. Gadd45a protein promotes skeletal muscle atrophy by forming a complex with the protein kinase MEKK4. J. Biol. Chem. 2016;291:17496–17509. doi: 10.1074/jbc.M116.740308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burniston J.G., Hoffman E.P. Proteomic responses of skeletal and cardiac muscle to exercise. Expert Rev. Proteomics. 2011;8:361–377. doi: 10.1586/epr.11.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Z., Shi T., Zhang L., Zhu P., Deng M., Huang C., Hu T., Jiang L., Li J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: a review of the past decade. Canc. Lett. 2016;370:153–164. doi: 10.1016/j.canlet.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 10.Choi S., Park K.A., Lee H.J., Park M.S., Lee J.H., Park K.C., Kim M., Lee S.H., Seo J.S., Yoon B.W. Expression of Cu/Zn SOD protein is suppressed in hsp 70.1 knockout mice. J. Biochem. Mol. Biol. 2005;38:111–114. doi: 10.5483/bmbrep.2005.38.1.111. [DOI] [PubMed] [Google Scholar]
- 11.Dodd S., Hain B., Judge A. Hsp70 prevents disuse muscle atrophy in senescent rats. Biogerontology. 2009;10:605–611. doi: 10.1007/s10522-008-9203-1. [DOI] [PubMed] [Google Scholar]
- 12.Ebert S.M., Dyle M.C., Kunkel S.D., Bullard S.A., Bongers K.S., Fox D.K., Dierdorff J.M., Foster E.D., Adams C.M. Stress-induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J. Biol. Chem. 2012;287:27290–27301. doi: 10.1074/jbc.M112.374777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falk D.J., Kavazis A.N., Whidden M.A., Smuder A.J., McClung J.M., Hudson M.B., Powers S.K. Mechanical ventilation-induced oxidative stress in the diaphragm: role of heme oxygenase-1. Chest. 2011;139:816–824. doi: 10.1378/chest.09-2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Febbraio M.A., Koukoulas I. HSP72 gene expression progressively increases in human skeletal muscle during prolonged, exhaustive exercise. J. Appl. Physiol. 1985;89:1055–1060. doi: 10.1152/jappl.2000.89.3.1055. 2000. [DOI] [PubMed] [Google Scholar]
- 16.Fujino H., Ishihara A., Murakami S., Yasuhara T., Kondo H., Mohri S., Takeda I., Roy R.R. Protective effects of exercise preconditioning on hindlimb unloading-induced atrophy of rat soleus muscle. Acta Physiol (Oxf) 2009;197:65–74. doi: 10.1111/j.1748-1716.2009.01984.x. [DOI] [PubMed] [Google Scholar]
- 17.Glover L.A., Lindsay J.G. Targeting proteins to mitochondria: a current overview. Biochem. J. 1992;284(Pt 3):609–620. doi: 10.1042/bj2840609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gollnick P.D., Struck P.J., Bogyo T.P. Lactic dehydrogenase activities of rat heart and skeletal muscle after exercise and training. J. Appl. Physiol. 1967;22:623–627. doi: 10.1152/jappl.1967.22.4.623. [DOI] [PubMed] [Google Scholar]
- 19.Goncalves R.L., Quinlan C.L., Perevoshchikova I.V., Hey-Mogensen M., Brand M.D. Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J. Biol. Chem. 2015;290:209–227. doi: 10.1074/jbc.M114.619072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall S.E., Smuder A.J., Hayward R. Effects of calorie restriction and voluntary exercise on doxorubicin-induced cardiotoxicity. Integr. Canc. Ther. 2019;18 doi: 10.1177/1534735419843999. 1534735419843999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hearn G.R., Wainio W.W. Succinic dehydrogenase activity of the heart and skeletal muscle of exercised rats. Am. J. Physiol. 1956;185:348–350. doi: 10.1152/ajplegacy.1956.185.2.348. [DOI] [PubMed] [Google Scholar]
- 22.Holloszy J.O. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 1967;242:2278–2282. [PubMed] [Google Scholar]
- 23.Hood D.A., Uguccioni G., Vainshtein A., D'Souza D. Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle: implications for health and disease. Comp. Physiol. 2011;1:1119–1134. doi: 10.1002/cphy.c100074. [DOI] [PubMed] [Google Scholar]
- 24.Hudson M.B., Smuder A.J., Nelson W.B., Wiggs M.P., Shimkus K.L., Fluckey J.D., Szeto H.H., Powers S.K. Partial support ventilation and mitochondrial-targeted antioxidants protect against ventilator-induced decreases in diaphragm muscle protein synthesis. PloS One. 2015;10 doi: 10.1371/journal.pone.0137693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hyldahl R.D., Chen T.C., Nosaka K. Mechanisms and mediators of the skeletal muscle repeated bout effect. Exerc. Sport Sci. Rev. 2017;45:24–33. doi: 10.1249/JES.0000000000000095. [DOI] [PubMed] [Google Scholar]
- 26.Ichikawa Y., Bayeva M., Ghanefar M., Potini V., Sun L., Mutharasan R.K., Wu R., Khechaduri A., Jairaj Naik T., Ardehali H. Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proc. Natl. Acad. Sci. U. S. A. 2012;109:4152–4157. doi: 10.1073/pnas.1119338109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ichikawa Y., Ghanefar M., Bayeva M., Wu R., Khechaduri A., Naga Prasad S.V., Mutharasan R.K., Naik T.J., Ardehali H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Invest. 2014;124:617–630. doi: 10.1172/JCI72931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanter M.M., Hamlin R.L., Unverferth D.V., Davis H.W., Merola A.J. Effect of exercise training on antioxidant enzymes and cardiotoxicity of doxorubicin. J. Appl. Physiol. 1985;59:1298–1303. doi: 10.1152/jappl.1985.59.4.1298. 1985. [DOI] [PubMed] [Google Scholar]
- 29.Kavazis A.N., Smuder A.J., Min K., Tumer N., Powers S.K. Short-term exercise training protects against doxorubicin-induced cardiac mitochondrial damage independent of HSP72. Am. J. Physiol. Heart Circ. Physiol. 2010;299:H1515–H1524. doi: 10.1152/ajpheart.00585.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kavazis A.N., Smuder A.J., Powers S.K. Effects of short-term endurance exercise training on acute doxorubicin-induced FoxO transcription in cardiac and skeletal muscle. J. Appl. Physiol. 1985;117:223–230. doi: 10.1152/japplphysiol.00210.2014. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korzeniewski B. 'Idealized' state 4 and state 3 in mitochondria vs. rest and work in skeletal muscle. PLoS One. 2015;10 doi: 10.1371/journal.pone.0117145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kunkel G.H., Chaturvedi P., Tyagi S.C. Mitochondrial pathways to cardiac recovery: TFAM. Heart Fail. Rev. 2016;21:499–517. doi: 10.1007/s10741-016-9561-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Locke M., Noble E.G., Atkinson B.G. Exercising mammals synthesize stress proteins. Am. J. Physiol. 1990;258:C723–C729. doi: 10.1152/ajpcell.1990.258.4.C723. [DOI] [PubMed] [Google Scholar]
- 34.Luck H. Methods of Enzymatic Analysis. Bergmeyer HAcademic Press; 1965. Catalase; pp. 885–894. [Google Scholar]
- 35.McClung J.M., Kavazis A.N., Whidden M.A., DeRuisseau K.C., Falk D.J., Criswell D.S., Powers S.K. Antioxidant administration attenuates mechanical ventilation-induced rat diaphragm muscle atrophy independent of protein kinase B (PKB Akt) signalling. J. Physiol. 2007;585:203–215. doi: 10.1113/jphysiol.2007.141119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McClung J.M., Van Gammeren D., Whidden M.A., Falk D.J., Kavazis A.N., Hudson M.B., Gayan-Ramirez G., Decramer M., DeRuisseau K.C., Powers S.K. Apocynin attenuates diaphragm oxidative stress and protease activation during prolonged mechanical ventilation. Crit. Care Med. 2009;37:1373–1379. doi: 10.1097/CCM.0b013e31819cef63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Min K., Kwon O.S., Smuder A.J., Wiggs M.P., Sollanek K.J., Christou D.D., Yoo J.K., Hwang M.H., Szeto H.H., Kavazis A.N., Powers S.K. Increased mitochondrial emission of reactive oxygen species and calpain activation are required for doxorubicin-induced cardiac and skeletal muscle myopathy. J. Physiol. 2015;593:2017–2036. doi: 10.1113/jphysiol.2014.286518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Min K., Smuder A.J., Kwon O.S., Kavazis A.N., Szeto H.H., Powers S.K. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J. Appl. Physiol. 1985;111:1459–1466. doi: 10.1152/japplphysiol.00591.2011. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Montalvo R.N., Doerr V., Min K., Szeto H.H., Smuder A.J. Doxorubicin-induced oxidative stress differentially regulates proteolytic signaling in cardiac and skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019;318(2) doi: 10.1152/ajpregu.00299.2019. [DOI] [PubMed] [Google Scholar]
- 40.Morton A.B., Mor Huertas A., Hinkley J.M., Ichinoseki-Sekine N., Christou D.D., Smuder A.J. Mitochondrial accumulation of doxorubicin in cardiac and diaphragm muscle following exercise preconditioning. Mitochondrion. 2019;45:52–62. doi: 10.1016/j.mito.2018.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morton A.B., Smuder A.J., Wiggs M.P., Hall S.E., Ahn B., Hinkley J.M., Ichinoseki-Sekine N., Huertas A.M., Ozdemir M., Yoshihara T., Wawrzyniak N.R., Powers S.K. Increased SOD2 in the diaphragm contributes to exercise-induced protection against ventilator-induced diaphragm dysfunction. Redox Biol. 2019;20:402–413. doi: 10.1016/j.redox.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Naito H., Powers S.K., Demirel H.A., Aoki J. Exercise training increases heat shock protein in skeletal muscles of old rats. Med. Sci. Sports Exerc. 2001;33:729–734. doi: 10.1097/00005768-200105000-00008. [DOI] [PubMed] [Google Scholar]
- 43.Nakamura K., Ohsawa I., Masuzawa R., Konno R., Watanabe A., Kawano F. Running training experience attenuates disuse atrophy in fast-twitch skeletal muscles of rats. J. Appl. Physiol. 1985;123:902–913. doi: 10.1152/japplphysiol.00289.2017. 2017. [DOI] [PubMed] [Google Scholar]
- 44.Parry T.L., Hayward R. Exercise training does not affect anthracycline antitumor efficacy while attenuating cardiac dysfunction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015;309:R675–R683. doi: 10.1152/ajpregu.00185.2015. [DOI] [PubMed] [Google Scholar]
- 45.Powers S.K., Criswell D., Lawler J., Ji L.L., Martin D., Herb R.A., Dudley G. Influence of exercise and fiber type on antioxidant enzyme activity in rat skeletal muscle. Am. J. Physiol. 1994;266:R375–R380. doi: 10.1152/ajpregu.1994.266.2.R375. [DOI] [PubMed] [Google Scholar]
- 46.Powers S.K., Criswell D., Lawler J., Martin D., Lieu F.K., Ji L.L., Herb R.A. Rigorous exercise training increases superoxide dismutase activity in ventricular myocardium. Am. J. Physiol. 1993;265:H2094–H2098. doi: 10.1152/ajpheart.1993.265.6.H2094. [DOI] [PubMed] [Google Scholar]
- 47.Powers S.K., Duarte J.A., Le Nguyen B., Hyatt H. Endurance exercise protects skeletal muscle against both doxorubicin-induced and inactivity-induced muscle wasting. Pflügers Archiv. 2019;471:441–453. doi: 10.1007/s00424-018-2227-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Powers S.K., Hudson M.B., Nelson W.B., Talbert E.E., Min K., Szeto H.H., Kavazis A.N., Smuder A.J. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit. Care Med. 2011;39:1749–1759. doi: 10.1097/CCM.0b013e3182190b62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Powers S.K., Jackson M.J. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008;88:1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Powers S.K., Ji L.L., Kavazis A.N., Jackson M.J. Reactive oxygen species: impact on skeletal muscle. Comp. Physiol. 2011;1:941–969. doi: 10.1002/cphy.c100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Powers S.K., Kavazis A.N., McClung J.M. Oxidative stress and disuse muscle atrophy. J. Appl. Physiol. 1985;102:2389–2397. doi: 10.1152/japplphysiol.01202.2006. 2007. [DOI] [PubMed] [Google Scholar]
- 52.Powers S.K., Morton A.B., Ahn B., Smuder A.J. Redox control of skeletal muscle atrophy. Free Radic. Biol. Med. 2016;98:208–217. doi: 10.1016/j.freeradbiomed.2016.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Samelman T.R., Shiry L.J., Cameron D.F. Endurance training increases the expression of mitochondrial and nuclear encoded cytochrome c oxidase subunits and heat shock proteins in rat skeletal muscle. Eur. J. Appl. Physiol. 2000;83:22–27. doi: 10.1007/s004210000241. [DOI] [PubMed] [Google Scholar]
- 54.Senf S.M., Dodd S.L., McClung J.M., Judge A.R. Hsp70 overexpression inhibits NF-kappaB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J. 2008;22:3836–3845. doi: 10.1096/fj.08-110163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smuder A.J. Exercise stimulates beneficial adaptations to diminish doxorubicin-induced cellular toxicity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019;317:R662–R672. doi: 10.1152/ajpregu.00161.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smuder A.J., Kavazis A.N., Min K., Powers S.K. Exercise protects against doxorubicin-induced markers of autophagy signaling in skeletal muscle. J. Appl. Physiol. 1985;111:1190–1198. doi: 10.1152/japplphysiol.00429.2011. 2011. [DOI] [PubMed] [Google Scholar]
- 57.Smuder A.J., Kavazis A.N., Min K., Powers S.K. Exercise protects against doxorubicin-induced oxidative stress and proteolysis in skeletal muscle. J. Appl. Physiol. 1985;110:935–942. doi: 10.1152/japplphysiol.00677.2010. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smuder A.J., Min K., Hudson M.B., Kavazis A.N., Kwon O.S., Nelson W.B., Powers S.K. Endurance exercise attenuates ventilator-induced diaphragm dysfunction. J. Appl. Physiol. 1985;112:501–510. doi: 10.1152/japplphysiol.01086.2011. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smuder A.J., Morton A.B., Hall S.E., Wiggs M.P., Ahn B., Wawrzyniak N.R., Sollanek K.J., Min K., Kwon O.S., Nelson W.B., Powers S.K. Effects of exercise preconditioning and HSP72 on diaphragm muscle function during mechanical ventilation. J Cachexia Sarcopenia Muscle. 2019;10:767–781. doi: 10.1002/jcsm.12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sollanek K.J., Burniston J.G., Kavazis A.N., Morton A.B., Wiggs M.P., Ahn B., Smuder A.J., Powers S.K. Global proteome changes in the rat diaphragm induced by endurance exercise training. PLoS One. 2017;12 doi: 10.1371/journal.pone.0171007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sorensen J.C., Cheregi B.D., Timpani C.A., Nurgali K., Hayes A., Rybalka E. Mitochondria: inadvertent targets in chemotherapy-induced skeletal muscle toxicity and wasting? Canc. Chemother. Pharmacol. 2016;78:673–683. doi: 10.1007/s00280-016-3045-3. [DOI] [PubMed] [Google Scholar]
- 62.Stevenson E.J., Giresi P.G., Koncarevic A., Kandarian S.C. Global analysis of gene expression patterns during disuse atrophy in rat skeletal muscle. J. Physiol. 2003;551:33–48. doi: 10.1113/jphysiol.2003.044701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tacar O., Sriamornsak P., Dass C.R. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013;65:157–170. doi: 10.1111/j.2042-7158.2012.01567.x. [DOI] [PubMed] [Google Scholar]
- 64.Talbert E.E., Smuder A.J., Min K., Kwon O.S., Szeto H.H., Powers S.K. Immobilization-induced activation of key proteolytic systems in skeletal muscles is prevented by a mitochondria-targeted antioxidant. J. Appl. Physiol. 1985;115:529–538. doi: 10.1152/japplphysiol.00471.2013. 2013. [DOI] [PubMed] [Google Scholar]
- 65.Thakur S.S., Swiderski K., Ryall J.G., Lynch G.S. Therapeutic potential of heat shock protein induction for muscular dystrophy and other muscle wasting conditions. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018;373 doi: 10.1098/rstb.2016.0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Theilen N.T., Jeremic N., Weber G.J., Tyagi S.C. Exercise preconditioning diminishes skeletal muscle atrophy after hindlimb suspension in mice. J. Appl. Physiol. 1985;125:999–1010. doi: 10.1152/japplphysiol.00137.2018. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Theilen N.T., Jeremic N., Weber G.J., Tyagi S.C. TFAM overexpression diminishes skeletal muscle atrophy after hindlimb suspension in mice. Arch. Biochem. Biophys. 2019;666:138–147. doi: 10.1016/j.abb.2018.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Theilen N.T., Kunkel G.H., Tyagi S.C. The role of exercise and TFAM in preventing skeletal muscle atrophy. J. Cell. Physiol. 2017;232:2348–2358. doi: 10.1002/jcp.25737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tupling A.R., Gramolini A.O., Duhamel T.A., Kondo H., Asahi M., Tsuchiya S.C., Borrelli M.J., Lepock J.R., Otsu K., Hori M., MacLennan D.H., Green H.J. HSP70 binds to the fast-twitch skeletal muscle sarco(endo)plasmic reticulum Ca2+ -ATPase (SERCA1a) and prevents thermal inactivation. J. Biol. Chem. 2004;279:52382–52389. doi: 10.1074/jbc.M409336200. [DOI] [PubMed] [Google Scholar]
- 70.Tyml K., Swarbreck S., Pape C., Secor D., Koropatnick J., Feng Q., Veldhuizen R.A.W., Gill S.E. Voluntary running exercise protects against sepsis-induced early inflammatory and pro-coagulant responses in aged mice. Crit. Care. 2017;21:210. doi: 10.1186/s13054-017-1783-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vincent H.K., Powers S.K., Demirel H.A., Coombes J.S., Naito H. Exercise training protects against contraction-induced lipid peroxidation in the diaphragm. Eur. J. Appl. Physiol. Occup. Physiol. 1999;79:268–273. doi: 10.1007/s004210050505. [DOI] [PubMed] [Google Scholar]
- 72.Whidden M.A., McClung J.M., Falk D.J., Hudson M.B., Smuder A.J., Nelson W.B., Powers S.K. Xanthine oxidase contributes to mechanical ventilation-induced diaphragmatic oxidative stress and contractile dysfunction. J. Appl. Physiol. 1985;106:385–394. doi: 10.1152/japplphysiol.91106.2008. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Whidden M.A., Smuder A.J., Wu M., Hudson M.B., Nelson W.B., Powers S.K. Oxidative stress is required for mechanical ventilation-induced protease activation in the diaphragm. J. Appl. Physiol. 1985;108:1376–1382. doi: 10.1152/japplphysiol.00098.2010. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yoshihara T., Tsuzuki T., Chang S.W., Kakigi R., Sugiura T., Naito H. Exercise preconditioning attenuates hind limb unloading-induced gastrocnemius muscle atrophy possibly via the HDAC4/Gadd45 axis in old rats. Exp. Gerontol. 2019;122:34–41. doi: 10.1016/j.exger.2019.04.010. [DOI] [PubMed] [Google Scholar]
- 75.Zhang Y.K., Wang Y.J., Gupta P., Chen Z.S. Multidrug resistance proteins (MRPs) and cancer therapy. AAPS J. 2015;17:802–812. doi: 10.1208/s12248-015-9757-1. [DOI] [PMC free article] [PubMed] [Google Scholar]