

Abstract
L-type amino acid transporter 1 (LAT1), which is encoded by solute carrier transporter 7a5 (Slc7a5), plays a crucial role in amino acid sensing and signaling in specific cell types, contributing to the pathogenesis of cancer and neurological disorders. Amino acid substrates of LAT1 have a beneficial effect on bone health directly and indirectly, suggesting a potential role for LAT1 in bone homeostasis. Here, we identified LAT1 in osteoclasts as important for bone homeostasis. Slc7a5 expression was substantially reduced in osteoclasts in a mouse model of ovariectomy-induced osteoporosis. The osteoclast-specific deletion of Slc7a5 in mice led to osteoclast activation and bone loss in vivo, and Slc7a5 deficiency increased osteoclastogenesis in vitro. Loss of Slc7a5 impaired activation of the mechanistic target of rapamycin complex 1 (mTORC1) pathway in osteoclasts, whereas genetic activation of mTORC1 corrected the enhanced osteoclastogenesis and bone loss in Slc7a5-deficient mice. Last, Slc7a5 deficiency increased the expression of nuclear factor of activated T cells, cytoplasmic 1 (Nfatc1) and the nuclear accumulation of NFATc1, a master regulator of osteoclast function, possibly through the canonical nuclear factor κB pathway and the Akt–glycogen synthase kinase 3β signaling axis, respectively. These findings suggest that the LAT1-mTORC1 axis plays a pivotal role in bone resorption and bone homeostasis by modulating NFATc1 in osteoclasts, thereby providing a molecular connection between amino acid intake and skeletal integrity.
INTRODUCTION
Amino acid transporters are transmembrane proteins that mediate the transfer of amino acids into and out of cells and are implicated in various cellular processes, including gene expression, protein synthesis, energy metabolism, and catabolic and anabolic reactions (1–3). Dysregulation or dysfunction of amino acid transporters contributes to various pathologies, such as cancer, neurodegenerative diseases, chronic kidney diseases, obesity, and diabetes, by inducing an imbalance in intracellular amino acid concentration (3–7). L-type amino acid transporter 1 (LAT1), encoded by solute carrier transporter 7a5 (Slc7a5), is a Na+-independent amino acid transporter that mediates cellular uptake of large neutral amino acids, including the essential amino acids (EAAs) Leu, Ile, and Val (8, 9). LAT1 forms a heterodimer with the heavy chain of 4F2, a cell surface antigen encoded by Slc3a2, through a disulfide bond (8, 10). The intracellular concentration of Leu is an essential signal for promoting the activity of the mechanistic target of rapamycin complex 1 (mTORC1) in specific cell types, and its homeostasis is, at least in part, dependent on the uptake of Leu by LAT1 (11–14). LAT1 is reported to be present in a limited number of normal tissues, including the blood-brain barrier, placenta, and testes; however, genetic studies in mice have revealed functional LAT1 in T cells and skeletal muscle (15–18).
The development and integrity of the skeleton, as well as bone homeostasis, are coregulated by two cell types, bone-forming osteoblasts and bone-resorbing osteoclasts (19, 20). An imbalance in the sophisticated mutual regulation between osteoclasts and osteoblasts leads to pathogenesis and underlies the etiology of certain metabolic bone diseases including osteoporosis and osteopetrosis (21, 22). Adequate dietary protein and amino acid intake are widely acknowledged to be essential for proper skeletal development and bone homeostasis. In addition, amino acid supplementation can modify the progression of skeletal manifestations of certain genetic disorders of the bone such as Coffin-Lowry syndrome (23). Amino acid substrates of LAT1 are suggested to have a beneficial impact on bone health directly and indirectly. For example, EAA supplements that include Leu and Ile increase bone strength through modification of bone mineral density and bone microarchitecture in ovariectomized osteoporotic rats fed a low-protein diet (24). Moreover, higher Leu intake is associated with greater bone mineral density in the spine and the forearm in a human study of female monozygotic twins discordant for amino acid intake (25).
Here, we found that the amino acid transport system involving LAT1 was active in both the osteoclasts and the osteoblasts of mice and that Slc7a5 expression was reduced in osteoclasts in a mouse model of postmenopausal osteoporosis. Mouse genetic studies revealed that Slc7a5 played a crucial role in bone resorption and bone homeostasis through its expression in osteoclasts. Subsequent analyses identified mTORC1 as a critical mediator of LAT1-dependent osteoclastogenesis, bone resorption, and bone homeostasis. Last, we found that the LAT1-mTORC1 axis controlled the nuclear accumulation of nuclear factor of activated T cells, cytoplasmic 1 (NFATc1), a master regulator of osteoclast differentiation, and Nfatc1 expression, likely through the Akt–glycogen synthase kinase 3β (GSK3β) axis and the canonical nuclear factor κB (NF-κB) pathway, respectively. Our results demonstrate that the LAT1-mTORC1 axis may be a pivotal player in bone resorption and bone homeostasis by modulating NFATc1 in osteoclasts, thereby suggesting LAT1 as a novel potential target for metabolic bone diseases.
RESULTS
LATs mediate amino acid uptake in bone cells
We first evaluated whether the functional amino acid transport system involving LATs was operational in osteoblasts and osteoclasts. To that end, we tested whether primary mouse osteoblasts and osteoclasts took up radiolabeled L-3-[125I]iodo-α-methyltyrosine ([125I]IMT), an artificial amino acid derived from the neutral amino acid Tyr (Fig. 1A), because its accumulation is mediated mainly by the LATs (26, 27). We synthesized [125I]IMT at a radiochemical purity of 99% by high-performance liquid chromatography (HPLC). [125I]IMT uptake was significantly higher at 37°C compared to 4°C in both primary osteoblasts and osteoclasts (Fig. 1B). Replacing the sodium chloride (NaCl) in the uptake buffer with choline chloride (ChoCl) reduced the uptake of [125I]IMT at 37°C to about 70% in osteoblasts but not in osteoclasts (Fig. 1, C and D). Moreover, JPH203, a specific inhibitor of LAT1 (28), modestly but significantly reduced [125I]IMT incorporation to about 60% in osteoblasts (Fig. 1E), whereas it reduced uptake to about 10% in osteoclasts (Fig. 1F). These results suggest that both osteoblasts and osteoclasts have a temperature-dependent, Na+-independent, and LAT1-dependent amino acid uptake system, whereas a Na+-dependent and JPH203-insensitive amino acid uptake system may be partially responsible for amino acid uptake in osteoblasts.
Fig. 1. A LAT-dependent amino acid uptake system operates in both osteoclasts and osteoblasts.

(A) Schematic diagram of the procedure for generating [125I]IMT. (B) Primary osteoblasts and osteoclasts cultured from wild-type (WT) mice were incubated with [125I]IMT at 4° or 37°C for 30 min in HBSS buffer. n = 4 cell cultures from different mice. (C to F) Primary cells were incubated with [125I]IMT at 37°C for 30 min in HBSS buffer in which NaCl was replaced with equimolar ChoCl (C and D) or in HBSS buffer containing JPH203 (E and F). n = 4 cell cultures from different mice. Data were analyzed by the two-tailed Student’s t test. *P < 0.05 and **P < 0.01, significantly different from the value obtained in cells incubated at 4°C (B), cells incubated in HBSS buffer with NaCl (C and D), or cells incubated in HBSS buffer with dimethyl sulfoxide (DMSO) (E and F).
Slc7a5 expression in preosteoclasts is reduced in ovariectomized mice
To determine whether LATs are involved in the pathogenesis of metabolic bone disease, we examined the expression of LAT-encoding genes in bone cells in a mouse model of postmenopausal osteoporosis. C57BL6 mice underwent ovariectomy (OVX) or sham operation, and CD11blow/−CX3CR1+Ly6Chi cells (preosteoclasts), which comprise most of the osteoclast precursor activity in the bone marrow (29), and CD45−CD51+Sca1− cells (osteoblasts), which represent a mixture of both immature and mature osteoblasts (30), were subsequently isolated by fluorescence-activated cell sorting (FACS) for mRNA expression analysis (Fig. 2A). Expression of Slc7a5, Slc7a8, Slc3a2, and Slc1a5 was higher in preosteoclasts than in osteoblasts relative to a reference RNA (Actb) (fig. S1). Among the transcripts tested, only Slc7a5 mRNA was significantly reduced in the preosteoclasts of ovariectomized mice compared to sham-operated controls (Fig. 2B). In contrast, no significant alterations in amino acid transporter mRNA abundance were observed in the osteoblasts of ovariectomized mice (Fig. 2C).
Fig. 2. Slc7a5 expression in preosteoclasts is reduced in ovariectomized mice.

(A) C57BL6 mice were subjected to ovariectomy (OVX) or sham operation and subsequent isolation of CD11blow/−CX3CR1+Ly6Chi cells (preosteoclasts) and CD45−CD51+Sca1− cells (osteoblasts) by flow cytometry. (B and C) Quantification of mRNAs encoding amino acid transporters in preosteoclasts (B) and osteoblasts (C) by qPCR. n = 5 mice. All data were analyzed by the two-tailed Student’s t test. **P < 0.01, significantly different from the value obtained for sham-operated mice. U.D., under detectable values.
Slc7a5 deficiency in osteoclasts causes bone loss and osteoclast activation
Our data revealed that LAT1 was produced and functional in osteoclasts and possibly associated with the pathogenesis of postmenopausal osteoporosis (Figs. 1 and 2); however, the physiological importance of LAT1 in the maintenance of bone homeostasis in vivo has not been demonstrated. Given that global Slc7a5 knockout in mice is lethal between embryonic days 9.5 and 11.5 (E9.5 and E11.5) (16, 31), we generated osteoclast-specific Slc7a5 knockout mice by crossing Slc7a5-floxed mice with Tnfrsf11a-Cre mice or Lyz2-Cre mice to determine whether LAT1 plays a role in bone homeostasis through Slc7a5 expression in osteoclasts (Fig. 3A). Slc7a5 mRNA abundance was decreased in osteoclasts cultured from mice lacking Slc7a5 in Tnfrsf11a-expressing cells, hereafter referred to as Tnfrsf11a-Cre;Slc7a5fl/fl mice, compared with that in the control mice not expressing Cre (Fig. 3B and fig. S2A). The bone volume–to–tissue volume (BV/TV) ratio of Tnfrsf11a-Cre;Slc7a5fl/fl mice was significantly lower than that of the control mice in both the femurs (Fig. 3, C and D) and the vertebrae (Fig. 3, E and F). Bone histomorphometric analyses revealed that bone formation indices, such as number of osteoblasts/tissue area (N.Ob/T.Ar) and bone formation rate (BFR), were comparable between Tnfrsf11a-Cre;Slc7a5fl/fl and control mice (Fig. 3, G and H), whereas the osteoclast surface/bone surface (Oc.S/BS), an index of osteoclastic function, was significantly increased in Tnfrsf11a-Cre;Slc7a5fl/fl mice (Fig. 3I). Moreover, serum biochemical analyses revealed significantly higher circulating concentrations of the bone resorption marker CTx but a normal abundance of the bone formation marker P1NP in Tnfrsf11a-Cre;Slc7a5fl/fl mice (Fig. 3, J and K).
Fig. 3. Slc7a5 in osteoclasts is essential for bone resorption and bone homeostasis in vivo.
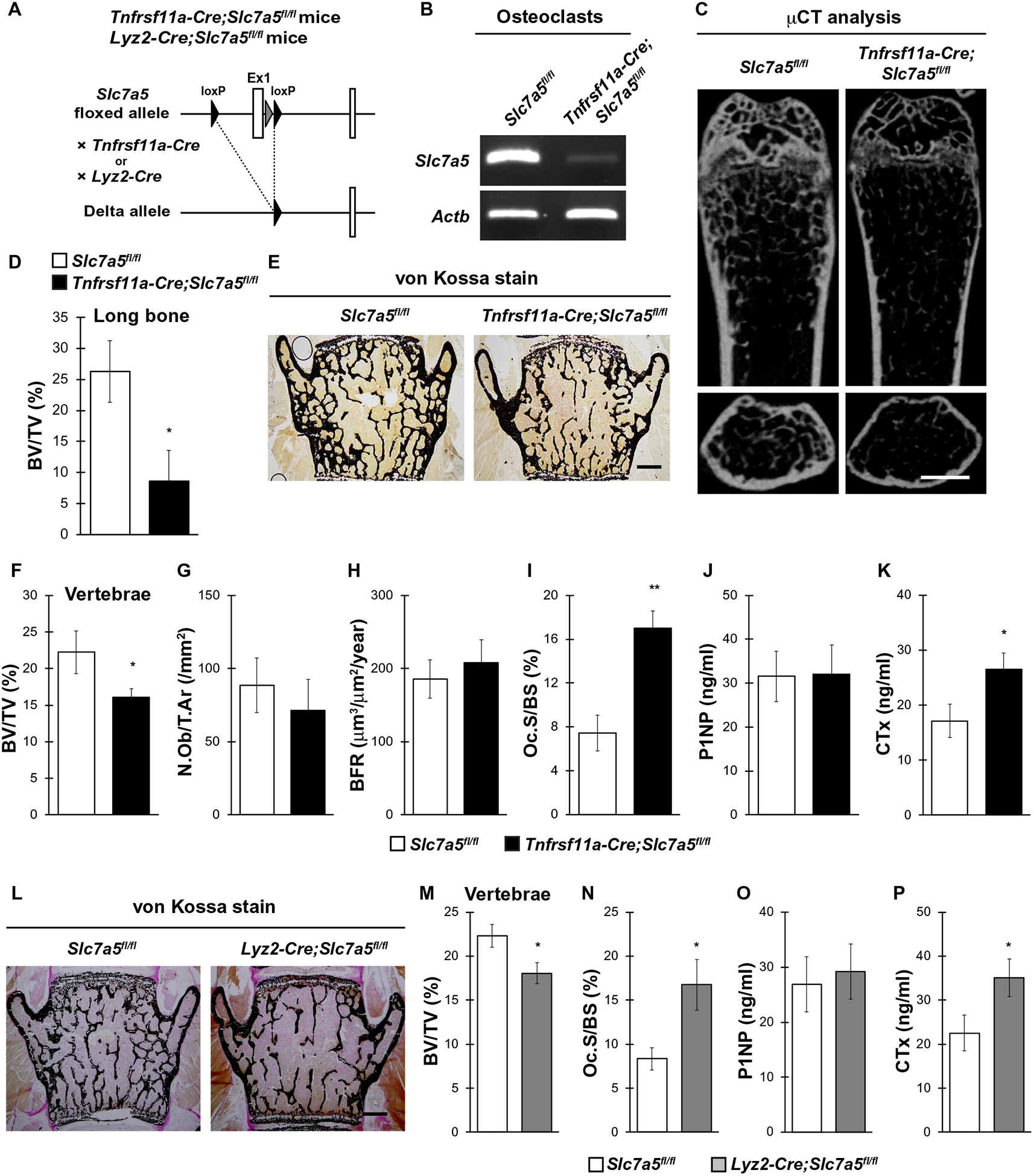
(A) Schematic diagram of the generation of osteoclast-specific Slc7a5 knockout mice using the osteoclast-specific Cre drivers Tnfrs11a and Lyz2. (B) Slc7a5 expression in cultured osteoclasts from Slc7a5fl/fl (control) and Tnfrsf11a-Cre;Slc7a5fl/fl mice as determined by PCR. Actb is a control. n = 3 mice per genotype. (C) μCT analysis and (D) bone volume–to–tissue volume (BV/TV) ratio measurements of femurs from Slc7a5fl/fl and Tnfrsf11a-Cre;Slc7a5fl/fl mice. (E) von Kossa staining, (F) BV/TV, (G) number of osteoblasts/tissue area (N.Ob/T.Ar), (H) bone formation rate (BFR), and (I) osteoclast surface/bone surface (Oc.S/BS) measurements from vertebrae and (J) P1NP and (K) CTx concentrations in serum of Slc7a5fl/fl and Tnfrsf11a-Cre;Slc7a5fl/fl mice at 12 weeks of age. n = 5 to 7 Slc7a5fl/fl mice; n = 6 to 9 Tnfrsf11a-Cre;Slc7a5fl/fl mice. (L) von Kossa staining, (M) BV/TV, and (N) Oc.S/BS of vertebrae and (O) P1NP and (P) CTx concentrations in serum from Slc7a5fl/fl and Lyz2-Cre;Slc7a5fl/fl mice at 12 weeks of age. n = 7 Slc7a5fl/fl mice; n = 8 Lyz2-Cre;Slc7a5fl/fl mice. All data were analyzed by the two-tailed Student’s t test. *P < 0.05 and **P < 0.01, significantly different from the value obtained for control mice. Scale bars, 1 mm (C) and 200 μm (E and L).
We used Lyz2-Cre transgenic mice to target Slc7a5 deletion to osteoclasts. Similar to Tnfrsf11a-Cre;Slc7a5fl/fl mice, Lyz2-Cre;Slc7a5fl/fl mice showed not only a significantly lower BV/TV ratio but also significantly higher osteoclast and bone resorption indices compared with those in control mice (Fig. 3, L to P). Collectively, these results suggest that LAT1 in osteoclasts might have an important role in bone resorption and bone homeostasis under physiological conditions in vivo.
Slc7a5 deficiency in osteoblasts does not affect the bone phenotype
We showed that the LAT1-dependent amino acid uptake system possibly could be functional in osteoblasts in addition to osteoclasts (Fig. 1). To determine the role of LAT1 in osteoblasts in bone homeostasis, we generated osteoblast-specific Slc7a5 knockout mice by crossing Slc7a5-floxed mice with Osterix (Osx)–Cre mice, also known as Osx-GFP::Cre, which expresses the GFP::Cre fusion protein (Fig. 4A). Slc7a5 expression was markedly decreased in the green fluorescent protein (GFP)–positive cells of osteoblast-specific Slc7a5 knockout mice, termed Osx-Cre;Slc7a5fl/fl mice (Fig. 4B and fig. S2B). The BV/TV ratio in femurs of Osx-Cre;Slc7a5fl/fl mice was indistin-guishable from that of control mice (Fig. 4, C to F). Bone histomorphometric analyses revealed that the bone resorption index (Oc.S/BS) and bone formation indices (BFR and N.Ob/T.Ar) were comparable between Osx-Cre;Slc7a5fl/fl and control mice (Fig. 4, G to K). These results indicate that LAT1 in osteoblasts is likely not required for bone formation and homeostasis under physiological conditions in vivo.
Fig. 4. Slc7a5 in osteoblasts is dispensable for bone formation and bone homeostasis in vivo.

(A) Schematic diagram of the generation of osteoblast-specific Slc7a5 knockout mice. (B) Slc7a5 expression in the GFP-positive cells of control and Osx-Cre;Slc7a5fl/fl mice as determined by PCR. n = 3 mice per genotype. (C) μCT analysis, (D) BV/TV ratio as determined by μCT, (E) von Kossa staining, (F) BV/TV as determined by von Kossa staining, (G) TRAP staining, (H) Oc.S/BS ratio, (I) calcein labeling, (J) BFR, and (K) N.Ob/T.Ar of femurs from control and Osx-Cre;Slc7a5fl/fl mice at 12 weeks of age. n = 6 Osx-Cre mice; n = 8 Osx-Cre;Slc7a5fl/fl mice. All data were analyzed by the two-tailed Student’s t test. The double-headed white arrow in (I) indicates the distance between calcein double labeling. Scale bars, 1 mm (C and E), 100 μm (G), and 10 μm (I).
Slc7a5 deficiency stimulates osteoclastogenesis in vitro
We evaluated whether Slc7a5 affected osteoclastogenesis in vitro. We infected bone marrow macrophages (BMMs) from Slc7a5fl/fl mice with a retrovirus expressing Cre recombinase to induce Slc7a5 knockout and differentiated the cells into osteoclasts by treating them with receptor activator of NF-κB ligand (RANKL). The cells were then subjected to tartrate-resistant acid phosphatase (TRAP) staining and pit formation assays (Fig. 5A). The number of TRAP-positive multinucleated cells and the area of pit formation were both significantly increased in Slc7a5-deleted BMMs, indicating an enhancement of osteoclast differentiation and function (Fig. 5, B and C). Moreover, the expression of the osteoclast differentiation and fusion markers, Nfatc1, transmembrane 7 superfamily member 4 (Dcstamp), cathepsin K (Ctsk), and matrix metalloproteinase 9 (Mmp9), was markedly increased in Slc7a5-deleted osteoclasts compared to controls (Fig. 5D). We further determined the effect of Slc7a5 deletion on proliferation and survival of osteoclasts. Flow cytometric measurements after BrdU incorporation revealed that the number of cells in S phase was comparable between control cells and Slc7a5-deficient BMMs, irrespective of RANKL treatment (Fig. 5E). Further-more, Slc7a5 deficiency did not significantly affect the number of apoptotic cells in BMM cultures irrespective of RANKL treatment (Fig. 5F). These results show that LAT1 negatively regulates osteoclastogenesis in vitro without affecting cell proliferation or cell survival.
Fig. 5. Slc7a5 deficiency stimulates osteoclastogenesis in vitro.
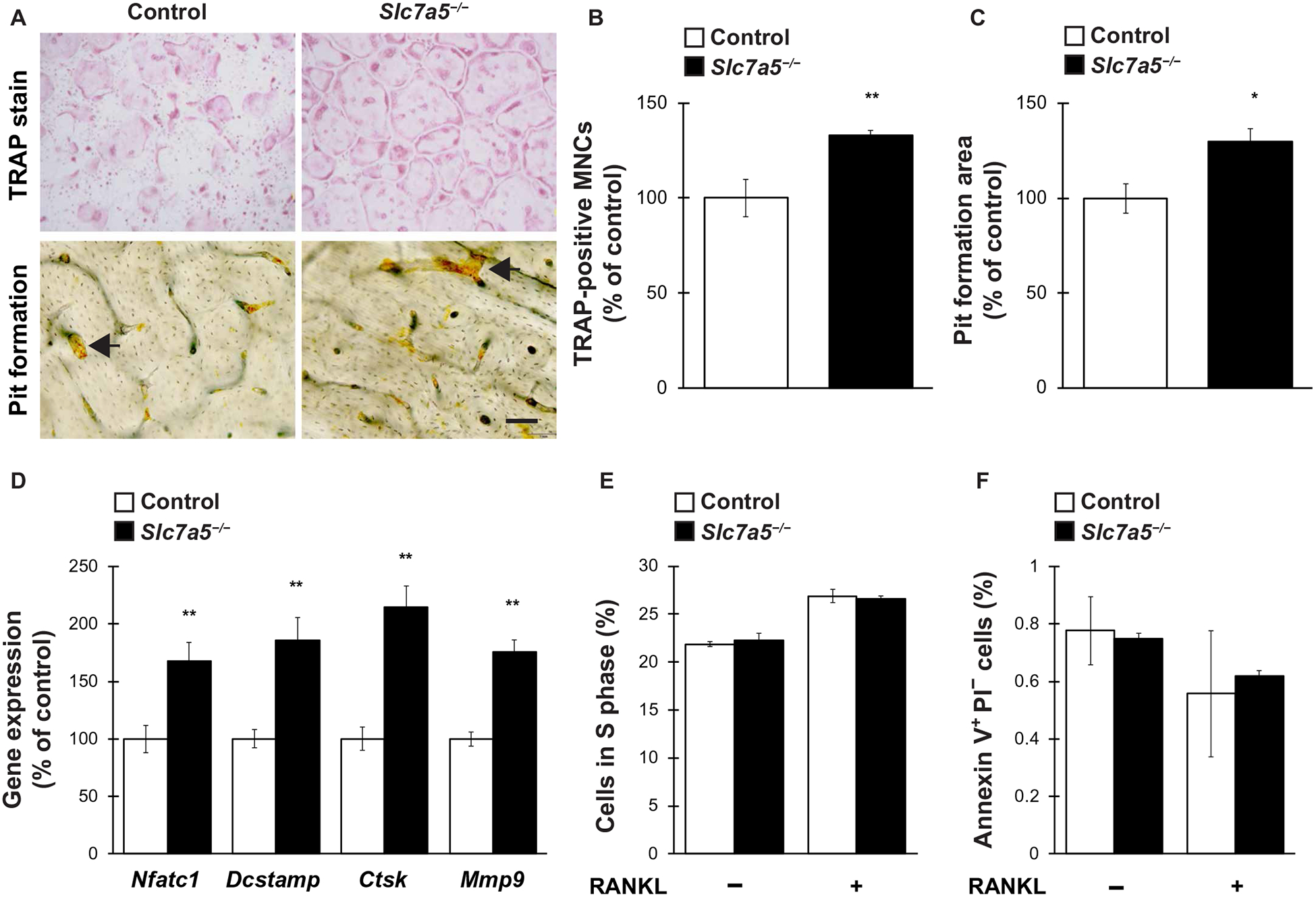
BMMs from Slc7a5fl/fl mice were retrovirally infected with Cre recombinase and subsequently stimulated with RANKL to induce osteoclast differentiation. Cells were then analyzed for (A to C) the number of TRAP-positive multinucleated cells MNCs and pit formation area, (D) expression of osteoclast marker genes, (E) cell proliferation ratio, and (F) cell survival. n = 4 to 6 mice per genotype. Data were analyzed by the two-tailed Student’s t test (B to D) or the two-way ANOVA with Bonferroni and/or Dunnett post hoc test (E and F). *P < 0.05 and **P < 0.01, significantly different from the value obtained in control cells. Arrows in (A) indicate representative resorption pits. Scale bar, 50 μm.
mTORC1 activation corrects bone loss and osteoclast activation in osteoclast-specific Slc7a5 knockout mice
LAT1 mediates the transport of large neutral amino acids, including Leu, in many cell types, leading to activation of the mTORC1 pathway (11, 12), and mTORC1 in osteoclasts is essential for maintaining bone homeostasis and bone resorption (32, 33). Thus, we next examined whether the mTORC1 pathway might be implicated in LAT1-dependent negative regulation of osteoclastogenesis, bone homeostasis, and bone resorption.
Phosphorylation of p70S6 kinase (p70S6K) and 4E-BP1, both of which are downstream effectors of mTORC1, was significantly decreased in Slc7a5-deficient BMMs, irrespective of RANKL treatment, indicating reduced mTORC1 activity in osteoclasts (Fig. 6, A to C). Moreover, RANKL-dependent p70S6K phosphorylation tended to decrease in osteoclasts when the cells were deprived of Val, Ile, or Leu (fig. S3), whereas RANKL did not alter [125I]IMT uptake in osteoclasts (fig. S4), indicating that Leu, Val, and Ile were all required for mTORC1 activation by RANKL. In contrast, phosphorylation of eukaryotic initiation factor-2α (eIF2α), which allows the preferential translation of the master regulator of amino acid metabolism activating transcription factor 4 (ATF4), was markedly increased in Slc7a5-deficient cells (fig. S5), indicating the activation of the general amino acid control (GAAC) pathway in osteoclasts.
Fig. 6. mTORC1 activation rescues Slc7a5 deficiency–induced osteoclast activation in vitro and in vivo.

(A to C) BMMs from Slc7a5fl/fl mice were retrovirally infected with Cre recombinase and subsequently stimulated with RANKL, followed by immunoblotting and densitometric quantification of phosphorylation of the indicated downstream effectors of mTORC1 signaling. β-Actin is a loading control. n = 4 mice per genotype. (D and E) BMMs from Slc7a5fl/fl mice were retrovirally infected with Cre recombinase and constitutively active (CA) Rheb, an upstream activator of mTORC1, and subsequently treated with RANKL, followed by determination of the number of TRAP-positive multinucleated cells. n = 5 mice per genotype. E.V., empty vector “pMX-Control”. (F) μCT analysis and (G) BV/TV ratio and Oc.S/BS ratio of femurs and P1NP and CTx concentrations in serum of Tnfrsf11a-Cre;Slc7a5fl/fl;Tsc1fl/+ mice at 12 weeks of age. n = 6 control mice (Slc7a5fl/fl;Tsc1fl/+ mice or Slc7a5fl/+;Tsc1fl/+ mice) and Tnfrsf11a-Cre;Slc7a5fl/fl mice; n = 7 Tnfrsf11a-Cre;Slc7a5fl/fl;Tsc1fl/+ mice. Data were analyzed by the two-way ANOVA with Bonferroni and/or Dunnett post hoc test (B, C, and E) or the one-way ANOVA with Bonferroni and/or Dunnett post hoc test (G). *P < 0.05 and ** P < 0.01, significantly different from control cells (B, C, and E) or control mice (G). #P < 0.05 and ##P < 0.01, significantly different from the value obtained for Slc7a5-deficient cells infected with control vector (E) or Tnfrsf11a-Cre;Slc7a5fl/fl mice (G). Scale bars, 50 μm (D) and 1 mm (F).
Tuberous sclerosis complex 1 (Tsc1), also known as hamartin, is a critical negative regulator of mTORC1 because it acts as a guano-sine triphosphatase–activating protein for the small G protein Ras homolog enriched in the brain (Rheb) (34, 35). Introduction of a constitutively active form of Rheb, the upstream activator of mTORC1, significantly inhibited the enhanced osteoclastogenesis observed in Slc7a5-deficient BMMs (Fig. 6, D and E). To confirm this finding in vivo, we generated Tnfrsf11a-Cre;Slc7a5fl/fl mice lacking one copy of the floxed Tsc1 allele, hereafter referred to as Tnfrsf11a-Cre;Slc7a5fl/fl; Tsc1fl/+ mice. The low bone mass phenotype of Tnfrsf11a-Cre;Slc7a5fl/fl mice was rescued in Tnfrsf11a-Cre;Slc7a5fl/fl;Tsc1fl/+ mice as a result of the normalization of osteoclast activation (Fig. 6, F and G). In line with our in vitro findings, these results suggest that LAT1, at least in part, negatively regulates bone resorption and bone homeostasis by modulating mTORC1 signaling in osteoclasts in vivo.
Slc7a5 deficiency increases nuclear accumulation of NFATc1 in osteoclasts
To elucidate the potential mechanisms underlying osteoclastogenesis inhibition by the LAT1-mTORC1 axis, we first examined the protein abundance of NFATc1, a master transcription factor required for osteoclastogenesis, in Slc7a5-deficient cells. The whole-cell abundance of NFATc1 was significantly increased in Slc7a5-deficient cells in the presence of RANKL (Fig. 7, A and B), in accordance with the observed increase in Nfatc1 mRNA abundance in Slc7a5-deficient cells (Fig. 5D). Moreover, the nuclear abundance of NFATc1 was also significantly increased in Slc7a5-deficient cells in the presence of RANKL (Fig. 7, A and C), suggesting that Slc7a5 deficiency leads to an increase in the nuclear accumulation of NFATc1, in addition to its transcriptional enhancement, in osteoclasts.
Fig. 7. Nuclear accumulation of NFATc1 increases in Slc7a5-deficient osteoclasts.

BMMs from Slc7a5fl/fl mice were retrovirally infected with Cre recombinase and subsequently stimulated with RANKL, followed by immunoblotting and densitometric analysis of (A to C) NFATc1 abundance in the nucleus and whole-cell extracts and (D to I) phosphorylation of Erk1/2, JNK1/2, and p38. n = 4 mice per genotype. All data were analyzed by the two-way ANOVA with Bonferroni and/or Dunnett post hoc test. **P < 0.01, significantly different from the value obtained in control cells.
Slc7a5 deficiency induces nuclear NFATc1 accumulation through the Akt-GSK3β axis and Nfatc1 expression through the canonical NF-κB pathway in osteoclasts
To determine which signaling pathways might be involved in the LAT1-dependent regulation of Nfatc1 expression and nuclear NFATc1 accumulation, we analyzed various signaling pathways known to regulate osteoclastogenesis and NFATc1 abundance. RANKL-induced phosphorylation of mitogen-activated protein kinase (MAPK) family members Erk1 and Erk2 (Erk1/2), Jun N-terminal kinases 1 and 2 (JNK1/2), and p38, all of which are immediately downstream of RANKL signaling in osteoclasts, was comparable between the control and Slc7a5-deficient cells (Fig. 7, D to I).
In contrast to the MAPK family members, Akt phosphorylation was markedly increased in Slc7a5-deficient cells irrespective of RANKL stimulation (Fig. 8, A and B). Akt phosphorylates and inhibits GSK3β, thus preventing GSK3β from promoting NFATc1 translocation from the nucleus into the cytoplasm, leading to enhanced osteoclastogenesis by nuclear NFATc1 (36). In accordance with an increase in nuclear NFATc1 accumulation (Fig. 7, A and C), GSK3β phosphorylation was also significantly increased in RANKL-stimulated Slc7a5-deficient cells compared to RANKL-treated control cells (Fig. 8, A and C), indicating an increase in nuclear NFATc1 accumulation by Slc7a5 deficiency, at least in part, through the Akt-GSK3β axis.
Fig. 8. The Akt-GSK3β and canonical NF-κB pathways are activated in Slc7a5-deficient osteoclasts.

BMMs from Slc7a5fl/fl mice were retrovirally infected with Cre recombinase and subsequently stimulated with RANKL, followed by immunoblotting and densitometric quantification of (A to C) phosphorylation of Akt and GSK3β, (D to F) phosphorylation of IKKα/β and IκBα, and (G and H) abundance of p65. n = 4 mice per genotype. (I and J) BMMs from Slc7a5fl/fl mice were retrovirally infected with Cre recombinase and subsequently stimulated with RANKL in the presence or absence of Akt inhibitor X (I) or the IKK inhibitor BMS-345541 (J), followed by TRAP staining. n = 4 mice per genotype. All data were analyzed by the two-way ANOVA with Bonferroni and/or Dunnett post hoc test. **P < 0.01, significantly different from the value obtained in control cells. ##P < 0.01, significantly different from the value obtained in Slc7a5-deficient cells without inhibitors. (K) Model for LAT1-mediated control of osteoclast activation. The LAT1-mTORC1 axis (red dashed square) inhibits the activation of Akt, thus preventing GSK3β from stimulating the translocation of NFATc1 from the nucleus to the cytoplasm. LAT1-mTORC1 signaling also inhibits the activation of IKK and IκBα in osteoclasts, thus preventing the degradation of IκB and the nuclear translocation of the p65 and p50 subunits that constitute NF-κB. These actions repress the nuclear accumulation of NFATc1 and Nfatc1 expression, respectively, leading to the inhibition of NFATc1-dependent osteoclastic gene expression, thereby impeding osteoclastogenesis.
RANKL stimulation causes the activation of inhibitor of κB (IκB) kinase (IKK), which induces phosphorylation and proteasomal degradation of IκB. This results in the nuclear translocation of NF-κB, composed of p65 and p50 subunits, and the transcription of target genes, including Nfatc1 (37–39). Nuclear accumulation of p65, as well as the phosphorylation of both IKKα/β and IκBα, was significantly increased in Slc7a5-deficient cells in the presence of RANKL (Fig. 8, D to H), indicating the activation of the canonical NF-κB pathway by Slc7a5 deficiency.
The enhanced osteoclastogenesis phenotype of Slc7a5-deficient cells was significantly inhibited by treatment with either the Akt inhibitor X or the IKK inhibitor BMS-345541, suggesting the functional importance of the Akt and NF-κB pathways in the LAT1-dependent inhibition of osteoclastogenesis (Fig. 8, I and J, and fig. S6,A and B). Collectively, these results suggest that the LAT1-mTORC1 axis negatively regulates not only the nuclear accumulation of NFATc1 through the Akt-GSK3β axis but also Nfatc1 expression through the canonical NF-κB pathway, thereby inhibiting osteoclastogenesis (Fig. 8K).
DISCUSSION
Appropriate dietary protein and amino acid intake is widely accepted as essential for adequate accumulation of bone tissue during growth and for maintenance of skeletal structural integrity throughout life, although a high-protein diet has long been considered to have a negative impact on bone health through an increase in bone breakdown, leading to an increase in urinary calcium (40, 41). Although amino acid substrates for LAT1 are suggested to have a beneficial impact on bone health directly and indirectly, a potential direct role of LAT1 in bone cells in the maintenance of bone homeostasis had not been previously described. The main relevance of the present findings is that the cell surface amino acid transporter LAT1 is present in both osteoblasts and osteoclasts and is functionally required in osteoclasts for proper amounts of bone resorption to maintain bone homeostasis. The fact that Slc7a5 transcripts decreased in osteoclasts of ovariectomized mice (Fig. 2) suggests a possible inverse association between osteoclast Slc7a5 expression and the pathogenesis of postmenopausal osteoporosis, although the mechanisms governing Slc7a5 expression in osteoclasts currently remain unclear. Given that Slc7a5 global knockout in mice causes early embryonic lethality at E9.5 to E11.5 (16, 31), the in vivo role of LAT1 (Slc7a5) on bone development is currently unknown. Slc7a5 deficiency in osteoblasts did not alter bone volume or bone formation parameters in vivo (Fig. 4), irrespective of the JPH203-sensitive LAT1-dependent amino acid uptake in osteoblasts (Fig. 1), suggesting that LAT1 might contribute to intracellular amino acid homeostasis and various cellular processes in osteoblasts to a lesser extent compared with other members of the LAT family. Alternatively, compensatory activation of other members of the LAT family might be occurring in Slc7a5-deficient osteoblasts, similar to that reported in muscle-specific Slc7a5 knockout mice (16). Although further examination is necessary to completely exclude the possible involvement of LAT1 in bone formation and bone remodeling through its activity in osteoblasts, LAT1 is likely the primary amino acid sensor involved in the regulation of bone resorption and bone remodeling in the osteoclast lineage.
There are two major intracellular amino acid sensing mechanisms: the GAAC pathway, which is mediated by general control nonderepressible 2 and the transcription factor ATF4, and the mTORC1 pathway (42–44). Various independent lines of investigation using pharmacological and genetic strategies demonstrate that mTORC1 signaling is important for osteoclast differentiation and function in vitro and in vivo; however, the results of these studies have been inconsistent (32, 33, 45, 46). For example, bone mass was increased or decreased by osteoclast-specific Raptor deletion depending on the driver that was used to generate the deficiency (Ctsk-Cre or Lyz2-Cre, respectively) (32, 33, 45). This discrepancy might be caused by Raptor deletion in different cell types—mature osteoclasts in the case of Ctsk-Cre and preosteoclasts with Lyz2-Cre. Given that genetic activation of mTORC1 almost completely corrects both the lower bone mass and the higher bone resorption phenotypes of Slc7a5-deficient mice (Fig. 6), our findings suggest that mTORC1 might be a pivotal downstream effector of LAT1 in osteoclasts. In contrast, ATF4, the major transcription factor in the GAAC pathway, activates osteoclastogenesis through the induction of Nfatc1 (47). We, therefore, cannot exclude the possibility that the GAAC pathway, which is a well-known alternative major amino acid sensing pathway, might also contribute to LAT1-dependent regulation of bone homeostasis. The increase in eIF2α phosphorylation that we observed upon Slc7a5 deficiency is consistent with the involvement of the GAAC pathway (fig. S5).
Our findings suggest that the LAT1-mTORC1 axis inhibits osteoclastogenesis through the negative control of the NF-κB-NFATc1 pathway and the Akt-GSK3β axis (Fig. 8); the latter enhances osteoclastogenesis by dephosphorylation and nuclear accumulation of NFATc1 (36). Given that mTORC1 directly phosphorylates NFATc1, leading to a decrease in its nuclear accumulation and therefore inhibiting osteoclastogenesis (33), the possibility that the LAT1-mTORC1 axis inhibits osteoclastogenesis through negative regulation of the NFATc1 function by promoting direct mTORC1-mediated phosphorylation of NFATc1 should not be ruled out.
In conclusion, the current findings indicate that the amino acid transporter LAT1 functions in osteoclasts to repress osteoclastogenesis during bone resorption and bone homeostasis, thereby providing a molecular connection between a nutritional signal and skeletal integrity. LAT1 has been proposed as a promising target for both diagnostic cancer imaging (for positron emission tomography or single-photon emission computed tomography) and anticancer therapeutics (48–51). Our findings suggest that LAT1 is also a potential target for the discovery and development of drugs for diagnostic and therapeutic approaches in various metabolic bone diseases relevant to abnormal osteoclastogenesis, such as osteoporosis, rheumatoid arthritis, and metastatic bone cancer in addition to congenital skeletal dysplasias such as Coffin-Lowry syndrome.
MATERIALS AND METHODS
Mice
The protocol used here meets the guidelines of the Japanese Society for Pharmacology and was approved by the Committee for Ethical Use of Experimental Animals at Kanazawa University. Tsc1fl/fl mice were obtained from the Jackson laboratory. Slc7a5fl/fl and Tsc1fl/fl mice were crossed with Osx-Cre, Tnfrsf11a-Cre, or Lyz2-Cre mice (52–55). These mutant mice were backcrossed more than five generations with C57BL/6J. Mice were bred under standard animal housing conditions at 23° ± 1°C with humidity of 55% and a 12-hour light/ 12-hour dark cycle, with free access to food and water. Osx-Cre mice were maintained in the absence of doxycycline. Genotyping was performed by polymerase chain reaction (PCR) using tail genomic DNA. The numbers of animals used per experiment are stated in the figure legends. For OVX, 8 -week-old mice were anesthetized by an intraperitoneal injection of pentobarbital and subjected to OVX or sham operation under aseptic environments as described previously (56). Mice were killed by decapitation 28 days after operation.
Cell sorting
Femur and tibia were crushed and digested with 0.2% collagenase in phosphate-buffered saline (PBS), followed by depletion of red blood cells by soaking in 0.15 M NH4Cl for 5 min. Cells were then resus-pended in PBS containing 2% fetal bovine serum and incubated for 30 min at 4°C with a cocktail of antibodies for preosteoclast-like cell markers [allophycocyanin-conjugated anti-CD11b antibody (M1/70) (BioLegend), fluorescein isothiocyanate (FITC)–conjugated anti-CX3CR1 antibody (SA011F11) (BioLegend), and phycoerythrin (PE)–Cy7–conjugated anti-Ly6C (AL-21) (BD Pharmingen)] or a cocktail of antibodies for osteoblast-like cell markers [FITC-conjugated anti-CD45 antibody (30-F11) (BD Pharmingen), PE-conjugated anti-CD51 antibody (RMV-7) (BioLegend), and PE-Cy7–conjugated anti-Sca1 antibody (D7) (BD Pharmingen)] and 7-Aminoactinomycin D for dead cell exclusion. Immunostained cells were washed and analyzed on a FACS Aria II cell sorter (BD Biosciences) and then sorted for CD11blow/−/CX3CR1+/Ly6Chi cells (preosteoclasts) and CD45−/CD51+/Sca-1− cells (osteoblasts) (29, 57, 58).
Preparation of [125I]IMT
[125I]NaI (629 GBq/mg as iodine) was obtained from PerkinElmer. No-carrier-added [125I]IMT was prepared using the conventional chloramine-T method from L-α-methyltyrosine as a precursor.Briefly, chloramine-T (Nacalai Tesque) aqueous solution (2 mg/ml) was added to a mixture of L-α-methyltyrosine (1.7 mg/ml) dissolved in 0.1 M phosphate buffer (pH 7.4) and carrier-free [125I]NaI (925 to 1100 kBq). After incubation for 10 min, the reaction was quenched by adding sodium bisulfite aqueous solution (2 mg/ml). [125I]IMT was identified by HPLC (Shimadzu), and IMT, which was prepared using nonradioactive iodine, was identified by mass spectrometry (JMS-T100TD, JEOL). HPLC was performed with a COSMOSIL 5C18-AR-II column (4.5 mm by 150 mm; Nacalai Tesque) at a flow rate of 1 ml/min with a gradient mobile phase of 20% methanol in water with 0.1% trifluoroacetic acid (TFA) to 40% methanol in water with 0.1% TFA for 20 min. The column temperature was maintained at 40°C.
[125I]IMT uptake assay
Cultured cells were washed twice with Hanks’ balanced salt solution (HBSS) [125 mM NaCl, 4.8 mM KCl, 1.3 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 25 mM Hepes, and 5.6 mM D-glucose (pH 7.4)] and subsequent incubation in HBSS at 37°C for 10 min in a 5% CO2 incubator. Cells were then incubated with [125I]IMT (176.5 pM) at 4° or 37°C for 30 min. Cells were treated with JPH203 at 30 μM. The reaction was terminated by the aspiration of buffer, followed by superficial rinsing with ice-cold HBSS containing 1 mM unlabeled L-α-methyltyrosine at 4°C three times to remove extracellular [125I]IMT. The cells were lysed using 0.1 M NaOH. The radioactivity was measured by a γ-counter (ARC-7010, Hitachi Ltd.). Protein concentration was determined with a Protein Assay Bicinchoninate Kit (Nacalai Tesque).
Bone histomorphometric analysis, x-ray micro–computed tomography (μCT) analysis, and serum biochemical analysis
Bone histomorphometric analyses were performed on uncalcified long bones and vertebrae in accordance with previously described methods (59). Briefly, the bone was fixed with 10% formalin, followed by dehydration in an ethanol series and subsequent embedding in methyl methacrylate resin, according to standard protocols. The BV/TV ratio was measured by von Kossa staining. Osteoblast and osteoclast parameters were analyzed by staining with toluidine blue and TRAP, respectively. Analyses were performed using the OsteoMeasure analysis system (OsteoMetrics), according to standard protocols (60). Trabecular architecture was assessed in long bones using a μCT system (Comscan), and the BV/TV ratio was measured using TRI/3D-BON software (RATOC) (61, 62). Calcein, which has a high calcium affinity, was injected twice into mice with an interval of 3 days, and then, mice were killed 2 days after the last injection. The new-forming bone area was marked with the calcein-calcium complex. Concentrations of CTx and P1NP in serum were determined by enzyme-linked immunosorbent assay kits according to the manufacturer’s instructions (Immunodiagnostic Systems).
Culture of osteoclasts, TRAP staining, pit formation assay, cell cycle assay, and cell death assay
BMMs were cultured in the presence of macrophage colony-stimulating factor (M-CSF) (20 ng/ml) and RANKL (20 ng/ml) for 5 days, and TRAP staining and the pit formation assay were performed as previously described (63, 64). Cells were treated with BMS-345541 (Abcam) at 0.16 μM or Akt inhibitor X (Cayman) at 0.2 μM followed by TRAP staining. Cells were cultured with 10 μM BrdU for 45 min and were prepared for analysis of BrdU incorporation using the FITC BrdU Flow Kit (BD Biosciences) by flow cytometric analysis using the FACSVerse (BD Biosciences). Cell death assay was performed with FITC–Annexin V (BD Biosciences) and propidium iodide by FACSVerse. Data were analyzed by FACSuite software (BD Biosciences).
Generation of retroviral vectors and infection
pMX-CA-Rheb was generated by subcloning into the pMX vector from the pcDNA3-FLAG-Rheb-N153 T vector (Addgene plasmid no. 19997). Retroviral vectors were transfected into Plat-E cells using the calcium carbonate method. Virus supernatants were collected 48 hours after transfection, and then, cells were infected with virus supernatants for 72 hours in the presence of polybrene (4 μg/ml). Cells were then subjected to selection by culture with puromycin (1 μg/ml) for 3 days before use in experiments.
Real-time qPCR
Total RNA was extracted from cells, followed by synthesis of complementary DNA (cDNA) with reverse transcriptase and oligo-dT primer. The cDNA samples were then used as templates for real-time PCR analysis, which was performed on an Mx3005P instrument (Agilent Technologies), by using specific primers for each gene (table S1). The levels of the genes examined were normalized using Actb as an internal control for each sample.
Immunoblotting analysis
Cultured cells were solubilized in lysis buffer containing 1% NP-40. Samples were then subjected to SDS–polyacrylamide gel electro-phoresis, followed by transfer to polyvinylidene difluoride membranes and subsequent immunoblotting assay. Quantification was performed by densitometry using ImageJ. The primary antibodies used were anti–phospho-p70S6k1 (no. 9205), anti-p70S6k1 (no. 2708), anti–phospho-4E-BP1 (no. 2855), anti–4E-BP1 (no. 9644), anti-eIF2α (no. 5324), anti–phospho-eIF2α (no. 9721), anti-NFATc1 (no. 8032), anti–phospho-Erk1/2 (no. 4370), anti-Erk1/2 (no. 4695), anti–phospho-JNK (no. 4668), anti-JNK (no. 9252), anti–phospho-p38 (no. 4511), anti-p38 (no. 8690), anti–phospho-Akt (no. 4060), anti-Akt (no. 4685), anti–phospho-GSK3β (no. 9323), anti-GSK3β (no. 9315), anti– phospho-IKKα/β (no. 2697), anti-IKKα (no. 2689), anti–phospho-IκBα (no. 2859), anti-IκBα (no. 4812), and anti-p65 (no. 8242) (Cell Signaling Technologies); anti–β-actin (no. sc-47778) (Santa Cruz Biotechnology); and anti-LaminB1 (MABS492) (EMD Millipore).
Data analysis
Results are all expressed as the means ± SE, and statistical significance was determined by the two-tailed Student’s t test or the one-way or two-way analysis of variance (ANOVA) with Bonferroni and/or Dunnett post hoc test.
Supplementary Material
Fig. S1. Expression of genes encoding amino acid transporters in preosteoclasts and osteoblasts.
Fig. S2. Expression of Slc7a5 mRNA in mutant mice.
Fig. S3. Phosphorylation of p70S6K1 by EAA deprivation in osteoclasts.
Fig. S4. Effect of RANKL on [125I]IMT incorporation in osteoclasts.
Fig. S5. Phosphorylation of eIF2α in Slc7a5-deficient cells.
Fig. S6. Effects of Akt and NF-κB inhibitors in Slc7a5-deficient cells.
Table S1. List of primers used for real-time PCR.
Acknowledgments:
We highly thank T. Kitamura (Tokyo University) and T. Nakanishi (Kanazawa University) for providing Plat-E cells and critical review of the manuscript, respectively.
Funding: This work was supported, in part, by the Japan Society for the Promotion of Science (16H05131, 17KT0051, and 18H04971 to E.H.) and the Japan Agency for Medical Research and Development (17824969 to E.H.).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data Availability: All the data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Hyde R, Taylor PM, Hundal HS, Amino acid transporters: Roles in amino acid sensing and signalling in animal cells. Biochem. J 373, 1–18 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor PM, Role of amino acid transporters in amino acid sensing. Am. J. Clin. Nutr 99, 223S–230S (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kandasamy P, Gyimesi G, Kanai Y, Hediger MA, Amino acid transporters revisited: New views in health and disease. Trends Biochem. Sci 43, 752–789 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Suviolahti E, Oksanen LJ, Öhman M, Cantor RM, Ridderstrale M, Tuomi T, Kaprio J, Rissanen A, Mustajoki P, Jousilahti P, Vartiainen E, Silander K, Kilpikari R, Salomaa V, Groop L, Kontula K, Peltonen L, Pajukanta P, The SLC6A14 gene shows evidence of association with obesity. J. Clin. Invest 112, 1762–1772 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drgonova J, Jacobsson JA, Han JC, Yanovski JA, Fredriksson R, Marcus C, Schiöth HB, Uhl GR, Involvement of the neutral amino acid transporter SLC6A15 and leucine in obesity-related phenotypes. PLOS ONE 8, e68245 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tărlungeanu DC, Deliu E, Dotter CP, Kara M, Janiesch PC, Scalise M, Galluccio M, Tesulov M, Morelli E, Sonmez FM, Bilguvar K, Ohgaki R, Kanai Y, Johansen A, Esharif S, Ben-Omran T, Topcu M, Schlessinger A, Indiveri C, Duncan KE, Caglayan AO, Gunel M, Gleeson JG, Novarino G, Impaired amino acid transport at the blood brain barrier is a cause of autism spectrum disorder. Cell 167, 1481–1494.e18 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bröer S, Palacin M, The role of amino acid transporters in inherited and acquired diseases. Biochem. J 436, 193–211 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Kanai Y, Segawa H, Miyamoto K, Uchino H, Takeda E, Endou H, Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J. Biol. Chem 273, 23629–23632 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Yanagida O, Kanai Y, Chairoungdua A, Kim DK, Segawa H, Nii T, Cha SH, Matsuo H, Fukushima J, Fukasawa Y, Tani Y, Taketani Y, Uchino H, Kim JY, Inatomi J, Okayasu I, Miyamoto K, Takeda E, Goya T, Endou H, Human L-type amino acid transporter 1 (LAT1): Characterization of function and expression in tumor cell lines. Biochim. Biophys. Acta 1514, 291–302 (2001). [DOI] [PubMed] [Google Scholar]
- 10.Mastroberardino L, Spindler B, Pfeiffer R, Skelly PJ, Loffing J, Shoemaker CB, Verrey F, Amino-acid transport by heterodimers of 4F2hc/CD98 and members of a permease family. Nature 395, 288–291 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Goberdhan DCJ, Wilson C, Harris AL, Amino acid sensing by mTORC1: Intracellular transporters mark the spot. Cell Metab. 23, 580–589 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dodd KM, Tee AR, Leucine and mTORC1: A complex relationship. Am. J. Physiol. Endocrinol. Metab 302, E1329–E1342 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Duan Y, Li F, Tan K, Liu H, Li Y, Liu Y, Kong X, Tang Y, Wu G, Yin Y, Key mediators of intracellular amino acids signaling to mTORC1 activation. Amino Acids 47, 857–867 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Fan X, Ross DD, Arakawa H, Ganapathy V, Tamai I, Nakanishi T, Impact of system L amino acid transporter 1 (LAT1) on proliferation of human ovarian cancer cells: A possible target for combination therapy with anti-proliferative aminopeptidase inhibitors. Biochem. Pharmacol 80, 811–818 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Gaccioli F, Aye IL, Roos S, Lager S, Ramirez VI, Kanai Y, Powell TL, Jansson T, Expression and functional characterisation of System L amino acid transporters in the human term placenta. Reprod. Biol. Endocrinol 13, 57 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poncet N, Mitchell FE, Ibrahim AFM, McGuire VA, English G, Arthur JS, Shi Y-B, Taylor PM, The catalytic subunit of the system L1 amino acid transporter (slc7a5) facilitates nutrient signalling in mouse skeletal muscle. PLOS ONE 9, e89547 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kageyama T, Nakamura M, Matsuo A, Yamasaki Y, Takakura Y, Hashida M, Kanai Y, Naito M, Tsuruo T, Minato N, Shimohama S, The 4F2hc/LAT1 complex transports L-DOPA across the blood-brain barrier. Brain Res. 879, 115–121 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Matsuo H, Tsukada S, Nakata T, Chairoungdua A, Kim DK, Cha SH, Inatomi J, Yorifuji H, Fukuda J, Endou H, Kanai Y, Expression of a system L neutral amino acid transporter at the blood-brain barrier. Neuroreport 11, 3507–3511 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Harada S.-i., Rodan GA, Control of osteoblast function and regulation of bone mass. Nature 423, 349–355 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Teitelbaum SL, Ross FP, Genetic regulation of osteoclast development and function. Nat. Rev. Genet 4, 638–649 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Karsenty G, Kronenberg HM, Settembre C, Genetic control of bone formation. Annu. Rev. Cell Dev. Biol 25, 629–648 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Feng X, McDonald JM, Disorders of bone remodeling. Annu. Rev. Pathol 6, 121–145 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elefteriou F, Benson MD, Sowa H, Starbuck M, Liu X, Ron D, Parada LF, Karsenty G, ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metab. 4, 441–451 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ammann P, Laib A, Bonjour JP, Meyer JM, Ruegsegger P, Rizzoli R, Dietary essential amino acid supplements increase bone strength by influencing bone mass and bone microarchitecture in ovariectomized adult rats fed an isocaloric low-protein diet. J. Bone Miner. Res 17, 1264–1272 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Jennings A, MacGregor A, Spector T, Cassidy A, Amino acid intakes are associated with bone mineral density and prevalence of low bone mass in women: Evidence from discordant monozygotic twins. J. Bone Miner. Res 31, 326–335 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawai K, Fujibayashi Y, Saji H, Yonekura Y, Konishi J, Kubodera A, Yokoyama A, A strategy for the study of cerebral amino acid transport using iodine-123-labeled amino acid radiopharmaceutical: 3-iodo-alpha-methyl-L-tyrosine. J. Nucl. Med 32, 819–824 (1991). [PubMed] [Google Scholar]
- 27.Jager PL, Vaalburg W, Pruim J, de Vries EG, Langen KJ, Piers DA, Radiolabeled amino acids: Basic aspects and clinical applications in oncology. J. Nucl. Med 42, 432–445 (2001). [PubMed] [Google Scholar]
- 28.Oda K, Hosoda N, Endo H, Saito K, Tsujihara K, Yamamura M, Sakata T, Anzai N, Wempe MF, Kanai Y, Endou H, L-type amino acid transporter 1 inhibitors inhibit tumor cell growth. Cancer Sci. 101, 173–179 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charles JF, Hsu L-Y, Niemi EC, Weiss A, Aliprantis AO, Nakamura MC, Inflammatory arthritis increases mouse osteoclast precursors with myeloid suppressor function. J. Clin. Invest 122, 4592–4605 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, Wagers AJ, Hsiao EC, Passegue E, Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 13, 285–299 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohgaki R, Ohmori T, Hara S, Nakagomi S, Kanai-Azuma M, Kaneda-Nakashima K, Okuda S, Nagamori S, Kanai Y, Essential roles of L-Type amino acid transporter 1 in syncytiotrophoblast development by presenting fusogenic 4F2hc. Mol. Cell. Biol 37, e00427–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Xu S, Li K, Tan K, Liang K, Wang J, Shen J, Zou W, Hu L, Cai D, Ding C, Li M, Xiao G, Liu B, Liu A, Bai X, mTORC1 inhibits NF-κB/nfatc1 signaling and prevents osteoclast precursor differentiation, in vitro and in mice. J. Bone Miner. Res 32, 1829–1840 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Huynh H, Wan Y , mTORC1 impedes osteoclast differentiation via calcineurin and NFATc1. Commun. Biol 1, 29 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K, Hara K, Tanaka N, Avruch J, Yonezawa K, The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem 278, 15461–15464 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J, Rheb binds and regulates the mTOR kinase. Curr. Biol 15, 702–713 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Moon JB, Kim JH, Kim K, Youn BU, Ko A, Lee SY, Kim N, Akt induces osteoclast differentiation through regulating the GSK3beta/NFATc1 signaling cascade. J. Immunol 188, 163–169 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Nakashima T, Hayashi M, Takayanagi H, New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol. Metab 23, 582–590 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Jimi E, Aoki K, Saito H, D’Acquisto F, May MJ, Nakamura I, Sudo T, Kojima T, Okamoto F, Fukushima H, Okabe K, Ohya K, Ghosh S, Selective inhibition of NF-κB blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat. Med 10, 617–624 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Abu-Amer Y, NF-kappaB signaling and bone resorption. Osteoporosis Int. 24, 2377–2386 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heaney RP, Layman DK, Amount and type of protein influences bone health. Am. J. Clin. Nutr 87, 1567S–1570S (2008). [DOI] [PubMed] [Google Scholar]
- 41.Rizzoli R, Nutritional aspects of bone health. Best Pract. Res. Clin. Endocrinol. Metab 28, 795–808 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Hay N, Sonenberg N, Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Chen R, Zou Y, Mao D, Sun D, Gao G, Shi J, Liu X, Zhu C, Yang M, Ye W, Hao Q, Li R, Yu L, The general amino acid control pathway regulates mTOR and autophagy during serum/glutamine starvation. J. Cell Biol 206, 173–182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bröer S, Broer A, Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem. J 474, 1935–1963 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dai Q, Xie F, Han Y, Ma X, Zhou S, Jiang L, Zou W, Wang J, Inactivation of regulatory-associated protein of mTOR (Raptor)/mammalian target of rapamycin complex 1 (mTORC1) signaling in osteoclasts increases bone mass by inhibiting osteoclast differentiation in mice. J. Biol. Chem 292, 196–204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen J, Long F, mTOR signaling in skeletal development and disease. Bone Res. 6, 1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao H, Yu S, Yao Z, Galson DL, Jiang Y, Zhang X, Fan J, Lu B, Guan Y, Luo M, Lai Y, Zhu Y, Kurihara N, Patrene K, Roodman GD, Xiao G, Activating transcription factor 4 regulates osteoclast differentiation in mice. J. Clin. Invest 120, 2755–2766 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nobusawa A, Kim M, Kaira K, Miyashita G, Negishi A, Oriuchi N, Higuchi T, Tsushima Y, Kanai Y, Yokoo S, Oyama T, Diagnostic usefulness of 18F-FAMT PET and L-type amino acid transporter 1 (LAT1) expression in oral squamous cell carcinoma. Eur. J. Nucl. Med. Mol. Imaging 40, 1692–1700 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Wiriyasermkul P, Nagamori S, Tominaga H, Oriuchi N, Kaira K, Nakao H, Kitashoji T, Ohgaki R, Tanaka H, Endou H, Endo K, Sakurai H, Kanai Y, Transport of 3-fluoro-L-alpha-methyl-tyrosine by tumor-upregulated L-type amino acid transporter 1: a cause of the tumor uptake in PET. J. Nucl. Med 53, 1253–1261 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Geier EG, Schlessinger A, Fan H, Gable JE, Irwin JJ, Sali A, Giacomini KM, Structure-based ligand discovery for the large-neutral amino acid transporter 1, LAT-1. Proc. Natl. Acad. Sci. U.S.A 110, 5480–5485 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singh N, Ecker GF, Insights into the structure, function, and ligand discovery of the large neutral amino acid transporter 1, LAT1. Int. J. Mol. Sci 19, 1278 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sinclair LV, Rolf J, Emslie E, Shi Y-B, Taylor PM, Cantrell DA, Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol 14, 500–508 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I, Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277 (1999). [DOI] [PubMed] [Google Scholar]
- 54.Rodda SJ, McMahon AP, Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 133, 3231–3244 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Maeda K, Kobayashi Y, Udagawa N, Uehara S, Ishihara A, Mizoguchi T, Kikuchi Y, Takada I, Kato S, Kani S, Nishita M, Marumo K, Martin TJ, Minami Y, Takahashi N, Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat. Med 18, 405–412 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Hinoi E, Takarada T, Uno K, Inoue M, Murafuji Y, Yoneda Y, Glutamate suppresses osteoclastogenesis through the cystine/glutamate antiporter. Am. J. Pathol 170, 1277–1290 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Winkler IG, Barbier V, Wadley R, Zannettino ACW, Williams S, Lévesque J-P, Positioning of bone marrow hematopoietic and stromal cells relative to blood flow in vivo: serially reconstituting hematopoietic stem cells reside in distinct nonperfused niches. Blood 116, 375–385 (2010). [DOI] [PubMed] [Google Scholar]
- 58.Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, Poulton IJ, van Rooijen N, Alexander KA, Raggatt LJ, Lévesque JP, Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 116, 4815–4828 (2010). [DOI] [PubMed] [Google Scholar]
- 59.Yamamoto T, Hinoi E, Fujita H, Iezaki T, Takahata Y, Takamori M, Yoneda Y, The natural polyamines spermidine and spermine prevent bone loss through preferential disruption of osteoclastic activation in ovariectomized mice. Br. J. Pharmacol 166, 1084–1096 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM, Standardized nomenclature, symbols, and units for bone histomorphometry: A 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res 28, 2–17 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R, Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Miner. Res 25, 1468–1486 (2010). [DOI] [PubMed] [Google Scholar]
- 62.Takarada T, Xu C, Ochi H, Nakazato R, Yamada D, Nakamura S, Kodama A, Shimba S, Mieda M, Fukasawa K, Ozaki K, Iezaki T, Fujikawa K, Yoneda Y, Numano R, Hida A, Tei H, Takeda S, Hinoi E, Bone resorption is regulated by circadian clock in osteoblasts. J. Bone Miner. Res 32, 872–881 (2017). [DOI] [PubMed] [Google Scholar]
- 63.Hinoi E, Ochi H, Takarada T, Nakatani E, Iezaki T, Nakajima H, Fujita H, Takahata Y, Hidano S, Kobayashi T, Takeda S, Yoneda Y, Positive regulation of osteoclastic differentiation by growth differentiation factor 15 upregulated in osteocytic cells under hypoxia. J. Bone Miner. Res 27, 938–949 (2012). [DOI] [PubMed] [Google Scholar]
- 64.Iezaki T, Fukasawa K, Park G, Horie T, Kanayama T, Ozaki K, Onishi Y, Takahata Y, Nakamura Y, Takarada T, Yoneda Y, Nakamura T, Vacher J, Hinoi E, Transcriptional modulator Ifrd1 regulates osteoclast differentiation through enhancing the NF-κB/ NFATc1 pathway. Mol. Cell. Biol 36, 2451–2463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Expression of genes encoding amino acid transporters in preosteoclasts and osteoblasts.
Fig. S2. Expression of Slc7a5 mRNA in mutant mice.
Fig. S3. Phosphorylation of p70S6K1 by EAA deprivation in osteoclasts.
Fig. S4. Effect of RANKL on [125I]IMT incorporation in osteoclasts.
Fig. S5. Phosphorylation of eIF2α in Slc7a5-deficient cells.
Fig. S6. Effects of Akt and NF-κB inhibitors in Slc7a5-deficient cells.
Table S1. List of primers used for real-time PCR.