

Abstract
Immune response is one of the functions that have been more strongly targeted by natural selection during human evolution. The evolutionary genetic dissection of the immune system has greatly helped to distinguish genes and functions that are essential, redundant or advantageous for human survival. It is also becoming increasingly clear that admixture between early Eurasians with now-extinct hominins such as Neanderthals or Denisovans, or admixture between modern human populations, can be beneficial for human adaptation to pathogen pressures. In this review we discuss how the integration of population genetics with functional genomics in diverse human populations can inform about the changes in immune functions related to major lifestyle transitions (e.g., from hunting and gathering to farming), the action of natural selection to the evolution of the immune system, and the history of past epidemics. We also highlight the need of expanding the characterization of the immune system to a larger array of human populations – particularly neglected human groups historically exposed to different pathogen pressures – in order to fully capture the relative contribution of genetic, epigenetic and environmental factors to immune response variation in humans.
Introduction
Over the last decade, it has become increasingly clear that understanding the different ways in which natural selection can act informs the biological relevance of immune genes, which can be essential, redundant or adaptable (Quintana-Murci, et al. 2007; Barreiro and Quintana-Murci 2010; Quintana-Murci and Clark 2013; Quintana-Murci 2019). Purifying selection removes deleterious alleles from the population, and genes evolving under this regime, e.g. STAT1, TRAF3, IFNG or TLR3, fulfill functions that are essential and non-redundant (Barreiro, et al. 2009; Manry, et al. 2011; Vasseur, et al. 2012; Deschamps, et al. 2016). Mutations in such highly-constrained genes usually lead to severe disorders, for example, HSV-1 encephalitis, pyogenic bacterial infections, or Mendelian susceptibility to mycobacterial disease (see (Casanova and Abel 2013, 2018) for extensive reviews). Selective constraints have relaxed for other genes such as some type-I IFNs, MBL2 or TLR5, in which loss-of-function variants can reach high population frequencies and evolve neutrally (or quasi-neutrally), reflecting higher immunological redundancy (Quintana-Murci and Clark 2013; Casanova and Abel 2018).
Advantageous alleles can increase in frequency in the population by positive or balancing selection, highlighting cases of genetic adaptation (Quintana-Murci and Clark 2013; Key, et al. 2014; Fan, et al. 2016). Signals of positive selection have been detected at gene variants associated with malaria resistance (DARC, G6PD, CD36, GYPA, GYPB, GYPE), weaker inflammation/NF-κB signaling (TLR10-TLR1-TLR6 cluster), antiviral responses (type-III IFNs), or immunity to Lassa virus infection (LARGE and IL21), among others infectious or immune phenotypes (see (Grossman, et al. 2013; Quintana-Murci and Clark 2013; Brinkworth and Barreiro 2014; Fumagalli and Sironi 2014; Karlsson, et al. 2014) and references therein).
Immune functions have been particularly affected by balancing selection (Andres, et al. 2009; Leffler, et al. 2013; DeGiorgio, et al. 2014; Teixeira, et al. 2015; Siewert and Voight 2017), a rare type of selection that can act at different time depths. It can maintain polymorphisms over millions of years, as is the case for vertebrate MHC or the ABO group in primates (Lawlor, et al. 1988; Klein, et al. 2007; Segurel, et al. 2012), but it can also act in recent times, as attested by the iconic sickle-cell mutation (HbS) (Allison 1954; Ackerman, et al. 2005). A recent report has estimated that HbS appeared ~20,000 years ago, informing the time at which malaria started to be a health burden in Africa (Laval, et al. 2019).
A subtler mechanism of adaptation is through polygenic selection, in which selection acts simultaneously on advantageous alleles at multiple loci (Pritchard, et al. 2010). Signals of this selective regime have been reported for various traits (Turchin, et al. 2012; Berg and Coop 2014; Field, et al. 2016; Gouy, et al. 2017; Racimo, et al. 2018), including gene sets and immune pathways associated with the IL-6 signaling pathway, malaria, cytokine–cytokine receptor interaction, and pathogenic Escherichia coli infection (Daub, et al. 2013).
In the following, we will not review how natural selection has targeted specific immune functions or pathways in humans, as comprehensive reviews of genes evolving under different selection types and the immunological and clinical consequences of such patterns can be found elsewhere (Barreiro and Quintana-Murci 2010; Casanova and Abel 2013; Quintana-Murci and Clark 2013; Brinkworth and Barreiro 2014; Fumagalli and Sironi 2014; Karlsson, et al. 2014; Casanova and Abel 2018; Quintana-Murci 2019). Instead, we will focus on the most recent data showing how ancient and modern admixture have participated in human adaptation to pathogen pressures, how human populations differ in the way they respond to infection, and on the genetic, epigenetic and evolutionary drivers of immune response variation in humans (Figure 1).
Figure 1.
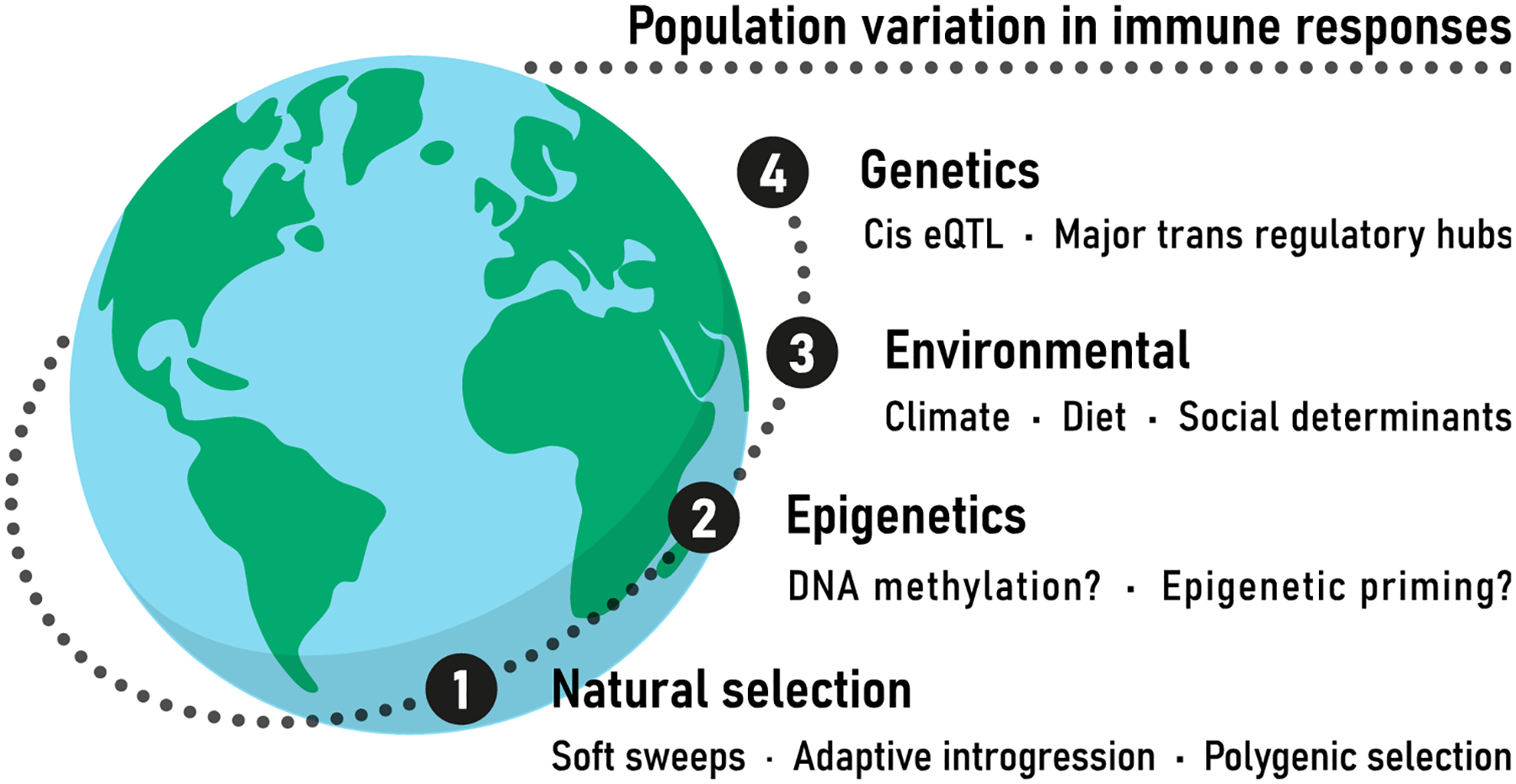
Summary of the different factors contributing to population variation in immune responses that are discussed in this review.
Borrowing beneficial immune alleles from archaic hominins
Advantageous alleles can be found in other species or populations, which can act as “donors” of adaptive variation. There is increasing evidence that archaic, now-extinct hominins with whom humans admixed served as donors (Gittelman, et al. 2016; Racimo, et al. 2017). The genomes of individuals of non-African ancestry carry today the legacy of ancient admixture, with ~2% Neanderthal ancestry in Eurasians, <1% Denisovan ancestry in East and South East Asians, and up to 6% Denisovan ancestry in some Oceanian populations (Dannemann and Racimo 2018) (Figure 2A). Although archaic introgression was generally selected against (Sankararaman, et al. 2014; Sankararaman, et al. 2016; Petr, et al. 2019), some cases of beneficial introgression, i.e. adaptive introgression, have been reported for genes relating to body morphology, metabolism, and responses to environmental conditions including pathogens (Gittelman, et al. 2016; Racimo, et al. 2017).
Figure 2. Ancestry-associated differences in immune response are under genetic control.
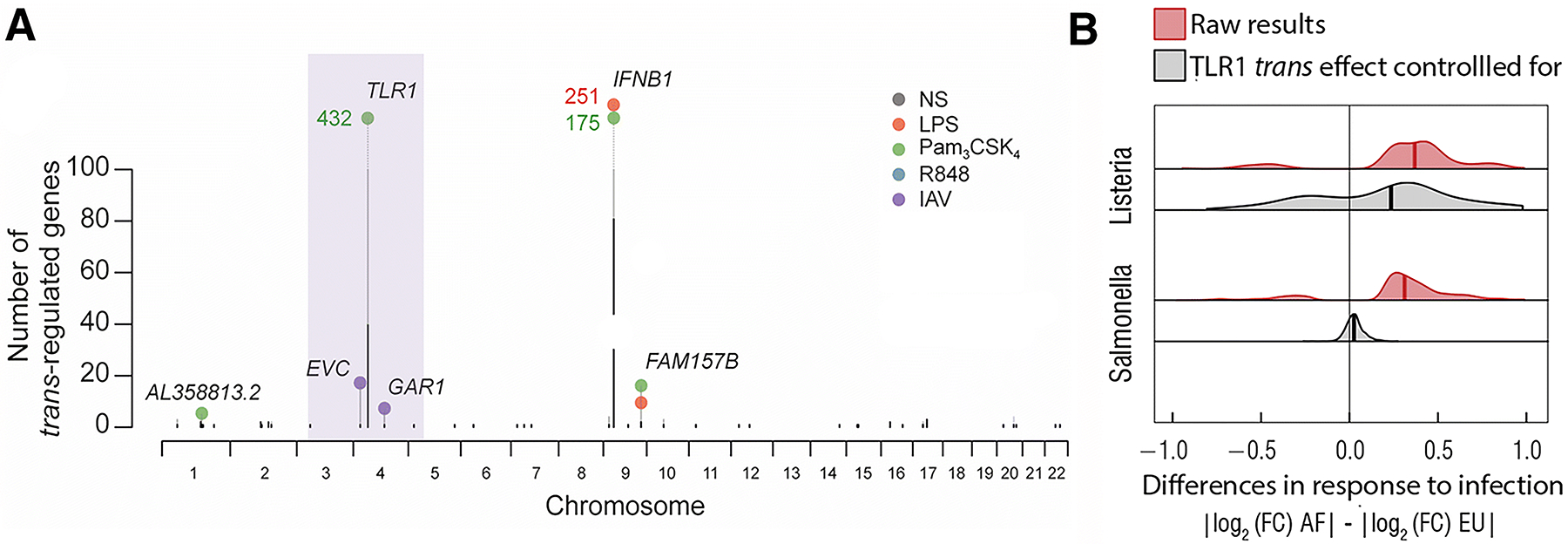
A) Manhattan plot representing the number of trans-regulated genes (y-axis) in monocytes. The different colors represent the conditions under which gene expression levels were measured: NS – non-stimulated; LPS – lipopolysaccharide (TLR4 agonist); Pam3CSK4 (TLR2 agonist); R848 (TLR7/8 agonist); IAV (flu virus). The purple box highlight the non-synonymous variant in TLR1 that regulates in trans over 400 genes in response to Pam3CSK4. B) Effect of the TLR1 trans-eQTL, rs5743618, on genome-wide distributions of inter-population response differences to Listeria, Salmonella infection of macrophages. The x-axis represents the absolute difference in the log2 fold change response to Listeria infection (top panel) and Salmonella (bottom panel) between European and African individuals. Positive values indicates that the transcriptional response was stronger among African ancestry individuals, whereas a negative value indicates a stronger response among European ancestry individuals. Regressing out the effect of rs5743618 (gray distributions) reduces differences between populations, suggesting that the weaker proinflammatory response observed in Europeans is in part explained by this single trans-eQTL.
An early study proposed that some HLA haplotypes of modern humans were acquired through admixture with Denisovans or Neanderthals (Abi-Rached, et al. 2011). Since then, multiple immune genes with signals of adaptive introgression have been reported: STAT2, the OAS1–3 (Figure 2B) and the TLR6-1-10 gene clusters or TNFAIP3 (Mendez, et al. 2012, 2013; Sankararaman, et al. 2014; Dannemann, et al. 2016; Deschamps, et al. 2016; Gittelman, et al. 2016; Sams, et al. 2016; Racimo, et al. 2017). Archaic variants at these genes can reach high population frequencies, as reported for TLR6-1-10 in Asia (~39%) or TNFAIP3 in Melanesia (~60%). These studies highlight the beneficial role of admixture between humans entering Eurasia ~60,000 years ago and ancient human forms who were present in the region and possibly adapted for a longer time period.
Modern admixture as a vehicle of rapid adaptation
The history of humans is also characterized by pervasive admixture between modern human populations, particularly over the past 4,000 years (Hellenthal, et al. 2014). In contrast with archaic introgression, the role of modern admixture in genetic adaptation, i.e. adaptive admixture, has only recently started to be explored. Admixture between different African populations has been accompanied by the exchange of adaptive alleles such as the lactase persistence allele or HLA variants (Busby, et al. 2017; Patin, et al. 2017). The role of HLA as a substrate for adaptive admixture is further attested by the excess of African ancestry at the HLA locus in recently admixed Hispanic/Latino groups (Tang, et al. 2007; Rishishwar, et al. 2015; Zhou, et al. 2016). Likewise, the Duffy-null allele at DARC, which protects against vivax malaria and reaches very high frequency in Africa, supports the beneficial role of admixture, as high African ancestry has been detected in admixed populations from Pakistan and Madagascar, where vivax malaria is endemic (Hodgson, et al. 2014; Laso-Jadart, et al. 2017; Pierron, et al. 2018).
Understanding the adaptive nature of admixture between populations with different modes of subsistence or exposed to different ecologies can also inform about both immune functions related to lifestyle transitions, and past epidemics. In this context, the study of hunter-gatherers and farmers from Africa — the continent that harbors the largest groups of active hunter-gatherers — has been particularly informative. For example, it has been shown that rainforest hunter-gatherers, who share an extensive history of admixture with farmers (Patin and Quintana-Murci 2018), acquired the HbS allele through adaptive admixture over the last 6,000 years only (Laval, et al. 2019). This suggests that rainforest hunter-gatherers started to be exposed to malarial pressures in recent times, alongside their first contacts with agrarian communities. Nonetheless, it has also been shown that selection has maintained adaptive variation in rainforest hunter-gatherers in the face of recent gene flow from farmers, as immune response genes are enriched in both selection signals and local hunter-gatherer ancestry in admixed populations (Lopez, et al. 2019). In southern Africa, San hunter-gatherers that have frequent contact with farmers present stronger selection signals at immune genes than those that are more isolated, supporting the notion that incoming groups can bring new pathogens to which local populations can rapidly adapt (Owers, et al. 2017).
Back to the past through ancient DNA to understand immune relevance
Sequencing the genome of individuals who lived at different time periods allows to measure the action of selection directly, and can inform the nature of genes that contributed to host adaptation during particular epochs, e.g. the Neolithic transition, before and after major epidemics, etc. (Skoglund and Mathieson 2018). A pioneering study compared allele frequency changes in individuals living in Europe between 8,500 and 2,300 years ago, and showed that immune genes such as the TLR6-1-10 cluster, the HLA region and SLC22A4 carry variants that have increased in frequency during this time period by positive selection (Mathieson, et al. 2015). Another study, which sequenced the genome of a 7,000-year-old individual, has concluded that pathogen-driven selection at genes such as TLR1, CD14, IFIH1, CASP12 and NOS2A occurred before changes in skin pigmentation in Mesolithic Europeans (Olalde, et al. 2014).
The arrival of Europeans to the Americas, which has been linked to the introduction of new pathogens (Thornton 1997), also represents an excellent model to understand rapid adaptation to environmental change. The sequencing of ancient samples of Native Americans dating from pre-Columbian times has revealed signals of positive selection at immune loci, the strongest being detected at the HLA-DQA1 (Lindo, et al. 2016). Yet, the selected alleles appear to have decreased in frequency, by purifying selection, in the corresponding modern population from the Northwest Coast of North America, suggesting that the European arrival triggered pathogen-related environmental changes that made these variants deleterious for Native Americans. Likewise, purifying selection targeting a deleterious allele has been detected by a recent study that has identified a variant in TYK2 that confers predisposition to tuberculosis (Boisson-Dupuis, et al. 2018), this variant having decreased in frequency in Europe from ~9% to 4% over the last 4,000 years.
Natural selection and population variation in immune response
The population genetic studies described above have helped to identify genes for which patterns of genetic diversity are not compatible with neutral evolution. However, in the absence of functional studies it remains unclear the nature of the immune phenotypes that have been selectively advantageous, and how they have diverged across populations. Recent studies have started to fill this gap by combining population genetics approaches with quantitative trait loci (eQTL) mapping, in innate immune cells exposed to different immune stimuli or live infectious agents (Nedelec, et al. 2016; Quach, et al. 2016). These studies have functionally defined the extent to which immune responses are differentiated across individuals of different genetic ancestries, the genetic variants that account for such differences, and the evolutionary mechanisms (neutral genetic drift vs. positive selection) that led to their establishment in modern human populations.
Hundreds of genes for which the transcriptional response of phagocytic cells varies significantly between European- and African-ancestry individuals have been identified. Interestingly, increased African ancestry has been found to be associated with a stronger transcriptional response to immune stimulation, particularly in response to immune stimuli that signals via TLR1 (Nedelec, et al. 2016; Quach, et al. 2016; Sanz, et al. 2018). This is largely explained by a non-synonymous variant in TLR1, shown to cause dampened inflammatory immune responses due to hindered NF-kB signalling and activation (Figure 3B) (Barreiro, et al. 2009; Quach, et al. 2016). The allele associated with reduced inflammation is almost absent in African populations but is found at very high frequency in European populations (Derived Allele Frequency (DAF)Africa = 0.04, DAFEurope = 0.67). In addition to this major trans-regulatory hub shown to control the expression levels of hundreds of genes (Figure 3A), cis-eQTLs were shown to explain, on average, ~30% and 50% of the ancestry-related differences in expression in macrophages and monocytes, respectively (Nedelec, et al. 2016; Quach, et al. 2016). Collectively, these findings highligh the key role played by host genetic factors to differences in immune response detected between populations.
Figure 3. The role of introgression in the evolution of the immune system.
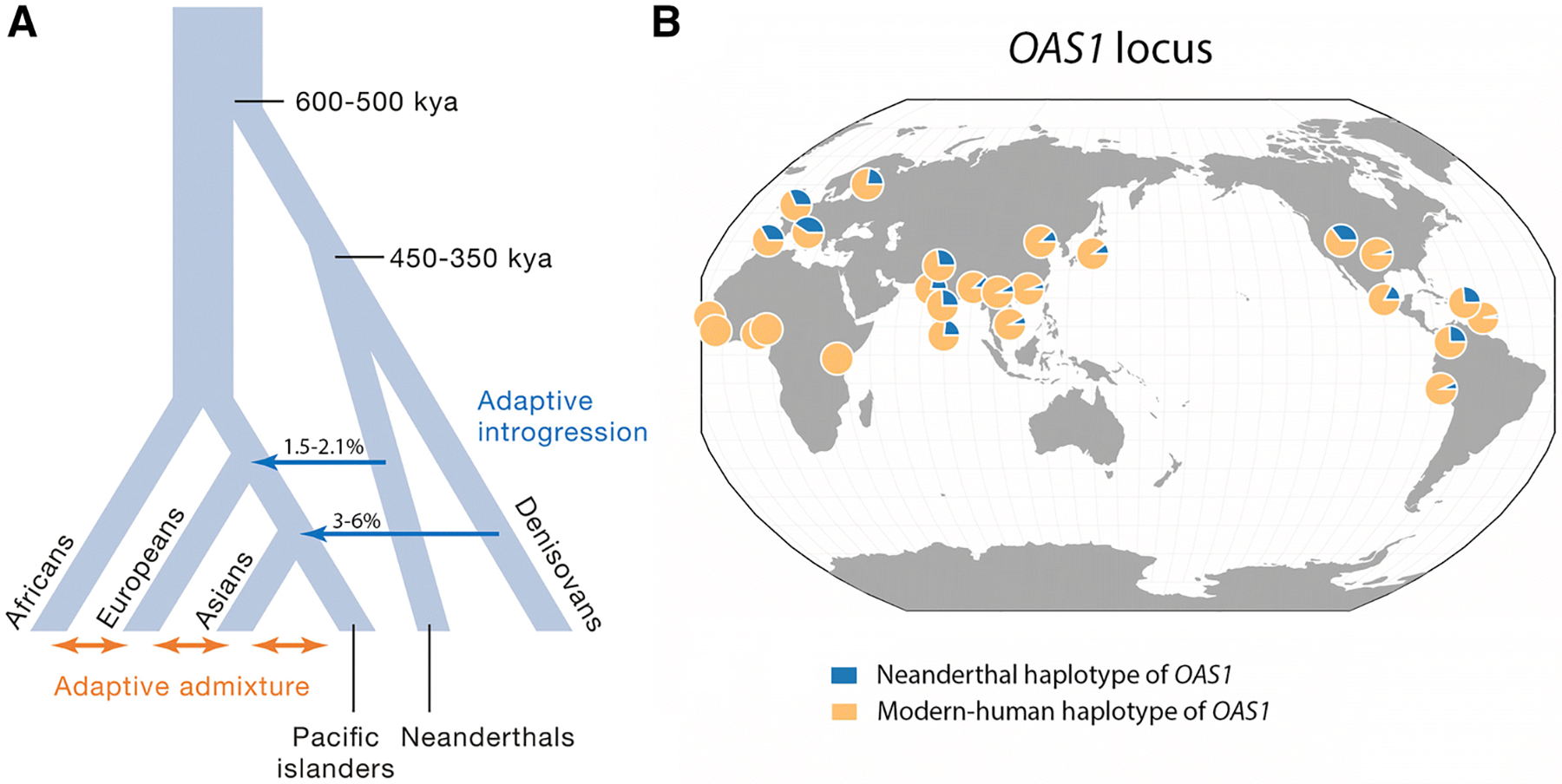
A) Beneficial genetic variation can be acquired from other species or populations through admixture. Adaptive introgression from archaic humans is represented by blue arrows, while adaptive admixture between modern human populations is represented by orange arrows. B) Worldwide distribution of Neanderthal vs modern human haplotypes for the OAS1 gene – one of the most emblematic cases of adaptive introgression in humans. Specifically, we show the allele frequencies for the two alleles of rs1557866, a SNP which derived allele is of Neandertal origin (blue) and that is a strong proxy of individuals harboring the Neandertal haplotype in the OAS region. The map was generated using the tool described by (Marcus and Novembre 2017).
Interestingly, immune response eQTLs have been shown to be strongly enriched for signatures of recent natural selection (Nedelec, et al. 2016; Quach, et al. 2016), suggesting that a significant fraction of population differences in responses to infection results from past events of local adaptation. Notably, the trans-eQTL at TLR1 shows some of the most compelling signatures of positive selection in the human genome, suggesting that selection has favored a lower inflammatory response in humans that left Africa (Barreiro, et al. 2009; Quach, et al. 2016). This observation highlights the possible evolutionary trade-off between mounting a strong inflammatory response to fight pathogens in a high-pathogen load environment (Guernier et al., 2004) while avoiding the detrimental consequences of acute and chronic inflammation, which can lead to tissue damage, inflammation and autoimmunity.
Recent studies have also identified several genes for which ancestry-associated differences in immune response can be attributed to alleles that were introgressed from Neanderthals to European-ancestry individuals – especially among genes involved in antiviral response (Nedelec, et al. 2016; Quach, et al. 2016; Sams, et al. 2016). The OAS gene cluster provides one particular example of a genomic region harboring adaptively-introgressed variants important for antiviral activity that reach frequencies above 40% in Eurasian populations while absent in Africans (Figure 2B) (Mendez, et al. 2013; Sams, et al. 2016). Positive selection of a Neanderthal haplotype has led to the reintroduction of an ancestral splice variant of OAS1 associated with higher enzymatic activity (Sams, et al. 2016). The adaptive potential of this variant is supported by its association with reduced infectivity with West Nile virus (Lim, et al. 2009), higher resistance to HCV infection (El Awady, et al. 2011; Kwon, et al. 2013); and variable symptomology of tick-borne encephalitis (TBE) virus-induced disease (Barkhash, et al. 2010). That West Nile, HCV, and TBE are all Flaviviruses suggests that this family of virus are the main selective drivers. Strikingly, RNA viruses such as Flaviviruses have also been proposed to be the main cause of adaptive introgression between modern humans and Neanderthals (Enard and Petrov 2018).
The impact of agricultural practices on the evolution of the immune system
The transition from hunter gathering to farming, which began ~12,000 years ago in Mesopotamia (Harris 1967; Diamond 2002; Zeder 2008; Vigne 2011), has been associated with profound changes in human ecology (Braidwood 1960; Diamond and Bellwood 2003). Such changes were hypothesized to have precipitated major new infectious disease burdens. With animal husbandry came the continuous, close proximity to livestock, providing opportunity for novel or expanded zoonotic transmission (Wolfe, et al. 2007) of pathogens potentially including rotavirus, measles virus, and influenza (Suzuki and Nei 2002; Matthijnssens, et al. 2008; Furuse, et al. 2010). With the transition to agriculture came a concomitant increase in population size (Gignoux, et al. 2011); high population sizes are critical requirements for several infectious agents (e.g. measles, rubella, etc.) to spread and persist (Anderson and May 1991). Finally, the physical changes to the environment enacted by humans through the course of agriculture (e.g., land clearing and irrigation) also may have precipitated increases in the number of vector-borne diseases (McNeill 1989), such as Plasmodium falciparum malaria (Sundararaman, et al. 2016; Otto, et al. 2018).
A recent study has reported the first functional comparison of immune responses between a rainforest hunter-gatherer population from southwest Uganda and their agriculturalist neighbors (Harrison, et al. 2019). While their results demonstrate that positive selection has contributed to differences in immune responses between the two groups, interestingly, they do not support the long-standing hypothesis that selective pressures imposed by pathogens were particularly acute for agriculturalist populations. Instead, most selection signatures accounting for population differences in immune responses were observed in the hunter-gatherer population. Thus, while the advent of agriculture likely led to the emergence of new pathogens, other infectious diseases might have simultaneously been a stronger selective pressure among hunter-gatherers.
Several studies suggest indeed that viral burden is higher in hunter-gatherer populations as compared to agriculturalist (Johnson, et al. 1993; Gonzalez, et al. 2000; Harrison, et al. 2019), which might account for the increased divergence in immune response observed between hunter-gatherers and farmers to viral as compared to bacterial stimulus (Harrison, et al. 2019). Importantly, these groups diverged more than 60,000 years ago (Patin and Quintana-Murci 2018), long before the agriculture emergence in Africa. A substantial proportion of the functional genetic divergences observed may thus reflect pre-agriculture responses to long-standing ecological differences facing each lineage. Future work on other pairs of populations differing in their subsistence patterns are needed to fully understand the impact of the agricultural revolution on the evolution of the human immune system.
Not all is about genetics: epigenetics of infection
Although genetics plays a significant role in driving inter-individual variation in immune responses, most of the variance observed at the population level remains unaccounted for when considering genetics alone (Aguirre-Gamboa, et al. 2016; Li, et al. 2016; Bakker, et al. 2018; Piasecka, et al. 2018). Other factors have been linked to variation in immune responses, including sex, age (Bakker, et al. 2018; Piasecka, et al. 2018), and gut microbiome diversity (Schirmer, et al. 2016). Although less studied, epigenetic variation is also likely to play an important role at explaining immune response variance. DNA methylation, in particular, has been shown to dynamically change in response to infection (Marr, et al. 2014; Zhang, et al. 2014; Ichiyama, et al. 2015; Pacis, et al. 2015; Sinclair, et al. 2015; Wiencke, et al. 2016; Pacis, et al. 2019). For example, the response of dendritic cells and macrophages to bacterial infection has been shown to be associated with rapid and active demethylation at thousands of loci, primarily at distal enhancer elements (Pacis, et al. 2015; Pacis, et al. 2019). Although the causal role of some of these epigenetic changes remain to be functionally defined (Martin, et al. 2019; Pacis, et al. 2019), some of them may be long-lasting – potentially irreversible – which might alter the ability of immune cells to respond to secondary immune challenges.
The idea of an epigenetically-driven innate immune memory is increasingly discussed (Netea, et al. 2016; Netea and van der Meer 2017). For example, monocytes stimulated with β-glucan are able to mount faster and stronger gene transcriptional responses upon re-stimulation with non-related immune stimuli (Netea, et al. 2016; Netea and van der Meer 2017). Interestingly, both BCG vaccination (Kaufmann, et al. 2018) and β-glucan (Mitroulis, et al. 2018) are able to epigenetically reprogram stem cells in the bone marrow leading to the generation of differentiated monocytes/macrophages with enhanced pro-inflammatory potential and increased ability to fight against subsequent bacterial infections. Thus, it is tempting to speculate that life-course variation in exposure to certain pathogens might lead to long-lasting epigenetic alterations that ultimately contribute to population variation in innate immune responses.
Population epigenomic studies are, however, still lagging behind, and most of these hypotheses remain to be formally tested. So far, the field of population epigenomics has been limited to the study of DNA methylation variation. These studies have globally shown that both genetic ancestry (Fraser, et al. 2012; Heyn, et al. 2013; Husquin, et al. 2018) and differences in lifestyle (e.g., farming vs hunting and gathering) (Fagny, et al. 2015) are associated with changes in DNA methylation at thousands of CpG sites. Interestingly, the bulk of CpG sites (~70%) showing differential methylation between individuals of African and European ancestry have been found to be associated with methylation quantitative trait loci (or meQTLs), indicating that populations differences in DNA methylation are simply a downstream consequence of divergence at the DNA sequence level. Future work will help to determine the extent to which variation in DNA methylation and other epigenetic marks across individuals have a direct impact on the ability of innate immune cells to response to a pathogenic encounter.
Conclusions and perspectives
Our increased ability to combine classical population genetics tools with immunogenomic approaches is providing unique insights into how natural selection has impacted the evolution and function of the human immune system. Yet, much more work is needed to fully comprehend the role of selection at shaping population variation in immune responses. For example, most population-level studies have focused only on individuals of European or African ancestry. Broadening the characterization of the immune system to a larger array of human populations — particularly neglected human groups historically exposed to different pathogen pressures — is required to capture the full extent of variation in immune responses across the globe. Moreover, one should move beyond the phenotypic characterization of immune responses based on gene expression to include other phenotypes, such as variation in the epigenome as well as more in-depth immune profiling of different cellular populations and their immune activation state based on multi-dimensional proteomic measurements.
From a theoretical standpoint, improved tools are needed for detecting alternative selection regimes, notably polygenic adaptation and adaptive admixture – whether archaic or modern. Limiting the search of selection signals to classical sweeps – as it has been done so far – will probably provide only a glimpse of the actual role played by natural selection to the evolution of the immune system. A perpetual challenge in the field has been the identification of the specific pathogen(s) that have exerted pressure at different time periods. Ancient DNA has the great potential to fill this gap. By analyzing ancient DNA samples corresponding to different time points across major historical events (e.g., the various outbreaks of plague), we might be able to shed light on the host and microbial factors underlying susceptibility to infectious disease and the effects of selection on these factors (Skoglund and Mathieson 2018)
Finally, most studies so far have characterized inter-individual variation in immune responses in isolated cell types. These studies, therefore, fail to capture the critical interactions observed in vivo between the many different immune cell types such as those found in circulating blood or complex tissues (Brodin, et al. 2019). By leveraging new technological advances in single-cell sequencing, we will be able to generate a more realistic landscape of the diversity of immune responses across human groups, what cell types contribute the most to such variation, and what role natural selection has had to the diversification of immune responses in humans.
Acknowledgements
This work has been supported by grants NIH R01-GM115656 and R01-GM134376 to L.B.B, and by the Institut Pasteur, the French Government’s Investissement d’Avenir program, Laboratoires d’Excellence “Integrative Biology of Emerging Infectious Diseases” (ANR-10-LABX-62-IBEID) and “Milieu Intérieur” (ANR-10-LABX-69-01), and the Fondation pour la Recherche Médicale (Equipe FRM DEQ20180339214) to L.Q.-M. We thank Maxime Rotival for help with Figure 1.
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest
References
- Abi-Rached L, Jobin MJ, Kulkarni S, McWhinnie A, Dalva K, Gragert L, Babrzadeh F, Gharizadeh B, Luo M, Plummer FA, et al. 2011. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science 334:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman H, Usen S, Jallow M, Sisay-Joof F, Pinder M, Kwiatkowski DP. 2005. A comparison of case-control and family-based association methods: the example of sickle-cell and malaria. Ann Hum Genet 69:559–565. [DOI] [PubMed] [Google Scholar]
- Aguirre-Gamboa R, Joosten I, Urbano PCM, van der Molen RG, van Rijssen E, van Cranenbroek B, Oosting M, Smeekens S, Jaeger M, Zorro M, et al. 2016. Differential Effects of Environmental and Genetic Factors on T and B Cell Immune Traits. Cell Rep 17:2474–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison AC. 1954. Protection afforded by sickle-cell trait against subtertian malareal infection. Br Med J 1:290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RM, May RM. 1991. Infectious Diseases of Humans: Dynamics and Control.
- Andres AM, Hubisz MJ, Indap A, Torgerson DG, Degenhardt JD, Boyko AR, Gutenkunst RN, White TJ, Green ED, Bustamante CD, et al. 2009. Targets of balancing selection in the human genome. Mol Biol Evol 26:2755–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker OB, Aguirre-Gamboa R, Sanna S, Oosting M, Smeekens SP, Jaeger M, Zorro M, Vosa U, Withoff S, Netea-Maier RT, et al. 2018. Integration of multi-omics data and deep phenotyping enables prediction of cytokine responses. Nat Immunol 19:776–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkhash AV, Perelygin AA, Babenko VN, Myasnikova NG, Pilipenko PI, Romaschenko AG, Voevoda MI, Brinton MA. 2010. Variability in the 2’−5’-oligoadenylate synthetase gene cluster is associated with human predisposition to tick-borne encephalitis virus-induced disease. J Infect Dis 202:1813–1818. [DOI] [PubMed] [Google Scholar]
- Barreiro LB, Ben-Ali M, Quach H, Laval G, Patin E, Pickrell JK, Bouchier C, Tichit M, Neyrolles O, Gicquel B, et al. 2009. Evolutionary dynamics of human Toll-like receptors and their different contributions to host defense. PLoS Genet 5:e1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro LB, Quintana-Murci L. 2010. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat Rev Genet 11:17–30. [DOI] [PubMed] [Google Scholar]
- Berg JJ, Coop G. 2014. A population genetic signal of polygenic adaptation. PLoS Genet 10:e1004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisson-Dupuis S, Ramirez-Alejo N, Li Z, Patin E, Rao G, Kerner G, Lim CK, Krementsov DN, Hernandez N, Ma CS, et al. 2018. Tuberculosis and impaired IL-23-dependent IFN-gamma immunity in humans homozygous for a common TYK2 missense variant. Sci Immunol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidwood RJ. 1960. The agricultural revolution. Scientific American 203:130–152. [PubMed] [Google Scholar]
- Brinkworth JF, Barreiro LB. 2014. The contribution of natural selection to present-day susceptibility to chronic inflammatory and autoimmune disease. Curr Opin Immunol 31:66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodin P, Duffy D, Quintana-Murci L. 2019. A Call for Blood-In Human Immunology. Immunity 50:1335–1336. [DOI] [PubMed] [Google Scholar]
- Busby GB, Christ R, Band G, Leffler EM, Le QS, Rockett K, Kwiatkowski D, Spencer C. 2017. Inferring adaptive gene-flow in recent African history. bioRxiv 10.1101/205252 [DOI] [Google Scholar]
- Casanova JL, Abel L. 2013. The genetic theory of infectious diseases: a brief history and selected illustrations. Annu Rev Genomics Hum Genet 14:215–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova JL, Abel L. 2018. Human genetics of infectious diseases: Unique insights into immunological redundancy. Semin Immunol 36:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannemann M, Andres AM, Kelso J. 2016. Introgression of Neandertal- and Denisovan-like Haplotypes Contributes to Adaptive Variation in Human Toll-like Receptors. Am J Hum Genet 98:22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannemann M, Racimo F. 2018. Something old, something borrowed: admixture and adaptation in human evolution. Curr Opin Genet Dev 53:1–8. [DOI] [PubMed] [Google Scholar]
- Daub JT, Hofer T, Cutivet E, Dupanloup I, Quintana-Murci L, Robinson-Rechavi M, Excoffier L. 2013. Evidence for polygenic adaptation to pathogens in the human genome. Mol Biol Evol 30:1544–1558. [DOI] [PubMed] [Google Scholar]
- DeGiorgio M, Lohmueller KE, Nielsen R. 2014. A model-based approach for identifying signatures of ancient balancing selection in genetic data. PLoS Genet 10:e1004561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschamps M, Laval G, Fagny M, Itan Y, Abel L, Casanova JL, Patin E, Quintana-Murci L. 2016. Genomic Signatures of Selective Pressures and Introgression from Archaic Hominins at Human Innate Immunity Genes. Am J Hum Genet 98:5–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond J 2002. Evolution, consequences and future of plant and animal domestication. Nature 418:700–707. [DOI] [PubMed] [Google Scholar]
- Diamond J, Bellwood P. 2003. Farmers and their languages: the first expansions. Science 300:597–603. [DOI] [PubMed] [Google Scholar]
- El Awady MK, Anany MA, Esmat G, Zayed N, Tabll AA, Helmy A, El Zayady AR, Abdalla MS, Sharada HM, El Raziky M, et al. 2011. Single nucleotide polymorphism at exon 7 splice acceptor site of OAS1 gene determines response of hepatitis C virus patients to interferon therapy. J Gastroenterol Hepatol 26:843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enard D, Petrov DA. 2018. Evidence that RNA Viruses Drove Adaptive Introgression between Neanderthals and Modern Humans. Cell 175:360–371 e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagny M, Patin E, MacIsaac JL, Rotival M, Flutre T, Jones MJ, Siddle KJ, Quach H, Harmant C, McEwen LM, et al. 2015. The epigenomic landscape of African rainforest hunter-gatherers and farmers. Nat Commun 6:10047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan S, Hansen ME, Lo Y, Tishkoff SA. 2016. Going global by adapting local: A review of recent human adaptation. Science 354:54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field Y, Boyle EA, Telis N, Gao Z, Gaulton KJ, Golan D, Yengo L, Rocheleau G, Froguel P, McCarthy MI, et al. 2016. Detection of human adaptation during the past 2000 years. Science 354:760–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB, Lam LL, Neumann SM, Kobor MS. 2012. Population-specificity of human DNA methylation. Genome Biol 13:R8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, Sironi M. 2014. Human genome variability, natural selection and infectious diseases. Curr Opin Immunol 30C:9–16. [DOI] [PubMed] [Google Scholar]
- Furuse Y, Suzuki A, Oshitani H. 2010. Origin of measles virus: divergence from rinderpest virus between the 11 th and 12 th centuries. Virology journal 7:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gignoux CR, Henn BM, Mountain JL. 2011. Rapid, global demographic expansions after the origins of agriculture. Proc Natl Acad Sci U S A 108:6044–6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittelman RM, Schraiber JG, Vernot B, Mikacenic C, Wurfel MM, Akey JM. 2016. Archaic Hominin Admixture Facilitated Adaptation to Out-of-Africa Environments. Curr Biol 26:3375–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez JP, Nakoune E, Slenczka W, Vidal P, Morvan JM. 2000. Ebola and Marburg virus antibody prevalence in selected populations of the Central African Republic. Microbes Infect 2:39–44. [DOI] [PubMed] [Google Scholar]
- Gouy A, Daub JT, Excoffier L. 2017. Detecting gene subnetworks under selection in biological pathways. Nucleic Acids Res 45:e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SR, Andersen KG, Shlyakhter I, Tabrizi S, Winnicki S, Yen A, Park DJ, Griesemer D, Karlsson EK, Wong SH, et al. 2013. Identifying recent adaptations in large-scale genomic data. Cell 152:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris DR. 1967. New Light on Plant Domestication and Origins of Agriculture - Review. Geographical Review 57:90–107. [Google Scholar]
- Harrison GF, Sanz J, Boulais J, Mina MJ, Grenier JC, Leng Y, Dumaine A, Yotova V, Bergey CM, Nsobya SL, et al. 2019. Natural selection contributed to immunological differences between hunter-gatherers and agriculturalists. Nat Ecol Evol 3:1253–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellenthal G, Busby GB, Band G, Wilson JF, Capelli C, Falush D, Myers S. 2014. A genetic atlas of human admixture history. Science 343:747–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn H, Moran S, Hernando-Herraez I, Sayols S, Gomez A, Sandoval J, Monk D, Hata K, Marques-Bonet T, Wang L, et al. 2013. DNA methylation contributes to natural human variation. Genome Res 23:1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson JA, Pickrell JK, Pearson LN, Quillen EE, Prista A, Rocha J, Soodyall H, Shriver MD, Perry GH. 2014. Natural selection for the Duffy-null allele in the recently admixed people of Madagascar. Proc Biol Sci 281:20140930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husquin LT, Rotival M, Fagny M, Quach H, Zidane N, McEwen LM, MacIsaac JL, Kobor MS, Aschard H, Patin E, et al. 2018. Exploring the genetic basis of human population differences in DNA methylation and their causal impact on immune gene regulation. Genome Biol 19:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiyama K, Chen T, Wang X, Yan X, Kim BS, Tanaka S, Ndiaye-Lobry D, Deng Y, Zou Y, Zheng P, et al. 2015. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 42:613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ED, Gonzalez JP, Georges A. 1993. Filovirus activity among selected ethnic groups inhabiting the tropical forest of equatorial Africa. Trans R Soc Trop Med Hyg 87:536–538. [DOI] [PubMed] [Google Scholar]
- Karlsson EK, Kwiatkowski DP, Sabeti PC. 2014. Natural selection and infectious disease in human populations. Nat Rev Genet 15:379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonca LE, Pacis A, Tzelepis F, Pernet E, Dumaine A, Grenier JC, et al. 2018. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 172:176–190 e119. [DOI] [PubMed] [Google Scholar]
- Key FM, Teixeira JC, de Filippo C, Andres AM. 2014. Advantageous diversity maintained by balancing selection in humans. Curr Opin Genet Dev 29C:45–51. [DOI] [PubMed] [Google Scholar]
- Klein J, Sato A, Nikolaidis N. 2007. MHC, TSP, and the origin of species: from immunogenetics to evolutionary genetics. Annu Rev Genet 41:281–304. [DOI] [PubMed] [Google Scholar]
- Kwon YC, Kang JI, Hwang SB, Ahn BY. 2013. The ribonuclease L-dependent antiviral roles of human 2’,5’-oligoadenylate synthetase family members against hepatitis C virus. FEBS Lett 587:156–164. [DOI] [PubMed] [Google Scholar]
- Laso-Jadart R, Harmant C, Quach H, Zidane N, Tyler-Smith C, Mehdi Q, Ayub Q, Quintana-Murci L, Patin E. 2017. The Genetic Legacy of the Indian Ocean Slave Trade: Recent Admixture and Post-admixture Selection in the Makranis of Pakistan. Am J Hum Genet 101:977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laval G, Peyregne S, Zidane N, Harmant C, Renaud F, Patin E, Prugnolle F, Quintana-Murci L. 2019. Recent Adaptive Acquisition by Rainforest Hunter-Gatherers of the Late Pleistocene Sickle-Cell Mutation Suggests Past Ecological Differences in Exposure to Malaria in Africa. Am J Hum Genet in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor DA, Ward FE, Ennis PD, Jackson AP, Parham P. 1988. HLA-A and B polymorphisms predate the divergence of humans and chimpanzees. Nature 335:268–271. [DOI] [PubMed] [Google Scholar]
- Leffler EM, Gao Z, Pfeifer S, Segurel L, Auton A, Venn O, Bowden R, Bontrop R, Wall JD, Sella G, et al. 2013. Multiple instances of ancient balancing selection shared between humans and chimpanzees. Science 339:1578–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Oosting M, Smeekens SP, Jaeger M, Aguirre-Gamboa R, Le KTT, Deelen P, Ricano-Ponce I, Schoffelen T, Jansen AFM, et al. 2016. A Functional Genomics Approach to Understand Variation in Cytokine Production in Humans. Cell 167:1099–1110 e1014. [DOI] [PubMed] [Google Scholar]
- Lim JK, Lisco A, McDermott DH, Huynh L, Ward JM, Johnson B, Johnson H, Pape J, Foster GA, Krysztof D, et al. 2009. Genetic variation in OAS1 is a risk factor for initial infection with West Nile virus in man. PLoS Pathog 5:e1000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindo J, Huerta-Sanchez E, Nakagome S, Rasmussen M, Petzelt B, Mitchell J, Cybulski JS, Willerslev E, DeGiorgio M, Malhi RS. 2016. A time transect of exomes from a Native American population before and after European contact. Nat Commun 7:13175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez M, Choin J, Sikora M, Siddle K, Harmant C, Costa HA, Silvert M, Mouguiama-Daouda P, Hombert JM, Froment A, et al. 2019. Genomic Evidence for Local Adaptation of Hunter-Gatherers to the African Rainforest. Curr Biol 29:2926–2935 e2924. [DOI] [PubMed] [Google Scholar]
- Manry J, Laval G, Patin E, Fornarino S, Itan Y, Fumagalli M, Sironi M, Tichit M, Bouchier C, Casanova JL, et al. 2011. Evolutionary genetic dissection of human interferons. J Exp Med 208:2747–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus JH, Novembre J. 2017. Visualizing the geography of genetic variants. Bioinformatics 33:594–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr AK, MacIsaac JL, Jiang R, Airo AM, Kobor MS, McMaster WR. 2014. Leishmania donovani infection causes distinct epigenetic DNA methylation changes in host macrophages. PLoS Pathog 10:e1004419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BJE, Brind’Amour J, Jensen KN, Kuzmin A, Liu ZC, Lorincz M, Howe LJ. 2019. The majority of histone acetylation is a consequence of transcription. bioRxiv. [Google Scholar]
- Mathieson I, Lazaridis I, Rohland N, Mallick S, Patterson N, Roodenberg SA, Harney E, Stewardson K, Fernandes D, Novak M, et al. 2015. Genome-wide patterns of selection in 230 ancient Eurasians. Nature 528:499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthijnssens J, Ciarlet M, Heiman E, Arijs I, Delbeke T, McDonald SM, Palombo EA, Iturriza-Gómara M, Maes P, Patton JT. 2008. Full genome-based classification of rotaviruses reveals a common origin between human Wa-Like and porcine rotavirus strains and human DS-1-like and bovine rotavirus strains. Journal of virology 82:3204–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill WH. 1989. Plagues and peoples. New York: Anchor Books. [Google Scholar]
- Mendez FL, Watkins JC, Hammer MF. 2012. A haplotype at STAT2 Introgressed from neanderthals and serves as a candidate of positive selection in Papua New Guinea. Am J Hum Genet 91:265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez FL, Watkins JC, Hammer MF. 2013. Neandertal origin of genetic variation at the cluster of OAS immunity genes. Mol Biol Evol 30:798–801. [DOI] [PubMed] [Google Scholar]
- Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, Eugster A, Troullinaki M, Palladini A, Kourtzelis I, et al. 2018. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 172:147–161 e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelec Y, Sanz J, Baharian G, Szpiech ZA, Pacis A, Dumaine A, Grenier JC, Freiman A, Sams AJ, Hebert S, et al. 2016. Genetic Ancestry and Natural Selection Drive Population Differences in Immune Responses to Pathogens. Cell 167:657–669 e621. [DOI] [PubMed] [Google Scholar]
- Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, O’Neill LA, Xavier RJ. 2016. Trained immunity: A program of innate immune memory in health and disease. Science 352:aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, van der Meer JW. 2017. Trained Immunity: An Ancient Way of Remembering. Cell Host Microbe 21:297–300. [DOI] [PubMed] [Google Scholar]
- Olalde I, Allentoft ME, Sanchez-Quinto F, Santpere G, Chiang CW, DeGiorgio M, Prado-Martinez J, Rodriguez JA, Rasmussen S, Quilez J, et al. 2014. Derived immune and ancestral pigmentation alleles in a 7,000-year-old Mesolithic European. Nature 507:225–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto TD, Gilabert A, Crellen T, Böhme U, Arnathau C, Sanders M, Oyola SO, Okouga AP, Boundenga L, Willaume E. 2018. Genomes of all known members of a Plasmodium subgenus reveal paths to virulent human malaria. Nature microbiology 3:687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owers KA, Sjodin P, Schlebusch CM, Skoglund P, Soodyall H, Jakobsson M. 2017. Adaptation to infectious disease exposure in indigenous Southern African populations. Proc Biol Sci 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacis A, Mailhot-Leonard F, Tailleux L, Randolph HE, Yotova V, Dumaine A, Grenier JC, Barreiro LB. 2019. Gene activation precedes DNA demethylation in response to infection in human dendritic cells. Proc Natl Acad Sci U S A 116:6938–6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacis A, Tailleux L, Morin AM, Lambourne J, MacIsaac JL, Yotova V, Dumaine A, Danckaert A, Luca F, Grenier JC, et al. 2015. Bacterial infection remodels the DNA methylation landscape of human dendritic cells. Genome Res 25:1801–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patin E, Lopez M, Grollemund R, Verdu P, Harmant C, Quach H, Laval G, Perry GH, Barreiro LB, Froment A, et al. 2017. Dispersals and genetic adaptation of Bantu-speaking populations in Africa and North America. Science 356:543–546. [DOI] [PubMed] [Google Scholar]
- Patin E, Quintana-Murci L. 2018. The demographic and adaptive history of central African hunter-gatherers and farmers. Curr Opin Genet Dev 53:90–97. [DOI] [PubMed] [Google Scholar]
- Petr M, Paabo S, Kelso J, Vernot B. 2019. Limits of long-term selection against Neandertal introgression. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piasecka B, Duffy D, Urrutia A, Quach H, Patin E, Posseme C, Bergstedt J, Charbit B, Rouilly V, MacPherson CR, et al. 2018. Distinctive roles of age, sex, and genetics in shaping transcriptional variation of human immune responses to microbial challenges. Proc Natl Acad Sci U S A 115:E488–E497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierron D, Heiske M, Razafindrazaka H, Pereda-Loth V, Sanchez J, Alva O, Arachiche A, Boland A, Olaso R, Deleuze JF, et al. 2018. Strong selection during the last millennium for African ancestry in the admixed population of Madagascar. Nat Commun 9:932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Pickrell JK, Coop G. 2010. The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Curr Biol 20:R208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach H, Rotival M, Pothlichet J, Loh YE, Dannemann M, Zidane N, Laval G, Patin E, Harmant C, Lopez M, et al. 2016. Genetic Adaptation and Neandertal Admixture Shaped the Immune System of Human Populations. Cell 167:643–656 e617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana-Murci L 2019. Human Immunology through the Lens of Evolutionary Genetics. Cell 177:184–199. [DOI] [PubMed] [Google Scholar]
- Quintana-Murci L, Alcais A, Abel L, Casanova JL. 2007. Immunology in natura: clinical, epidemiological and evolutionary genetics of infectious diseases. Nat Immunol 8:1165–1171. [DOI] [PubMed] [Google Scholar]
- Quintana-Murci L, Clark AG. 2013. Population genetic tools for dissecting innate immunity in humans. Nat Rev Immunol 13:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racimo F, Berg JJ, Pickrell JK. 2018. Detecting Polygenic Adaptation in Admixture Graphs. Genetics 208:1565–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racimo F, Marnetto D, Huerta-Sanchez E. 2017. Signatures of Archaic Adaptive Introgression in Present-Day Human Populations. Mol Biol Evol 34:296–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishishwar L, Conley AB, Wigington CH, Wang L, Valderrama-Aguirre A, Jordan IK. 2015. Ancestry, admixture and fitness in Colombian genomes. Sci Rep 5:12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sams AJ, Dumaine A, Nedelec Y, Yotova V, Alfieri C, Tanner JE, Messer PW, Barreiro LB. 2016. Adaptively introgressed Neandertal haplotype at the OAS locus functionally impacts innate immune responses in humans. Genome Biol 17:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankararaman S, Mallick S, Dannemann M, Prufer K, Kelso J, Paabo S, Patterson N, Reich D. 2014. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507:354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankararaman S, Mallick S, Patterson N, Reich D. 2016. The Combined Landscape of Denisovan and Neanderthal Ancestry in Present-Day Humans. Curr Biol 26:1241–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz J, Randolph HE, Barreiro LB. 2018. Genetic and evolutionary determinants of human population variation in immune responses. Curr Opin Genet Dev 53:28–35. [DOI] [PubMed] [Google Scholar]
- Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA, Horst RT, Jansen T, Jacobs L, Bonder MJ, et al. 2016. Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell 167:1897. [DOI] [PubMed] [Google Scholar]
- Segurel L, Thompson EE, Flutre T, Lovstad J, Venkat A, Margulis SW, Moyse J, Ross S, Gamble K, Sella G, et al. 2012. The ABO blood group is a trans-species polymorphism in primates. Proc Natl Acad Sci U S A 109:18493–18498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siewert KM, Voight BF. 2017. Detecting Long-Term Balancing Selection Using Allele Frequency Correlation. Mol Biol Evol 34:2996–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair SH, Yegnasubramanian S, Dumler JS. 2015. Global DNA methylation changes and differential gene expression in Anaplasma phagocytophilum-infected human neutrophils. Clin Epigenetics 7:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoglund P, Mathieson I. 2018. Ancient Genomics of Modern Humans: The First Decade. Annu Rev Genomics Hum Genet 19:381–404. [DOI] [PubMed] [Google Scholar]
- Sundararaman SA, Plenderleith LJ, Liu W, Loy DE, Learn GH, Li Y, Shaw KS, Ayouba A, Peeters M, Speede S. 2016. Genomes of cryptic chimpanzee Plasmodium species reveal key evolutionary events leading to human malaria. Nature communications 7:11078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Nei M. 2002. Origin and evolution of influenza virus hemagglutinin genes. Molecular biology and evolution 19:501–509. [DOI] [PubMed] [Google Scholar]
- Tang H, Choudhry S, Mei R, Morgan M, Rodriguez-Cintron W, Burchard EG, Risch NJ. 2007. Recent genetic selection in the ancestral admixture of Puerto Ricans. Am J Hum Genet 81:626–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira JC, de Filippo C, Weihmann A, Meneu JR, Racimo F, Dannemann M, Nickel B, Fischer A, Halbwax M, Andre C, et al. 2015. Long-Term Balancing Selection in LAD1 Maintains a Missense Trans-Species Polymorphism in Humans, Chimpanzees, and Bonobos. Mol Biol Evol 32:1186–1196. [DOI] [PubMed] [Google Scholar]
- Thornton R 1997. Aboriginal North American Population and Rates of Decline, ca. a.d. 1500–1901. Curr Anthropol 38:310–315. [Google Scholar]
- Turchin MC, Chiang CW, Palmer CD, Sankararaman S, Reich D, Hirschhorn JN. 2012. Evidence of widespread selection on standing variation in Europe at height-associated SNPs. Nat Genet 44:1015–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasseur E, Boniotto M, Patin E, Laval G, Quach H, Manry J, Crouau-Roy B, Quintana-Murci L. 2012. The evolutionary landscape of cytosolic microbial sensors in humans. Am J Hum Genet 91:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigne JD. 2011. The origins of animal domestication and husbandry: a major change in the history of humanity and the biosphere. C R Biol 334:171–181. [DOI] [PubMed] [Google Scholar]
- Wiencke JK, Butler R, Hsuang G, Eliot M, Kim S, Sepulveda MA, Siegel D, Houseman EA, Kelsey KT. 2016. The DNA methylation profile of activated human natural killer cells. Epigenetics 11:363–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe ND, Dunavan CP, Diamond J. 2007. Origins of major human infectious diseases. Nature 447:279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeder MA. 2008. Domestication and early agriculture in the Mediterranean Basin: Origins, diffusion, and impact. Proc Natl Acad Sci U S A 105:11597–11604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ulm A, Somineni HK, Oh S, Weirauch MT, Zhang HX, Chen X, Lehn MA, Janssen EM, Ji H. 2014. DNA methylation dynamics during ex vivo differentiation and maturation of human dendritic cells. Epigenetics Chromatin 7:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Zhao L, Guan Y. 2016. Strong Selection at MHC in Mexicans since Admixture. PLoS Genet 12:e1005847. [DOI] [PMC free article] [PubMed] [Google Scholar]