

Abstract
Objective
Macrophages have been described in calcific aortic valve disease, but it is unclear if they promote or counteract calcification. We aimed to determine how macrophages are involved in calcification using the Notch1+/− model of calcific aortic valve disease.
Approach and Results
Macrophages in wild-type and Notch1+/− murine aortic valves were characterized by flow cytometry. Macrophages in Notch1+/− aortic valves had increased expression of MHCII. We then used bone marrow transplants to test if differences in Notch1+/− macrophages drive disease. Notch1+/− mice had increased valve thickness, macrophage infiltration, and proinflammatory macrophage maturation regardless of transplanted bone marrow genotype. In vitro approaches confirm that Notch1+/− aortic valve cells promote macrophage invasion as quantified by migration index and proinflammatory phenotypes as quantified by Ly6C and CCR2 positivity independent of macrophage genotype. Finally, we found that macrophage interaction with aortic valve cells promotes osteogenic, but not dystrophic, calcification and decreases abundance of the STAT3β isoform.
Conclusions
This study reveals that Notch1+/− aortic valve disease involves increased macrophage recruitment and maturation driven by altered aortic valve cell secretion, and that increased macrophage recruitment promotes osteogenic calcification and alters STAT3 splicing. Further investigation of STAT3 and macrophage-driven inflammation as therapeutic targets in calcific aortic valve disease is warranted.
Keywords: calcific aortic valve disease, macrophages, STAT3 transcription factor, Valvular Heart Disease, Mechanisms, Inflammation
Graphical Abstract
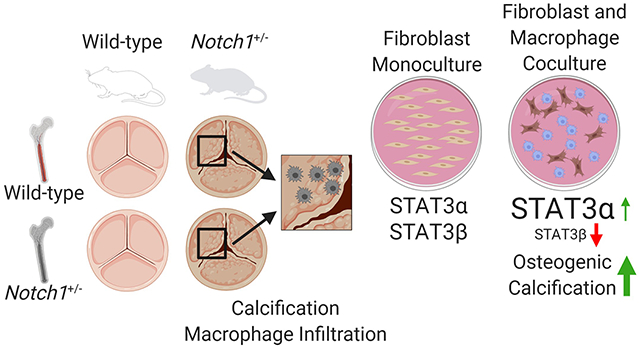
Introduction
Calcific aortic valve disease (CAVD) affects one in four people over 65 years of age and is the primary cause of aortic stenosis.1 This prevalent and insidious disease inevitably leads to surgical or transcatheter replacement of the valve, as there are currently no pharmaceutical treatments. Understanding the cellular and molecular pathophysiology of CAVD may lead to pharmaceutical approaches for patients who are not optimal surgical candidates or prevent prosthetic valve recalcification.
CAVD studies have traditionally focused on aortic valve interstitial cells (AVICs),2 yet no successful pharmacological strategies have emerged from this approach.1 Recent studies have shown that inflammation and immunomodulation may play a key role in determining the calcification potential of these cells,3–5 suggesting that immune signaling may be a viable target for therapeutic intervention. Immune cells are linked to CAVD,6 and up to 10-15% of cells in the healthy murine valve express CD45, a hematopoietic lineage marker.7,8 These cells are primarily major histocompatibility complex II positive macrophages.7,9 Macrophages with MHCII positivity (MHCII+) are more metabolically active, direct a proinflammatory immune response, and are increased in CAVD.4,10,11 As part of this proinflammatory response, MHCII+ macrophages secrete interleukin-6 (IL-6) and tumor necrosis factor α (TNFα), both of which promote calcification of AVICs.4 However, macrophage depletion with liposomal clodronate increases disease as measured by aortic valve (AV) thickening in mice.12 Thus, it is unclear if macrophages drive CAVD, inhibit CAVD, or respond to calcification.
Moving from cellular to molecular inflammation, STAT3 signaling is linked to both the activity of the osteogenic transcription factor RUNX2 and fibrotic inflammation in the heart.13–16 These activities reflect the two primary pathways of AV calcification: osteogenic and dystrophic calcification, respectively.17,18 Additionally, single nucleotide polymorphisms in the IL-6 receptor (a major contributor to STAT3 activation) are associated with decreased severity of CAVD,19 whereas transforming growth factor beta 1 (TGF-β1; another direct activator of STAT3) signaling is increased in CAVD and leads to calcification of AV cells in vitro.20,21 Adding to the evidence, Tsai, et al. reported increased STAT3 phosphorylation in human CAVD.22 This confluence of findings suggests that STAT3-mediated inflammation, potentially driven by macrophage-secreted factors, may promote CAVD and serve as a pharmacological target.
In order to determine the role of immune cells in CAVD, we utilized the Notch1+/− murine CAVD model; human families with NOTCH1 mutations have increased incidence of both CAVD and congenital bicuspid AV disease.23 Mice with Notch1 haploinsufficiency have increased AV calcification,24,25 while AVICs with a Notch1 mutation have increased calcification potential in vitro.26 Interestingly, Notch1 has long been known to play a significant role in the differentiation and maturation of hematopoietic lineages—including specific inhibition of myeloid development—thus highlighting the potential for haploinsufficiency to affect valve calcification through infiltrating macrophages.27–29 After assaying macrophage phenotypes in the Notch1+/− model, we utilized bone marrow transplants and in vitro coculture models to assess the contribution of Notch1+/− immune cells to AV calcification. Finally, we assessed and manipulated STAT3 activity using overexpression plasmids and phosphorylation blockade to investigate the contribution of macrophage-mediated changes in STAT3 to AVIC calcification. We found that Notch1+/− AVICs increase recruitment and inflammatory maturation of macrophages, and that macrophages promote AVIC calcification and alter STAT3 splicing.
Methods
Animal Studies
All animal experiments used C57BL/6J mus musculus animals. Bone marrow transplant experiments included both sexes, while experiments with less than 8 mice per group included only male mice due to the increased prevalence of CAVD in male patients. In total, 135 mice were used for this study. All experimental protocols were approved by the Institutional Animal Care and Use Committee at Vanderbilt University.
Flow cytometry
AVs were isolated from littermate wild-type (WT) and Notch1+/− mice. Cells were isolated from the AV by nine, seven-minute collagenase digestions at 37°C.30 After each digestion, the supernatant was removed and diluted into FC buffer (PBS, 3% FBS). The cell pellet was then subjected to red blood cell lysis buffer for five minutes before quenching with FC buffer. For in vitro assays, BMMs and/or AVICs were lifted with Accutase. Cells were then blocked in Fc Block for 10 minutes at room temperature before staining with conjugated antibody for 30 minutes at 4°C (please see the Major Resources Tables in the Supplemental Materials).
Bone marrow transplants
8- to 12-week-old WT or Notch1+/− C57BL/6J mice were given a split 12 Gy dose of radiation from a Cs137 source followed by retro-orbital administration of 3×106 bone marrow cells isolated from a sex-matched WT or Notch1+/− donor. Mice were allowed six weeks for bone marrow reconstitution before aging on high-fat diet. After six months, mice were euthanized and bone marrow and AVs were isolated. Incidence of unanticipated death was similar between transplant groups and are noted in the Supplemental Materials. Bone marrow was digested in rat tail lysis buffer overnight and genotyped for Notch1 and the Notch1del cassette using polymerase chain reaction (PCR) to confirm successful transplants.
Histology and Immunofluorescence
Murine AVs were frozen in OCT and sectioned at 10 μm thickness. Von Kossa staining was performed by incubating with 3% Ag2NO3 for 40 minutes under a UV lamp, followed by incubation with 5% sodium thiosulfate for five minutes. Slides were counterstained with Nuclear Fast Red. Leaflet thickness was measured using a semi-automated MATLAB script to calculate leaflet area divided by leaflet length, resulting in average leaflet width. For immunofluorescence, slides were fixed and permeabilized with 10% formalin and 0.1% Triton-X for 15 minutes, followed by epitope blockade for one hour with 1% BSA in PBS. Primary antibody staining was performed in blocking solution overnight at 4°C. When applicable, secondary antibody staining was performed for one hour at room temperature. Slides were mounted in ProLong Gold with DAPI and imaged at 4X magnification. CD68+ and MHCII+ macrophages were counted manually and normalized to the area of the leaflet DAPI mask, giving macrophages/mm2.
Aortic valve interstitial cells
AVICs were isolated from WT or Notch1+/− C57BL/6J mice as previously described.31 Briefly, AVs were digested in 2 mg/mL collagenase in HBSS for 30 minutes at room temperature and placed in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin (pen/strep), and 10 μg/mL recombinant murine interferon-γ to induce activation of the simian virus 40 T antigen. Cells were allowed to adhere to 0.1% gelatin-coated six-well tissue culture-treated plates and expanded. To allow for sustained immortal growth, cells were cultured at 33°C and 5% CO2 when not plated for experiments. At least 12 hours prior to experiments (overnight), AVICs were incubated at 37°C and 5% CO2 in DMEM supplemented with 10% FBS and 1% pen/strep (complete media), wherein the immortalization element degrades due to temperature changes. AVICs were seeded at 20,000 cells/cm2 in all experiments unless otherwise noted.
Bone marrow-derived macrophages
Macrophages (BMMs) were generated from the bone marrow of WT or Notch1+/− C57BL/6J mice using M-CSF.32 BMM generation was verified by flow cytometry for CD11b and F4/80 (Supp Fig I). Following differentiation, all experiments were carried out without M-CSF supplementation.
Coculture design
BMMs and AVICs were seeded at a 1:7 physiologic ratio7 in RPMI supplemented with 10% FBS and 1% pen/strep and cultured for 48 hours before harvesting for various experiments. Transwell cocultures were seeded at the same ratio with AVICs seeded on the tissue culture-treated plate and BMMs seeded on a 0.4 μm-pore Transwell insert (Corning, Corning, NY). AVIC monoculture controls for all coculture experiments were also performed in supplemented RPMI.
Cultured media
Media was harvested from cultures after 24 hours and filtered using 0.45 μm sterile filters before use. In all cultured media experiments, RPMI supplemented with 10% FBS and 1% pen/strep was used.
Migration assay
Using a modified Transwell migration protocol,33 10,000 BMMs were seeded in 100 μL of uncultured media on an 8 μm-pore Transwell insert and incubated for 10 minutes at 37°C. 600 μL of cultured media was then added to the well below each insert and cells were allowed to migrate for 3 hours. Transwell inserts were then removed and fixed in 70% ethanol for 10 minutes prior to mounting in ProLong Gold with DAPI. All cells that migrated through the membrane were counted based on visualization of DAPI staining. Migration index was defined as the number of migrated cells divided by the number of migrated cells into a control condition of uncultured media.
Microarray
The Proteome Profiler Mouse Cytokine Array Kit, Panel A (R&D Systems, Minneapolis, MN) was used per the manufacturer’s instructions. Briefly, protein from either cell lysates or conditioned media was incubated with an antibody mixture and allowed to bind to the patterned membrane overnight. Antibodies were then conjugated and the membrane was imaged using an Odyssey Classic imager (Li-Cor, Lincoln, NE).
Calcific nodule assay
Cultures were treated with 5 ng/mL TGF-β1 for 24 hours followed by 24 hours of cyclic biaxial 10% mechanical strain at 1 Hz on BioFlex plates coated with Pronectin, using a FlexCell 3000 machine (FlexCell, Burlington, NC).26 Cultures were stained for calcification using Alizarin Red S and calcific nodules were manually counted in each well by visual inspection.
Magnetic-activated cell sorting
Cells were lifted with Accutase, incubated for 15 minutes with anti-CD45 MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany) to allow for magnetic labeling, and resuspended in MACS buffer (PBS, 0.5% BSA, 2 mM EDTA), followed by positive selection of CD45+ BMMs. Further downstream analysis was conducted on the unperturbed CD45− AVICs.
Quantitative real time-polymerase chain reaction (RT-qPCR)
AVIC mRNA was isolated using Trizol (Life Technologies, Carlsbad, CA) and cDNA libraries were produced using the Superscript IV Reverse Transcriptase kit with oligo(dT) primer, as per manufacturer protocols (ThermoFisher Scientific, Waltham, MA). Quantitative real time polymerase chain reaction for all targets was performed on the CFX-96 Real Time System using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA) (please see the Major Resources Table in the Supplemental Materials for primer sequences). Products were confirmed by gel electrophoresis. Gapdh was used as a housekeeping gene. All statistics were performed on untransformed ΔCt values (“gene of interest” Ct – Gapdh Ct), but for clarity, gene expression was normalized and displayed as 2ΔΔCt.
Cell proximity analysis
Cocultures were performed on glass coverslips and stained by immunofluorescence for CD68 and either RUNX2 or αSMA. Immunofluorescence staining was performed as described above (see Histology and Immunofluorescence). Immunofluorescence images were analyzed using a custom algorithm designed to determine whether the proximity of activated AVICs—as identified by RUNX2 or αSMA staining—to CD68+ macrophages is closer or further than expected based on Monte Carlo simulations of random macrophage placement. Additional details are included in the Supplemental Materials (Supp Fig II).
Western blotting
AVICs and human AVs were lysed in RIPA buffer or PBS, respectively, supplemented with benzonase, sodium orthovanadate, and protease inhibitor. Lysates were denatured using SB at 100°C for 5 minutes, then 10-15 μg was loaded into 15 cm 10% acrylamide gels and run at 150V for 1 hour and 45 minutes. Membrane transfer was performed at 80V for 1 hour and 45 minutes. Membranes were blocked in TBST + 5% BSA and stained in primary antibody overnight at 4°C. Membranes were then stained with fluorescent secondary antibody and imaged on an Odyssey Classic imager (Li-Cor). Quantification was performed in Image Studio Lite (Li-Cor).
Human aortic valve samples
AV samples were collected at the time of replacement and separated into calcified and non-calcified tissue based on the sample location relative to apparent calcification before being flash frozen in liquid nitrogen and stored at −80°C. Samples were mechanically digested with a bead homogenizer (BioSpec Products, Bartlesville, OK) in PBS supplemented with benzonase, sodium orthovanadate, and protease inhibitor. Written informed consent was obtained from patients and tissue sample collection was approved by the institutional review board at Washington University in St. Louis.
Plasmid transfection
Prior to transfection, AVICs were serum-starved in 1 mL of DMEM with 1% FBS overnight in 12-well plates. Lipofectamine 2000 (ThermoFisher) and concentrated STAT3α, STAT3β, or vector control plasmids (Genscript, Piscataway, NJ) were diluted in Opti-MEM media (ThermoFisher) and 200 μL of Opti-MEM containing 4 μL of Lipofectamine and 1 μg of plasmid DNA was added to each well. After 4 hours, media was replaced with complete media. In coculture models, macrophages were added 24 hours after transfection initiation, and in all experiments AVICs were harvested at 48 hours.
Micropipette aspiration
Micropipette aspiration was used to determine the elastic modulus of AVICs as reported previously.21,34–36 Additional details are included in the Supplemental Materials (Supp Fig III).
STAT3 blockade
Stattic (MilliporeSigma), a STAT3 tyrosine (Y705) phosphorylation inhibitor, was used to block STAT3 activity. Stattic was solubilized in DMSO and added to cells in complete media.
Statistics
All data points are shown throughout the manuscript in addition to mean ± standard error of the mean (s.e.m.) or boxplots signifying median and first and third quartiles for non-normal data. Comparisons between normal data were performed by ANOVA followed by Student’s t-test with Holm-Sidak adjustment for multiple comparisons; non-normal data were analyzed using either the Kruskal-Wallis or Mann-Whitney U test. Murine data were analyzed by aligned rank transformed ANOVA37 to allow for two- and three-way non-parametric comparisons. All statistical analyses were performed using the statistical programming language R, version 3.5.2.38 The authors declare that all supporting data are available within the article [and its online supplementary files].
Results
Notch1+/− Aortic Valves Have an Altered Myeloid Profile
We first assessed macrophage phenotypes in the AVs of both WT and Notch1+/− mice in young adulthood (10–12 weeks), prior to disease onset. Hematopoietic cells make up similar proportions of the AV in WT (7.1%) and Notch1+/− (7.0%) mice (Fig 1A). Among hematopoietic cells, there is a majority myeloid fraction (CD11b+) that is greater in the Notch1+/− valve (Fig 1B) and comprised primarily of F4/80+ macrophages in both genotypes (Fig 1C). Valvular macrophages are majority CX3CR1high/Ly6C-/CCR2- in both genotypes (Fig 1D–F), but in the Notch1+/− valve, macrophages show increased CX3CR1 and MHCII expression, suggesting an enhanced migratory and proinflammatory phenotype (Fig 1F, G).39 When analyzing all hematopoietic cells, non-myeloid cell types include CD11b-/MHCII+ antigen-presenting cells, similar to previous reports (Fig 1H, I).7 Simultaneously, BMMs were generated from the same WT and Notch1+/− mice and no differences were observed (Supp Fig IV). In summary, macrophages make up the majority of hematopoietic cells in the AV and are different in the valves of Notch1+/− vs WT mice.
Figure 1. Notch1+/− murine valves have increased macrophage polarization.
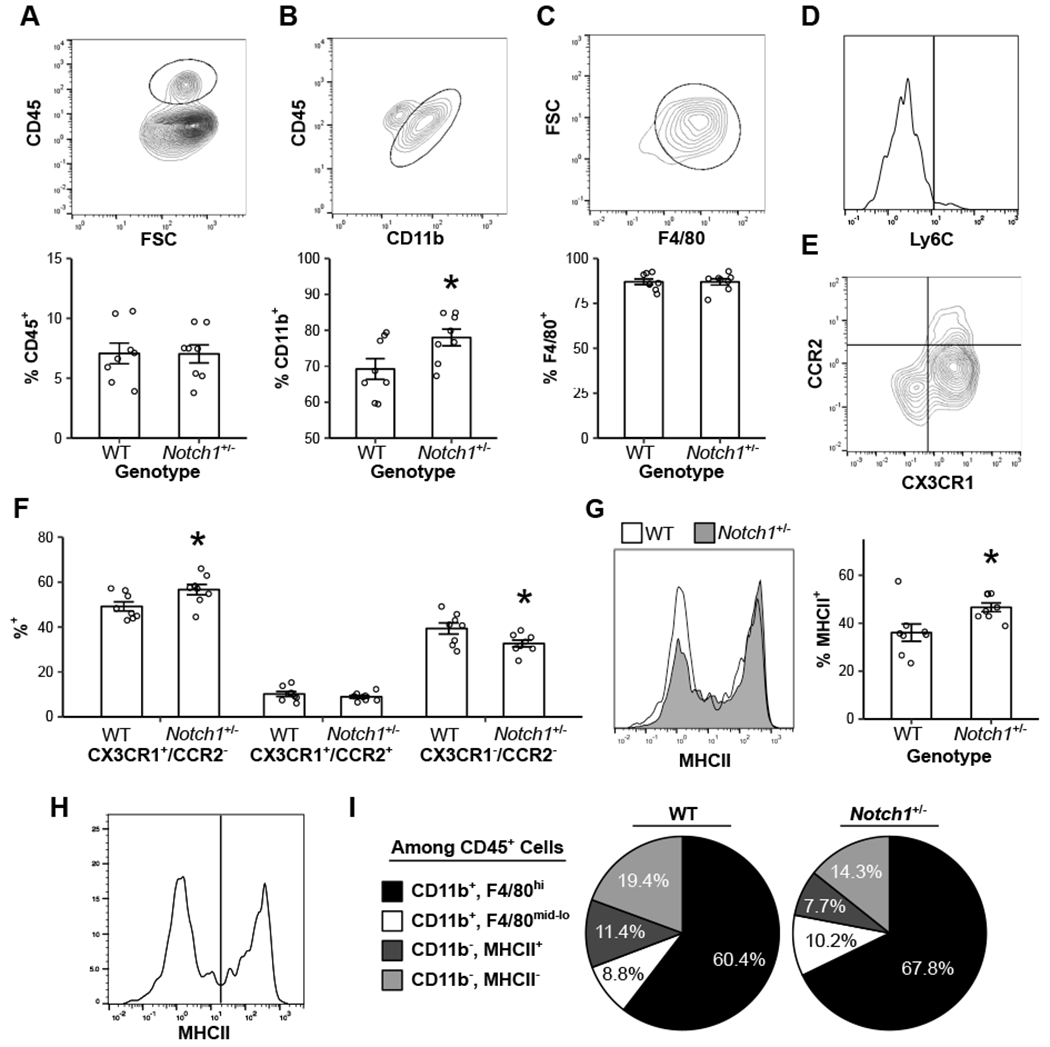
Flow cytometry was performed on both WT and Notch1+/− aortic valves for CD45 (A), followed by CD11b (B), then F4/80 (C). Macrophages were characterized by Ly6C expression (D), CX3CR1 and CCR2 expression (E, F), and MHCII expression (G, outline = WT; gray fill = Notch1+/−). Non-myeloid hematopoietic cells were grouped by MHCII expression (H), and all cell types were plotted as average percentage of the total CD45+ population (I). Bar plots represent mean ± s.e.m (A-C, F, G). Representative flow plots are of WT animals (A-E, H). N = 8 biological replicates. *P < 0.05 by two-tailed t test.
Notch1 Haploinsufficiency in Aortic Valve Cells Drives Disease and Macrophage Recruitment
Considering the increased macrophage infiltration in Notch1+/− mice previously reported,24 and the altered macrophage phenotypes observed, we performed bone marrow transplants to identify if underlying differences in hematopoietic cells were driving the macrophage changes found in the Notch1+/− CAVD model. WT and Notch1+/− mice were transplanted with WT or Notch1+/− bone marrow and aged for 6 months on high-fat diet to allow for disease progression. After aging, leaflet calcification was visualized by von Kossa and Alizarin Red staining (Fig 2A, B, Supp Fig V). Leaflet thickness was measured and body genotype, but not bone marrow genotype, was significantly associated with leaflet thickness (Fig 2C). The same pattern was seen with immunofluorescence staining for CD68+ macrophage infiltration (Fig 2D, E). There was no change in valve phenotype by echocardiography (Supp Fig VI). Valves were additionally stained for MHCII to detect differences observed by flow cytometry (Fig 2F, G). Valves of Notch1+/− mice have an increased prevalence of MHCII+ macrophages and a lesser increase in MHCII- macrophages, leading to a higher MHCII+ macrophage fraction (Fig 2H, I, Supp Fig VII). Thus, Notch1 haploinsufficient valve phenotypes, including differences in hematopoietic cell recruitment, are mediated by valvular cells.
Figure 2. Notch1+/− valve cells drive aortic valve disease and macrophage infiltration and maturation in vivo.
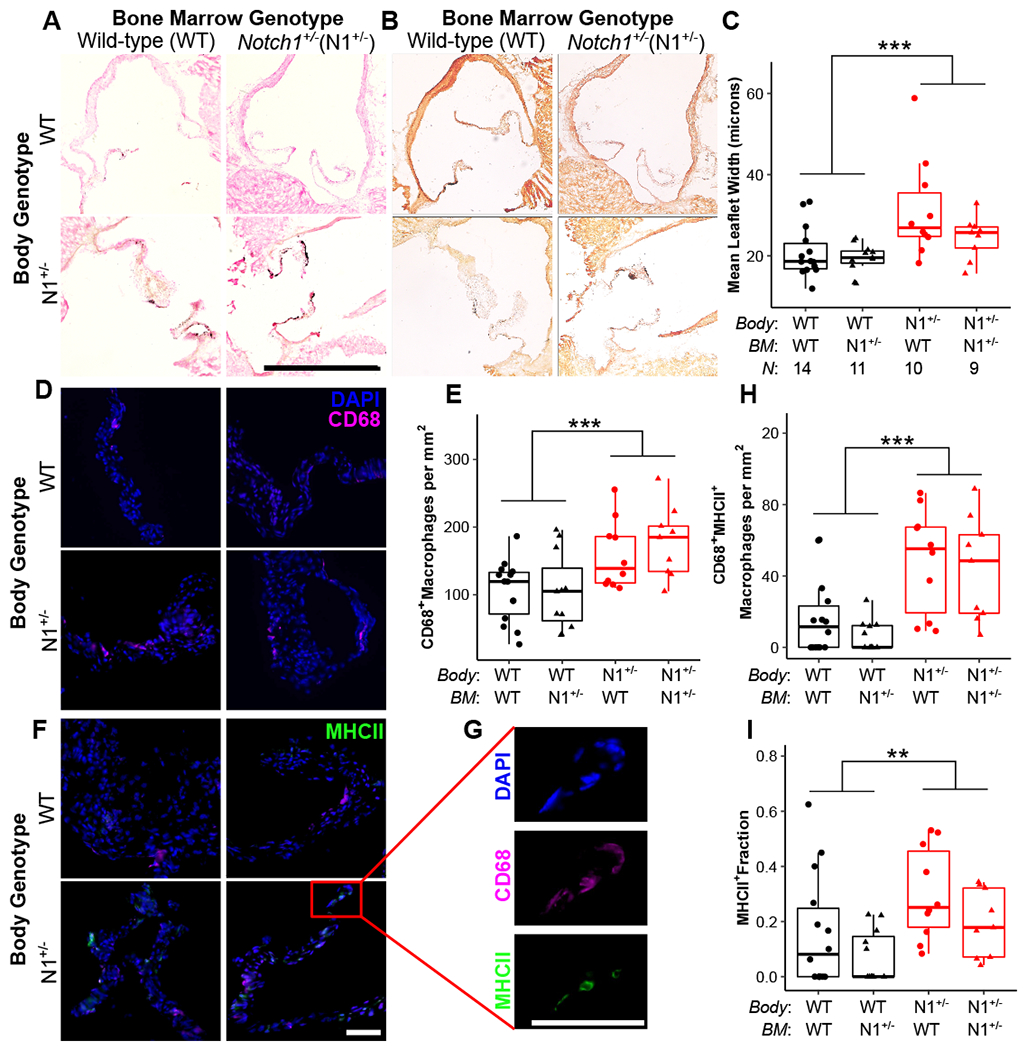
Following bone marrow transplant, aortic valves from Notch1+/− (N1+/−) and wild-type (WT) mice were assessed for calcification (A, B), thickness (A, C), macrophage infiltration (D, E), and macrophage maturation, as measured by MHCII positivity (F, G, H, I). (A) Aortic valves are stained for histology and calcification by von Kossa and (B) Alizarin Red; scale bar = 1 mm. (C) Leaflet thickness is plotted as mean width across the entire section. (D, F, G) Aortic valves are stained by immunofluorescence for DAPI (blue), CD68 (pink), and MHCII (green); scale bar = 100 μm. (E, H) Macrophage data is plotted as cells per mm2 of tissue. Boxplots represent 25th, 50th, and 75th percentiles. All data were analyzed by two-way aligned rank transformed ANOVA.37 **P < 0.01, ***P < 0.001. N = biological replicates, and is the same across panels. BM = bone marrow.
Notch1+/− AVICs Drive Calcification and Macrophage Phenotypes In Vitro
With Notch1 haploinsufficiency acting through AVICs but accompanied by a clear difference in macrophage infiltration and phenotype, we used in vitro coculture models to explore this relationship. First, we replicated the previous bone marrow transplant experiment in vitro. WT and Notch1+/− AVICs and BMMs were cocultured and assayed for common transcriptional calcification markers. Notch1 haploinsufficiency altered coculture calcification genes only when carried in the AVICs (Fig 3A–D). Assessing macrophage phenotypes, BMMs cultured with Notch1+/− AVICs had increased CCR2 and Ly6C expression, while macrophage genotype had no effect (Fig 3E, F). Additionally, both WT and Notch1+/− macrophages increased migration towards media cultured by Notch1+/− AVICs compared to WT AVICs (Fig 3G, Supp Fig VIII). Microarray analysis of secreted factors and lysate from Notch1+/− and WT AVICs revealed an increase in cytokines that induce proinflammatory macrophage differentiation and migration (Fig 3H, Supp Fig IX). 40,41 The highest observed fold-change was that of IL-6, and immunofluorescence staining of 8- to 12-week-old WT and Notch1+/− mice recapitulated this pattern of increased IL-6 expression in Notch1+/− mice (Fig 3I).
Figure 3. Notch1+/− aortic valve interstitial cells promote calcification and macrophage maturation in vitro through altered cytokine secretion.
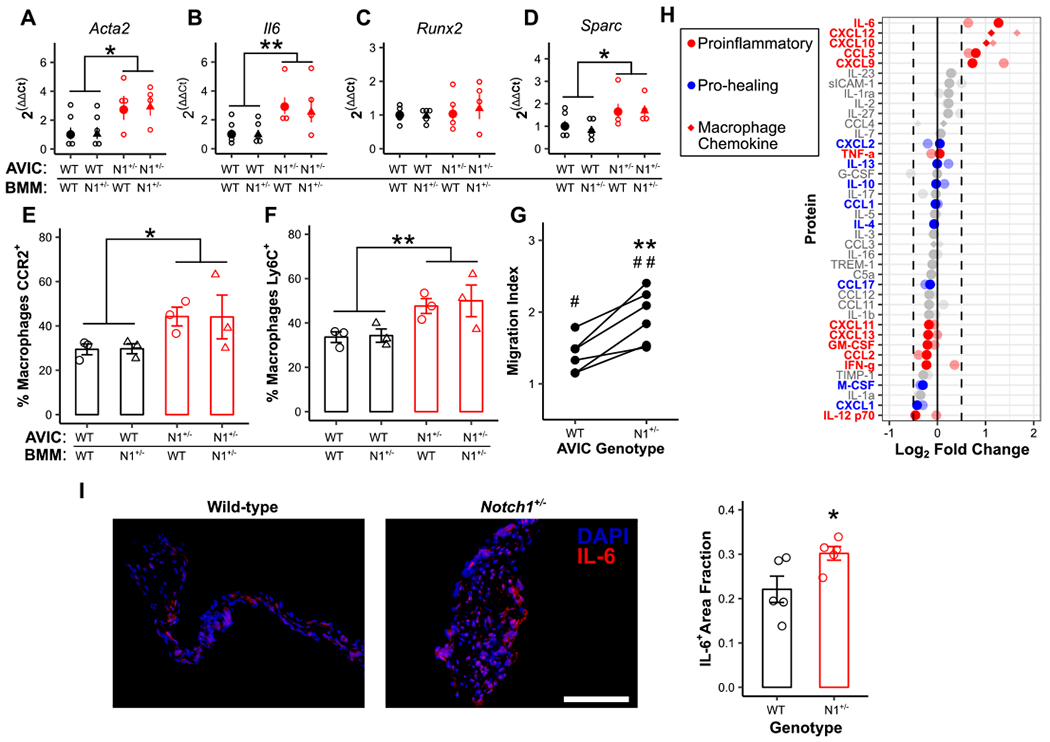
Wild-type (WT) and Notch1+/− (N1+/−) aortic valve interstitial cells (AVICs) were cultured with WT and N1+/− bone marrow-derived macrophages (BMMs) and assayed for transcription of common markers of dystrophic (A, B) and osteogenic (C, D) calcification (N = 4). Flow cytometry for CCR2 (E) and Ly6C (F) expression was performed on BMMs (black = WT AVICs; red = N1+/− BMMs; circle = WT BMMs, triangle = N1+/− BMMs, N = 3). (G, N = 6) Migration of WT macrophages in the presence of either WT or N1+/− AVIC-cultured media normalized to uncultured media controls. (H) Cytokine microarray analysis of secreted media from WT and N1+/− AVICs. Data is plotted as fold change in N1+/− AVIC-cultured media compared to WT control. Full color points are cultured media; faded points are cell lysates. Points represent an average of two experiments with 3 pooled samples each. (I, N = 5) IL-6 (red) expression in WT and N1+/− aortic valves; scale bar = 200 μm. All summary data represent mean ± s.e.m. Data were analyzed by two-way ANOVA on untransformed ΔCt values (A-D) or flow populations (E, F), or one-way ANOVA followed by paired, two-tailed t tests with Holm-Sidak corrections (G), or two-tailed t test (I). *P < 0.05 from wild-type AVICs, **P < 0.01 from wild-type AVICs, #P < 0.05 from monoculture control, ##P < 0.01 from monoculture control. N = biological replicates.
Macrophages Promote Osteogenic, and not Dystrophic, Calcification of AVICs
Given the observation of increased macrophage recruitment and proinflammatory maturation in the Notch1+/− model, we sought to determine how macrophages affect AVIC calcification. When cultured with macrophages, AVICs formed more calcific nodules in vitro (Fig 4A). We then cultured AVICs either in monoculture (Mono), Transwell culture with macrophages (TW), or direct coculture with macrophages (CC) (Fig 4B). RT-qPCR revealed increases in osteogenic calcification transcripts in both Transwell and, more profoundly, direct coculture as compared to monoculture (Fig 4C–E). There was no change in dystrophic calcification markers (Fig 4F–H). AVIC-specific expression was confirmed by immunofluorescent staining for CD68 (macrophages) and RUNX2 (Fig 4I). To further assess this relationship, macrophage proximity to RUNX2-positive and αSMA-positive AVICs was calculated (Fig 4I, Supp Fig X). RUNX2-positive AVICs (osteoblasts, osteogenic calcification) were closer to macrophages than expected, while αSMA-positive AVICs (myofibroblasts, dystrophic) were normally distributed as expected (Fig 4J). Additionally, Notch1+/− animals with increased macrophage recruitment (Fig 2) were stained for RUNX2 and αSMA expression in the AV. RUNX2 expression alone was increased (Fig 4K, Supp Fig XI). These data conclude that increased macrophage recruitment promotes osteogenic, but not dystrophic, AVIC calcification.
Figure 4. Macrophages promote osteogenic but not dystrophic calcification of aortic valve interstitial cells.
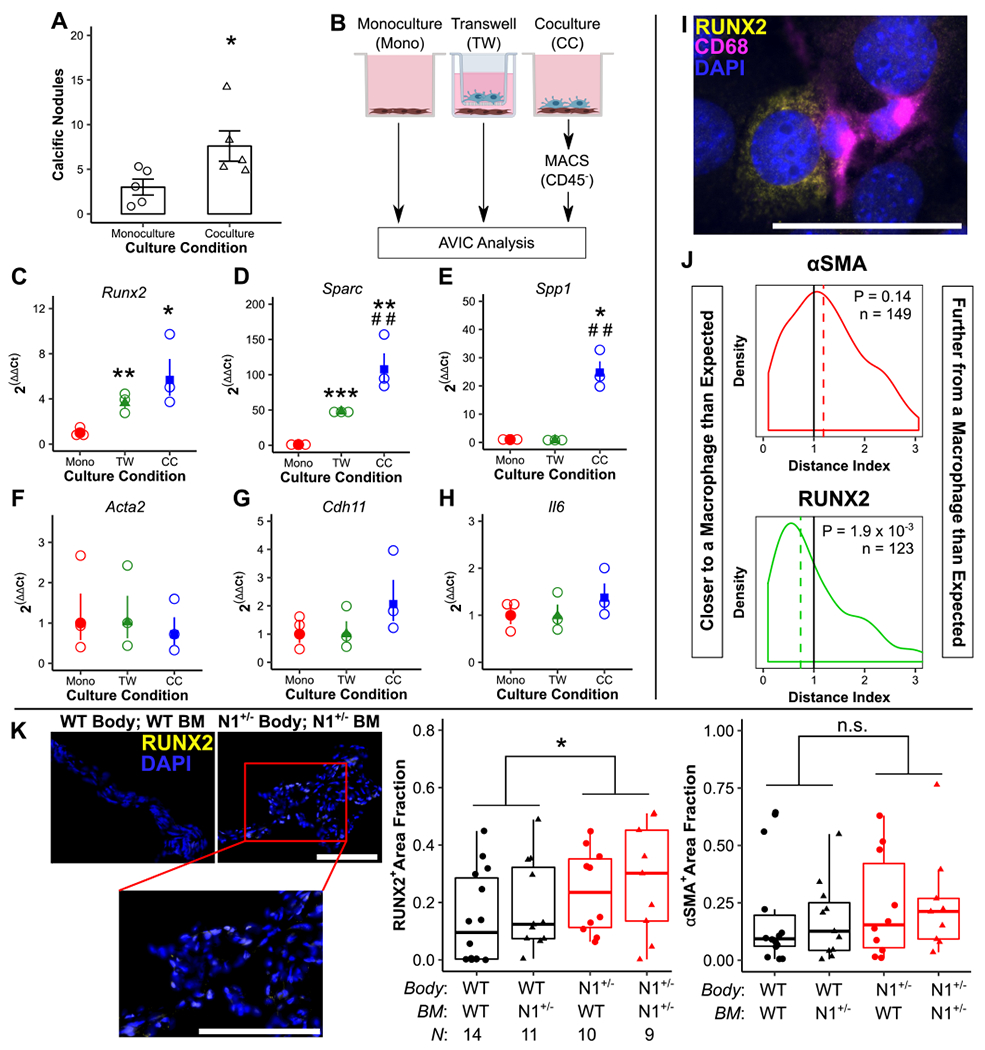
Cocultures of aortic valve interstitial cells (AVICs) with bone marrow-derived macrophages (BMMs) were assayed for calcific nodule formation (A, N = 5). AVICs cultured in monoculture (Mono), Transwell culture with BMMs (TW), or direct coculture with BMMs (CC) (B) were assayed for transcription of markers of osteogenic (C-E) and dystrophic (F-H) calcification (N = 3). Images of cocultures stained for RUNX2, CD68, and DAPI (I) were analyzed by a Monte Carlo-assisted simulation to calculate expected distance and distance index between activated AVICs and BMMs (J). Bone marrow transplanted wild-type (WT) and Notch1+/− (N1+/−) mice were stained for RUNX2 and αSMA expression (K). Scale bars = 200 μm (A, K) and 1 mm (I). All summary data represent mean ± s.e.m. Data were analyzed by Mann Whitney U test (A), one-way ANOVA followed by two-tailed t tests with Holm-Sidak corrections on untransformed ΔCt values (C-H), one sample Wilcoxon Signed-Rank test on log-transformed data (L), or two-way aligned rank transformed ANOVA (K). *P < 0.05, **P < 0.01, ***P < 0.001 from monoculture AVICs (A-H) or between genotypes (K); ##P < 0.01 from Transwell AVICs. N = biological replicates (B-K) or activated AVICs across 4 biological replicates (L).
Altered STAT3 Splicing is Present in Both In Vitro Calcification and Human Calcified Valves
In addition to canonical osteogenic signaling, we further hypothesized that STAT3-mediated inflammation played a role in RUNX2 activation based on previous studies in CAVD22 and the role of STAT3 in other fibrotic inflammatory diseases.14–16 AVICs cultured with macrophages had no increase in STAT3 phosphorylation or total STAT3 but did show a marked decrease in STAT3β expression (Fig 5A–E, Supp Fig XII). STAT3β is an alternative splice product of the STAT3 gene that inhibits canonical STAT3 signaling mediated through STAT3α.42,43 RT-qPCR of STAT3 transcriptional targets Icam1 and Vegfa confirmed an increase in STAT3 activity (Fig 5F). Adar1 transcription increased with decreasing expression of STAT3β, opposing a previously proposed mechanism for altered STAT3 splicing (Supp Fig XIII).44 Notably, STAT3 splicing was not impacted by interferons-α or -γ (Supp Fig XIV), which can induce Adar1 activity.45 To assess the role of STAT3β as an anti-calcification signaling molecule in human disease, excised AVs from patients undergoing AVR were analyzed. Leaflet tissue involved in disease (calcified) had decreased STAT3β expression and increased RUNX2 expression compared to adjacent uninvolved tissue (non-calcified) from the same patients (Fig 5G, H, Supp Fig XV, XVI). Across all samples, RUNX2 expression negatively correlated with the STAT3β fraction (Fig 5I).
Figure 5. STAT3 splicing is altered in calcified human aortic valves and murine aortic valve cells.
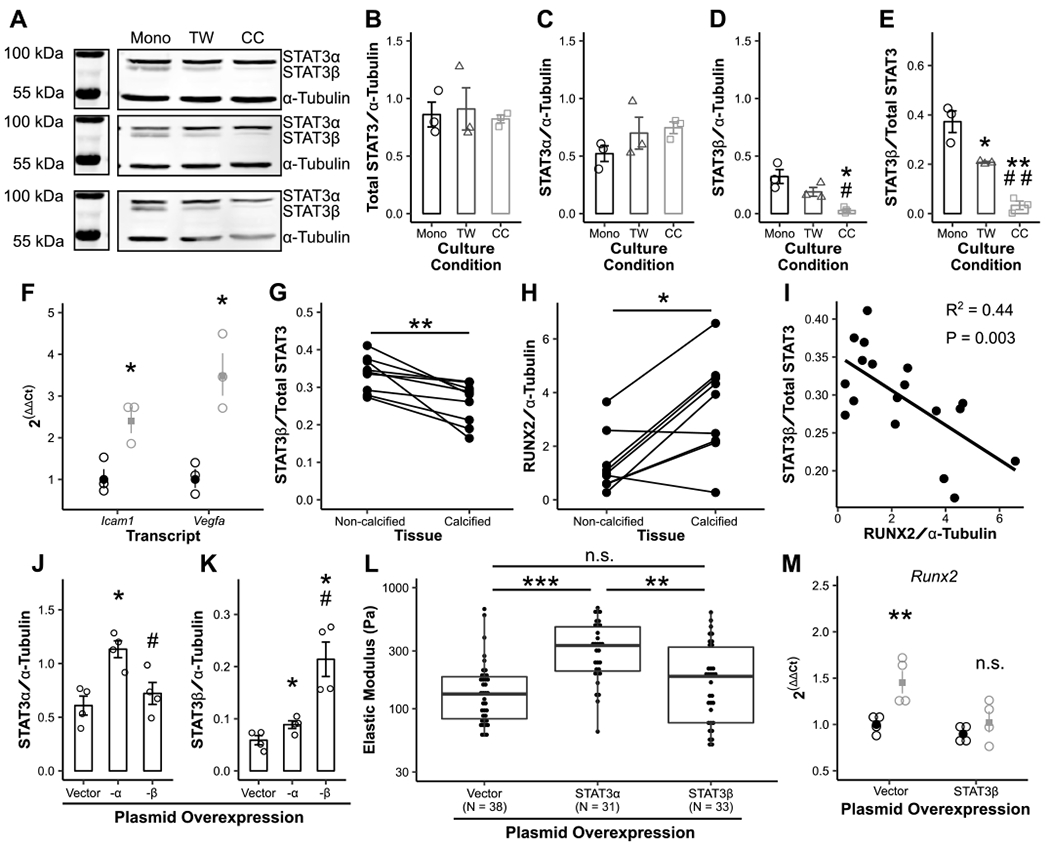
Overall STAT3 expression (A, B), STAT3α expression (C), STAT3β expression (D, E), and expression of STAT3-associated transcripts (F) across 3 biological coculture replicates (black = monoculture [Mono], gray = Transwell [TW], light gray = coculture [CC]). STAT3β and RUNX2 expression was assayed by Western blot in human aortic valves divided into calcified and non-calcified tissue (G-I, N = 9). Plasmid overexpression of STAT3α and β was performed (J, K, N = 4), and cellular stiffness measured by micropipette (L). Overexpression of STAT3β was performed prior to coculture and cocultures assayed for Runx2 transcription (M, N = 4). Bars and dot plots represent mean ± s.e.m. Boxplots represent 25th, 50th, and 75th percentiles. Data were analyzed by one-way ANOVA followed by two-tailed t tests with Holm-Sidak corrections on densitometry data (B-E, J, K) or untransformed ΔCt values (F, M); paired Mann Whitney U tests (G, H); linear regression (I); or Kruskal-Wallis followed by Mann Whitney U tests with Holm-Sidak corrections (L). *P < 0.05, **P < 0.01, ***P < 0.001 from monoculture AVICs (D-F, M), non-calcified aortic valve tissue (G, H) or vector control (J, K); #P < 0.05, ##P < 0.01 from Transwell AVICs (D-E) or STAT3α transfection (J, K). N = biological replicates (B-K, M) or tests of individual cells across 3 biological replicates in 2 independent experiments (L).
Two STAT3 blockade strategies were assessed for efficacy in mitigating calcification. First, AVICs were treated with Stattic, a STAT3 phosphorylation inhibitor, and assayed for cellular stiffness and Runx2 transcription. Stattic treatment decreased cellular stiffness but increased Runx2 transcription in monoculture (Supp Fig XVII). Separately, STAT3α and -β plasmids were used to artificially manipulate STAT3 splicing (Fig 5J, K, Supp Fig XVIII). Such transfections had no effect on calcification-associated transcripts in monoculture AVICs, but STAT3α overexpression increased cellular stiffness (Fig 5L). In the coculture model, STAT3β overexpression rescued Runx2 transcription (Fig 5M).
Discussion
While the cardiovascular immunology field has developed at a rapid pace, the role of immune cells in CAVD has remained unclear.8 Here, we have focused on macrophages, which make up the majority of hematopoietic cells in both diseased human and healthy murine valves.7,46 We have shown that Notch1+/− AVICs promote macrophage maturation and infiltration, and that macrophages promote AVIC calcification and alter STAT3 splicing (Fig 6).
Figure 6. Proposed mechanism for macrophage-associated calcification in Notch1+/− calcific aortic valve disease.
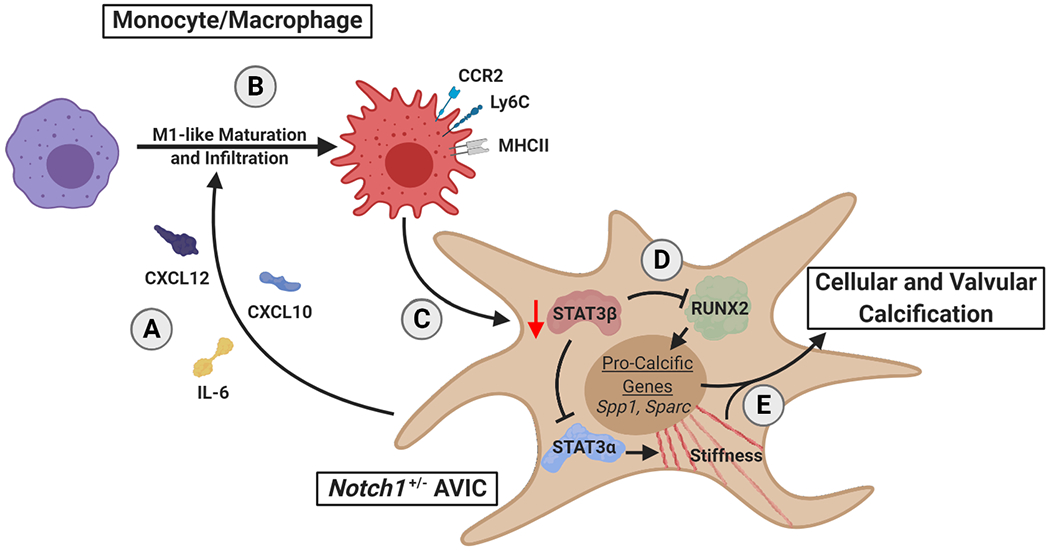
Notch1+/− aortic valve interstitial cells (AVICs) secrete pro-inflammatory factors (A) leading to increased macrophage infiltration and maturation to a proinflammatory phenotype (B). These infiltrating macrophages alter STAT3 splice products to decrease STAT3β (C). Decrease of STAT3β removes inhibition of STAT3α and RUNX2 (D), promoting cellular stiffening and expression of osteogenic transcripts, respectively, and leading to valvular calcification (E).
First, we used flow cytometry to show that the Notch1+/− model has an increased myeloid compartment in the AV with increased CX3CR1 and MHCII positivity prior to disease progression (Fig 1). This aligns with previous data that murine aging and human disease correlate with proinflammatory maturation,4,7,47 and that proinflammatory phenotypes themselves promotes cellular calcification.4,48 This suggested that perhaps differences in macrophage phenotype at baseline in Notch1+/− mice are driving AV phenotypes. In addition, Notch1 is known to inhibit myeloid cell maturation, reflected in our data by an increased myeloid compartment in the Notch1+/− valve.28,29 Together, these data strengthened our hypothesis that altered hematopoietic cells drive disease in the Notch1+/− model. However, bone marrow transplant experiments show that Notch1+/− AV cells promote macrophage infiltration regardless of macrophage genotype (Fig 2). Contrary to our hypothesis, it seems that hematopoietic cells play their role in response to altered valve cell phenotypes. Independent of bone marrow genotype, Notch1+/− mice had increased infiltration of MHCII+ macrophages. Thus, Notch1+/− valve cells are likely the instigating force behind valve pathology by both driving traditional disease markers and recruiting hematopoietic cells that then promote disease.
We built off of these findings with in vitro studies to explore how Notch1+/− AVICs alter macrophage phenotypes and infiltration (Fig 3). Conditioned media from Notch1+/− AVICs promotes increased migration and proinflammatory maturation of WT macrophages, relative to media from WT AVICs. Reinforcing the importance of Notch1 haploinsufficiency in AVICs specifically, Notch1+/− macrophages responded similarly. Notably, the proinflammatory phenotype induced in vitro is characterized by Ly6C and CCR2 positivity and no change in MHCII, whereas the in vivo data instead showed an increase in MHCII+ macrophages but no change in Ly6C or CCR2. It is possible that this difference is due to the timelines involved. High Ly6C expression defines inflammatory monocytes and macrophages,49,50 and CCR2 is necessary for recruitment of such Ly6Chi monocytes.51,52 Thus, the roles of Ly6C and CCR2 are in the recruitment and egress of monocytes and macrophages, but expression is variable and can decrease after extravasation and tissue residency.53,54 Alternatively, development of MHCII expression occurs after macrophage extravasation and allows for antigen presentation and generation of an adaptive immune response.53,55 This would explain the observation of increased MHCII expression in macrophages within murine valves.
Cytokine microarrays on AVIC secreted media and immunofluorescence of murine AVs confirmed an increase in factors that induce migration and proinflammatory maturation. Indeed, Notch1 signaling has previously been shown to inhibit NF-κB activity and inflammatory cytokine production.56,57 Together, these in vivo and in vitro phenomena provide a mechanism for macrophage involvement in Notch1-associated CAVD. They also highlight an additional lens for the interpretation of transcriptomic and proteomic datasets like that reported by Schlotter, et al.58 Our results contribute to the body of literature suggesting that the effects of these secreted factors on immune cell recruitment and activation may also play a significant role in CAVD pathophysiology.
Our remaining studies focused on how macrophages alter AVIC phenotypes. We utilized a Transwell model to show that not only does the macrophage secretome promote calcification, but that physical interaction increases this effect (Fig 4). This is perhaps due to a macrophage-to-AVIC signal, but considering the findings that AVICs promote macrophage activation, it is also possible that physical interactions with AVICs can induce a further activated macrophage state and secretion of pro-calcification cytokines. It has also been suggested that extracellular vesicles may mediate cardiovascular calcification, and macrophage-derived vesicles may play a similar role here.59,60 Unintuitively, macrophages promoted osteogenic calcification and not dystrophic calcification, which is characterized by cytokine production and myofibroblast transition. To test the hypothesis that myofibroblast transition was not increased in toto, but that myofibroblast activation was occurring closer to macrophages, we developed an image analysis algorithm. The results instead confirmed the above findings: αSMA-positive myofibroblasts were normally distributed around their expected distance, while RUNX2-positive osteoblast-like cells were significantly closer to macrophages than expected. Finally, we stained AVs from bone marrow transplanted WT and Notch1+/− mice for these same markers, and saw an increase in RUNX2 alone in the mice with increased macrophage recruitment. Thus, we show–both in vitro and in vivo–that increased exposure to macrophages is associated with osteogenic and not dystrophic calcification.
We then tested the hypothesis that STAT3 was mediating the connection between macrophage-secreted factors and RUNX2 expression. We observed a drastic shift in STAT3 splicing, resulting in a decrease in the inhibitory STAT3β splice product and an increase in the canonical STAT3α splice product (Fig 5). We confirmed an associated increase in canonical STAT3 signaling as measured through increased Vegfa and Icam1 transcription–two signaling markers previously described in CAVD.61,62 These phenomena translated to human AVs. STAT3β decreases in calcified regions of diseased AVs and negatively correlates with RUNX2 expression. We then used overexpression models to manipulate STAT3 splicing directly. In AVIC monoculture, manipulation of STAT3 splicing ratios altered cellular stiffness, a disease marker,21 and in coculture this manipulation mitigated increased RUNX2 transcription.
We attempted to understand the mechanism of STAT3β rescue by blocking STAT3 activity with Stattic, a STAT3 phosphorylation inhibitor. Stattic treatment decreased cellular stiffness but increased RUNX2 expression. This leads to the conclusion that STAT3β is functioning to inhibit calcification through its own unique characteristics, perhaps requiring phosphorylation, rather than solely through an auto-inhibitory function against canonical STAT3α signaling. The ability of STAT3β to bind RUNX2 and inhibit its function as a transcription factor may be a key step in its calcification-mitigating capabilities shown here.63
Limitations
We have used primarily murine data throughout this manuscript. This has allowed us both to study the Notch1+/− CAVD model, and to use coculture models with syngeneic macrophages. Our in vitro models also focused on AVICs and not aortic valve endothelial cells. It is possible that Notch1 haploinsufficiency may similarly contribute to macrophage recruitment through endothelial cell phenotypes. Second, it is possible in this murine model that there are resident hematopoietic progenitor cells in the AV that have persisted through irradiation and proliferated. However, literature in this mouse model has shown that all hematopoietic cells in the valve are perpetually recruited, rather than existing as resident cells,64 and we have used a high radiation dose to minimize this risk. The murine model of CAVD used here is subject to relatively large variance, making some studies underpowered for phenotype detection by echocardiography. Thus, we have focused on quantitative histological and immunofluorescence methods that we believe capture with integrity the extent of disease in mice.
Conclusions
Herein, we report heightened macrophage infiltration and maturation in Notch1-associated CAVD driven by altered cytokine secretion of AVICs. This increased interaction between macrophages and AVICs promotes AVIC calcification and altered STAT3 splicing. Altered STAT3 splicing is found in calcified human AVs, and splicing manipulation opposes macrophage-induced calcification. These findings suggest that cellular inflammation and the STAT3 axis may play a targetable role in CAVD.
Supplementary Material
Highlights.
Notch1+/− valve disease involves increased inflammatory macrophage infiltration, driven by altered cytokine secretion of aortic valve interstitial cells.
Macrophages promote osteogenic, but not dystrophic, calcification of aortic valve interstitial cells.
Macrophages promote aortic valve cell disease phenotypes in part through altered STAT3 splicing leading to a decrease in the STAT3β splice product.
STAT3β is decreased in calcified human aortic valve tissue.
Acknowledgments
The authors would like to thank Caleb Snider for technical assistance with murine irradiation and Ethan Joll for discussion of image processing and data analysis.
Sources of Funding
This work was funded by the National Institutes of Health (F30-HL147464, R35-HL135790, and T32-GM007347) and Fondation Leducq.
Disclosures
Dr. Lindman has received research grants from Edwards Lifesciences and Roche Diagnostics; served on scientific advisory boards for Roche Diagnostics; and has been a consultant to Medtronic and Roche Diagnostics.
Nonstandard Abbreviations and Acronyms
- αSMA
alpha smooth muscle actin
- AV
aortic valve
- AVIC
aortic valve interstitial cell
- AVR
aortic valve replacement
- BMM
bone marrow-derived macrophage
- CAVD
calcific aortic valve disease
- IL
interleukin
- MACS
magnetic-assisted cell sorting
- MHCII
major histocompatibility complex II
- N1+/−
Notch1+/−
- RT-qPCR
quantitative real time-polymerase chain reaction
- STAT3
signal transducer and activator of transcription 3
- TGF-β1
transforming growth factor beta 1
- TNFα
tumor necrosis factor alpha
- WT
wild-type
References
- 1.Lindman BR, Clavel M-A, Mathieu P, et al. Calcific aortic stenosis. Nat Rev Dis Prim. 2016;2(1):16006. doi: 10.1038/nrdp.2016.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bowler MA, Merryman WD. In vitro models of aortic valve calcification: solidifying a system. Cardiovasc Pathol. 2015;24(1):1–10. doi: 10.1016/j.carpath.2014.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aguado BA, Schuetze KB, Grim JC, et al. Transcatheter aortic valve replacements alter circulating serum factors to mediate myofibroblast deactivation. Sci Transl Med. 2019;11(509):eaav3233. doi: 10.1126/scitranslmed.aav3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li G, Qiao W, Zhang W, Li F, Shi J, Dong N. The shift of macrophages toward M1 phenotype promotes aortic valvular calcification. J Thorac Cardiovasc Surg. 2017;153(6):1318–1327.e1. doi: 10.1016/j.jtcvs.2017.01.052 [DOI] [PubMed] [Google Scholar]
- 5.Isoda K, Matsuki T, Kondo H, Iwakura Y, Ohsuzu F. Deficiency of Interleukin-1 Receptor Antagonist Induces Aortic Valve Disease in BALB/c Mice. Arterioscler Thromb Vasc Biol. 2010;30(4):708–715. doi: 10.1161/ATVBAHA.109.201749 [DOI] [PubMed] [Google Scholar]
- 6.Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O’Brien KD. Characterization of the early lesion of “degenerative” valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 1994;90(2):844–853. http://www.ncbi.nlm.nih.gov/pubmed/7519131. Accessed April 24, 2018. [DOI] [PubMed] [Google Scholar]
- 7.Hulin A, Anstine LJ, Kim AJ, et al. Macrophage Transitions in Heart Valve Development and Myxomatous Valve Disease. Arterioscler Thromb Vasc Biol. 2018:ATVBAHA.117.310667. doi: 10.1161/ATVBAHA.117.310667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raddatz MA, Madhur MS, Merryman WD. Adaptive immune cells in calcific aortic valve disease. Am J Physiol Circ Physiol. 2019;317(1):H141–H155. doi: 10.1152/ajpheart.00100.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi J, Do Y, Cheong C, et al. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J Exp Med. 2009;206(3):497–505. doi: 10.1084/jem.20082129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazzone A, Epistolato MC, De Caterina R, et al. Neoangiogenesis, T-lymphocyte infiltration, and heat shock protein-60 are biological hallmarks of an immunomediated inflammatory process in end-stage calcified aortic valve stenosis. J Am Coll Cardiol. 2004;43(9):1670–1676. doi: 10.1016/j.jacc.2003.12.041 [DOI] [PubMed] [Google Scholar]
- 11.Skowasch D, Schrempf S, Wernert N, et al. Cells of primarily extravalvular origin in degenerative aortic valves and bioprostheses. Eur Heart J. 2005;26(23):2576–2580. doi: 10.1093/eurheartj/ehi458 [DOI] [PubMed] [Google Scholar]
- 12.Calin MV, Manduteanu I, Dragomir E, et al. Effect of depletion of monocytes/macrophages on early aortic valve lesion in experimental hyperlipidemia. Cell Tissue Res. 2009;336(2):237–248. doi: 10.1007/s00441-009-0765-2 [DOI] [PubMed] [Google Scholar]
- 13.Nicolaidou V, Wong MM, Redpath AN, et al. Monocytes induce STAT3 activation in human mesenchymal stem cells to promote osteoblast formation. PLoS One. 2012;7(7). doi: 10.1371/journal.pone.0039871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chakraborty D, Šumová B, Mallano T, et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat Commun. 2017;8(1). doi: 10.1038/s41467-017-01236-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurdi M, Zgheib C, Booz GW. Recent Developments on the Crosstalk Between STAT3 and Inflammation in Heart Function and Disease. Front Immunol. 2018;9(December):1–10. doi: 10.3389/fimmu.2018.03029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loperena R, Van Beusecum JP, Itani HA, et al. Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: roles of STAT3, interleukin 6 and hydrogen peroxide. Cardiovasc Res. 2018;(July):1–17. doi: 10.1093/cvr/cvy112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohler ER, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation. 2001;103(11):1522–1528. doi: 10.1161/01.CIR.103.11.1522 [DOI] [PubMed] [Google Scholar]
- 18.Chen J- H, Simmons CA. Cell–Matrix Interactions in the Pathobiology of Calcific Aortic Valve Disease. Circ Res. 2011;108(12):1510–1524. doi: 10.1161/CIRCRESAHA.110.234237 [DOI] [PubMed] [Google Scholar]
- 19.Wypasek E, Potaczek DP, Lamplmayr M, Sadowski J, Undas A. Interleukin-6 receptor Asp358Ala gene polymorphism is associated with plasma C-reactive protein levels and severity of aortic valve stenosis. Clin Chem Lab Med. 2014;52(7). doi: 10.1515/cclm-2013-0606 [DOI] [PubMed] [Google Scholar]
- 20.Merryman WD, Lukoff HD, Long RA, Engelmayr GC, Hopkins RA, Sacks MS. Synergistic effects of cyclic tension and transforming growth factor-β1 on the aortic valve myofibroblast. Cardiovasc Pathol. 2007;16(5):268–276. doi: 10.1016/J.CARPATH.2007.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Merryman WD, Youn I, Lukoff HD, et al. Correlation between heart valve interstitial cell stiffness and transvalvular pressure: Implications for collagen biosynthesis. Am J Physiol - Hear Circ Physiol. 2006;290(1):224–231. doi: 10.1152/ajpheart.00521.2005 [DOI] [PubMed] [Google Scholar]
- 22.Tsai C-L, Chiu Y-M, Lee Y-J, et al. Interleukin-32 plays an essential role in human calcified aortic valve cells. Eur Cytokine Netw. 2018;29(1):36–47. doi: 10.1684/ECN.2018.0407 [DOI] [PubMed] [Google Scholar]
- 23.Garg V, Muth AN, Ransom JF, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437(7056):270–274. doi: 10.1038/nature03940 [DOI] [PubMed] [Google Scholar]
- 24.Nus M, MacGrogan D, Martínez-Poveda B, et al. Diet-induced aortic valve disease in mice haploinsufficient for the notch pathway effector RBPJK/CSL. Arterioscler Thromb Vasc Biol. 2011;31(7):1580–1588. doi: 10.1161/ATVBAHA.111.227561 [DOI] [PubMed] [Google Scholar]
- 25.Clark CR, Bowler MA, Snider JC, David Merryman W. Targeting Cadherin-11 Prevents Notch1-Mediated Calcific Aortic Valve Disease. Circulation. 2017;135(24):2448–2450. doi: 10.1161/CIRCULATIONAHA.117.027771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J, Ryzhova LM, Sewell-Loftin MK, et al. Notch1 Mutation Leads to Valvular Calcification Through Enhanced Myofibroblast Mechanotransduction. Arterioscler Thromb Vasc Biol. 2015;35(7):1597–1605. doi: 10.1161/ATVBAHA.114.305095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohishi K, Katayama N, Shiku H, Varnum-Finney B, Bernstein ID. Notch signalling in hematopoiesis. Semin Cell Dev Biol. 2003;14(2):143–150. doi: 10.1016/S1084-9521(02)00183-0 [DOI] [PubMed] [Google Scholar]
- 28.Stier S, Cheng T, Dombkowski D, Carlesso N, Scadden DT. Notch1 activation increases hematopoietic stem cell self-renewal in vivo and favors lymphoid over myeloid lineage outcome. Blood. 2002;99(7):2369–2378. doi: 10.1182/blood.V99.7.2369 [DOI] [PubMed] [Google Scholar]
- 29.Bigas A, Martin DIK, Milner LA. Notch1 and Notch2 Inhibit Myeloid Differentiation in Response to Different Cytokines. Mol Cell Biol. 1998;18(4):2324–2333. doi: 10.1128/mcb.18.4.2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller LJ, Lincoln J. Isolation of Murine Valve Endothelial Cells. J Vis Exp. 2014;(90). doi: 10.3791/51860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bowler MA, Bersi MR, Ryzhova LM, Jerrell RJ, Parekh A, Merryman WD. Cadherin-11 as a regulator of valve myofibroblast mechanobiology. Am J Physiol Circ Physiol. 2018;315(6):H1614–H1626. doi: 10.1152/ajpheart.00277.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008;(SUPPL. 83):1–14. doi: 10.1002/0471142735.im1401s83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Justus CR, Leffler N, Ruiz-Echevarria M, Yang LV. In Vitro Cell Migration and Invasion Assays. J Vis Exp. 2014;(88):1–8. doi: 10.3791/51046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trickey WR, Vail TP, Guilak F. The role of the cytoskeleton in the viscoelastic properties of human articular chondrocytes. J Orthop Res. 2004;22(1):131–139. doi: 10.1016/S0736-0266(03)0150-5 [DOI] [PubMed] [Google Scholar]
- 35.Hochmuth RM. Micropipette aspiration of living cells. J Biomech. 2000;33(1):15–22. doi: 10.1016/S0021-9290(99)00175-X [DOI] [PubMed] [Google Scholar]
- 36.Theret DP, Levesque MJ, Sato M, Nerem RM, Wheeler LT. The Application of a Homogeneous Half-Space Model in the Analysis of Endothelial Cell Micropipette Measurements. J Biomech Eng. 1988;110(3):190–199. doi: 10.1115/1.3108430 [DOI] [PubMed] [Google Scholar]
- 37.Wobbrock JO, Findlater L, Gergle D, Higgins JJ. The Aligned Rank Transform for nonparametric factorial analyses using only ANOVA procedures. Conf Hum Factors Comput Syst - Proc. 2011;(May):143–146. doi: 10.1145/1978942.1978963 [DOI] [Google Scholar]
- 38.R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2018. [Google Scholar]
- 39.Lee M, Lee Y, Song J, Lee J, Chang SY. Tissue-specific role of CX3CR1 expressing immune cells and their relationships with human disease. Immune Netw. 2018;18(1):1–19. doi: 10.4110/in.2018.18.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murray PJ. Macrophage Polarization. Annu Rev Physiol. 2017;79(1):541–566. doi: 10.1146/annurev-physiol-022516-034339 [DOI] [PubMed] [Google Scholar]
- 41.Xuan W, Qu Q, Zheng B, Xiong S, Fan G-H. The chemotaxis of M1 and M2 macrophages is regulated by different chemokines. J Leukoc Biol. 2015;97(1):61–69. doi: 10.1189/jlb.1A0314-170R [DOI] [PubMed] [Google Scholar]
- 42.Caldenhoven E, van Dijk TB, Solari R, et al. STAT3β, a Splice Variant of Transcription Factor STAT3, Is a Dominant Negative Regulator of Transcription. J Biol Chem. 1996;271(22):13221–13227. doi: 10.1074/jbc.271.22.13221 [DOI] [PubMed] [Google Scholar]
- 43.Maritano D, Sugrue ML, Tininini S, et al. The STAT3 isoforms α and β have unique and specific functions. Nat Immunol. 2004;5(4):401–409. doi: 10.1038/ni1052 [DOI] [PubMed] [Google Scholar]
- 44.Goldberg L, Abutbul-Amitai M, Paret G, Nevo-Caspi Y. Alternative Splicing of STAT3 Is Affected by RNA Editing. DNA Cell Biol. 2017;36(5):367–376. doi: 10.1089/dna.2016.3575 [DOI] [PubMed] [Google Scholar]
- 45.Lamers MM, van den Hoogen BG, Haagmans BL. ADAR1: “Editor-in-Chief” of Cytoplasmic Innate Immunity. Front Immunol. 2019;10(July):1–11. doi: 10.3389/fimmu.2019.01763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coté N, Mahmut A, Bosse Y, et al. Inflammation is associated with the remodeling of calcific aortic valve disease. Inflammation. 2013;36(3):573–581. doi: 10.1007/s10753-012-9579-6 [DOI] [PubMed] [Google Scholar]
- 47.Anstine LJ, Horne TE, Horwitz EM, Lincoln J. Contribution of extra-cardiac cells in murine heart valves is age-dependent. J Am Heart Assoc. 2017;6(10):1–13. doi: 10.1161/JAHA.117.007097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li X, Wang Y, Zheng D, et al. M1 macrophages promote aortic valve calcification mediated by microRNA-214/TWIST1 pathway in valvular interstitial cells. Am J Transl Res. 2016;8(12):5773–5783. http://www.ncbi.nlm.nih.gov/pubmed/28078049. [PMC free article] [PubMed] [Google Scholar]
- 49.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–455. doi: 10.1038/nature12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science (80- ). 2010;327(5966):656–661. doi: 10.1126/science.1178331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsou C- L, Peters W, Si Y, et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117(4):902–909. doi: 10.1172/JCI29919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Geissmann F, Jung S, Littman DR. Blood Monocytes Consist of Two Principal Subsets with Distinct Migratory Properties. Immunity. 2003;19(1):71–82. doi: 10.1016/S1074-7613(03)00174-2 [DOI] [PubMed] [Google Scholar]
- 53.Crane MJ, Daley JM, Van Houtte O, Brancato SK, Henry WL, Albina JE. The monocyte to macrophage transition in the murine sterile wound. PLoS One. 2014;9(1). doi: 10.1371/journal.pone.0086660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boniakowski AE, Kimball AS, Joshi A, et al. Murine macrophage chemokine receptor CCR2 plays a crucial role in macrophage recruitment and regulated inflammation in wound healing. Eur J Immunol. 2018;48(9):1445–1455. doi: 10.1002/eji.201747400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang J, Zhang L, Yu C, Yang XF, Wang H. Monocyte and macrophage differentiation: Circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res. 2014;2(1):1–9. doi: 10.1186/2050-7771-2-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J, Shelly L, Miele L, Boykins R, Norcross MA, Guan E. Human Notch-1 Inhibits NF-κB Activity in the Nucleus Through a Direct Interaction Involving a Novel Domain. J Immunol. 2001;167(1):289–295. doi: 10.4049/jimmunol.167.1.289 [DOI] [PubMed] [Google Scholar]
- 57.Zhang Q, Wang C, Liu Z, et al. Notch signal suppresses toll-like receptor-triggered inflammatory responses in macrophages by inhibiting extracellular signal-regulated kinase 1/2-mediated nuclear factor κB activation. J Biol Chem. 2012;287(9):6208–6217. doi: 10.1074/jbc.M111.310375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schlotter F, Halu A, Goto S, et al. Spatiotemporal Multi-omics Mapping Generates a Molecular Atlas of the Aortic Valve and Reveals Networks Driving Disease. Circulation. 2018;138(4):377–393. doi: 10.1161/CIRCULATIONAHA.117.032291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blaser MC, Aikawa E. Roles and Regulation of Extracellular Vesicles in Cardiovascular Mineral Metabolism. Front Cardiovasc Med. 2018;5(December). doi: 10.3389/fcvm.2018.00187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cui L, Rashdan NA, Zhu D, et al. End stage renal disease-induced hypercalcemia may promote aortic valve calcification via Annexin VI enrichment of valve interstitial cell derived-matrix vesicles. J Cell Physiol. 2017;232(11):2985–2995. doi: 10.1002/jcp.25935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perrotta I, Moraca FM, Sciangula A, Aquila S, Mazzulla S. HIF-1α and VEGF: Immunohistochemical Profile and Possible Function in Human Aortic Valve Stenosis. Ultrastruct Pathol. 2015;39(3):198–206. doi: 10.3109/01913123.2014.991884 [DOI] [PubMed] [Google Scholar]
- 62.Zeng Q, Jin C, Ao L, et al. Cross-talk between the toll-like receptor 4 and notch1 pathways augments the inflammatory response in the interstitial cells of stenotic human aortic valves. Circulation. 2012;126(11 SUPPL.1):1–20. doi: 10.1161/CIRCULATIONAHA.111.083675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ziros PG, Georgakopoulos T, Habeos I, Basdra EK, Papavassiliou AG. Growth hormone attenuates the transcriptional activity of Runx2 by facilitating its physical association with Stat3β. J Bone Miner Res. 2004;19(11):1892–1904. doi: 10.1359/JBMR.040701 [DOI] [PubMed] [Google Scholar]
- 64.Hajdu Z, Romeo SJ, Fleming PA, Markwald RR, Visconti RP, Drake CJ. Recruitment of bone marrow-derived valve interstitial cells is a normal homeostatic process. J Mol Cell Cardiol. 2011;51(6):955–965. doi: 10.1016/j.yjmcc.2011.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.