

Abstract
Background & Aims:
Non-alcoholic fatty liver disease (NAFLD) is a multifactorial condition and the most common liver disease worldwide, affecting more than one-third of the population. So far there have been no reports on mendelian inheritance in families with NAFLD.
Methods:
We performed whole-exome or targeted next-generation sequencing on patients with autosomal dominant NAFLD.
Results:
We report a heritable form of NAFLD and/or dyslipidemia due to monoallelic ABHD5 mutations, with complete clinical expression after the fourth decade of life, in 7 unrelated multiplex families encompassing 39 affected individuals. The prevalence of ABHD5-associated NAFLD was estimated to be 1 in 1,137 individuals in a normal population.
Conclusion:
We associate a Mendelian form of NAFLD and/or dyslipidemia with monoallelic ABHD5 mutations.
Keywords: Non-alcoholic fatty liver disease, ABHD5, CGI-58, Mendelian, NAFLD, Familial aggregation, Dyslipidemia, Inheritance
GRAPHICAL ABSTRACT
Lay summary:
Non-alcoholic fatty liver disease (NAFLD) is a common multifactorial disorder with a strong genetic component. Inherited forms of NAFLD have been suspected but, their molecular pathogenesis has not been disclosed. Here we report a heritable form of NAFLD with clinical expression after 40 years of age, associated with monoallelic ABHD5 mutations.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is an increasingly common disorder that is strongly associated with the metabolic syndrome, and which may progress from simple steatosis to non-alcoholic steatohepatitis (NASH), cirrhosis, hepatic failure, and hepatocellular carcinoma. NAFLD is a major health issue worldwide and is associated with significant morbidity and mortality. The prevalence of this condition reaches 12–18% in the European countries and 27–38% in the US.1,2 NAFLD is a multifactorial disease and up to 50% of its relative risk has been attributed to genetic susceptibility, with evidence coming from familial aggregation, twin studies, and differential ethnic pre-disposition.3 Genome-wide association studies identified a number of variants that are associated with NAFLD, but in most cases the odds ratios were relatively small. However, in large-scale population studies, variants in the PNPLA3, TM6SF2, LYPLAL1, and GCKR genes were associated with elevated liver fat levels.4 The existence of inherited forms of NAFLD has been suspected, but neither a specific causal gene or a susceptibility locus has been identified.5 We hypothesized that genetic alterations might account for some cases of NAFLD, especially those that exhibit Mendelian inheritance.
Patients and methods
After obtaining written informed consent to perform genomic studies, we performed whole-exome sequencing (WES) or targeted next-generation sequencing (NGS) of genomic DNA from NAFLD cases in 7 unrelated multiplex families (Fig. 1A; Supplementary materials and methods).
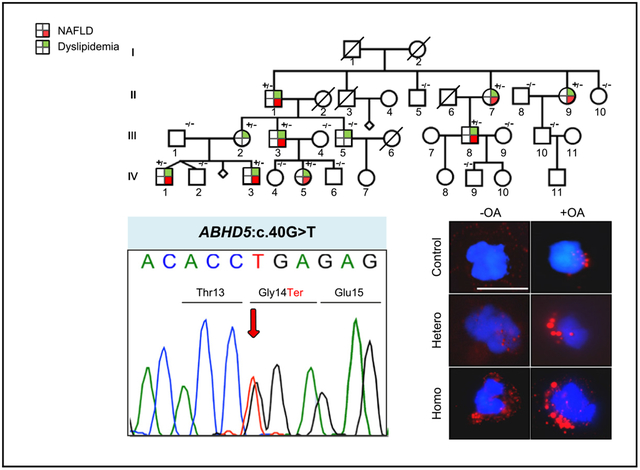
Fig. 1. Pedigree structures and clinical findings in NAFLD families with ABHD5 mutations.
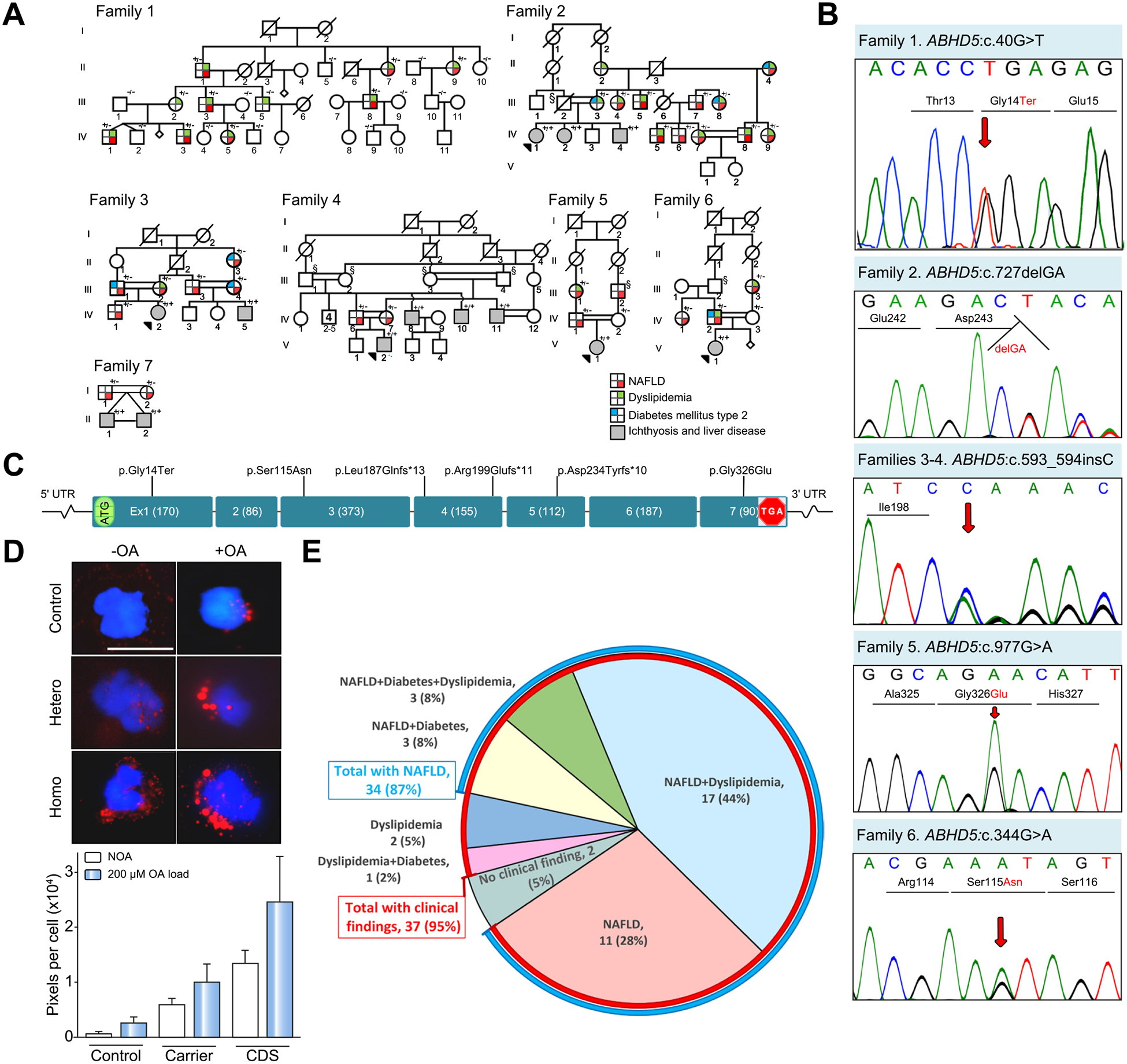
(A) Family 1 is of Italian ancestry with a monoallelic mutation in ABHD5. Note the lack of consanguinity in this family. Families 2–7 of Iranian ancestry show extensive consanguinity. Heterozygous carriers (+/−)show evidence of NAFLD and/or dyslipidemia and type 2 diabetes mellitus, and patients with bi-allelic mutations (+/+) manifest with CDS with neonatal ichthyosis and NAFLD. Individuals marked with § are presumed obligatory carriers of the mutation. For the presence of clinical manifestations in individuals tested, see the color code. (B) Sanger sequencing of mutations in Families 1–6. The mutation of Family 7, ABHD5:c.560_578 del19 was published previously.17 (C) Positions of the distinct mutations along the ABHD5 consisting of 7 exons drawn to scale; the introns are not in scale. (D) Presence of lipid droplets (red) in leukocytes from a control (upper panels), a heterozygous carrier (middle panel), and a homozygous individual (lower panel) after incubation without (-OA, left) or with (+OA, right) 200 lM OA. The lipid content was quantitated by assay of pixel density of Oil red O and DAPI stained cells (bar graph). The values represent the mean ± SD of 105–125 cells for each sample. (E) Relative distribution of clinical findings and their combinations in 39 ABHD5 carriers. Note that 35 individuals had NAFLD (87%, blue line) while 37 had clinical findings (95%, red line). CDS, Chanarin-Dorfman syndrome; NAFLD, non-alcoholic fatty liver disease; OA, oleic acid; UTR, untranslated region.
Results
In a large non-consanguineous family of Italian ancestry (Family 1-F1; Fig. 1A), 9 affected members were diagnosed with NAFLD and/or dyslipidemia, based on blood chemical analyses and liver ultrasound findings (Table 1). In subjects F1, III-3 and II-1 with NAFLD, WES was performed and bioinformatics analysis identified a heterozygous nonsense mutation (c.40G>T; p.Gly14Ter) in exon 1 of ABHD5 (OMIM #604780), confirmed by Sanger sequencing (Fig. 1B). Segregation analysis of the pedigree revealed that all 9 affected patients carry the same ABHD5 mutation, while 13 members without evidence of NAFLD have a wild-type ABHD5 genotype, among which only 1 patient (F1, III-5) presented with dyslipidemia. Jordans’ bodies (abnormal lipid droplets [LDs] accumulation)6 were evident in leukocytes from ABHD5 mutation carriers after in vitro oleic acid loading (Fig. 1D). None of the tested individuals demonstrated homozygosity for the ABHD5 mutation. Thus, these findings suggested that monoallelic mutations in ABHD5 predispose individuals to NAFLD.
Table 1.
Clinical manifestations, and genotypes of individuals with monoallelic ABHD5 mutations.
| No. | Patient | Phenotype | Age (yrs) | Gender | Levels of Ch, TG (mg/dl) | Liver enzymes levels | NAFLD grades+ | FIB-4 score | Weight (kg) | Height (m) | BMI | Other diseases |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family 1: c.40G>T, p.G14* | ||||||||||||
| 1 | II:1* | NAFLD, DL | 76 | M | TG:284 CH:248 |
High | 1 to 2 | 64 | 1.78 | 20.2 | ||
| 2 | II:7 | NAFLD, DL | 67 | F | TG:256 CH:235 |
High | 2 | 76 | 1.71 | 26.0 | Hypertension, heart arrhythmia | |
| 3 | II:9 | NAFLD, DL | 64 | F | TG:188–212, Ch:210–244 |
High | 1 | 55 | 1.63 | 20.7 | ||
| 4 | III:2 | DL | 51 | F | TG:179 CH:255 |
Moderately raised | – | 63 | 1.59 | 24.9 | ||
| 5 | III:3 | NAFLD, DL | 48 | M | TG:287–324, Ch:237–269 |
High | 2 | 82 | 1.75 | 26.8 | Hypertension | |
| 6 | III:8 | NAFLD, DL | 42 | M | TG:195 CH:230 |
High | 2 | 57 | 1.65 | 20.9 | ||
| 7 | IV:1 | NAFLD, DL | 26 | M | TG:171–188, Ch: 218–240 |
High | 1 | 59 | 1.74 | 19.5 | Left varicocele (grade II) | |
| 8 | IV:3 | NAFLD, DL | 23 | M | TG:165, Ch:215 |
Moderately raised | 1 | 61 | 1.58 | 24.4 | ||
| 9 | IV:5 | NAFLD, DL | 21 | F | TG:155–171, Ch:205–226 |
Moderately raised | 0 to 1 | 66 | 1.79 | 20.6 | Pollen allergy, chronic fatigue | |
| Family 2: c.727delGA, p.D243Y*fs10 | ||||||||||||
| 10 | II-2 | DL | 77 | F | TG:235 Ch:249 |
Normal | – | 2.54 | 45 | 1.55 | 18.7 | |
| 11 | II-4 | NAFLD, DL, DM | 57 | F | LDL:190 Ch:285 |
Normal | 1 | 60 | 1.63 | 22.6 | Skin cancer | |
| 12 | III-3 | DL, DM | 46 | F | TG:224 Ch:231 |
Normal | −(S0) | 60 | 1.6 | 23.4 | ||
| 13 | III-4 | NAFLD, DL | 38 | F | TG: 457, CH:259 | High | 1 to 2 | 0.87 | 60 | 1.6 | 23.4 | Hypothyroidism |
| 14 | III-5 | NAFLD, DL | 44 | M | Ch:240 | Normal | 1 | 70 | 1.9 | 19.4 | ||
| 15 | III-7 | NAFLD | 59 | M | Normal | Normal | 1 | 0.82 | 73 | 1.78 | 23 | |
| 16 | III-8 | NAFLD, DL, DM | 47 | F | Ch:285 | Normal | 1 | 90 | 1.58 | 36.1 | Focal nodular hyperplasia | |
| 17 | IV-5 | NAFLD, DL | 32 | M | Ch:254 | High | 1 | 0.77 | 89 | 1.8 | 27.5 | |
| 18 | IV-6 | NAFLD | 29 | M | Normal | High | 1 | 0.74 | 85 | 1.81 | 25.9 | |
| 19 | IV-7 | NAFLD, DL | 28 | F | LDL:108 | Normal | 1 | 63 | 1.6 | 24.6 | ||
| 20 | IV-8 | NAFLD, DL | 40 | M | LDL:126 | Normal | 2 | 80 | 1.83 | 23.9 | ||
| 21 | IV-9 | NAFLD, DL | 42 | F | LDL:117 | Normal | 1 | 72 | 1.65 | 26.4 | Hypertension, Infertility |
|
| Family 3: c.593_594insC, p.R199Qfs*11 | ||||||||||||
| 22 | II-3 | NAFLD, DM |
70 | F | Normal | Normal | 1 | 62 | 1.65 | 22.8 | ||
| 23 | III-1 | NAFLD, DM |
57 | M | Normal | Normal | 1 | 80 | 1.85 | 23.4 | ||
| 24 | III-2 | NAFLD, DL | 48 | F | Ch:230 | Normal | 1 | 0.81 | 68 | 1.74 | 22.5 | |
| 25 | III-3 | NAFLD | 57 | M | Normal | Normal | 2 | 0.88 | 74 | 1.7 | 25.6 | |
| 26 | III-4 | NAFLD, DM |
47 | F | Normal | Normal | 1 | 85 | 1.65 | 31.2 | Hypothyroidism | |
| 27 | IV-1 | NAFLD | 31 | M | Normal | High | 1 | 0.24 | 100 | 1.89 | 28 | Immune thrombocytopenic purpura |
| Family 4: c.593_594insC, p.R199Qfs*11 | ||||||||||||
| 28 | IV-6 | NAFLD | 36 | M | Normal | Normal | 1 (S2) | 0.88 | 100 | 1.9 | 27.7 | |
| 29 | IV-7 | NAFLD | 38 | F | Normal | Normal | − (S2) | 0.52 | 60 | 1.56 | 24.7 | |
| Family 5: c.977G>A, p.G326E | ||||||||||||
| 30 | III-1 | NAFLD, DL | 59 | F | LDL: 136 (<130) Ch:224 | Normal | 1 (S2) | 1.11 | 85 | 1.7 | 31.2 | Hypothyroidism |
| 31 | IV-1 | NAFLD | 36 | M | Normal | Normal | − (S2) | 80 | 1.83 | 23.9 | ||
| 32 | IV-2 | Normal | 29 | F | Normal | Normal | 0 to 1 (S2) |
73 | 1.68 | 25.9 | ||
| 33 | III-2 | NAFLD | 64 | M | Normal | n.a. | (S3) | 100 | 1.78 | 31.6 | ||
| Family 6: c.344G>A, p.S115N | ||||||||||||
| 34 | III-3 | NAFLD, DL | 60 | F | Ch:218 | Normal | 1 | 1.79 | 63 | 1.6 | 24.6 | (Atorvastatin 20 mg/day) |
| 35 | IV-1 | NAFLD | 35 | F | Normal | Normal | 2 | 75 | 1.7 | 26 | ||
| 36 | IV-2 | NAFLD, DL, DM | 43 | M | TG:356 | High | 1 (S3) | 0.94 | 76 | 1.73 | 25.4 | |
| 37 | IV-3 | Normal | 40 | F | Normal | Normal | – | 1.41 | 60 | 1.6 | 23.4 | |
| Family 7: c.560_578 delTTGCTGATCAAGACAGACC | ||||||||||||
| 38 | I-1 | NAFLD | 26 | F | Normal | Normal | 1 | 68 | 1.62 | 25.91 | ||
| 39 | I-2 | NAFLD | 28 | M | Normal | Normal | 1 | 72 | 1.75 | 23.5 | ||
BMI, body mass index; Ch, cholesterol; DL, dyslipidemia; DM, diabetes mellitus; LDL, low-density lipoproteins; n.a., not available; NAFLD, non-alcoholic fatty liver disease; TG, triglyceride.
No more than 1 glass of wine or beer twice a day, during lunch and dinner (no alcohol intake outside meals).
The values were obtained by ultrasound, while those in parenthesis were acquired by FiboScan.
While individuals with NAFLD in Family 1 presented with a monoallelic loss-of-function ABHD5 mutation that predisposed them to liver disease with no apparent multisystemic involvement, biallelic mutations in the same gene encoding for the LD-binding protein CGI-58 have been reported in patients with Chanarin-Dorfman syndrome (CDS; OMIM #275630). This rare autosomal recessive neutral lipid storage disorder is characterized by an excessive accumulation of LDs in multiple tissues and is more frequent in Mediterranean, Middle Eastern and Indian inbred populations.7,8 The clinical hallmark of CDS is congenital generalized dry and scaly skin (ichthyosis). Further clinical features include hepatomegaly with late fatty degeneration of the liver, myopathy, cataracts and/or ectropion, progressive hearing loss, developmental delay, and cognitive impairment.9 Vacuolated leukocytes are present in patients and are occasionally identified in non-ichthyotic carriers.
To explore the potential presence of NAFLD in heterozygous carriers of ABHD5 mutations in CDS families, we investigated 6 independent, highly consanguineous Iranian families with ichthyosis suggestive of CDS (Fig. 1a: F2–F7) by a 38-gene targeted NGS panel for hereditary ichthyoses including ABHD5.10,11 In all 6 families, comprising 13 patients with ichthyosis and liver disease, CDS was confirmed by the identification of 5 distinct biallelic ABHD5 mutations (Fig. 1B, C). We next genotyped non-CDS family members who were either proven or obligate heterozygous carriers of the ABHD5 mutation, did not consume alcohol, and provided their informed consent to the study. We found that 28 of the 30 (see Fig. 1A, E) Iranian participants had features of NAFLD, dyslipidemia, or type 2 diabetes, as ascertained by blood chemical analyses, hepatic ultrasound findings, and FibroScan® (transient elastography) (Table 1). The age of the 2 individuals without clinical findings (F5, IV-2, and F6, IV-3), who did not display features of NAFLD, was 29 and 40 years, respectively. Calculation of Fibrosis-4 scores in 14 individuals from whom necessary data were available, revealed scores ranging from 0.24 to 2.54, and 2 of them were above the lower cut-off value, 1.45, indicating risk for advanced fibrosis.12
Overall, 37 out of 39 ascertained ABHD5 mutation carriers showed evidence of NAFLD and/or dyslipidemia (Fig. 1E; estimated penetrance: 95%), and 15 of those with NAFLD could be graded as ≤2 and 2 of them ≥3 (Table 1). All individuals >40 years of age with monoallelic mutations showed evidence of liver involvement and/or dyslipidemia. The youngest ABHD5 carrier (F1, IV-5; 21 years of age) showed only initial signs of NAFLD, suggesting an age-dependent development of this inherited form of NAFLD, with complete clinical expression after the fourth decade of life. Interestingly, the 2 individuals without NAFLD are carriers of missense ABHD5 mutations (p.Ser115Asn and p.Gly326Glu) which, in contrast to nonsense or frameshift mutations, could retain some residual activity that may contribute to reduced penetrance or delayed onset in families 5 and 6. Liver histopathology was not available because of local Institutional Review Boards’ refusal to approve liver biopsy from patients with monoallelic mutations. However, patients with CDS and biallelic mutations have previously been shown to have liver fibrosis in biopsy13. In our series, FibroScan of 6 patients with biallelic ABHD5 mutations revealed evidence of fibrosis in 3 of them: In 1 patient who was 17 years of age (Family 6, V-1), the F score was F4, indicating cirrhosis, and in 2 other patients who were 10 and 30 years of age (Family 2, IV-1 and IV-4, respectively), the F score F3 implied moderate fibrosis. Three patients had an F score of F0/1, all of whom were relatively young (3–20 years of age).
In an attempt to estimate the frequency of potentially NAFLD-causing ABHD5 deleterious alleles, we analyzed ABHD5 variants from 188,951 unrelated individuals of diverse ethnic backgrounds reported in the Exome Aggregation Consortium (ExAC), Genome Aggregation (gnomAD) and Greater Middle East Variome Project (GME) databases (Fig. S1).14 The reported 1,149 variants in ABHDs were annotated (ANNOVAR software), with exclusion of synonymous, intronic, 3ʹ and 5ʹ UTR variants, and removal of those predicted to be benign by in silico analysis. The surviving 77 variants (Fig. S1) were present in a total of 167 individuals, predicting a prevalence of ABHD5-associated NAFLD in the general population of 1 in 1,131 individuals, a figure that is <0.001 if the genetic penetrance is assumed to be 95%.
Discussion
The specific metabolic and cellular mechanism(s) leading to NAFLD are still under investigation, but there is strong evidence that increased hepatic deposition of triglycerides resulting from an imbalance in lipid storage and lipolysis or secretion plays an important role in the establishment and progression of NAFLD to NASH and its inflammatory, organ-failure and neoplastic sequelae.15 CGI-58, the protein encoded by ABHD5, is a highly conserved regulator of adipose triglyceride lipase and is involved in lipid metabolism, tumor progression, viral replication, and skin barrier formation. While the latter role requires at least 1 functional ABHD5 allele as evidenced by the autosomal recessive mode of inheritance of CDS, the hypothesis of a haplo-insufficiency effect on LD homeostasis in the liver, with incomplete penetrance and progressive, age-dependent expressivity of dysfunctional ABHD5 variants, is consistent with our clinical, chemical, and genetic observations and previous animal studies. Specifically, ABHD5 complete knockout mice die shortly after birth due to a skin barrier defect that closely resembles ichthyosis in patients with CDS, while a liver-specific ABHD5 knockout mouse shows steatohepatitis and fibrosis.16
In conclusion, our findings disclose the existence of a rare heritable form of NAFLD associated with monoallelic mutations in, ABHD5, involved in neutral lipid metabolism, and highlight the importance of further studies on the role of LD disorders in the liver pathology.
Supplementary Material
Highlights.
Non-alcoholic fatty liver disease (NAFLD) is the most common liver disease in the world.
We identified monoallelic ABHD5 mutations in 7 families with NAFLD.
ABHD5 is involved in neutral lipid metabolism, highlighting the role of lipid disorders in NAFLD.
Acknowledgments
We would like to thank Kaveh Khademhossein, Drs. Shahrbanoo Nakaei, Seyedeh Maryam Tara and Maryam Abiri for assistance in sample collection. Drs. Andrew Touati, Farzaneh Abbasi and Roshanak Abbasi assisted in clinical evaluation of patients. Ali Jazayeri and Gaurav Kumar assisted in bioinformatics analysis, and Carol Kelly provided assistance in manuscript preparation. This study is in partial fulfillment of the PhD Thesis of Leila Youssefian.
Financial support
The authors received no financial support to produce this manuscript.
Footnotes
Conflict of interest
The authors declare no conflicts of interest that pertain to this work.
Please refer to the accompanying ICMJE disclosure forms for further details.
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jhep.2019.03.026.
References
- [1].Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018;15:11–20. [DOI] [PubMed] [Google Scholar]
- [2].Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science 2011;332:1519–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: clinical impact. J Hepatol 2018;68:268–279. [DOI] [PubMed] [Google Scholar]
- [4].Caussy C, Soni M, Cui J, Bettencourt R, Schork N, Chen CH, et al. Nonalcoholic fatty liver disease with cirrhosis increases familial risk for advanced fibrosis. J Clin Invest 2017;127:2697–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lefevre C, Jobard F, Caux F, Bouadjar B, Karaduman A, Heilig R, et al. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am J Hum Genet 2001;69:1002–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jordans GH. The familial occurrence of fat containing vacuoles in the leukocytes diagnosed in two brothers suffering from dystrophia musculorum progressiva (ERB). Acta Med Scand 1953;145:419–423. [DOI] [PubMed] [Google Scholar]
- [7].Dorfman ML, Hershko C, Eisenberg S, Sagher F. Ichthyosiform dermatosis with systemic lipidosis. Arch Dermatol 1974;110:261–266. [PubMed] [Google Scholar]
- [8].Chanarin I, Patel A, Slavin G, Wills EJ, Andrews TM, Stewart G, et al. Neutral-lipid storage disease: a new disorder of lipid metabolism. Br Med J 1975;1:553–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Williams ML, Koch TK, O’Donnell JJ, Frost PH, Epstein LB, Grizzard WS, et al. Ichthyosis and neutral lipid storage disease. Am J Med Genet 1985;20:711–726. [DOI] [PubMed] [Google Scholar]
- [10].Vahidnezhad H, Youssefian L, Saeidian AH, Zeinali S, Mansouri P, Sotoudeh S, et al. Gene-targeted next generation sequencing identifies PNPLA1 mutations in patients with a phenotypic spectrum of autosomal recessive congenital ichthyosis: The impact of consanguinity. J Invest Dermatol 2017;137:678–685. [DOI] [PubMed] [Google Scholar]
- [11].Youssefian L, Vahidnezhad H, Saeidian AH, Sotoudeh S, Mahmoudi H, Mansouri P, et al. Autosomal recessive congenital ichthyosis: Genomic landscape and phenotypic spectrum in a cohort of 125 cnsanguineous families. Hum. Mutat 2019;40:288–298. [DOI] [PubMed] [Google Scholar]
- [12].Sterling RK, Lissen E, Clumeck N, Sola R, Correa MC, Montaner J, et al. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology 2006;43:1317–1325. [DOI] [PubMed] [Google Scholar]
- [13].Demerjian M, Crumrine DA, Milstone LM, Williams ML, Elias PM. Barrier dysfunction and pathogenesis of neutral lipid storage disease with ichthyosis (Chanarin-Dorfman syndrome). J Invest Dermatol 2006;126:2032–2038. [DOI] [PubMed] [Google Scholar]
- [14].Karczewski KJ, Weisburd B, Thomas B, Solomonson M, Ruderfer DM, Kavanagh D, et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res 2017;45: D840–D845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gluchowski NL, Becuwe M, Walther TC, Farese RV Jr. Lipid droplets and liver disease: from basic biology to clinical implications. Nat Rev Gastroenterol Hepatol 2017;14:343–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Guo F, Ma Y, Kadegowda AK, Betters JL, Xie P, Liu G, et al. Deficiency of liver Comparative Gene Identification-58 causes steatohepatitis and fibrosis in mice. J Lipid Res 2013;54:2109–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nakhaei S, Heidary H, Rahimian A, Vafadar M, Rohani F, Bahoosh GR, et al. A new case of Chanarin-Dorfman Syndrome with a novel deletion in ABHD5 gene. Iran Biomed J 2018;22:415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.