

Summary
Focal copy number increases (genomic amplification) pinpoint oncogenic driver genes and therapeutic targets in cancer genomes. With the advent of genomic technologies, recurrent genomic amplification has been mapped throughout the genome. Recurrent amplification could be solely due to the positive selection for the tumor-promoting effects of amplified gene products. Alternatively, recurrence could result from the susceptibility of the loci to amplification. Distinguishing between these possibilities requires a full understanding of amplification mechanisms. Two mechanisms, the formation of double minute chromosomes and breakage-fusion-bridge cycles, have been repeatedly linked to genomic amplification, and the impact of both mechanisms has been confirmed in cancer genomics data. Here we review the details of these mechanisms and discuss the mechanisms underlying recurrence.
Gene amplification in biology and cancer
A gene can express its product robustly by increasing the copy number (gene amplification) and accelerate a particular cellular activity. In somatic cell development, gene amplification is a regulated process to meet the increasing demands of protein synthesis (e.g. ribosomal RNA gene in Xenopus oocytes and ciliated protozoa) or of a particular protein necessary for a specific developmental stage (e.g. chorion gene in ovarian follicle cells in Drosophila) [1–3]. Gene amplification can also be an adaptive process to the environment. For instance, both bacteria and unicellular eukaryotes develop resistance to antibiotics by amplifying genes that efflux drugs from the cell [4]. By far the most studied cases of gene amplification are its occurrence in tumors. The adaptive aspect of gene amplification is also a focused issue in tumors, as gene amplification is a measure to antagonize anti-cancer treatments by directly increasing the dosage of target proteins [5] or by activating an alternative cell-proliferation pathway [6]. More commonly, genes promoting tumorigenesis and progression employ gene amplification to increase their protein level. MYC at chromosome 8q24.21, a basic helix-loop-helix transcription factor is amplified frequently across tumor types and influences various cellular processes by modulating global transcriptome [7]. ERBB2 (see Glossary) at 17q12-21, a gene encoding HER2, a member of epidermal growth factor receptors, is amplified in 15-20% of breast tumors and marks a distinct subtype that is treated differently from other breast tumors [8].
The process of identifying amplified genes has been greatly accelerated since the advent of genomic technologies. The Cancer Genome Atlas (TCGA, see Glossary) project has cataloged the amplified regions (cytoband, see Glossary) across tumor types [9–14] (Table 1). Among the adult epithelial tumors from six organs, 8q24.21 stands out as it is amplified in all tumor types. 8q24.21 spans 800 kb regions and harbors the MYC oncogene, which confirms the finding in the earlier studies. Cytobands 1q21.3, 8p11.21, 8p11.23, 10q22.3, 11q13.3, 12q15 and 15q26.3 are amplified in more than three tumor types, and in some case harbor validated targets. CCND1 at 11q13.3 encodes a Cyclin D1 that promotes G1/S transitions of cell cycles. MDM2 at 12q15 encodes an E3 ubiquitin ligase that degrades tumor suppressor protein TP53. IGF1R at 15q26.3 is a gene for a receptor tyrosine kinase that binds to the insulin-like growth factor. MCL1 at 1q21.3 encodes a protein for an anti-apoptotic function. There are also cytobands that are amplified in a particular tumor lineage. 3q26.33 is frequently amplified in squamous cell lung tumors. Among the 19 genes within the amplified region, SOX2, a gene encoding the transcription factor for genes that maintain pluripotency, is a candidate driver of amplification [15]. In lung adenocarcinoma, 14q13.3 is frequently amplified, and a gene for homeobox transcription factor NKX2-1 at 14q13.3 was shown to promote the survival of lung adenocarcinoma cells [16, 17]. Interestingly, while tumor-type specific amplicons (see Glossary) harbor lineage-specific transcription factors, pan-cancer amplicons have proteins that promote basic cellular processes for proliferation.
Table 1.
Recurrent genomic amplification in human tumors
| Breast | Colorec | Lung sq | Lung ad | Prostate | Ovarian | |
|---|---|---|---|---|---|---|
| 1q21.2 | ||||||
| 1q21.3 | ||||||
| 1p22.3 | ||||||
| 3q26.2 | ||||||
| 4q13.3 | ||||||
| 5p15.33 | ||||||
| 6q21 | ||||||
| 7p11.2 | ||||||
| 8p11.21 | ||||||
| 8p11.23 | ||||||
| 8q24.21 | ||||||
| 10q22.3 | ||||||
| 11q13.3 | ||||||
| 11q14.1 | ||||||
| 12p12.1 | ||||||
| 12p13.33 | ||||||
| 12q15 | ||||||
| 14q21.1 | ||||||
| 15q26.3 | ||||||
| 17q12 | ||||||
| 19q12 | ||||||
| 20q11.21 | ||||||
| 20q13.12 | ||||||
| 20q13.2 | ||||||
| 20q13.33 |
From TCGA data. Recurrently amplified cytobands (GISTIC score <0.01) are highlighted.
With the TCGA data, gene amplification is now viewed as genomic amplification in which copy number breakpoints have been determined with the nucleotide-level resolution, where copy number of the genome traverses from normal copy number to the increased copy number. Such resolution provides essential information for the mechanisms underlying genomic amplification and raises new questions. For a given driver gene, the locations of copy number breakpoints and amplified regions (amplicons) vary between individual tumors. Such variable breakpoints may indicate that amplification mechanisms are different between tumors. Alternatively, those variable breakpoints are the products of secondary rearrangements that mask the initiating breakpoints, and underlying mechanisms may be common between tumors. Second, considering the remarkable built-in redundancy of the human genome, genes with similar functions are elsewhere in the genome (Fig.1, Key Figure). For example, MYC has closely related bHLH transcription factors MYCL and MYCN at 1q34.2 and 2p24.3, respectively, both of which have oncogenic potential and can rescue proliferation defects of MYC-null cells [18, 19]. Although MYC is amplified frequently (15-40%) across tumor types, MYCL and MYCN are amplified much less frequently, in 2-8% of tumors. Another example is MDM2. MDM2 has a structurally related protein MDM4 (MDMX) at 1q32.1. Both MDM2 and MDM4 knockout mice are embryonic lethal but the lethality is rescued by the depletion of TP53 [20–23], indicating that both MDM2 and MDM4 are the regulators of TP53. While MDM2 is within a pan-cancer recurrent amplicon, MDM4 is not. Similarly, among the three D-type cyclins that bind to Cdk4 and promote G1/S transition of cell cycles [24–26], only CCND1 is frequently amplified across tumor types. Therefore, among genes with closely-related functions, a particular one may be more susceptible to amplification than others.
Figure 1 (Key Figure).
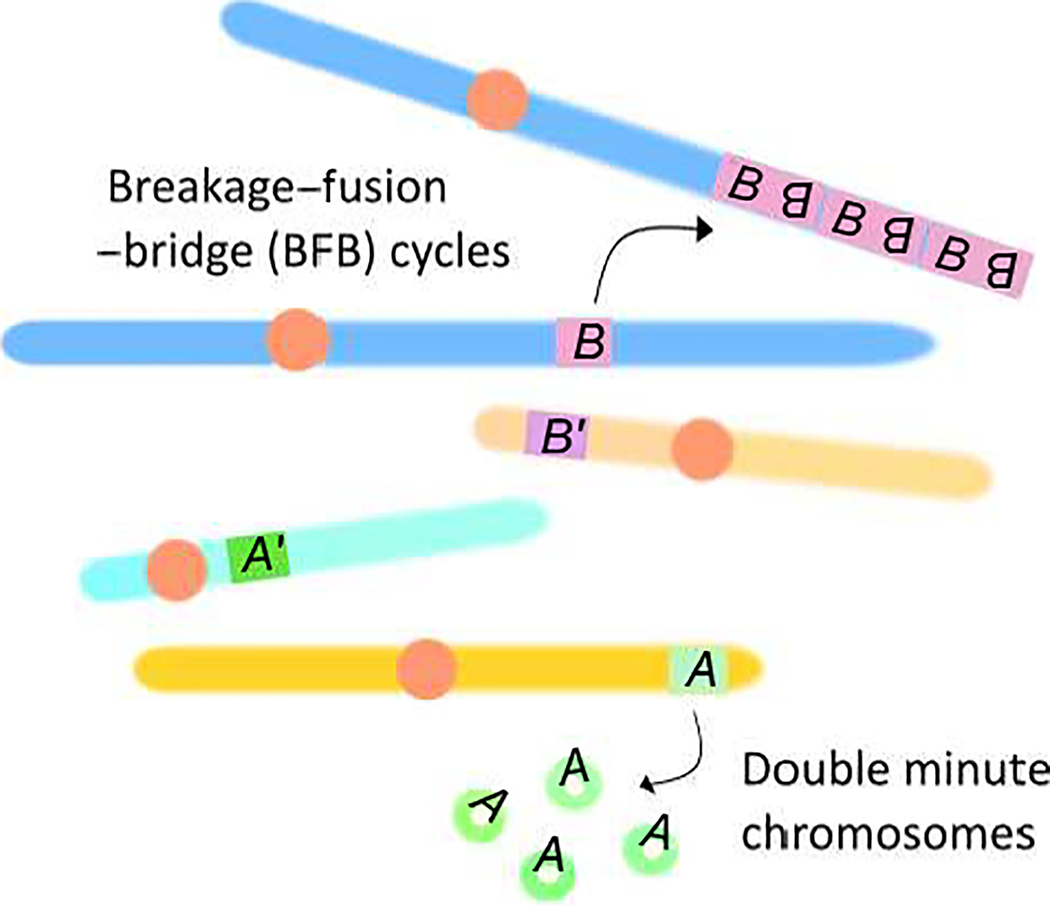
Local genomic environment defines the susceptibility to genomic amplification by promoting a specific amplification mechanism (model). Two mechanisms, Breakage-Fusion-Bridge (cycles) and the formation of double minute chromosomes (DM) are shown. Gene A is associated with the formation of DMs, whereas gene B is preferentially amplified by BFB cycles. Genes A’ and B’, although encoding proteins with similar functions to A and B, respectively, are located in different genomic environments and are thus not amplified recurrently.
A comprehensive review of the mechanisms of genomic amplification and genome instability could help us to gain insight. Historical studies are rich sources of conceptual frameworks, and recent studies, including studies from model organisms, provide detailed molecular processes that could underlie the susceptibility of genomic amplification.
Quests for the underlying amplification mechanisms using mammalian cell models
Gene amplification in mammalian cells was first described as a cellular phenomenon that counteracts the increasing dosage of clastogenic drugs. The enzyme dihydrofolate reductase (DHFR, see Glossary) increases dramatically in mouse cells during the stepwise selection with methotrexate (MTX), the inhibitor of DHFR. The increase of DHFR protein is associated with the multiplication (amplification) of the endogenous DHFR gene in a mouse sarcoma cell line [27]. This amplification was unstable and was lost without MTX selection. In contrast, the amplification of the DHFR gene was stable in Chinese hamster ovary and lung cells. The differing stability of DHFR amplification and MTX resistance was peculiar and soon was found to be associated with the chromosomal localization of the DHFR gene. In situ hybridization in a metaphase spread revealed that DHFR genes were present on extrachromosomal elements called double minute chromosomes (DMs, see Glossary) in the cell lines with unstable DHFR amplification [28, 29] (BOX1). In the stable cell lines, the amplified DHFR genes were localized to one or a few chromosomes in the form of locally expanded chromosomal region, called a homogeneously staining region (HSR, see Glossary) (BOX1) [30]. DMs lack functional centromeres [31] and thus can be distributed to daughter cells unequally at mitosis. In the absence of MTX, cells without DMs became dominant in the population, as cells with fewer DMs proliferated better [32]. In contrast, chromosomally integrated HSRs segregated equally into daughter cells in the absence of MTX, and therefore, amplification was stable. Of note is the strong association between the forms of DHFR amplification (DM or HSR) and species (mouse or Chinese hamster) under the MTX selection. The difference in the chromosomal environment surrounding the DHFR gene may dictate how it is amplified.
BOX 1: Double Minute Chromosomes and Homogenously Staining Regions.
A technical breakthrough in cytology in early 1960 is the trigger of the chromosome analysis in metaphase tumor cells under the microscope, and researchers began to characterize the chromosome numbers and morphologies rigorously. It was evident that tumor chromosomes are abnormal in both the numbers and morphologies. The peculiar finding in cancer chromosomes is the sister (double) small chromatin bodies [127] (Fig. I, left). These substances were initially called as minute chromosomes and minute chromatin bodies but later was referred to double minute chromosomes (DMs). DMs exhibit distinct behaviors during metaphase/anaphase and segregate into daughter nuclei in a spindle pole-independent manner, indicating that DMs do not carry centromere [128]. Instead, the DMs attach to the periphery of the chromosomes and move to daughter nuclei as passengers [129]. Because “sister” DMs often move with the same chromosome, the genetic material in the DMs is distributed unequally. Unequal segregation of genetic material could lead to gene amplification. Indeed, it was shown later that drug (methotrexate, MTX)-resistant cells carry the numerous copies of the drug target gene (Dhfr) in tiny chromosomes (presumably DMs). Another important feature of the DMs is that DMs are closed circular chromosomes [130].
Homogenously staining regions (HSR) are first reported for cells that exhibit MTX resistance and neuroblastoma cells by karyotype analysis [127] (Fig. I, right). HSRs lack the trypsin-Giemsa bands, and a chromosome carrying an HSR shows an increase in the length. The association with gene amplification was demonstrated later by the in situ hybridization that amplified Dhfr genes are specifically localized to the HSR.
The invention of fluorescence in situ hybridization in the mid-1980s [33] enabled scientists to resolve the locations and numbers of drug resistance genes reliably in metaphase at a single-cell level. The focus of the studies moved forward to understand the mechanisms underlying gene amplification. This subject was explored by examining the chromosomal structures of multiplied genes at a very early step of amplification. Hamster cell lines were a preferred model, given their long metaphase chromosomes offer better resolutions for each gene copy. A dominant location of multiplied Carbamoyl-Phosphate Synthetase 2, Aspartate Transcarbamylase, And Dihydroorotase (CAD) genes is within the body of chromosomes when Syrian hamster cells were selected with N-phosphonacetyl-L-asparate (PALA) [34]. Ladder-like, multiple hybridization signals were developed at the telomeric end of the chromosome arm from the single-copy CAD gene. This chromosomal structure was preceded by the recombination/fusion between the sister chromatids that could produce a giant inverted duplication and a chromosome with two centromeres (di-centric chromosome) [35]. Similarly, a giant inverted duplication was a very frequent event for the amplification of DHFR and adenylate deaminase 2 (AMPD2) genes in Chinese hamster ovary cells [36, 37]. The inverted duplication and dicentric chromosome formation are consistent with gene amplification by breakage-fusion-bridge (BFB) cycles (Box 2, see Glossary) (Fig. 2). BFB cycles were originally described by Barbara McClintock in 1939 for the fate of a dicentric chromosome during meiotic mitosis and endosperm development in maize [38]. In another study, the DHFR gene was shown to be amplified in extrachromosomal DMs at very early passages of MTX-resistant cell population, and with continuous passages, clones with chromosomal amplification became dominant [39]. This observation suggests that DMs were prevalent at the early stage of MTX-resistant cells, but later became integrated into chromosomes. It was also shown that AMPD2 amplification in Chinese hamster cells also occurred in the form of DMs [40]. In this study, the cells harboring DMs were shown to be associated with the loss of AMPD2 gene in the native chromosomal locus, indicating that the excision of native locus could precede the appearance of DMs. Thus, the formation of DM and BFB cycles seemed common in the amplification of drug-resistance genes in hamster cells, although the inter-dependence between two processes is still in debate [41, 42].
BOX 2: Breakage-Fusion Bridge cycles.
The BFB cycle was initially described by Barbara McClintock in 1939 as a fate of a dicentric chromosome during meiotic mitosis and endosperm development in maize [38]. The key observations include (i) ‘breakage’ of a dicentric chromosome in anaphase when the two centromeres pass to opposite poles, (ii) ‘fusion’ at the breakage site between two sister halves of the broken chromatid resulting in a duplicated chromatid with two centromeres, and (iii) the formation of a chromatid ‘bridge’ in the following anaphase. The bridge eventually ruptures, and broken chromatids enter into each daughter nuclei. Because the rupture can occur at any site between the two centromeres, the broken chromatids can inherit unequal amounts of genetic material: a partial inverted (palindromic) duplication in one chromatid and a partial deletion in the other (Fig. 2). The resulting broken chromatids repeat the cycle in following mitotic divisions, and, as a result, a particular segment between two centromeres becomes amplified in some cell descendants. The amplification was observed phenotypically in kernels with an extremely dark color because the gene required for pigment production was between two centromeres.
A critical step for continuing BFB cycles is the fusion of sister chromatids. How sister chromatids fuse in tumor cells remains elusive. However, recent reveal a plausible mechanism with experimental evidence [132–133]. A broken end in mitosis would undergo end resection and leave a 3’ single-stranded DNA (ssDNA) tail. The ssDNA would fold back and anneal using homologies. DNA synthesis would fill the gap and complete the end-capping. The entire chromosome would duplicate in the S-phase of the next cell cycle to produce chromatids fused at the broken end.
Figure 2.
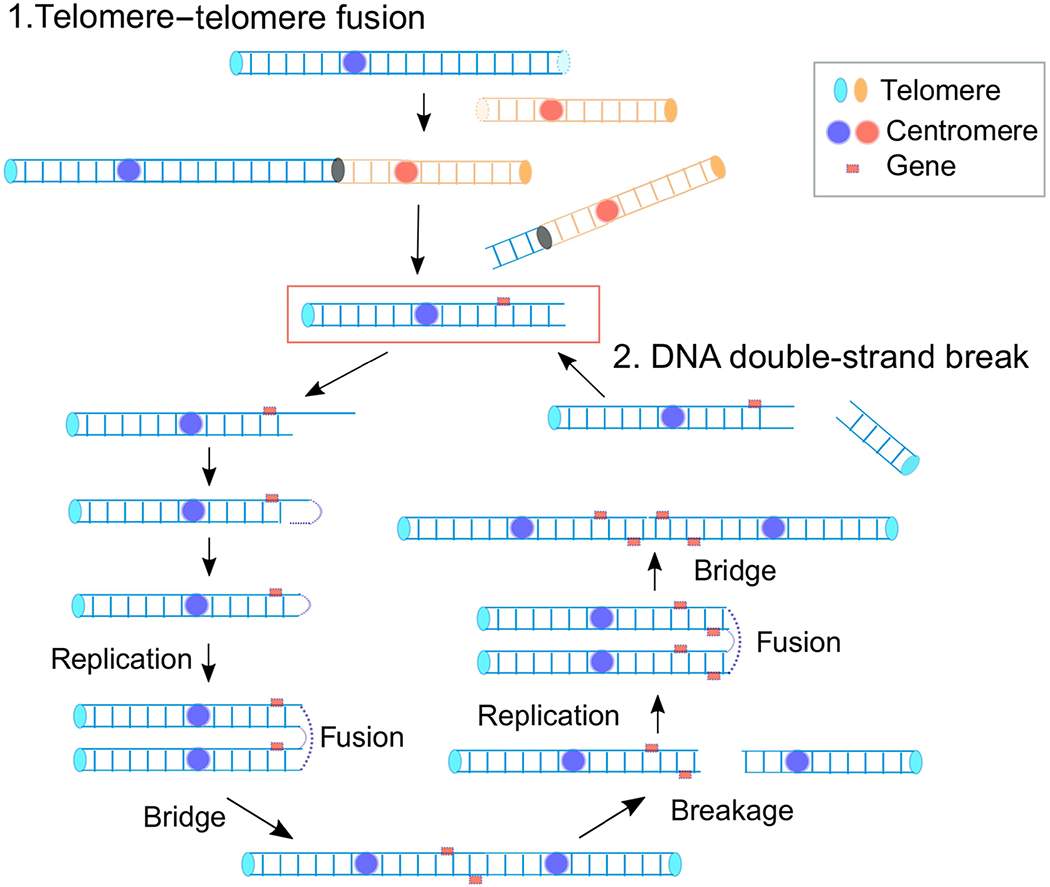
Molecular mechanisms underlying BFB cycles. A broken chromosome (red rectangle), either from the fusions of critically short telomeres or directly from DNA breaks, goes into the cycle. An initial step is the formation of a hairpin capped chromosome following the resection of broken and intra-strand annealing, which is converted into a dicentric chromosome after replication. The segregation of each centromere into different daughter nuclei results in a break and an unequal distribution of genetic material.
Clinical impact of amplification mechanisms
Having discovered two prevalent forms for the amplification of drug resistance genes in vitro, chromosomal locations of naturally amplified oncogenes have been of great interest. In the 1980s, metaphases obtained from tumor cell lines were subject to cytogenetic analysis and probed with known oncogenes. MYCN was the first gene to be demonstrated as an amplified gene in tumor cell lines. MYCN has homology to the MYC oncogene and was shown to be amplified in neuroblastomas in both DMs and HSRs [43, 44]. MYC was also shown to be in both DMs and HSRs [45, 46]. Later, gliomas were analyzed for the amplification of the EGFR oncogene and found to harbor EGFR in DMs [47]. This analysis was done using primary tumor cells obtained from xenografts in mice, so the information could be more relevant to primary tumors than cell lines after extensive passage. Notably, EGFR was retained in chromosome 7 in all tumors, suggesting that the excision and circularization mechanism is unlikely for this amplification. The extent of amplicons, determined by quantitative real-time PCR showed the amplification of contiguous genomic segments, suggesting the DMs with a simple structure of mono-locus origin. The same group later showed that DMs can also have complex structures originated from multi-loci [48]. In this glioma, some of the DMs harbored a single EGFR locus, while another one had genomic segments from six different loci.
An important clinical implication of genomic amplification is therapy-resistance. It was shown recently that anti-EGFR therapy selects against glioblastoma cells harboring the DMs with the hyper-active, mutant form of EGFR (EGFRvIII) [49]. Upon the withdrawal of an anti-EGFR drug, the number of mutant EGFR-DMs per cell increases, indicating a dynamic and adaptive route by which cancer cells evade targeted therapies. This process could be facilitated by shuttling EGFR amplicons between DMs and chromosomal HSRs. HSR configuration seems to suppress the expression of mutant EGFR, which renders cells being less dependent on mutant EGFR and escaping from anti-EGFR therapy. In this scenario, HSR could serve as a reservoir of mutant EGFR.
Representative oncogenes are amplified solely in DMs or concurrently in DMs and HSRs [50]. However, whether the formation of DMs is the exclusive gateway for oncogene amplification remains elusive. Oncogenes such as ERBB2 are amplified solely in HSRs or concurrently in DMs and HSRs. In such a case, it is plausible that chromosomal amplification could be the initial form and breaks into DMs [41]. A mechanism leading to the formation of HSR is BFB cycles. BFB cycles involve the inverted fusion between sister chromatids and leave a breakpoint signature called a fold-back inversion. Indeed, fold-back inversions constitute a significant class of structural alterations in cancer genomes [51, 52]. A recent genomic study uncovered the link between the prevalence of fold-back inversions and poor prognosis in patients with ovarian cancer [53]. Finally, direct evidence of BFB cycles underlying oncogene amplification was recently provided. Genomic data provides information on copy number alterations and breakpoint sequences that could apply to construct the trajectory of rearrangements leading to genomic amplification [54]. Using this approach, the trajectory of ERBB2 amplification in breast tumors was shown to indicate the history of BFB cycles [55]. Furthermore, fold-back inversions, identified by a technique for enriching inverted duplications (DNA palindromes), are shown to cluster within the ERBB2 locus in ERBB2-amplified breast tumors [56]. The prevalence of BFB driven mechanisms for ERBB2 amplification could explain why DMs always co-exist with HSRs in ERBB2-amplified tumors [50].
Cellular processes underlying genomic amplification
The formation of DMs and BFB cycles play crucial roles in both experimental gene amplification and cancer-driving gene amplification. An advantage in both mechanisms is that, once they are initiated, gaining additional gene copies is streamlined. Cells can accumulate additional DMs at each cell division by the unequal segregation of DMs. BFB cycles continue to distribute genetic material unequally to daughter nuclei by asymmetrical breaks, one of which would gain additional copies. Thus, the focus of this review will be these two mechanisms.
Also, because genomic amplification frequently occurs in tumor cells but not in normal cells, tumor cells are considered to be permissive to gene amplification. The permissive state is a recessive trait in tumor cells because gene amplification in tumor cells can be suppressed when tumor cells are fused with normal diploid fibroblasts [57]. This recessive trait should underlie both the formation of DMs and the initiation/progression of BFB cycles. The trait is most likely the checkpoint against damaged DNA that is very often dysfunctional in cancer cells. In normal cells, broken DNA activates checkpoints to halt cell proliferation and triggers cell death, so that cells cannot proliferate with aberrant DNA. However, with the dysfunctional checkpoint, cells with broken DNA could survive and develop DNA rearrangements and genomic amplification. TP53 tumor suppressor enforces the G1/S checkpoint to prevent cells with damaged DNA from entering into S phase [58]. TP53 dysfunction leads to cells with aberrant DNA to proliferate. The dysfunction of TP53 also prevents cells from apoptosis. Consistent with these ideas, TP53 inactivation was shown to be a prerequisite of genomic amplification in vitro [59, 60]. In vivo evidence includes the complex amplification of the MYC oncogene in mice lacking TP53 and non-homologous end-joining repair genes [61, 62].
Mechanisms of genomic amplification – BFB cycles
A classic model of genomic amplification mechanism is that a single DNA lesion initiates the process and additional copies accumulate progressively over time in a stepwise manner. BFB cycles fit into this model. The initiating mechanisms of BFB cycles include dysfunctional telomeres [63], endonuclease-induced DSBs [62, 64] and drug-induced fragile sites [65] (Fig. 2).
Telomere shortening
Telomeres are nucleoprotein structures at the termini of eukaryotic chromosomes. Long tandem repeats (TTAGGG/CCCTAA) of double-stranded DNA with a single-stranded DNA overhang at the end forms a higher-order structure with the protein complex called Shelterin (see Glossary) [66]. Such capping structures differentiate the natural chromosome ends from broken DNA and protect telomeres from DNA damage signaling and repair activities.
Shelterin cannot protect telomeres when telomeres become too short. Each time cells replicate their DNA, lagging strand synthesis cannot initiate replication from the end and instead primes DNA synthesis internally using a short RNA primer, resulting in the loss of telomeric repeats distal from the RNA primer in the newly synthesized strand. This process repeatedly occurs in proliferating cells. Progressively shortened telomeres and an altered state of the Shelterin complex lead to dysfunctional telomeres that recruit DNA damage response (DDR) proteins and eventually terminates cell proliferation (replicative senescence) [67]. Cells lacking both TP53 and retinoblastoma (RB) tumor suppressor proteins can escape replicative senescence by fusing two dysfunctional telomeres by non-homologous end-joining (NHEJ, see Glossary) [68]. End-to-end fusions can occur between different chromosomes or between sister chromatids and create dicentric chromosomes. The resulting chromosomes with two centromeres can initiate genomic amplification through BFB cycles. TP53 mutant mice lacking the telomerase RNA component (mTerc) undergo telomere crisis and develop tumors with numerous genomic alterations, including genomic amplification [63].
Replication Stress
Mounting evidence indicates that DNA replication errors are a major cause of genome instability in human tumors. Maintaining genome integrity requires the faithful duplication of chromosomal DNA. DNA replication is challenged in many ways, in both global DNA metabolism and local obstacles. Consequently, replication machinery stalls. Persistent stalled forks can collapse and be processed into DSBs [69] that could initiate genomic amplification. Alternatively, stalled forks restart erroneously to initiate DNA rearrangements [70] that could initiate the formation of dicentric chromosomes.
A sufficient level of DNA precursors (nucleotide pools) is crucial for faithful DNA replication. Premature S-phase entry with reduced nucleotide pools results in increased stalled forks and genomic instability [71]. The depletion of nucleotide pools by hydroxyurea results in the global increase of ssDNA, the protection of which requires single-strand DNA binding protein RPA [72]. In cells with reduced RPA levels, unprotected ssDNA undergoes massive DNA breakage. Endonucleases Mus81, Artemis and XPF have been shown to cause DNA breaks in hydroxyurea-treated cells [73, 74]. These cleavages provide substrates for fork restart and prevent the missegregation of under-replicated chromosomal regions [75, 76]; however, DNA ends could restart erroneously and initiate rearrangements. Indeed, the depletion of DNA precursors by hydroxyurea has previously shown to increase gene amplification frequency in rodent cells [77].
Replication forks could stall in a site-specific manner during normal replication processes. These “replication fork barriers (RFB, see Glossary)” have been studied extensively in simple organisms to clarify their biological roles and underlying mechanisms [78]. These barriers often consist of specific cis-acting DNA elements and binding proteins. Although natural barriers are necessary to maintain genome integrity, they could also pose a risk. A well-characterized example is the rDNA barriers in budding yeast Saccharomyces cerevisiae. rDNA encodes ribosomal RNA, the RNA component of ribosomes that is essential for protein synthesis in living organisms. Because large amounts of ribosomes are needed for sustaining cellular activities, rDNA genes exist in the genome at high copy. In S. cerevisiae, rRNA genes are located on the right arm of chromosome XII in a tandem array of 150–200 9-kb units. Each unit contains the RNA Polymerase I (PolI)-transcribed 35S rRNA gene, the RNA Polymerase III-transcribed 5S rRNA gene and a replication origin. A replication barrier is mediated by the binding of Fob1 protein to cis-acting RFB elements and prevents the head-on collision between 35S rDNA transcribing PolI and replication machinery. Prolonged fork stalling at RFBs causes rearrangements, resulting in the contraction, expansion and extrachromosomal circularization of rDNA [79]. Although less understood, large homopolymeric (dA/dT) sites are preferential sites of breaks within RFBs of rDNA in the mammalian genome [80]. It is thus plausible that such tightly-bound proteins at specific DNA sites can initiate DNA rearrangements in cancer cells. Such a possibility was demonstrated elegantly by using an E.coli replication termination protein-DNA complex Tus-Ter in mouse embryonic stem cells [81]. In this system, tethering Tus protein to a chromosomally-inserted Ter element induces stalled forks that initiate DNA rearrangements.
The conflict between transcription and replication is a common issue throughout the kingdom of life, as replication and transcription machineries compete for the same templates. Transcription activities can oscillate during the cell cycle and could be low in S-phase, but a subset of genes is shown to be induced in S phase [82, 83]. In such a setting, the collisions between two types of machinery seem unavoidable. Indeed, highly transcribed genes are potential impediments of replication forks in yeast [84]. Collisions can occur in either head-on or co-directional manner, and head-on collisions are considered more deleterious than co-directional collisions. To curtail such unwanted events, replication fork movements are biased towards the same orientation with transcription [85–87]. Although head on-collisions are thought to be more harmful than co-directional collisions, both collisions could activate DNA damage checkpoints [88] and thus, with impaired checkpoints, can cause adverse effects. In bacteria, co-directional collisions provoke the restart of replication forks and exhibit distinct mutation spectrums from co-directional collisions, with insertions and deletions more common in co-directional than head-on collisions [89, 90]. Consistently, natural co-directional collisions create stalled forks, the restart of which could lead to replication template switching events in human cells [91]. In addition to the collisions between replication and transcription machineries, RNA transcripts can form hybrids with DNA (R-loop, a three-strand nucleic acid structure formed by an RNA:DNA hybrid plus a displaced ssDNA strand, see Glossary) that could promote DNA damage [92, 93]. R-loops are particularly abundant in head-on collisions in both bacteria and replicating plasmids in human cells [88, 94]. R-loops have physiological functions, including transcriptional regulation and termination, programmed DNA rearrangements and homology-directed DNA repair [95–98]. Physiological R-loops are delicately regulated by an enzyme that degrades the RNA parts of R-loops (RNaseH) and a helicase that unwinds R-loops (Senetaxin). Persistent R-loops become irreparable DNA damages in S phase cells [99], indicating that R-loop prevents the progress of replication machinery. On the other hand, only a subset of persistent R-loops can cause irreparable DNA damages. Another factor, such as a particular histone modification (H3S10P) that is associated with condensed chromatin, could be crucial to cause the impediment of replication forks at the R-loops [100].
Fragile sites
The mechanisms described above can cause fragility in the specific regions of the genome and fragility near the oncogenes may initiate genomic amplification. Cytogenetically, fragile sites have been examined extensively and defined as specific chromosomal loci that preferentially exhibit gaps and breaks on metaphase chromosomes following partial inhibition of DNA synthesis (common fragile site, CFS, see Glossary) [101]. There are more than 70 aphidicolin-induced CFS mapped throughout the chromosomes. CFSs cover over 100-kb long regions and share both epigenetic and genetic features that together contribute to the fragility, including late replication, the paucity of replication origins, and co-localization with giant genes [101]. The mechanisms underlying the fragility is well-defined for a subset of CFSs. Because CFS is late-replicating, even a mild replication inhibition leads to the unfinished replication in S-phase. With giant genes being transcribed throughout the cell cycle, CFS could remain decondensed in metaphase. In such a condition, unfinished replication can be seen as a gap in a chromatid.
In hamster cells, clastogenic drugs-induced fragile sites initiate BFB cycles and genomic amplification [102]. Further evidence was provided in human gastric tumor cells in which a CFS on chromosome 7 is involved in the amplification of MET oncogene possibly through BFB cycles [103].
Mechanisms of genomic amplification – the formation of DMs
DMs of mono-locus origin
Because the formation of DMs requires DNA rearrangements, initiating mechanisms for BFB cycles discussed above, such as replication stress could underlie the formation of DMs as well. Several models have been proposed to explain how DMs arise (Fig. 3). Earlier molecular and cytogenetic studies revealed that a chromosome break triggers the deletion of a chromosomal region that becomes a circular DNA [40, 104]. In contrast, glioma cells harboring EGFR-DMs retain the EGFR gene copies in the native chromosomal locus [47]. The replication-associated model could explain the co-existence of chromosomal copies and DMs: a sister chromatid from the replication bubble is excised during replication and forks emanating from both sides complete replication. These mechanisms predict that DMs have simple structures of mono-locus origin. Recent genomic data for the DMs arising from EGFR-loci in glioblastomas may support this model [105, 106].
Figure 3.
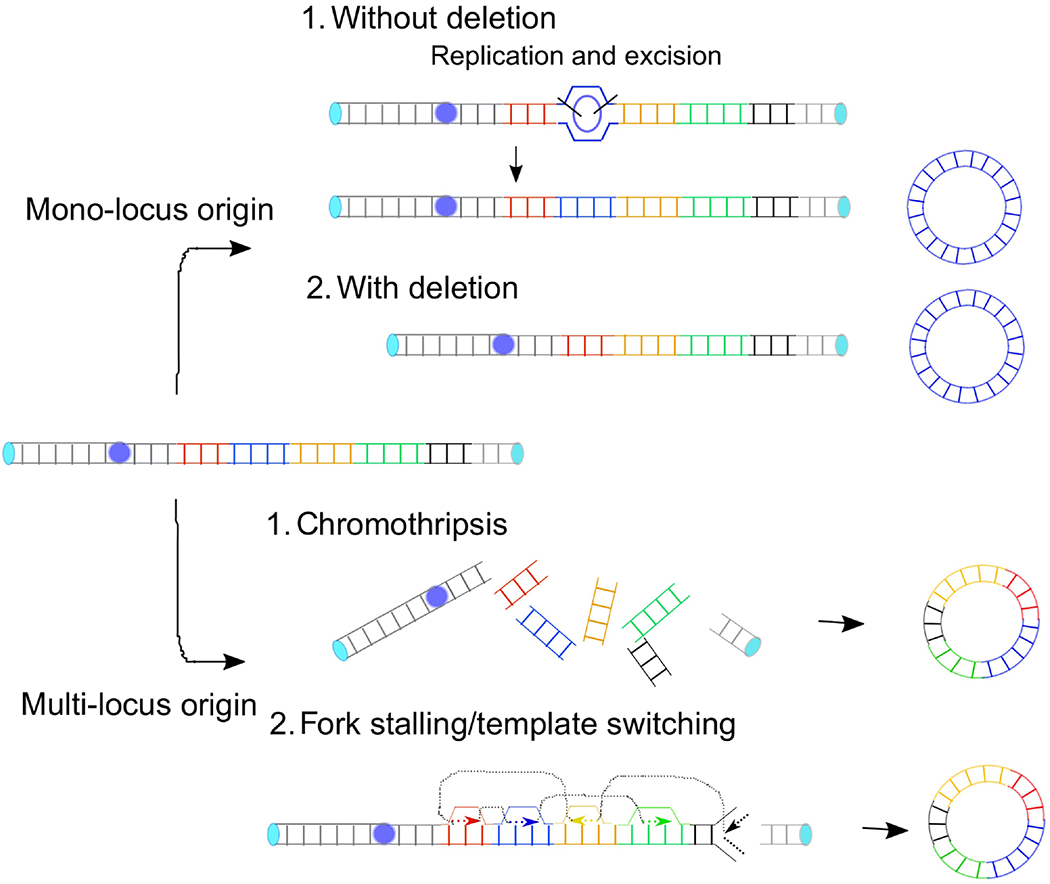
Models for the formation of DMs. For a DM of mono-locus origin, a DNA segment can be circularized from the excised fragment of the replication intermediate (top) or directly from a chromosomal locus (bottom). Multi-loci DM can arise from either the erroneous repair of fragmented chromosomes (top) or repeated template switching of a replication fork (bottom).
Complex DMs with a multi-loci origin
The advent of massively parallel sequencing for cancer genome analysis has transformed our thinking of how cancer genomes evolve. Paired-end sequencing allows us to search for DNA rearrangements in an unprecedented scale and resolution. A horrifying pattern of rearrangements emerged in chronic lymphocytic leukemia (CLL); several tens of rearrangements confined to a region of a chromosome [107]. Copy number profiles associated with the rearrangements are also unconventional and alternate just two states, namely one or two copies [108]. The copy number loss is not only mediated by simple “head-to-tail” deletions but also exhibits other types of rearrangements, including head-to-head and tail-to-tail inverted orientations. This phenomenon can be explained by a single catastrophic event and subsequent illegitimate repair – a restricted region of a chromosome broken into pieces, followed by the pieces randomly stitched together. The term chromothripsis (Greek, chromos for chromosome and thripsis for shuttering into pieces) is adopted to illustrate the phenomena, and how such a horrifying pattern of rearrangements arise becomes a center of the topic. A plausible mechanism has been proposed from the studies that followed the fate of a lagging chromosome, chromosomes that fail to move into daughter nuclei during mitosis [109, 110]. A lagging chromosome can be confined to micronuclei where defective DNA replication takes place to cause DNA damage and chromosome fragmentation. Importantly, micronuclei can fuse back to the main nuclei. As a result, the fragmented chromosome can become nuclear DNA and the fragmented chromosome becomes the feature of cancer genomes. How DNA fragments are woven to each other remains elusive and may depend on multiple repair processes. In human cancer cells, targeted inactivation of Y chromosome centromere led to the segregation errors and confinement of the Y chromosome into micronuclei [111, 112]. The fragmentation of Y chromosome is more common when NHEJ proteins are depleted than when either HDR or alternative end-joining is inhibited, providing direct evidence for an involvement of NHEJ in the repair in micronuclei. In another study, tumors developed from either NHEJ- or HDR-deficient mice display complex rearrangements reminiscent of chromothripsis [113].
In addition to the defective DNA replication in micronuclei, a nuclease that attacks a restricted region of a chromosome can cause chromothripsis [114]. Cells with defective telomeres exhibit telomere-telomere fusions and di-centric chromosomes. A chromatin bridge formed between daughter nuclei during mitosis exposes a restricted chromosomal region for a single-stranded DNA nuclease, TREX1. TREX1 activity results in the loading of a single-stranded DNA binding protein RPA and resolves chromatin bridges. Sequencing analysis revealed that half of the clones underwent telomere crisis in these experiments exhibited the localized massive genome rearrangements. How such extensive ssDNA leads to complex rearrangements is unknown. ssDNA of a broken chromosome end can successively invade into more than one donor segment and create the fusion between donor sequences (multi-invasion induced rearrangement) [115]. ssDNA from the TREX1-processed chromatin bridges could invade into multiple donor sequences and create fusions between discontinuous genomic segments.
The link between chromothripsis and genomic amplification came from the fact that some of the DMs display very complex structures and consist of large genomic segments from discontinuous multiple loci. In a small lung cancer cell line, 170 breakpoints cover the entire chromosome 8. Some of the breakpoints flank the large genomic regions (larger than 100-kb), including the fragments harboring the oncogene MYC, that are interwoven to each other to create a circular chromosome [51]. Cytogenetic analysis showed that the region is amplified extra-chromosomally, indicating that fragmented regions are assembled to DMs. Also, a subset of medulloblastoma amplifies MYC very frequently, and chromothripsis is a likely trigger by forming DMs [116]. Similar MYC-DMs are identified in esophageal adenocarcinoma [117]. The multi-loci DM was successfully created from the micronuclei-confined Y chromosome [112], demonstrating a chromosome segregation error as a process leading to the formation of DMs.
The majority of the fusion points of rearrangements harbor microhomology [51]. Microhomology was also seen for DMs of mono-locus origins harboring EGFR oncogene [118]. Thus, DNA repair mechanisms using microhomology play a significant role in the formation of DMs. A non-canonical NHEJ mechanism called microhomology-mediated end joining (MMEJ, see Glossary) could underlie the formation. However, microhomology-mediated fusions can occur through the replication-based, FoSTeS/MMBIR (fork stalling template switching /microhomology-mediated break-induced replication, see Glossary) mechanism [119, 120]. It is also noted that in some mono-locus DMs, fusion points are complex and contain several small fragments from discontinuous genomic regions [121]. Such a complex fusion point is reminiscent of repeated template switching events from restarted stalled forks, in which each fusion event is mediated by microhomology [91]. The underlying mechanism of repeated template switching remains elusive but may involve a specialized polymerase Polη that is recruited to the stalled forks [91]. In yeast, microhomology-mediated template switching depends on the specialized polymerase ζ [122]. Thus, specialized polymerases appear to be crucial mediators of template switching that could result in DNA rearrangements and the formation of DMs.
Concluding remarks
Here we focus on two amplification mechanisms – the formation of DMs and BFB cycles – that have been the centers of the topic and thus are strong candidate mechanisms for recurrent genomic amplification. A common feature between the two mechanisms is that, once they are initiated, the subsequent increase in copy number is streamlined. These mechanisms could also share the initiating events. For example, telomere-telomere fusion results in the formation of dicentric chromosomes that can provoke BFB cycles and chromothripsis [114]. Chromothripsis could underlie the formation of multi-loci DMs [107]. Stalled forks can lead to the formation of dicentric chromosomes and DMs [70, 123].
Both telomere dysfunction and chromothripsis in a chromosome in the micronucleus can occur for any chromosomes and can initiate the formation of DMs and BFB cycles at random loci. Therefore, these mechanisms unlikely specify where to amplify, but rather are consistent with the random amplification of loci followed by positive selection (Outstanding Questions Box). A paralogue-specific amplification can be a result of a positive selection for a function specific to a particular paralogous gene, for instance, a function in MYC but not in MYCN, that is necessary for tumorigenesis in specific cell lineages. The random model is supported by the fact that representative oncogenes are amplified in both DMs and HSRs [50]. However, the interdependence between these two mechanisms complicates the interpretation of such observation. For example, a dicentric chromosome by telomere-telomere fusion can lead to chromosthripsis that could generate DMs. It was also shown that HSR can be a precursor of extrachromosomal amplicons [41].
Outstanding questions.
Is recurrent amplification solely driven by the positive selection for the amplification target proteins? Or are any loci in the genome susceptible to genomic amplification?
Is chromosome fragility a determinant for the susceptibility to genomic amplification?
What are the underlying mechanisms of chromosome fragility that confer the susceptibility to genomic amplification?
Are there any areas in the genome that can be fragile but are not amenable to study? How can we address the fragility in those areas?
On the other hand, endogenous DNA damage can be loci-specific and could support the non-random model. One candidate includes chromosome fragile sites, although a strong link between common fragile sites and recurrent amplification in tumors is lacking. Other candidates include proteins that induce breaks at certain sequencing features in the genome, such as Topoisomerase 2, RAG1/RAG2 and AID. These enzymes mediate recurrent translocations in prostate cancer and lymphoid malignancies [124, 125]. Particular sequence features could also specify the break sites by perturbing replication. In addition to the well-characterized features such as AT-rich sequences [80], there are complex regions in the genome that contain many duplicated sequences [126]. These regions are very large (>100 kb) and can be very challenging for replication and be prone to break. Indeed, these regions are known to be highly variable between individuals, suggesting that these regions are fragile in the germline. Due to the variability between individuals, these regions remain ambiguous in the reference genome and are excluded from genomic analysis. Due to the variations and fragility, these regions would determine who gets genomic amplification and who does not.
Figure I (BOX 1).
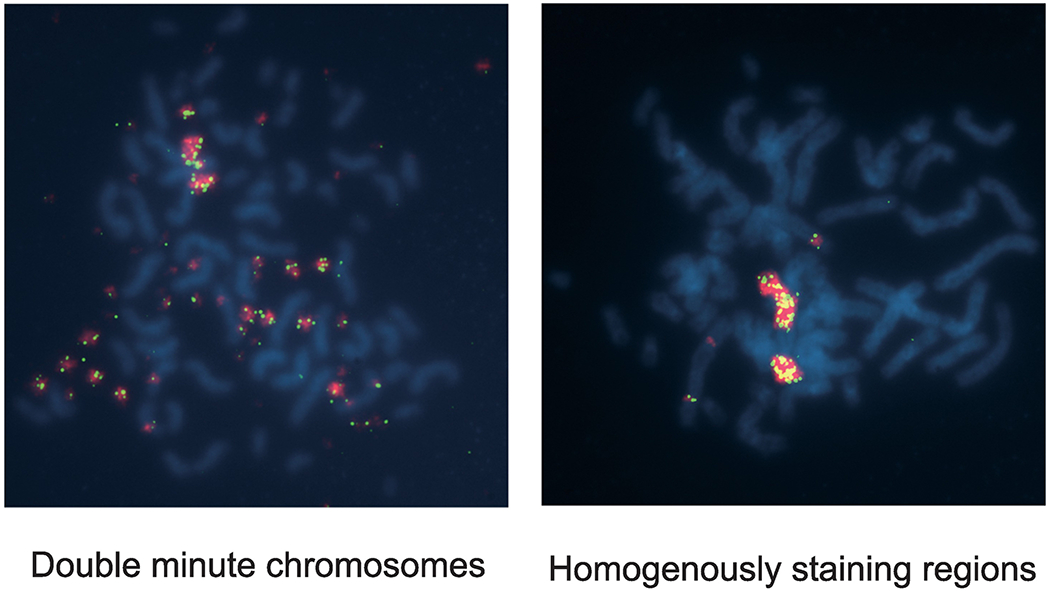
Extrachromosomal DMs (left) and intrachromosomal HSR (right). Metaphases of Colo320DM cells (left) and Colo320HSR cells (right) were examined using Fluorescence in situ hybridization (FISH) with the genomic probes for MYC (red) and for 8q24.21 (green). Please note that in DM cells, there are some signals in a chromosome in addition to DMs.
Highlights.
With the advent of genomic technologies, genomic regions of recurrent amplification in tumors have been mapped throughout the human genome. The mechanisms underlying the recurrence are of great interest.
Two mechanisms, breakage-fusion-bridge (BFB) cycles and the formation of double minute chromosome (DM) has been and continue to be the center of the middleic for the last three decades. A common feature of these mechanisms is that, once the process is initiated, additional copy number gain is streamlined.
BFB cycles can be initiated by the fusion of critically short telomeres and DNA breaks. A significant source of DNA breaks is replication stress.
DMs can be both single-locus and multi-loci origin. Chromothripsis and erroneous DNA repair is a likely underlying mechanism.
The implications of both BFB cycles and DMs in the management of cancer patients have recently emerged.
Acknowledgment
I would like to thank Drs. Ryusuke Suzuki, Neeraj Joshi, and Michael Murata, Ms. Lila Mouakkad for comments and edits of the manuscript. This work is supported by the NIH NCI R01CA149385, Department of Defense W81XWH-18-1-0058, Margie and Robert E. Petersen Foundation, and The Fashion Footwear Charitable Foundation of New York, Inc.
Glossary
- Amplicons
a segment of DNA that is amplified as a result of genomic amplification
- BFB cycles
breakage-fusion-bridge cycles
- CFS
Common fragile site
- Cytobands
Cytogenetic bands. Each human chromosome has a short arm (“p” for “petit”) and long arm (“q” for “queue”), separated by a centromere. Each chromosome arm is divided into distinct cytogenetic bands, that can be seen using a microscope and special stains.
- DHFR
dihydrofolate reductase
- DMs
double minute chromosomes
- ERBB2
erb-b2 receptor tyrosine kinase 2, a gene encoding HER2, a member of epidermal growth factor receptors
- FoSTeS/MMBIR
fork stalling template switching /microhomology-mediated break-induced replication. A replication-based mechanism of genomic rearrangements originally proposed for the complex genomic rearrangements associated with a central nervous system disorder in humans. Break-induced replication is a pathway that repairs one-ended DNA double-strand breaks (DSBs). One-ended DSBs can arise from stalled forks processed by nucleases and invade into ectopic template DNA using homology.
- GISTIC score
Genomic Amplification of Significant Targets in Cancer. GISTIC considers the frequency of Somatic Copy Number Alterations within tumors and the degree of amplification to define true-positive amplification events.
- HSR
homogeneously staining region
- MMEJ
microhomology-mediated end joining. Also known as alternative NHEJ (Alt-NHEJ). A non-canonical mechanism of NHEJ that directly ligates two DSB ends using a short stretch of homologous sequences.
- NHEJ
non-homologous end-joining. A DNA double-strand break pathway that directly ligates two DSB ends without the need for homology.
- R-loop
a three-strand nucleic acid structure formed by an RNA:DNA hybrid plus a displaced ssDNA strand
- RFB
replication fork barriers
- Shelterin
a six protein complex consisting of TRF1, TRF2, POT1, TPP1, TIN2, and Rap1 at mammalian telomeres.
- TCGA
The Cancer Genome Atlas. National Cancer Institute (USA) initiative of a landmark cancer genomics program that molecularly characterized over 20,000 primary cancer and matched normal samples spanning 33 cancer types.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brown DD and Dawid IB, Specific gene amplification in oocytes. Oocyte nuclei contain extrachromosomal replicas of the genes for ribosomal RNA. Science, 1968. 160(3825): p. 272–80. [DOI] [PubMed] [Google Scholar]
- 2.Tanaka H and Yao M-C, Palindromic gene amplification — an evolutionarily conserved role for DNA inverted repeats in the genome. Nature Reviews Cancer, 2009. 9: p. 216. [DOI] [PubMed] [Google Scholar]
- 3.Spradling AC, The organization and amplification of two chromosomal domains containing Drosophila chorion genes. Cell, 1981. 27(1 Pt 2): p. 193–201. [DOI] [PubMed] [Google Scholar]
- 4.Sandegren L and Andersson DI, Bacterial gene amplification: implications for the evolution of antibiotic resistance. Nature Reviews Microbiology, 2009. 7: p. 578. [DOI] [PubMed] [Google Scholar]
- 5.Gorre ME, et al. , Clinical Resistance to STI-571 Cancer Therapy Caused by BCR-ABL Gene Mutation or Amplification. Science, 2001. 293(5531): p. 876–880. [DOI] [PubMed] [Google Scholar]
- 6.Engelman JA, et al. , MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science, 2007. 316(5827): p. 1039–1043. [DOI] [PubMed] [Google Scholar]
- 7.Beroukhim R, et al. , The landscape of somatic copy-number alteration across human cancers. Nature, 2010. 463: p. 899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slamon D, et al. , Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science, 1987. 235(4785): p. 177–182. [DOI] [PubMed] [Google Scholar]
- 9.The Cancer Genome Atlas Research, N., et al. , Integrated genomic analyses of ovarian carcinoma. Nature, 2011. 474: p. 609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The Cancer Genome Atlas, N., et al. , Comprehensive molecular characterization of human colon and rectal cancer. Nature, 2012. 487: p. 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The Cancer Genome Atlas, N., et al. , Comprehensive molecular portraits of human breast tumours. Nature, 2012. 490: p. 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.The Cancer Genome Atlas Research, N., et al. , Comprehensive genomic characterization of squamous cell lung cancers. Nature, 2012. 489: p. 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The Cancer Genome Atlas Research, N., et al. , Comprehensive molecular profiling of lung adenocarcinoma. Nature, 2014. 511: p. 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abeshouse A, et al. , The Molecular Taxonomy of Primary Prostate Cancer. Cell, 2015. 163(4): p. 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hussenet T, et al. , SOX2 is an oncogene activated by recurrent 3q26.3 amplifications in human lung squamous cell carcinomas. PLoS One, 2010. 5(1): p. e8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamaguchi T, et al. , NKX2-1/TTF-1: an enigmatic oncogene that functions as a double-edged sword for cancer cell survival and progression. Cancer Cell, 2013. 23(6): p. 718–23. [DOI] [PubMed] [Google Scholar]
- 17.Winslow MM, et al. , Suppression of lung adenocarcinoma progression by Nkx2-1. Nature, 2011. 473(7345): p. 101–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berns K, et al. , A genetic screen to identify genes that rescue the slow growth phenotype of c-myc null fibroblasts. Oncogene, 2000. 19(29): p. 3330–4. [DOI] [PubMed] [Google Scholar]
- 19.Landay M, et al. , Promotion of growth and apoptosis in c-myc nullizygous fibroblasts by other members of the myc oncoprotein family. Cell Death Differ, 2000. 7(8): p. 697–705. [DOI] [PubMed] [Google Scholar]
- 20.Finch RA, et al. , mdmx is a negative regulator of p53 activity in vivo. Cancer Res, 2002. 62(11): p. 3221–5. [PubMed] [Google Scholar]
- 21.Parant J, et al. , Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet, 2001. 29(1): p. 92–5. [DOI] [PubMed] [Google Scholar]
- 22.Jones SN, et al. , Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature, 1995. 378(6553): p. 206–8. [DOI] [PubMed] [Google Scholar]
- 23.Montes de Oca Luna R, Wagner DS, and Lozano G, Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature, 1995. 378(6553): p. 203–6. [DOI] [PubMed] [Google Scholar]
- 24.Takaki T, et al. , The structure of CDK4/cyclin D3 has implications for models of CDK activation. Proc Natl Acad Sci U S A, 2009. 106(11): p. 4171–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z, et al. , Migratory localization of cyclin D2-Cdk4 complex suggests a spatial regulation of the G1-S transition. Cell Struct Funct, 2008. 33(2): p. 171–83. [DOI] [PubMed] [Google Scholar]
- 26.Xiong Y, Zhang H, and Beach D, D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell, 1992. 71(3): p. 505–14. [DOI] [PubMed] [Google Scholar]
- 27.Alt FW, et al. , Selective multiplication of dihydrofolate reductase genes in methotrexate-resistant variants of cultured murine cells. J Biol Chem, 1978. 253(5): p. 1357–70. [PubMed] [Google Scholar]
- 28.Kaufman RJ, Brown PC, and Schimke RT, Amplified dihydrofolate reductase genes in unstably methotrexate-resistant cells are associated with double minute chromosomes. Proc Natl Acad Sci U S A, 1979. 76(11): p. 5669–73.293670 [Google Scholar]
- 29.Haber DA and Schimke RT, Unstable amplification of an altered dihydrofolate reductase gene associated with double-minute chromosomes. Cell, 1981. 26(3 Pt 1): p. 355–62. [DOI] [PubMed] [Google Scholar]
- 30.Nunberg JH, et al. , Amplified dihydrofolate reductase genes are localized to a homogeneously staining region of a single chromosome in a methotrexate-resistant Chinese hamster ovary cell line. Proc Natl Acad Sci U S A, 1978. 75(11): p. 5553–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levan A and Levan G, Have double minutes functioning centromeres? Hereditas, 1978. 88(1): p. 81–92. [DOI] [PubMed] [Google Scholar]
- 32.Kaufman RJ, Brown PC, and Schimke RT, Loss and stabilization of amplified dihydrofolate reductase genes in mouse sarcoma S-180 cell lines. Mol Cell Biol, 1981. 1(12): p. 1084–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pinkel D, Straume T, and Gray JW, Cytogenetic analysis using quantitative, high-sensitivity, fluorescence hybridization. Proc Natl Acad Sci U S A, 1986. 83(9): p. 2934–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith KA, et al. , Distinctive chromosomal structures are formed very early in the amplification of CAD genes in Syrian hamster cells. Cell, 1990. 63(6): p. 1219–27. [DOI] [PubMed] [Google Scholar]
- 35.Smith KA, et al. , Fusions near telomeres occur very early in the amplification of CAD genes in Syrian hamster cells. Proc Natl Acad Sci U S A, 1992. 89(12): p. 5427–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma C, et al. , Sister chromatid fusion initiates amplification of the dihydrofolate reductase gene in Chinese hamster cells. Genes Dev, 1993. 7(4): p. 605–20. [DOI] [PubMed] [Google Scholar]
- 37.Toledo F, et al. , Co-amplified markers alternate in megabase long chromosomal inverted repeats and cluster independently in interphase nuclei at early steps of mammalian gene amplification. EMBO J, 1992. 11(7): p. 2665–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McClintock B, The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics, 1941. 26(2): p. 234–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Windle B, et al. , A central role for chromosome breakage in gene amplification, deletion formation, and amplicon integration. Genes Dev, 1991. 5(2): p. 160–74. [DOI] [PubMed] [Google Scholar]
- 40.Toledo F, Buttin G, and Debatisse M, The origin of chromosome rearrangements at early stages of AMPD2 gene amplification in Chinese hamster cells. Curr Biol, 1993. 3(5): p. 255–64. [DOI] [PubMed] [Google Scholar]
- 41.Singer MJ, et al. , Amplification of the human dihydrofolate reductase gene via double minutes is initiated by chromosome breaks. Proc Natl Acad Sci U S A, 2000. 97(14): p. 7921–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.George DL and Powers VE, Amplified DNA sequences in Y1 mouse adrenal tumor cells: association with double minutes and localization to a homogeneously staining chromosomal region. Proceedings of the National Academy of Sciences, 1982. 79(5): p. 1597–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kohl NE, et al. , Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell, 1983. 35(2 Pt 1): p. 359–67. [DOI] [PubMed] [Google Scholar]
- 44.Schwab M, et al. , Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature, 1983. 305(5931): p. 245–8. [DOI] [PubMed] [Google Scholar]
- 45.Alitalo K, et al. , Homogeneously staining chromosomal regions contain amplified copies of an abundantly expressed cellular oncogene (c-myc) in malignant neuroendocrine cells from a human colon carcinoma. Proc Natl Acad Sci U S A, 1983. 80(6): p. 1707–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collins S and Groudine M, Amplification of endogenous myc-related DNA sequences in a human myeloid leukaemia cell line. Nature, 1982. 298(5875): p. 679–81. [DOI] [PubMed] [Google Scholar]
- 47.Vogt N, et al. , Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proc Natl Acad Sci U S A, 2004. 101(31): p. 11368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gibaud A, et al. , Extrachromosomal amplification mechanisms in a glioma with amplified sequences from multiple chromosome loci. Hum Mol Genet, 2010. 19(7): p. 1276–85. [DOI] [PubMed] [Google Scholar]
- 49.Nathanson DA, et al. , Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science, 2014. 343(6166): p. 72–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turner KM, et al. , Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature, 2017. 543(7643): p. 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campbell PJ, et al. , The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature, 2010. 467(7319): p. 1109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hillmer AM, et al. , Comprehensive long-span paired-end-tag mapping reveals characteristic patterns of structural variations in epithelial cancer genomes. Genome Res, 2011. 21(5): p. 665–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang YK, et al. , Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat Genet, 2017. 49(6): p. 856–865. [DOI] [PubMed] [Google Scholar]
- 54.Zakov S, Kinsella M, and Bafna V, An algorithmic approach for breakage-fusion-bridge detection in tumor genomes. Proc Natl Acad Sci U S A, 2013. 110(14): p. 5546–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ferrari A, et al. , A whole-genome sequence and transcriptome perspective on HER2-positive breast cancers. Nat Commun, 2016. 7: p. 12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marotta M, et al. , Palindromic amplification of the ERBB2 oncogene in primary HER2-positive breast tumors. Sci Rep, 2017. 7: p. 41921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tlsty T, White A, and Sanchez J, Suppression of gene amplification in human cell hybrids. Science, 1992. 255(5050): p. 1425–1427. [DOI] [PubMed] [Google Scholar]
- 58.Paulson TG, et al. , Gene Amplification in a p53-Deficient Cell Line Requires Cell Cycle Progression under Conditions That Generate DNA Breakage. Molecular and Cellular Biology, 1998. 18(5): p. 3089–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yin Y, et al. , Wild-type p53 restores cell cycle control and inhibits gene amplification in cells with mutant p53 alleles. Cell, 1992. 70(6): p. 937–48. [DOI] [PubMed] [Google Scholar]
- 60.Livingstone LR, et al. , Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell, 1992. 70(6): p. 923–35. [DOI] [PubMed] [Google Scholar]
- 61.Difilippantonio MJ, et al. , DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature, 2000. 404: p. 510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu C, et al. , Unrepaired DNA Breaks in p53-Deficient Cells Lead to Oncogenic Gene Amplification Subsequent to Translocations. Cell, 2002. 109(7): p. 811–821. [DOI] [PubMed] [Google Scholar]
- 63.O’Hagan RC, et al. , Telomere dysfunction provokes regional amplification and deletion in cancer genomes. Cancer Cell, 2002. 2(2): p. 149–155. [DOI] [PubMed] [Google Scholar]
- 64.Tanaka H, et al. , Short inverted repeats initiate gene amplification through the formation of a large DNA palindrome in mammalian cells. Proc Natl Acad Sci U S A, 2002. 99(13): p. 8772–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coquelle A, et al. , Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell, 1997. 89(2): p. 215–25. [DOI] [PubMed] [Google Scholar]
- 66.de Lange T, Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev, 2005. 19(18): p. 2100–10. [DOI] [PubMed] [Google Scholar]
- 67.Karlseder J, Smogorzewska A, and de Lange T, Senescence Induced by Altered Telomere State, Not Telomere Loss. Science, 2002. 295(5564): p. 2446–2449. [DOI] [PubMed] [Google Scholar]
- 68.Maciejowski J and de Lange T, Telomeres in cancer: tumour suppression and genome instability. Nature Reviews Molecular Cell Biology, 2017. 18: p. 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Petermann E, et al. , Hydroxyurea-Stalled Replication Forks Become Progressively Inactivated and Require Two Different RAD51-Mediated Pathways for Restart and Repair. Molecular Cell, 2010. 37(4): p. 492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mizuno KI, et al. , Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature, 2012. 493: p. 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bester Assaf C., et al. , Nucleotide Deficiency Promotes Genomic Instability in Early Stages of Cancer Development. Cell, 2011. 145(3): p. 435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Toledo Luis I., et al. , ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell, 2013. 155(5): p. 1088–1103. [DOI] [PubMed] [Google Scholar]
- 73.Betous R, et al. , DNA replication stress triggers rapid DNA replication fork breakage by Artemis and XPF. PLoS Genet, 2018. 14(7): p. e1007541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hanada K, et al. , The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat Struct Mol Biol, 2007. 14(11): p. 1096–104. [DOI] [PubMed] [Google Scholar]
- 75.Naim V, et al. , ERCC1 and MUS81–EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nature Cell Biology, 2013. 15: p. 1008. [DOI] [PubMed] [Google Scholar]
- 76.Minocherhomji S, et al. , Replication stress activates DNA repair synthesis in mitosis. Nature, 2015. 528: p. 286. [DOI] [PubMed] [Google Scholar]
- 77.Brown PC, Tlsty TD, and Schimke RT, Enhancement of methotrexate resistance and dihydrofolate reductase gene amplification by treatment of mouse 3T6 cells with hydroxyurea. Molecular and cellular biology, 1983. 3(6): p. 1097–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Labib K and Hodgson B, Replication fork barriers: pausing for a break or stalling for time? EMBO reports, 2007. 8(4): p. 346–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kobayashi T, et al. , SIR2 Regulates Recombination between Different rDNA Repeats, but Not Recombination within Individual rRNA Genes in Yeast. Cell, 2004. 117(4): p. 441–453. [DOI] [PubMed] [Google Scholar]
- 80.Tubbs A, et al. , Dual Roles of Poly(dA:dT) Tracts in Replication Initiation and Fork Collapse. Cell, 2018. 174(5): p. 1127–1142 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Willis NA, et al. , BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature, 2014. 510: p. 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cho RJ, et al. , Transcriptional regulation and function during the human cell cycle. Nature Genetics, 2001. 27(1): p. 48–54. [DOI] [PubMed] [Google Scholar]
- 83.Liu Y, et al. , Transcriptional landscape of the human cell cycle. Proceedings of the National Academy of Sciences, 2017. 114(13): p. 3473–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Azvolinsky A, et al. , Highly Transcribed RNA Polymerase II Genes Are Impediments to Replication Fork Progression in Saccharomyces cerevisiae. Molecular Cell, 2009. 34(6): p. 722–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen Y-H, et al. , Transcription shapes DNA replication initiation and termination in human cells. Nature Structural & Molecular Biology, 2019. 26(1): p. 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Petryk N, et al. , Replication landscape of the human genome. Nature Communications, 2016. 7: p. 10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang JD, Berkmen MB, and Grossman AD, Genome-wide coorientation of replication and transcription reduces adverse effects on replication in Bacillus subtilis. Proceedings of the National Academy of Sciences, 2007. 104(13): p. 5608–5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hamperl S, et al. , Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell, 2017. 170(4): p. 774–786.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sankar TS, et al. , The nature of mutations induced by replication–transcription collisions. Nature, 2016. 535: p. 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Merrikh H, et al. , Co-directional replication–transcription conflicts lead to replication restart. Nature, 2011. 470: p. 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Watanabe T, et al. , Impediment of Replication Forks by Long Non-coding RNA Provokes Chromosomal Rearrangements by Error-Prone Restart. Cell Reports, 2017. 21(8): p. 2223–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Aguilera A and García-Muse T, R Loops: From Transcription Byproducts to Threats to Genome Stability. Molecular Cell, 2012. 46(2): p. 115–124. [DOI] [PubMed] [Google Scholar]
- 93.Crossley MP, Bocek M, and Cimprich KA, R-Loops as Cellular Regulators and Genomic Threats. Molecular Cell, 2019. 73(3): p. 398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lang KS, et al. , Replication-Transcription Conflicts Generate R-Loops that Orchestrate Bacterial Stress Survival and Pathogenesis. Cell, 2017. 170(4): p. 787–799.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ohle C, et al. , Transient RNA-DNA Hybrids Are Required for Efficient Double-Strand Break Repair. Cell, 2016. 167(4): p. 1001–1013.e7. [DOI] [PubMed] [Google Scholar]
- 96.Ginno Paul A., et al. , R-Loop Formation Is a Distinctive Characteristic of Unmethylated Human CpG Island Promoters. Molecular Cell, 2012. 45(6): p. 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Skourti-Stathaki K, Kamieniarz-Gdula K, and Proudfoot NJ, R-loops induce repressive chromatin marks over mammalian gene terminators. Nature, 2014. 516: p. 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yu K and Lieber MR, Nucleic acid structures and enzymes in the immunoglobulin class switch recombination mechanism. DNA Repair, 2003. 2(11): p. 1163–1174. [DOI] [PubMed] [Google Scholar]
- 99.Costantino L and Koshland D, Genome-wide Map of R-Loop-Induced Damage Reveals How a Subset of R-Loops Contributes to Genomic Instability. Molecular Cell, 2018. 71(4): p. 487–497.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.García-Pichardo D, et al. , Histone Mutants Separate R Loop Formation from Genome Instability Induction. Molecular Cell, 2017. 66(5): p. 597–609.e5. [DOI] [PubMed] [Google Scholar]
- 101.Glover TW, Wilson TE, and Arlt MF, Fragile sites in cancer: more than meets the eye. Nature Reviews Cancer, 2017. 17: p. 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Coquelle A, et al. , Expression of Fragile Sites Triggers Intrachromosomal Mammalian Gene Amplification and Sets Boundaries to Early Amplicons. Cell, 1997. 89(2): p. 215–225. [DOI] [PubMed] [Google Scholar]
- 103.Hellman A, et al. , A role for common fragile site induction in amplification of human oncogenes. Cancer Cell, 2002. 1(1): p. 89–97. [DOI] [PubMed] [Google Scholar]
- 104.Storlazzi CT, et al. , MYC-containing double minutes in hematologic malignancies: evidence in favor of the episome model and exclusion of MYC as the target gene. Hum Mol Genet, 2006. 15(6): p. 933–42. [DOI] [PubMed] [Google Scholar]
- 105.Brennan CW, et al. , The somatic genomic landscape of glioblastoma. Cell, 2013. 155(2): p. 462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sanborn JZ, et al. , Double minute chromosomes in glioblastoma multiforme are revealed by precise reconstruction of oncogenic amplicons. Cancer Res, 2013. 73(19): p. 6036–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stephens PJ, et al. , Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell, 2011. 144(1): p. 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Campbell PJ, et al. , Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nature Genetics, 2008. 40: p. 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Crasta K, et al. , DNA breaks and chromosome pulverization from errors in mitosis. Nature, 2012. 482: p. 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang C-Z, et al. , Chromothripsis from DNA damage in micronuclei. Nature, 2015. 522: p. 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ly P, et al. , Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nature cell biology, 2017. 19(1): p. 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ly P, et al. , Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nature genetics, 2019. 51(4): p. 705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ratnaparkhe M, et al. , Defective DNA damage repair leads to frequent catastrophic genomic events in murine and human tumors. Nature Communications, 2018. 9(1): p. 4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Maciejowski J, et al. , Chromothripsis and Kataegis Induced by Telomere Crisis. Cell, 2015. 163(7): p. 1641–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Piazza A, Wright WD, and Heyer W-D, Multi-invasions Are Recombination Byproducts that Induce Chromosomal Rearrangements. Cell, 2017. 170(4): p. 760–773.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Northcott PA, et al. , Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature, 2012. 488: p. 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nones K, et al. , Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nature Communications, 2014. 5: p. 5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vogt N, et al. , Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proceedings of the National Academy of Sciences of the United States of America, 2004. 101(31): p. 11368–11373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yang L, et al. , Diverse Mechanisms of Somatic Structural Variations in Human Cancer Genomes. Cell, 2013. 153(4): p. 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lee JA, Carvalho CM, and Lupski JR, A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell, 2007. 131(7): p. 1235–47. [DOI] [PubMed] [Google Scholar]
- 121.Sanborn JZ, et al. , Double Minute Chromosomes in Glioblastoma Multiforme Are Revealed by Precise Reconstruction of Oncogenic Amplicons. Cancer Research, 2013. 73(19): p. 6036–6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sakofsky Cynthia J., et al. , Translesion Polymerases Drive Microhomology-Mediated Break-Induced Replication Leading to Complex Chromosomal Rearrangements. Molecular Cell, 2015. 60(6): p. 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Watanabe T, et al. , Impediment of Replication Forks by Long Non-coding RNA Provokes Chromosomal Rearrangements by Error-Prone Restart. Cell Rep, 2017. 21(8): p. 2223–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gómez-Herreros F, et al. , TDP2 suppresses chromosomal translocations induced by DNA topoisomerase II during gene transcription. Nature Communications, 2017. 8(1): p. 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lieber MR, Mechanisms of human lymphoid chromosomal translocations. Nature reviews. Cancer, 2016. 16(6): p. 387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jiang Z, et al. , Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nature Genetics, 2007. 39(11): p. 1361–1368. [DOI] [PubMed] [Google Scholar]
- 127.Biedler J and Spengler B, Metaphase chromosome anomaly: association with drug resistance and cell-specific products. Science, 1976. 191(4223): p. 185–187. [DOI] [PubMed] [Google Scholar]
- 128.LEVAN G, et al. , Double minute chromosomes are not centromeric regions of the host chromosomes. Hereditas, 1976. 83(1): p. 83–90. [DOI] [PubMed] [Google Scholar]
- 129.Kanda T, Otter M, and Wahl GM, Mitotic segregation of viral and cellular acentric extrachromosomal molecules by chromosome tethering. Journal of Cell Science, 2001. 114(1): p. 49–58. [DOI] [PubMed] [Google Scholar]
- 130.Wu S, et al. , Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature, 2019. 575(7784): p. 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Biedler J and Spengler B, Metaphase chromosome anomaly: association with drug resistance and cell-specific products. Science, 1976. 191(4223): p. 185–187. [DOI] [PubMed] [Google Scholar]
- 132.Marotta M, et al. , Homology-mediated end-capping as a primary step of sister chromatid fusion in the breakage-fusion-bridge cycles. Nucleic acids research, 2013. 41(21): p. 9732–9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tanaka H, et al. , Intrastrand annealing leads to the formation of a large DNA palindrome and determines the boundaries of genomic amplification in human cancer. Mol Cell Biol, 2007. 27(6): p. 1993–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]