

Abstract
Five cyclitol analogues of SL0101 with variable substitution at the C-4′ position (i.e., OH, Cl, F, H, OMe) were synthesized. The series of analogues were evaluated for their ability to inhibit p90 ribosomal S6 kinase (RSK) activity. The study demonstrated the importance of the B-ring C-4′ hydroxy group for RSK1/2 inhibition.
The Ser/Thr protein kinase family, RSK, are downstream effectors of ERK1/2.1,2 RSK activity has been correlated with a number of disease etiologies, including cancer, but no RSK inhibitor has yet transitioned to the clinic.3 SL0101 (1) is a flavonoid glycoside natural product that has been identified as a selective inhibitor of RSK1/2 (Ki ~ 1 μm).4 To date, SL0101 (1) is the only RSK1/2 inhibitor available, which is advantageous as RSK3/4 inhibitors may act to suppress tumor formation.3 The high affinity binding of SL0101 (1) for the N-terminal kinase domain of RSK is dependent on a conformation change in the protein. This conformational change generates a unique binding pocket for the inhibitor and may explain the specificity of SL0101 for RSK1/2.5 In this work we investigated modifications to the B-ring in an effort to further identify the regions of SL0101 that are critical for RSK1/2 interaction and to improve bioavailability (Scheme 1).5,6
Scheme 1.
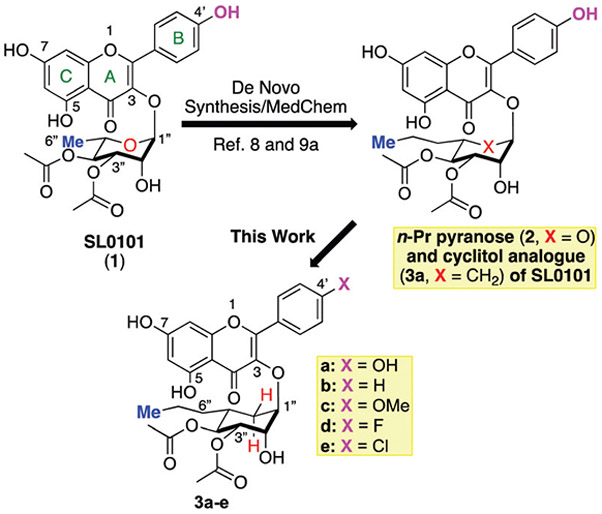
SL0101 structure activity relationship studies.
This combined medicinal chemistry/structural biology effort7 led to the discovery of a C-6″-substituted pyranose analogue (2)8 that possessed improved in vitro kinase inhibitory and anti-cancer activity.9 In an effort to find improved inhibitors with improved bioavailability, we identified a C-6″-substituted carbasugar (3a) analogue10 that retained the RSK-kinase inhibitory activity and demonstrated in vivo efficacy.11,12
The cyclitol variants of SL0101 (3) were designed to be isosteres of improved SL0101 analogue 2 with a pseudo-anomeric bond that is resistant to acid or enzyme catalysed SN1/SN2 hydrolysis.10 In a continued effort to find analogues with improved bioavailability, we became interested in a possible oxidative hydrolysis mechanism that could lead to net hydrolysis of the cyclitol anomeric bond (Scheme 2). Specifically, if the B-ring C-4′ phenol was oxidized in vivo it could lead to net hydrolysis via addition of water to the C-3 position of the A-ring to yield an oxidized aglycon 5 and the free carbasugar 6. The vinylogous quinone functionality of 5 could be biologically reduced to give the aglycon.
Scheme 2.
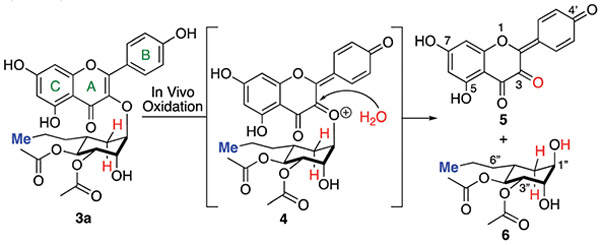
Proposed oxidative mechanism of aglycon hydrolysis.
Therefore, we became interested in finding structural congeners (3b─e) with C-4′ B-ring substitution that would impart resistance to metabolic B-ring oxidation. Previously we found that removal of the C-4′ OH group in SL0101 (1) led to analogues with reduced RSK inhibitory activity.13 Our modeling based upon crystallographic structure of SL0101 bound to the RSK2 NTKD suggested the C-6″ substitution in 2 and 3 would lead to rotation of the B-ring out of plane with the A-ring, which in turn could affect the hydrogen bonding of the C-4′ OH-group. Thus, in addition to the deoxy-variant 3b, we also targeted electronically similar analogues (3c─e) with hydrogen bond accepting methoxy group (3c) and variably sized halogens (i.e., the smaller fluorine 3d and larger chlorine 3e).
Retrosynthetically, we envisioned the synthesis of a small library of five B-ring analogues 3b─e to follow our previously reported synthesis of 3a.7a Thus, analogues 3a─e could be prepared from 7a─e which could result from a Pd-catalyzed cyclitolization reaction between 8 and 9b─f.14 Previously, we described the synthesis of 8 from quinic acid 12 and the aglycon with a free C-3 alcohol could be prepared in 5-steps from benzoic acids 10b─f and acetophenone 11, which can be prepared in two additional steps from phloroglucinol. Herein we disclose the synthesis of cyclitol analogues 3a─e as well as their relative RSK2 in vitro and cell-based inhibitory activity (Schemes 3 and 4).
Scheme 3.
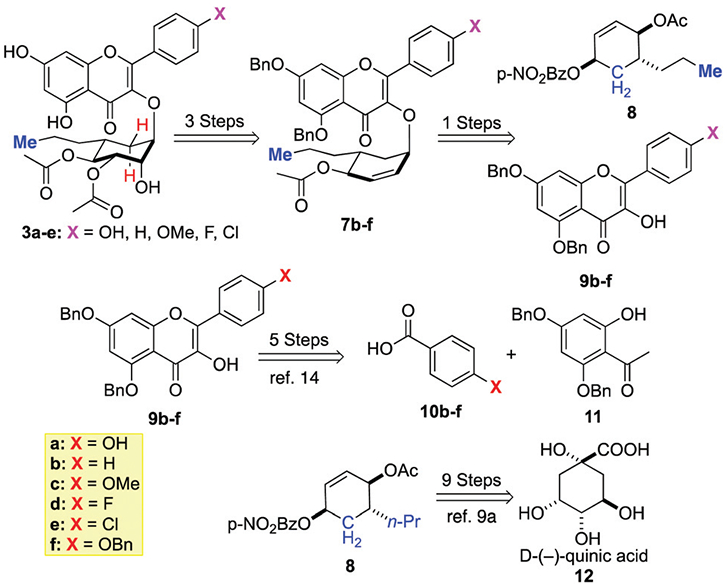
De novo approach to SL0101 cyclitol B-ring analogues.
Scheme 4.
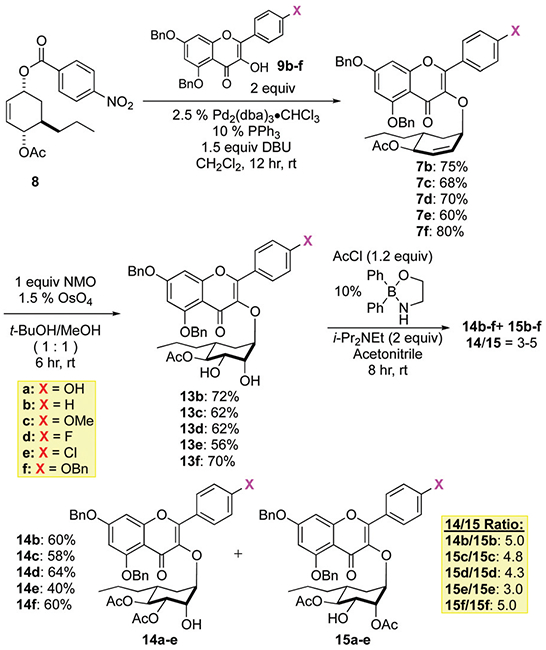
Synthesis of protected analogues.
The synthesis of the analogues begins with a Pd-π-allyl coupling between 8 and the various C-4″ substituted aglycons 9b─f to give 7b─f in generally good yield (60–80% yield). The alkenes in 7b─f was then diastereoselectively dehydroxylated to give diols 13b─f with rhamno-stereochemistry in 56 to 72% yields. We then looked into the regioselective introduction of the C-3″-acetates to form the C-3″/4″ diacetate 14b─f from diols 13b─f. This step was most easily accomplished by using Taylor’s borinate catalyst (10% Ph2BOCH2NH2, AcCl/Hünig’s base).15 This reaction selectively gave 14b─f, with a C-3″ equatorial acetate over 15b─f with a C-2″ axial acetate in a 3 : 1 to 5 : 1 ratio. These compounds were then regioselectively acylated at the C-3″ position via a borinate catalysis with AcCl. With the exception of chloride 14d (40% yield), the remaining diols 14b─c and 14e─f were isolated in good yield after flash chromatography (58 to 64%).
Finally, the benzyl protecting groups were selectively removed from 14b─f under hydrogenolysis condition to give the desired analogues 3a─e (Scheme 5). For the tris-benzyl substrate 14b this occurred, 1 atm H2 using 5% Pd/C to cleanly give 3a in a 70% yield. Similarly, the bis-benzyl substrates 14b─d reacted under identical conditions to give analogues 3b─d in good yields (71–72%). Unfortunately, under the same conditions (1 atm H2 using 5% Pd/C), the chlorine substituted analogue 14e occurred with a significant amount of concomitant reduction of the C-4′ chloride to afford 3b. Careful monitoring of the reaction conditions by lowering the amount of catalyst and reaction times increased the amount of desired analogue 3e being produced. Thus, under these optimized conditions, the desired Cl-substituted analogue 3e could be isolated in a 40% yield along with 30% of 3b.
Scheme 5.
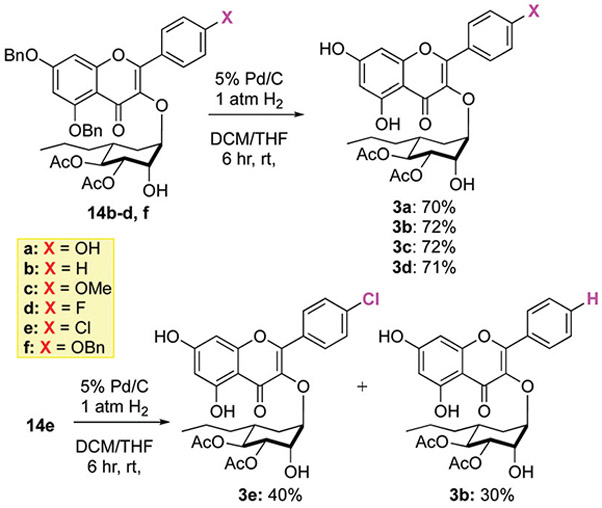
Global deprotection of analogues.
With synthetic access to the series of C-4′-substituted SL0101 cyclitol analogues 3a─e, we evaluated them as RSK2 inhibitors in an in vitro kinase assay. The results are outlined in Table 1. Interestingly, loss or replacement of the hydroxyl group in the B-ring with any other substitutient dramatically decreased the affinity for RSK2. To investigate whether the C-4′ series had potential anti-cancer targets independent of RSK their ability to inhibit the proliferation of the breast cancer cell line, MCF-7, was evaluated. In this analysis 3b and 3e had an IC50 ~ 25 μM and 3d an IC50 ~ 15 μM compared to 3a with an IC50 ~ 10 μM. The compound 3c had minimal inhibitory activity at the highest soluble concentration (50 μM), which may be due to poor cell permeability. These data indicate that C-4′ substitutions inhibit MCF-7 proliferation through a pathway independent of RSK1/2 activity.
Table 1.
In vitro RSK inhibitory activity of B-ring analogues
| Analogue | X = | RSK2 inhibition IC50, μMa | |
|---|---|---|---|
| 3a | OH | 0.58 | ±0.2 |
| 3b | H | 8.23 | ±2.7 |
| 3c | OMe | ND | |
| 3d | F | 15.6 | ±3.5 |
| 3e | Cl | ND |
RSK2 IC50: concentration needed for 50% RSK2 inhibition (n > 3; quadruplicate: mean, 95% confidence interval. ND = IC50 could not be determined because at the maximum soluble concentration in the kinase buffer (30 μM) only 30% inhibition was achieved. The IC50 is a relative value and to facilitate comparisons 3a was included in each assay as a positive control. The value for 3a is significantly higher than in other reports11a,12 and this variation is due to batch-to-batch differences.
In conclusion the asymmetric synthesis of a series of C-4′ substituted cyclitol analogues 3a─e was described. The five syntheses were accomplished in 13 longest linear steps (from d-quinic acid) and 20 total steps (from two commercially available starting materials; quinic acid and phloroglucinol). The convergent nature of the synthesis and the late stage point of divergence significantly reduced the impact of the number of synthetic steps. Thus, final products 3a─e were prepared for 8 and 9a─e in five unique 4-step syntheses. This synthetic effort provided access to four novel SL0101 analogues, which allowed the effects of the C-4″ B-ring substitution to be evaluated. Specifically, the importance of the B-ring phenol OH group was revealed to be essential for the high affinity interaction with RSK.
Acknowledgments
The authors gratefully acknowledge the National Science Foundation (CHE-1565788 (G. A. O.)), and the National Institutes of Health (AI146485 (G. A. O.), AI144196 (G. A. O.), AI142040 (G. A. O.) and CA213201 (D. A. L.)) for their support of this work.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc00128g
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Eisinger-Mathason TS, Andrade J and Lannigan DA, Steroids, 2010, 75, 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2(a).Doehn U, Hauge C, Frank SR, Jensen CJ, Duda K, Nielsen JV, Cohen MS, Johansen JV, Winther BR, Lund LR, Winther O, Taunton J, Hansen SH and Frödin M, Mol. Cell, 2009, 35, 511–522 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Larrea MD, Hong F, Wander SA, da Silva TG, Helfman D and Lannigan D, et al. , Proc. Natl. Acad. Sci. U. S. A, 2009, 106, 9268–9273 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Vial D and McKeown-Longo PJ, J. Biol. Chem, 2012, 287, 40371–40380 [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gawecka JE, Young-Robbins SS, Sulzmaier FJ, Caliva MJ, Heikkila MM, Matter ML and Ramos JW, J. Biol Chem, 2012, 287, 43424–43437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ludwik KA and Lannigan DA, Ribosomal S6 kinase (RSK) modulators: a patent review, Expert Opin. Ther. Pat, 2016, 26, 1061–1078. [DOI] [PubMed] [Google Scholar]
- 4.Smith JA, Poteet-Smith CE, Xu Y, Errington TM, Hecht SM and Lannigan DA, Cancer Res, 2005, 65, 1027–1034. [PubMed] [Google Scholar]
- 5.Utepbergenov D, Derewenda U, Olekhnovich N, Szukalska G, Banerjee B, Hilinski MK, Lannigan DA, Stukenberg PT and Derewenda ZS, Biochemistry, 2012, 51, 6499–6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mrozowski RM, Vemula R, Wu B, Zhang Q, Schroederd BR, Hilinski MK, Clarke DE, Hecht SM, O’Doherty GA and Lannigan DA, ACS Med. Chem. Lett, 2012, 4, 175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7(a).Maloney DJ and Hecht SM, Org. Lett, 2005, 7, 1097–1099; [DOI] [PubMed] [Google Scholar]; (b) Shan M and O’Doherty GA, Org. Lett, 2006, 8, 5149–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8(a).Wang HY, Wu B, Zhang Q, Kang S-W, Rojanasakul Y and O’Doherty GA, ACS Med. Chem. Lett, 2011, 2, 259–263 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Aljahdali AZ, Shi P, Zhong Y and O’Doherty GA, Adv. Carbohydr. Chem. Biochem, 2013, 69, 55–123. [DOI] [PubMed] [Google Scholar]
- 9(a).Li M, Li Y, Mrozowski RM, Sandusky ZM, Shan M, Song X, Wu B, Zhang Q, Lannigan DA and O’Doherty GA, ACS Med. Chem. Lett, 2015, 16, 95–99 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mrozowski RMM, Sandusky ZM, Vemula R, Wu B, Zhang Q, Deborah A, Lannigan DA and O’Doherty GA, Org. Lett, 2014, 16, 5996–5999 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mrozowski RM, Vemula R, Wu B, Zhang Q, Schroederd BR, Hilinski MK, Clarke DE, Hecht SM, O’Doherty GA and Lannigan DA, ACS Med. Chem. Lett, 2013, 4, 175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10(a).Shan M and O’Doherty GA, Org. Lett, 2010, 12, 2986–2989; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shan M and O’Doherty GA, Synthesis, 2008, 3171–3179; [Google Scholar]; (c) Shan M and O’Doherty GA, Org. Lett, 2008, 10, 3381–3384; [DOI] [PubMed] [Google Scholar]; (d) Shan M, Sharif EU and O’Doherty GA, Angew. Chem., Int. Ed, 2010, 49, 9492–9495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11(a).Li M, Li Y, Ludwik KA, Sandusky ZM, Lannigan DA and O’Doherty GA, Org. Lett, 2017, 19, 2410–2413 [DOI] [PubMed] [Google Scholar]; (b) Mrozowski RM, Vemula R, Wu B, Zhang Q, Schroederd BR, Hilinski MK, Clarke DE, Hecht SM, O’Doherty GA and Lannigan DA, ACS Med. Chem. Lett, 2013, 4, 175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For detailed in vivo studies of 3a see: Ludwik KA, Campbell JP, Li M, Li Y, Sandusky ZM, Pasic L, Sowder ME, Brenin DR, Pietenpol JA, O’Doherty GA and Lannigan DA, Mol. Cancer Ther, 2016, 15, 2598–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith JA, Maloney DJ, Sidney M, Hecht SM and Lannigan DA, Bioorg. Med. Chem, 2007, 15, 5018–5034. [DOI] [PubMed] [Google Scholar]
- 14.(a) The synthesis of compounds 9a─e is described in a manuscript under review, the synthesis follows a protocol described in, see: Yang W, Sun J, Lu W, Li Y, Shan L, Han W, Zhang W-D and Yu B, J. Org. Chem, 2010, 75, 6879–6888 [DOI] [PubMed] [Google Scholar]; (b) Li Y, Yang W, Ma Y, Sun J, Shan L, Zhang WD and Yu B, Synlett, 2011, 0915–0918. [Google Scholar]
- 15(a).Chan L and Taylor MS, Org. Lett, 2011, 13, 3090–3093; [DOI] [PubMed] [Google Scholar]; (b) Lee D, Williamson CL, Chan L and Taylor MS, J. Am. Chem. Soc, 2012, 134, 8260–8267 [DOI] [PubMed] [Google Scholar]; (c) Taylor MS, Acc. Chem. Res, 2015, 48, 295–305 [DOI] [PubMed] [Google Scholar]; (d) Bajaj SO, Sharif EU, Akhmedov NG and O’Doherty GA, Chem. Sci, 2014, 5, 2230–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]