

Abstract
Our nation is becoming increasingly diverse, however, few autopsy studies examine multiple ethnoracial groups, especially Hispanics. We examined differences in neuropathological diagnoses of 435 deceased participants with dementia from 3 ethnoracial groups (35 Black, 28 Hispanic, and 360 non-Hispanic White) evaluated at the University of California Davis Alzheimer’s Disease Center. We used novel applications of bootstrap resampling and logistic regression standardization to project neuropathological diagnostic rates for non-Hispanic Whites to minority sample characteristics to improve inference of findings. Alzheimer’s disease (AD) without significant Cerebrovascular disease (CVD) or other dementia-related pathologies (AD (non-mixed)) was present in 15 Black (43%), 4 Hispanic (14%) and 156 (43%) non-Hispanic Whites. CVD sufficient to contribute to dementia was confirmed in 14 Black (40%), 16 Hispanic (54%) and 101 (28%) non-Hispanic White decedents. The observed CVD prevalence of 40% in Blacks exceeded the predicted 29% [95% CI: 22%-36%]. Despite being outside the 95% confidence interval, the difference between observed and predicted was not statistically significant after bootstrap testing. Conversely, for Hispanics, the observed proportion at 54% exceeded significantly the predicted prevalence of 24% from non-Hispanic Whites [95% CI: 16%-34%], avg. p =0.008). An identical analysis using AD (non-mixed) as the outcome predicted AD (non-mixed) in Blacks averaging 41% [95% CI:34%-48%], nearly equal to observed prevalence. For Hispanics, however, the observed proportion at 14%, was well below predictions (mean=42%, 95% CI:32%-53%], avg. p =0.008). We conclude mixed diagnoses and CVD are more common in Hispanic and Black decedents than Non-Hispanic Whites with dementia in our cohort. The increased prevalence of vascular co-morbidity may be a potential opportunity to intervene more effectively in dementia treatment of those individuals.
Keywords: Alzheimer Disease, Neuropathology, Cohort Studies, minority, Dementia, Vascular, Brain, Autopsy, Cognitive Aging, Minority Groups
INTRODUCTION
Our nation’s population is increasingly older and more diverse[1]. The number of Americans with dementia, including those from ethnoracial minorities, is expected to rise to nearly 14 million by midcentury [2]. Although Alzheimer’s disease (AD) continues to be the major pathological cause of dementia, studies find dementia pathology is multifactorial, mostly due to co-occurrence of AD and vascular disease[3], and risk prediction scales emphasize the impact of vascular risk on incident dementia [4, 5].
Examination of diverse cohorts is advantageous as they can enhance understanding of the spectrum of pathological substrates of dementia, potentially leading to refined diagnosis and treatment options. Much of the field of dementia is dominated by studies involving non-Hispanic Whites [6–13]. This poses various challenges when comparing dementia risk and pathology to other ethnoracial groups[12, 14]. Having a diverse cohort, with inclusion of multiple ethnoracial groups in neuropathological studies, therefore, is needed to understand the spectrum of dementia pathophysiology. Studying mostly non-Hispanic Whites potentially diminishes diversity among various dementia risk factors including those related to cultural, economic, social, and behavioral characteristics. This is particularly true of cardiovascular risk factors which are significantly more common among Black and Hispanic individuals [2, 15–17]. Furthermore, it is hypothesized Hispanics and Blacks are more likely to have mixed pathological processes leading to dementia [18]. Published literature supports this hypothesis among Black decedents [19], however, there is a gap in neuropathological assessment of Hispanic cohorts despite their increased prevalence in the US and increased risk of developing clinical dementia [20]. For example, a recent publication found that although the sensitivity for AD diagnosis was high for Hispanics (comparing clinical to neuropathological diagnosis), specificity at 58% was lower than that commonly found for non-Hispanic Whites[21]. One must note there are certain biases that can arise within cohort studies, especially those with mixed ethnoracial groups [12]. However, diversity in cohort studies is important for identification of the spectrum of underlying pathology of clinical dementia as this is crucial for developing prevention, tailored treatments [22], and future health policies seeking to mitigate risk factors and facilitate early detection for all individuals.
Over the past 15 years, the UC Davis Alzheimer’s Disease Center (UCD ADC) has developed a subject cohort diverse in race/ethnicity, education, and medical comorbidities [23]. To understand the underlying substrates of dementia across ethnoracial groups in our cohort, we studied demented individuals from the UCD ADC research cohort comparing neuropathological diagnoses of non-Hispanic White, Black, and Hispanic decedents. In the current study, we tested two main hypotheses; 1) that a cerebrovascular (CVD) pathology is less common in the non-Hispanic White group than minority participants with dementia, and 2) that AD (non-mixed) was more common in non-Hispanics Whites than minority participants with dementia. For these purposes AD (non-mixed) was defined as having a clinicopathological diagnosis of AD, without the presence of CVD or other dementia related pathologies of sufficient degree to contribute to the dementia syndrome found at autopsy [24–26]. Furthermore, studies of group differences in post-mortem neuropathological diagnoses are often faced with statistical challenges inherent to small sample sizes. A common technique to overcome these challenges is to use a method known as ‘matching’ when analyzing differences between groups. This statistical method however, can have several disadvantages. Matching analyses can result in limiting information as an analysis can only use cases for which there exist matched controls. To avoid limiting information, reducing the number of matching variables can also be implemented, however, this approach also has certain limitations. We hoped to avoid these pitfalls by implementing bootstrap resampling and logistic regression standardization to project neuropathological diagnostic rates for non-Hispanic Whites to minority sample characteristics.
Although this is a small study and should not be considered to be generalizable to large populations due to certain cohort bias, this study examines the neuropathological diagnoses of a cohort of demented participants of mixed ethnoracial backgrounds, thereby further characterizing the influence of diversity on the neuropathology of clinical dementia.
MATERIALS & METHODS
Participants
Participants included 423 deceased persons of three ethnic groups from the UCD ADC Brain Donation Program, diagnosed with clinical dementia prior to death: 360 non-Hispanic White, 35 Black and 28 Hispanic decedents. This study was approved by the Institutional Review Board (IRB) of the University of California Davis, and written consent was obtained for each participant for repeated evaluations during life and for autopsy. Details of this program have been previously published [23]. As pathological diagnostic criteria have changed over the years, to decrease potential cohort effects yet still obtain enough subjects to conduct proper analysis, we limited participants to those who had dementia at their last visit before death and went to autopsy between 01/01/2000 and 01/14/2017 (details of cases located in Table 1). Furthermore, neuropathology reports were retrospectively examined on all participants for whom there was missing pathological data to confirm overall neuropathological diagnoses.
Table 1.
Demographics and participant characteristics. BRD: Blessed Roth Dementia Scale
| Non-Hispanic White N = 360 |
Black N = 35 |
Hispanic N = 28 |
Total N = 423 |
|
|---|---|---|---|---|
| Age at death, mean (SD) | 81.38 (9.43) | 84.11 (8.49) | 82.04 (11.63) | 81.65 (9.52) |
| Education (years), mean (SD) | 14.36 (3.18)A,B | 11.71 (2.83)B | 9.14 (4.31) | 13.8 (3.53) |
| BRD, nearest to death, mean (SD) | 11.61 (4.95) | 11.41 (4.90) | 10 (5.39) | 11.48 (4.99) |
| Time from last BRD (yrs), Mean (SD) | 1.06 (1.25) | 1.19 (1.17) | 1.42 (1.28) | 1.08 (1.25) |
| Apolipoprotein E genotype, n (%) | 282 (78%) | 24 (69%) | 22 (79%) | 328 (78%) |
| Male, n (%) | 194 (54%) | 16 (46%) | 12 (43%) | 222 (52%) |
| Cardiovascular Measures | ||||
| Heart Disease, n (%) | 82 (23%) | 8 (23%) | 4 (14%) | 94 (22%) |
| Diabetes, n (%) | 31 (9%) | 7 (20%) | 3 (11%) | 41 (10%) |
| Trans ischemic attack, n (%) | 15 (4%) | 3 (9%) | 0 (0%) | 18 (4%) |
| Hypertension, n (%) | 138 (38%)A | 22 (63%) | 11 (39%) | 171 (40%) |
| High Cholesterol, n (%) | 89 (25%) | 8 (23%) | 7 (25%) | 104 (25%) |
| Stroke, n (%) | 25 (7%) | 3 (9%) | 4 (14%) | 32 (8%) |
Statistically different than Black decedents
Statistically different than Hispanic decedents
Study Population Genetic Admixture
Studies of racially diverse populations often run the risk of disparities between self-reported race/ethnicity and genetic ancestry [12]. To validate the self-reported race/ethnicity of our ADC cohort, we assessed the correlation between self-reported race/ethnicity and ancestry for a separate group of 786 individuals evaluated by the UCD ADC, including 313 individuals who self-identified as non-White. Using standard genetic statistical approaches, we found a 95% concordance between self-identified race/ethnicity and genetic ancestry. Importantly, the genetic admixture for our Hispanic cohort was European and American Indian, consistent with peoples emigrating from Latin American countries. Similarly, the genetic admixture of our Black cohort was European and African (see Supplement Figure 1) supporting the veracity of the self-reported race and ethnicity of our participants.
Given the potential selection biases in brain autopsy samples, particularly among minorities [27, 28], we also characterized the entire group of 1082 demented subjects enrolled in our ADC longitudinal cohort by age, gender, APOE status (presence of the ε4 allele), vascular risk factor, and vascular disease prevalence (See Supplemental Materials for details). This larger group also includes all the autopsied decedents in this report.
Clinical Assessment
Only demented subjects were utilized for this study. To determine dementia, all subjects received multidisciplinary diagnostic evaluations through the UCD ADC at enrollment and approximately annual intervals thereafter until death, loss to follow-up or inability to return for reassessment due to disability. Baseline and follow-up evaluations followed the same protocol and included a detailed medical history, a physical and neurological exam. Participants who spoke only Spanish were examined by a physician fluent in Spanish. Information about change in the participant’s cognitive and functional status prior to each evaluation was assessed by independent interviews with the participant and the informant. Clinical neuropsychological evaluation using standardized neuropsychological tests [29, 30] was performed at enrollment and each follow-up. Routine laboratory tests were obtained at enrollment and when clinically indicated at follow-up. Diagnosis of cognitive syndrome (normal, mild cognitive impairment (MCI), dementia) and (for individuals with dementia) underlying etiology was made according to standardized criteria and methods [29]. Under this protocol, each subject was initially diagnosed at a consensus conference by the clinical team evaluating the participant and re-reviewed at a second, multidisciplinary UCD-ADC-wide case adjudication conference. The same approach was used for follow-up diagnoses. Dementia was diagnosed using criteria for dementia from the DSM-III-R (American Psychiatric Association, 1987), modified such that dementia could be diagnosed in the absence of memory impairment if there was significant impairment of two or more other cognitive domains. All decedents in this report received the clinical diagnosis of dementia prior to death. Individuals no longer able to follow-up in clinic due to illness or severe degree of cognitive impairment were regularly followed through phone contact. The Blessed Roth Dementia Scale[31] was used to assess basic activities of daily living.
Neuropathological evaluation
Following autopsy and neuropathological examination conducted by a neuropathologist blinded to all demographic and clinical data, diagnoses were given to each case according to published consensus criteria [32–34]. Hippocampal sclerosis associated with vascular disease or with aging was diagnosed as previously described [35, 36]. If routine pathological examination or clinical information revealed at the Clinical Pathology Conference (see details in section below) raised a suspicion of frontotemporal lobar degeneration (FTLD), the case was further studied by more extensive sampling and immunostaining to classify the case according to currently accepted guidelines [37–39]. Lewy body disease (LBD) was defined according to the consensus guideline published by McKeith et al [33]. TDP-43 was not evaluated; as this was a retrospective analysis, many of these individuals had neuropathologic diagnosis at a time when TDP43 antibodies were not routinely available.
Additional details for select neuropathologies including neurofibrillary tangles (NFTs), plaques, and CVD lesions were analyzed. As there were differences in the neuropathology data forms over the collection periods [40], derived variables were used to harmonize datasets. Furthermore, due to the retrospective nature of the study with a cohort spanning nearly 20 years, CVD lesion data was available for a subset of cases and total samples examined are indicated within the tables. For NFTs and plaques, Braak Neurofibrillary Tangle Stage and the Consortium to Establish a Registry for Alzheimer’s disease (CERAD) were used respectively [7, 41]. As peri-mortem infarcts could confound results, they were excluded- only subacute and chronic infarcts were evaluated. Macroscopic infarcts were denoted as present if they were visualized grossly and confirmed in microscopic evaluation. The presence of microscopic infarcts were assessed in the frontal, temporal, parietal, and occipital cortices as well as within the amygdala, hippocampus, striatum, pons, cerebellum, and frontal, parietal and occipital white matter. White matter rarefaction (WMR) was assessed as present if there were changes in pallor, moderate loss of myelin and/or axonal loss in the frontal, parietal and/or occipital white matter. Arteriolosclerosis was graded on a 4-tiered semi-quantitative scale (none, mild, moderate, and severe) based on narrowing of the lumen and hyalinized thickening of small arteriole walls. Atherosclerosis was assessed in the circle of Willis and graded in a semiquantitative manner similar to methods previously reported [42].
In the current study, ‘AD (non-mixed)’ was defined as having a clinicopathological diagnosis of AD, without the presence of CVD or other dementia related pathologies of sufficient degree to contribute to the dementia syndrome found at autopsy. Other dementia related pathologies included LBD, FTLD and hippocampal sclerosis. Pathological diagnoses by ethnoracial category are presented in Tables 2 and 3.
Table 2.
Pathological data across ethnoracial groups. Abbreviations CVD=Cerebrovascular disease, AD=Alzheimer’s disease, LBD=Lewy body disease, FTLD=Frontotemporal lobar degeneration
| Pathological Diagnosis of Dementia* | ||||
|---|---|---|---|---|
| Non-Hispanic White N = 360 |
Black N = 35 |
Hispanic N = 28 |
Total N = 423 |
|
| AD (non-mixed), n (%) | 156, (43 %) | 15, (43 %) | 4, (14 %) | 175, (41 %) |
| CVD (non-mixed) | 15 (4 %) | 4 (11 %) | 6 (21 %) | 25 (6 %) |
| LBD (non-mixed) | 13 (4 %) | 1 (3 %) | 1 (4 %) | 15 (4 %) |
| AD (mixed), n (%) | 134, (37 %) | 13, (37 %) | 15, (54 %) | 162, (38 %) |
| +CVD, n (%)** | 101, (28 %) | 14, (40 %) | 15, (54 %) | 130, (31 %) |
| +LBD, n (%)** | 65, (18%) | 4, (11%) | 7, (25%) | 76, (18%) |
| FTLD, n (%) | 30, (8 %) | 1, (3 %) | 1, (4 %) | 32, (8 %) |
| Hippocampal sclerosis, n (%) | 4, (1 %) | 1, (3 %) | 1, (4 %) | 6, (1 %) |
| Normal or no definite pathology, n (%) | 10, (3 %) | 1, (3 %) | 0, (0 %) | 11, (1 %) |
| CVD Pathologies# | ||||
| Macroscopic infarcts, n (%) | 96 (30.4%) | 8 (33.3%) | 9 (47.4%) | 113 (31.5%) |
| Total sample | N=316 | N=24 | N=19 | N=359 |
| Microscopic infarcts | 109 (35.5%) | 12 (54.5%) | 9 (52.9%) | 103 (37.6%) |
| Total sample | N=307 | N=22 | N=17 | N=346 |
| White matter rarefaction | 50 (15.9%)A | 9 (39.1%) | 3 (15.8%) | 62 (17.4%) |
| Total sample | N=314 | N=23 | N=19 | N=356 |
| Braak Neurofibrillary Tangle Stage | ||||
| BRAAK 0-II | 51 (14%) | 3 (9%) | 4 (14%) | 58 (14%) |
| BRAAK III-IV | 73 (20%) | 6 (17%) | 9 (32%) | 88 (21%) |
| BRAAK V-VI | 214 (59%)B | 26 (74%) | 12 (43%) | 252 (60%) |
| Missing/Unknown | 22 (6%) | 0 | 3 (11%) | 23 (5%) |
| CERAD Neuritic Plaque Score | ||||
| CERAD None | 53 (15%) | 1 (3%) | 4 (14%) | 58 (14%) |
| CERAD Sparse | 33 (9%) | 7 (20%) | 5 (18%) | 45 (11%) |
| CERAD Moderate | 60 (17%) | 6 (17%) | 9 (32%) | 75 (18%) |
| CERAD Frequent | 203 (56%) | 21 (60%) | 10 (36%) | 234 (55%) |
| Missing/Unknown | 11 (3%) | 0 (0%) | 0 (0%) | 11 (3%) |
Except for AD (non-mixed) and Normal or no definite pathology, diagnostic groups are not mutually exclusive.
(excluding amygdala predominant)
detailed data on CVD pathologies was not available for all cases, hence total sample are listed below each assessed pathology
Statistically different than Black decedents
Statistically different than Hispanic decedents
Table 3.
Severity of arteriolosclerosis, atherosclerosis, and cerebral amyloid angiopathy by ethnoracial group. NHW=Non-Hispanic White group.
| Arteriolosclerosis | Atherosclerosis | CAA | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Severity | White | Black | Hispanic | White | Black | Hispanic | White | Black | Hispanic |
| None | 18.4% | 13.0% | 21.1% | 12.7% | 4.8% | 10.5% | 36.1% | 27.3% | 38.9% |
| Mild | 40.8% | 26.1% | 36.8% | 46.3% | 33.3% | 31.6% | 36.1% | 27.3% | 38.9% |
| Moderate | 33.9% | 47.8% | 21.1% | 34.3% | 38.1% | 36.8% | 22.9% | 27.3% | 11.1% |
| Severe | 7.0% | 13.0% | 21.1% | 6.7% | 23.8% | 21.1% | 4.8% | 18.2% | 11.1% |
| Total persons assessed | 316 | 23 | 19 | 315 | 21 | 19 | 310 | 22 | 18 |
Clinical Pathological Correlations
Clinical pathological correlations for each individual were determined through presentations at monthly clinical pathological correlation conferences (CPC). The attendees at the CPC included all UCD ADC key personnel. Clinical, neuropsychological, and neuroimaging findings were presented without knowledge of the neuropathological results to render a final clinical diagnosis based on total clinical information available. The clinical summary was followed by presentation of neuropathological findings that were identified blinded to the clinical information. Detailed discussion of the clinical and pathological correlates led to a final consensus diagnosis where various pathological diagnoses were ranked in order of likelihood to contribute to the dementia syndrome. This included factors such as symptoms present and temporal onset, and degree and severity of the pathologic hallmarks of the neurodegenerative disease. For example, a slowly progressive memory predominant memory disorder with “high-likelihood” AD ranking according to current neuropathological criteria would be considered the primary cause of dementia, even in the presence of Lewy Body or CVD pathology sufficient to contribute to the dementia based on current neuropathological criteria for these disorders [25, 26, 33, 43] and the individual would be ascribed as having “AD mixed” dementia and the presence of AD and the comorbid disease would be separately recorded. Conversely, the presence of modest amounts of CVD or Lewy Bodies localized solely to the brain stem would not be considered contributory to the dementia and the diagnosis of “AD non-mixed” would be made.
Statistical Methods
To assess initial group differences, analyses of the frequency of each diagnosis and their combinations as well as frequency of data of Braak NFT stage, CERAD, infarctions, and white matter rarefaction across ethnoracial groups was assessed with a chi-square test of independence. For semiquantitative data of arteriolosclerosis and atherosclerosis, we dichotomized severity into less severe (0,1) and more severe (2,3) and assessed differences using Fisher exact tests. When simple tests found differences between groups, we ran additional analyses that controlled for age, APOE status, gender and education. Statistical significance was set at α=0.05.
Given recent literature suggesting mixed pathologies are more common among under-represented minorities, we sought to compare the prevalence of CVD and AD (non-mixed) pathology among three groups: Non-Hispanic White, Black, and Hispanics decedents by extending a previously described statistical approach [44, 45] to a logistic regression setting.
We began with a simple, unadjusted test of the null hypothesis of equal prevalence in Blacks or Hispanics with the prevalence in non-Hispanic Whites, via Fisher’s exact test comparing the observed proportions with CVD pathology. This simple comparison, however, did not account for observed differences between minority groups and non-Hispanic Whites in factors known to influence AD risk (i.e. age, gender, education and APOE ε4 allele presence). To adjust for the potential impact of these group differences in prevalent dementia risk factors, we employed a more sophisticated approach, formally testing the above null hypothesis. In brief, we estimated a logistic regression model to predict prevalence of CVD in non-Hispanic Whites, and then incorporated demographics and risk factor variables of Blacks and Hispanics into this model to test whether minorities with the same predictor profiles as non-Hispanic Whites have different predicted prevalence of CVD than were observed in their respective groups.
This approach had three components: in the first step, we fitted a logistic regression model in the sample for the non-Hispanic White group alone, including age, gender, education, recruitment source, and APOE genotype as predictors of CVD pathology as the outcome. Model validation checked assumptions of logit-linearity in age and years of education. We then applied this model to each of the individuals in the Black or Hispanic population, obtaining a predicted likelihood of CVD pathology for each individual. We summed these likelihoods to obtain a predicted total number of CVD cases under the null hypothesis that the minority group prevalence of CVD reflected exactly the relationship in the non-Hispanic White sample. In the second step, we compared the predicted total number of CVD cases in the Black or Hispanic subgroup to the actual observed number of CVD cases, using an exact probability calculation to obtain the chance of an observed number at least that extreme (two-sided p value.)
The first two steps treated the prediction based on the logistic regression as if that were known without error. To account for the sampling variation from the estimated logistic regression model, we further carried out a bootstrap replication of the first and second steps, 10,000 times. In each of the bootstrap replicates, we obtained a sample with replacement, of the same sample size, from the original non-Hispanic White sample. We then repeated the first step for each bootstrap replicate, fitting a new logistic regression model to the new sample of non-Hispanic Whites and predicting the total number of cases in the original Black or Hispanic groups. We followed by repeating the second step for each replicate, obtaining an exact calculation of the new p-value comparing the new prediction to the actual observed number of cases.
This bootstrap process provided 10,000 estimated p-values testing the null hypothesis that the Black or Hispanic population had a total number of CVD cases comparable to a non-Hispanic White group with similar predictor characteristics. The average across these 10,000 p-values provides an estimate of the probability of having a value as extreme as the observed prevalence in the Blacks or Hispanics, under the null hypothesis, accounting for the differences in predictors, and the sampling variation in both the smaller and larger groups. For visual reference, we illustrated with histograms that represent the distribution of the 10,000 bootstrap prevalences of CVD in the non-Hispanic White group and the 10,000 predicted prevalences of CVD in the Black and Hispanic groups, with the actual observed prevalence of CVD in each of the three groups shown for comparison.
As a secondary analysis and a means of further supporting our hypothesis, we ran an identical analysis that used AD (non-mixed) as an outcome. Recall that for the purposes of this analysis, ‘AD (non-mixed)’ was defined as having an AD related pathology, without presence of CVD or other dementia-related pathologies sufficient to contribute to dementia found at autopsy. For our analyses to be consistent with our hypothesis, we would expect an underestimation of CVD pathology in minority groups compared to what was observed, accompanied by an over estimation of the AD (non-mixed) outcome in minority groups. All analyses were carried out in R version 3.3.2.
RESULTS
Demographics
Neuropathological assessment was available for 360 non-Hispanic White, 35 Black, and 28 Hispanic decedents. Table 1 summarizes demographic and risk factor characteristics. Subjects were 81.65 ± 9.52 years on average at the time of their deaths and 52% were male. The mean level of educational attainment was 13.8 ± 3.5 years. Overall APOE ε4 prevalence was 78% and varied only slightly among groups (78% in non-Hispanic Whites, 69% in Blacks and 79% in Hispanics). Participants enrolled in the study who originally sought clinical evaluation for cognitive complaints constituted 91% of the decedents in this study and all decedents were severely cognitively impaired prior to death (Table 1). Group differences were apparent, both in demographics and in risk factors. The Hispanic cohort had less educational achievement than both non-Hispanic Whites and Blacks (p<0.005), and Blacks had significantly less education than non-Hispanic Whites (p<0.005). Blacks had the highest prevalence of hypertension (63%), and this was significantly higher than prevalence in non-Hispanic Whites (p<0.01).
To further examine the representativeness of this select autopsy sample, we also computed the prevalence of similar factors among all subjects enrolled in the UCD ADC longitudinal cohort with the diagnosis of dementia at final evaluation but without autopsy. The demographics of this cohort are summarized in detail in the supplemental table. There were no statistical differences between decedents in age and education when compared to demented individuals of the UCD ADC longitudinal cohort seen at final or most recent examination. In the longitudinal cohort, Black and Hispanic decedents were less likely to be male (37% and 36% versus 46% and 43%) when compared to our autopsied cohort. There were no statistical differences in APOE ε4 prevalence among autopsied decedents compared to the longitudinal cohort with dementia in Blacks (69% versus 62%) and non-Hispanic Whites (78% versus 60%), however, the APOE ε4 prevalence was significantly higher among Hispanic decedents than total Hispanic participants (79% versus 32%, P<0.001). The prevalence of vascular risk factors also tended to be lower among autopsied individuals than those enrolled in the parent cohort (see Supplementary Table).
Neuropathology
The breakdown of pathologies contributing to dementia is summarized in Tables 2 and 3, and Figure 1. The most common underlying neuropathology of dementia for all cases was AD (79.7%), with 80.5% among non-Hispanic Whites, 80% among Blacks, and 67.9% among Hispanics regardless of concomitant diagnosis. The second most common neuropathology contributing to dementia was CVD, with 31% of all cases, and 28% among non-Hispanic Whites, 37% among Black, and 54% among Hispanics. Importantly, no case was determined to have significant CVD due solely to the presence of hippocampal sclerosis.
Figure 1.
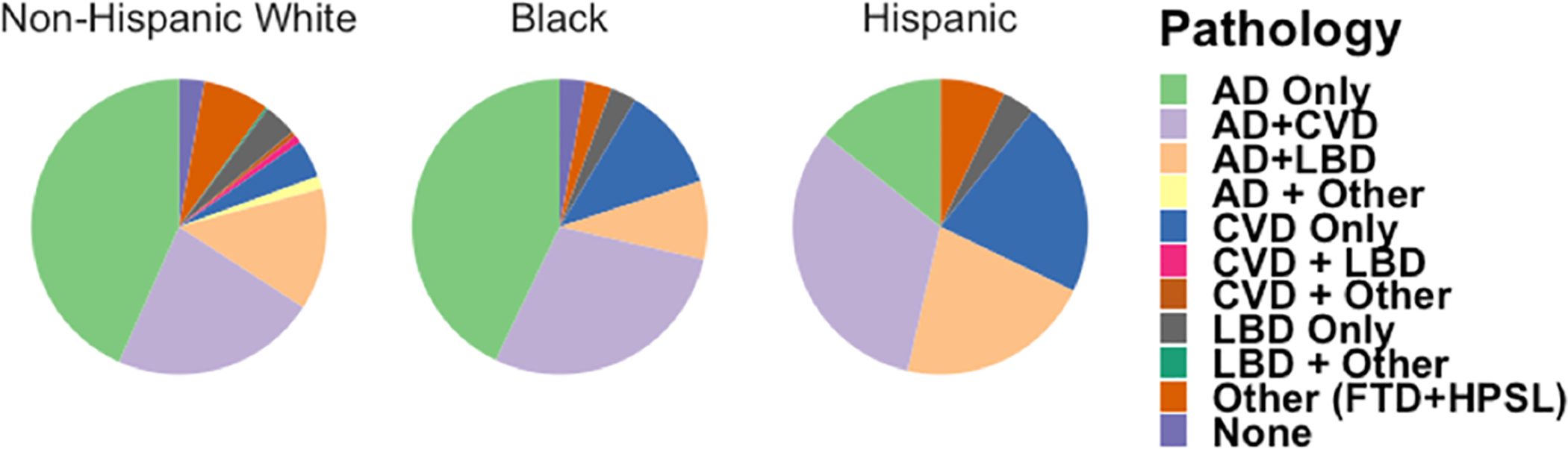
Ethnoracial differences in mixed pathology. Pie chart shows proportions of individual and mixed pathologies in Non-Hispanic White, Black and Hispanic decedents with dementia.
LBD was the least common pathology of the 3 major disease contributing to dementia (AD, CVD, and LBD) making up 18% of all cases; 18% of non-Hispanic Whites, 11% Black, and 25% Hispanics. Other diagnoses included FTLD (8%, 32 cases), hippocampal sclerosis (1%, 6 cases) and a normal (or no definite pathology) diagnosis (3%, 11 cases) despite the presence of clinical defined dementia. Using Fisher’s exact test to compare the observed proportions with CVD among the 3 groups, we found both Hispanic and Black participants had a higher observed proportion of CVD pathology (54% and 40% respectively) but only the Hispanic group had a significant difference from non-Hispanic Whites (28%), (p-values =0.003 and 0.08 respectively, Fisher exact test adjusted for multiple comparisons). There were no statistical differences in overall LBD pathology across groups (P values > 0.7). Fisher’s exact test to compare the observed proportions of macro and micro infarctions found no statistically significant differences in the presence of these pathologies in the 3 groups (P values > 0.3). However, Fisher’s exact test to compare the observed proportions of WMR found a statistically significant difference between non-Hispanic White and Black decedents (P value = 0.028); WMR was present in nearly 40% of Blacks compared to 15% in non-Hispanic Whites and Hispanics. This difference remained significant after controlling for age at death, APOE status, gender and education (OR = 4.8, 95% CI = 1.8–12.3). Black and Hispanic decedents also had more severe large and small vessel disease (atherosclerosis and arteriolosclerosis, respectively. Table 3) compared with non-Hispanic White decedents, though these differences were not statistically significant (P values > 0.2). Statistical testing of the various categories of AD pathology (i.e. CERAD and Braak Neurofibrillary Tangle Stage) was unstable due to the low number of observations in certain cells. However, the proportion of demented Hispanics in CERAD frequent (36% vs 56% and 60%) and Braak Stage V-VI (43% vs 59% and 74%) categories was smaller than their non-Hispanic or Black counterparts (Table 2).
Overall, more than 90% of cases had one or more of the following clinicopathological diagnosis: LBD, CVD, and/or AD. A breakdown of each mixed pathology is located in Table 2 and Figure 1. As there were many categories of mixed pathologies and many were unstable due to the low number of observations in certain cells, we collapsed data. Using Fisher’s exact test to compare the observed proportions with AD (non-mixed) among the 3 groups, we found Hispanics had a markedly lower observed proportion of AD (non-mixed) diagnoses (14%) than both non-Hispanic Whites (43%) and Blacks (43%), (P’s = 0.04 and 0.05 respectively, Fisher exact test adjusted for multiple comparisons). The prevalence of AD (non-mixed) among Blacks was 43% and not significantly different from non-Hispanic Whites (P = 0.47, Fisher exact test adjusted for multiple comparisons).
Bootstrapping and logistic regression of CVD and AD (non-mixed)
To provide more adequate interpretation, although based on a small sample size, group differences in the distribution of potential dementia risk factors were next addressed through bootstrap estimation of the predicted proportion of CVD contributing to the dementia for a comparable group of non-Hispanic Whites (Figure 2). The histograms in Figure 2 (top) represent the distribution of predicted prevalence of CVD for each minority group, if their likelihood were identical to that based on a (randomly resampled) similar group of non-Hispanic Whites. Similar figures for the AD (non-mixed) pathology are presented in Figure 2 (bottom).
Figure 2.
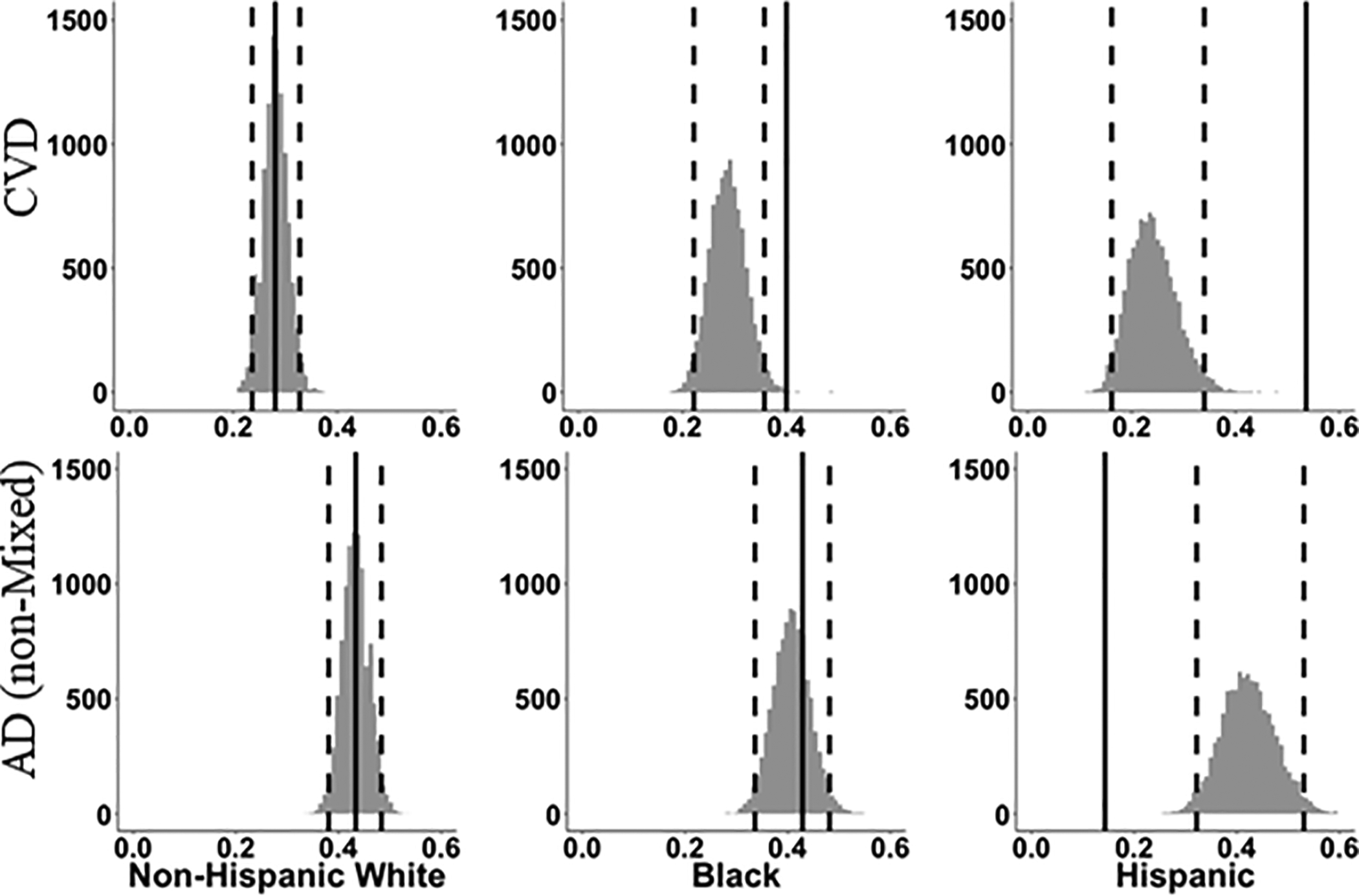
Predicted prevalence of CVD (top) and AD (non-mixed) (bottom). Histograms representing the distribution of predicted prevalence of CVD (top) and AD (non-mixed) (bottom) for each ethnoracial group if their likelihood were identical to that based on a (resampled) similar group of non-Hispanic Whites decedents using predictor variables of age at death, gender, education, recruitment source and APOE genotype. Dotted vertical lines include 95% of bootstrap resamples, while solid vertical red line represents the observed prevalence.
For Blacks, the prevalence estimates of CVD pathology contributing to dementia based on a non-Hispanic White sample with similar predictor values were centered at 28% with 95% of estimates between 22% and 36%. The average p-value, comparing the Black observed prevalence of 40% to these null-hypothesis estimates, was 0.20, suggesting that while the proportion of Blacks with CVD pathology, exceeded 95% of the bootstrap estimates, we cannot rule out that this was due to chance variation for that sample size when adjusting for age at death, sex, education, APOE status, and recruitment source. For Hispanics, the prevalence estimates of CVD pathology based on non-Hispanic White samples were all less than the actual observed value of 54%; the mean prediction was 24% with 95% of the bootstrap estimates between 16% and 34%. The mean of 2-sided p-values for comparisons of observed prevalence to bootstrap-sample predicted was 0.005, suggesting that this finding was not due to chance variation.
In summary, we could conclude with high confidence the observed proportion of CVD contributing to dementia in the Hispanics was different from the predicted proportions from the non-Hispanic White models within the UCD-ADC research cohort. We can also conclude that, although the observed proportion of CVD contributing to dementia in the Blacks was outside 95% of the bootstrap estimates, there is a possibility this finding may have occurred by chance given the small sample size of Black decedents.
Group differences in the distribution of potential dementia risk factors were next addressed through bootstrap estimation of the predicted proportion of AD (non-mixed). For Blacks, the prevalence estimates of AD (non-mixed) pathology based on a non-Hispanic White sample with similar predictor values were centered at 41% with 95% of estimates between 33% and 48%. The average p-value, comparing the Black observed prevalence of 43% to these null-hypothesis estimates, was 0.74, suggesting that the proportion of Blacks with AD (non-mixed) pathology, was within chance variation for that sample size when adjusting for age at death, sex, education, APOE status, and recruitment source.
For Hispanics, the prevalence estimates of AD (non-mixed) pathology based on non-Hispanic White samples were all more than the actual observed value of 14%; the mean prediction was 42% with 95% of the bootstrap estimates between 32% and 53%. The mean of 2-sided p-values for comparisons of observed prevalence to bootstrap-sample predicted was 0.008.
In summary, we could not conclusively conclude the observed proportion of AD (non-mixed) in the Blacks was different from the predicted proportions from the non-Hispanic White models, however, confidence of a difference between the non-Hispanic White and the Hispanic groups was very high in our UCD-ADC cohort.
DISCUSSION
Our findings from this research-based autopsy cohort suggest the prevalence of AD (non-mixed) in decedents with dementia is substantially lower among Hispanics than non-Hispanic White decedents, even after carefully accounting for group differences in known AD and CVD risk factors. The difference may be accounted for by a higher rate of CVD pathology, either as mixed dementia with AD or CVD alone (54% in Hispanics versus 28% in non-Hispanic Whites). Our findings among Black decedents are also generally consistent with Barnes et al.[19] where differences between Blacks and non-Hispanic Whites were less extreme but suggesting a higher prevalence of mixed pathologies, particularly CVD (40% in Blacks versus 28% in non-Hispanic Whites). These findings are also consistent with the generally higher prevalence of stroke risk factors for Blacks and Hispanics [46, 47]. Importantly, these differences were found in a relatively healthier group of decedents (i.e. less hypertension, hyperlipidemia and diabetes) than predicted from participants in our parent cohort, suggesting that our autopsy findings may underestimate the true effects of CVD on cognition in Black and Hispanic decedents involved in the larger UCD ADC research program. Caution should be taken, however, as this is a research-based cohort and cannot be considered a representative community sample.
Our analysis contributes to current AD research in four distinct ways. First, it is the only brain autopsy study to our knowledge carried out in a Hispanic cohort and across a non-Hispanic White, Black, and Hispanic sample using detailed participant demographic and health characteristics. Neuropathological evaluations were conducted at the same institution and by pathologists blinded to all demographic variables reducing potential bias in these evaluations. Second, as there may be discrepancies between self-identified race and genetic ancestry, we employed standard genetic statistical approaches, finding a 95% concordance between the two measures. Third, the sample from the UCD ADC is unique in that participants were not excluded based on prior stroke or CVD risk factors (as is often the case in AD cohort studies). Finally, the analytic methodology is novel and robust. Sample size and corresponding lack of power are often a concern in such studies. Our approach addresses these concerns by utilizing a bootstrap procedure that accounts for the differences in demographic and major gene effects between sample cohorts and for randomness and uncertainty both in the larger non-Hispanic White sample and in our relatively small minority samples.
Recent neuropathological studies between Black and non-Hispanic White participants with clinical AD have revealed differences in the prevalence of various pathologies[19, 48]. In the study by Barnes, et al.[19] 41 Black autopsy patients diagnosed clinically as having probable or possible AD were matched by age, gender, education and last Mini-Mental State Exam (MMSE) with 81 White autopsies. While AD pathology was common, AD as the single dementia pathology was considerably less common among Black participants (19.5%) as compared to their non-Hispanic White counterparts (42%), whereas AD mixed with an additional pathology (Lewy bodies or vascular brain injury) was substantially more common (70.7% vs 50.6%). Graff-Radford et al.[48] utilizing data from the National Alzheimer’s Coordinating Center’s (NACC), examined neuropathological differences between Black and non-Hispanic White decedents. After controlling for APOE ε4 allele presence, the difference in AD pathology between Blacks and non-Hispanic Whites was not significant.
An unexpected finding of our study, however, was the much higher prevalence of the APOE ε4 allele among the Hispanic decedents when compared to our overall Hispanic dementia sample. Sampling bias due to small sample size as well as volunteerism in a research cohort could be potential confounders. APOE ε4, well-established as the major genetic risk factor for late-onset AD, may also increase the risk for dementia through multiple pathways including vascular disease and inflammation [49]. The interaction between vascular risk and APOE ε4 on dementia, including dementia prevalence in Hispanic populations, has been reported [16, 50–53]. APOE ε4 may have pleiotropic effects that leads to increased likelihood of concurrent vascular disease. At least some of these effects may be due to increased deposition of amyloid [52], but the presence of the APOE ε4 allele may also be associated with vascular cognitive impairment [54] or increased brain injury due to vascular disease [55], however these studies had small sample sizes and many co-pathologies were overlooked. As a consequence, the prevalence of APOE ε4 in our Hispanic decedents may reflect greater risk for dementia due to factors beyond increasing AD pathology, however, more research is needed to test this hypothesis. As autopsy studies, especially with Hispanics are lacking, more research also is needed to understand the significance of these results as our current cohort of convenience may not generalizable to most Hispanics. Finally, these data must be interpreted carefully given the small sample size and potential cohort bias, and further, collaborative efforts on larger, well-characterized Hispanic/Latino cohorts is necessary to confirm or refute these findings.
When examining ethnoracial differences many caveats and limitations can exist as discussed by Ighodaro et al. [12]. A key limitation of this and other studies of minority cohorts [19, 48] as well as those involving autopsies, is that they are based on samples of convenience, consisting of volunteers (those who have agreed to have an autopsy), in addition to recruitment methods that can introduce biases. The UCD ADC does not exclude participants on the basis of prior or current cardiovascular risk factors or disease, and therefore when compared to national averages, rates of key risk factors in our sample (Table 1) are similar to those of the general population [47]. However, cardiovascular risk factors within the UCD ADC autopsy cohort were lower than that of demented individuals of an ethnic/racial minority enrolled in the parent cohort, suggesting that the effects found in the decedents should not be extrapolated to larger populations. In addition, the UCD ADC emphasizes recruitment of diverse populations as compared to recruitment of more specialized cases such as individuals with LBD or FTLD. This difference in recruitment efforts may explain the low frequencies of LBD and differences in our results compared to that of Barnes et al.[18]. Also, TDP-43 was not evaluated and previous studies have demonstrated differences in FTLD-TDP-43 [48]. We aim to evaluate TDP-43 in future studies. Lastly, as there are select biases within cohort studies results should not be considered generalizable to populations.
In the study, analyses were controlled for education, age at death, and APOE status. However, as stated above, there are many factors that may contribute to underlying differences in pathology which could not be adequately evaluated in this current study. Although numbers are small in the current study, which may limit the ability to detect subtle differences, we employed robust statistical techniques to aid in analysis. This study focused on major dementia diagnoses and did not thoroughly evaluate the contributions of other pathologies found in elderly cohorts (such as argyrophilic grains and TDP-43 deposition in the setting of AD) as well as the location and density of these pathologies. We aim to pursue these aspects in future studies to give a more detailed neuropathological landscape. If neuropathological differences in decedents with dementia truly do exist between ethnoracial groups, it is important to fully understand the underlying disease mechanisms and further research is warranted.
Our approach can be viewed as an enhanced version of a match study. In match studies (e.g., Barnes et al.[19]), participants across distinct groups are ‘matched’ according to key demographic/health related characteristics. Matching participants across groups helps to ensure key characteristics are accounted for (and therefore not the reason for) differences found between groups. This method helps to account for relevant predictors without directly including them into the model, which due to sample sizes would not be advisable. A drawback of this method, however, is all available data are often not used as those who do not ‘match’ on all characteristics are excluded from the analysis. In contrast, our method utilizes all available information. Similar to a propensity score regression approach, our method uses the non-Hispanic White group to estimate a probability of CVD and AD (non-mixed) pathology given relevant characteristics (i.e., propensity score). Propensity scores are then obtained for each participant in the minority groups, summed across the group, and compared to the actual proportions of CVD and AD (non-mixed) pathology observed. The bootstrap procedure allows one to generate the distribution of these propensity scores and therefore address issues of randomness and uncertainty in the propensity estimation process from the larger sample, while addressing sampling variation in the smaller sample through direct calculation of the exact p-value conditional on the current bootstrap estimate. Averaging across bootstrap samples uses a basic property of conditional expectation to obtain an overall p-value.
Our results indicate that, controlling for age, gender, education, APOE status, and recruitment source, Hispanic decedents with dementia prior to death in a research-based autopsy cohort are more likely to have clinicopathologically defined CVD contributing to their dementia than non-Hispanic White decedents. While our results do not rule out the possibility that the same may be true for the Black decedent cohort, they suggest any difference may be smaller and larger more community-based autopsy samples are required to further test this hypothesis. In addition, our results indicate that, controlling for age, gender, education, APOE status and recruitment source, non-Hispanic White decedents with dementia prior to death are more likely to have AD (non-mixed) pathology than Hispanics. If this hypothesis holds true in other cohorts, these findings could aid in enhancement of public health endeavors. First, the higher prevalence of CVD pathology in non-White decedents offers promise that alternative dementia treatment strategies may be more successful in individuals with similar characteristics. Second, given that the US older population is becoming more diverse and multiple pathologies are being recognized as contributing to the dementia syndrome [3], even if the clinical phenotype is that of AD [56], a precision medicine approach taking into account many factors that can have associations with ethnoracial groups may become increasingly important to the diagnosis and management of late-life dementia [57]. Future studies containing a more representative population sample having data on social, economic, cultural, and behavioral characteristics are needed to further understand the complexity of dementia in all individuals.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank the families and participants of the UCD ADC for their generous donations. This study was funded by the NIH [P30 AG010129], which had no role in any aspect of the study, including study design, data collection, analysis or writing.
Footnotes
CONFLICT OF INTEREST/DISCLOSURE STATEMENT
With the exception of Drs. Charles DeCarli and Brittany Dugger, the authors have no conflict of interest to report. Dr. DeCarli is a consultant to Novartis. Dr. Dugger has received previous funding from Daiichi Sankyo unrelated to this project. This article was prepared while Dr. Reed was employed at U.C. Davis. The opinions expressed in this article are the author’s own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States government.
REFERENCES
- [1].Ortman JM, Velkoff VA, H H (2014) in Current Population Reports U.S. Census Bureau, Washington, D.C., pp. p25–1140. [Google Scholar]
- [2].Alzheimer’s A (2016) 2016 Alzheimer’s disease facts and figures. Alzheimers Dement 12, 459–509. [DOI] [PubMed] [Google Scholar]
- [3].Schneider JA, Arvanitakis Z, Bang W, Bennett DA (2007) Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69, 2197–2204. [DOI] [PubMed] [Google Scholar]
- [4].Kivipelto M, Ngandu T, Laatikainen T, Winblad B, Soininen H, Tuomilehto J (2006) Risk score for the prediction of dementia risk in 20 years among middle aged people: a longitudinal, population-based study. Lancet Neurol 5, 735–741. [DOI] [PubMed] [Google Scholar]
- [5].Exalto LG, Quesenberry CP, Barnes D, Kivipelto M, Biessels GJ, Whitmer RA (2014) Midlife risk score for the prediction of dementia four decades later. Alzheimers Dement 10, 562–570. [DOI] [PubMed] [Google Scholar]
- [6].Thal DR, Rub U, Orantes M, Braak H (2002) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. [DOI] [PubMed] [Google Scholar]
- [7].Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259. [DOI] [PubMed] [Google Scholar]
- [8].Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24, 197–211. [DOI] [PubMed] [Google Scholar]
- [9].Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L, Liesinger AM, Petersen RC, Parisi JE, Dickson DW (2016) Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol 131, 571–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Alafuzoff I, Arzberger T, Al-Sarraj S, Bodi I, Bogdanovic N, Braak H, Bugiani O, Del-Tredici K, Ferrer I, Gelpi E, Giaccone G, Graeber MB, Ince P, Kamphorst W, King A, Korkolopoulou P, Kovacs GG, Larionov S, Meyronet D, Monoranu C, Parchi P, Patsouris E, Roggendorf W, Seilhean D, Tagliavini F, Stadelmann C, Streichenberger N, Thal DR, Wharton SB, Kretzschmar H (2008) Staging of neurofibrillary pathology in Alzheimer’s disease: a study of the BrainNet Europe Consortium. Brain Pathol 18, 484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, Sasse J, Boyer S, Shirohi S, Brooks R, Eschbacher J, White CL 3rd, Akiyama H, Caviness J, Shill HA, Connor DJ, Sabbagh MN, Walker DG (2009) Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117, 613–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ighodaro ET, Nelson PT, Kukull WA, Schmitt FA, Abner EL, Caban-Holt A, Bardach SH, Hord DC, Glover CM, Jicha GA, Van Eldik LJ, Byrd AX, Fernander A (2017) Challenges and Considerations Related to Studying Dementia in Blacks/African Americans. J Alzheimers Dis 60, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Breitkopf CR (2009) Attitudes, beliefs and behaviors surrounding organ donation among Hispanic women. Curr Opin Organ Transplant 14, 191–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vega IE, Cabrera LY, Wygant CM, Velez-Ortiz D, Counts SE (2017) Alzheimer’s Disease in the Latino Community: Intersection of Genetics and Social Determinants of Health. J Alzheimers Dis 58, 979–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sacco RL, Boden-Albala B, Abel G, Lin IF, Elkind M, Hauser WA, Paik MC, Shea S (2001) Race-ethnic disparities in the impact of stroke risk factors: the northern Manhattan stroke study. Stroke 32, 1725–1731. [DOI] [PubMed] [Google Scholar]
- [16].Haan MN, Mungas DM, Gonzalez HM, Ortiz TA, Acharya A, Jagust WJ (2003) Prevalence of dementia in older latinos: the influence of type 2 diabetes mellitus, stroke and genetic factors. J Am Geriatr Soc 51, 169–177. [DOI] [PubMed] [Google Scholar]
- [17].Froehlich TE, Bogardus ST Jr., Inouye SK (2001) Dementia and race: are there differences between African Americans and Caucasians? J Am Geriatr Soc 49, 477–484. [DOI] [PubMed] [Google Scholar]
- [18].Barnes LL, Leurgans S, Aggarwal NT, Shah RC, Arvanitakis Z, James BD, Buchman AS, Bennett DA, Schneider JA (2015) Mixed pathology is more likely in black than white decedents with Alzheimer dementia. Neurology 85, 528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Barnes LL, Leurgans S, Aggarwal NT, Shah RC, Arvanitakis Z, James BD, Buchman AS, Bennett DA, Schneider JA (2015) Mixed pathology is more likely in black than white decedents with Alzheimer dementia. Neurology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tang MX, Stern Y, Marder K, Bell K, Gurland B, Lantigua R, Andrews H, Feng L, Tycko B, Mayeux R (1998) The APOE-epsilon4 allele and the risk of Alzheimer disease among African Americans, whites, and Hispanics. Jama 279, 751–755. [DOI] [PubMed] [Google Scholar]
- [21].Soria JA, Huisa BN, Edland SD, Litvan I, Peavy GM, Salmon DP, Hansen LA, Galasko DR, Brewer JB, Gonzalez HM, Rissman RA (2018) Clinical-Neuropathological Correlations of Alzheimer’s Disease and Related Dementias in Latino Volunteers. J Alzheimers Dis. [DOI] [PubMed] [Google Scholar]
- [22].Barnes DE, Yaffe K (2011) The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol 10, 819–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hinton L, Carter K, Reed BR, Beckett L, Lara E, DeCarli C, Mungas D (2010) Recruitment of a community-based cohort for research on diversity and risk of dementia. Alzheimer Dis Assoc Disord 24, 234–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Roman GC, Tatemichi TK, Erkinjuntti T, Cummings JL, Masdeu JC, Garcia JH, Amaducci L, Orgogozo JM, Brun A, Hofman A, et al. (1993) Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology 43, 250–260. [DOI] [PubMed] [Google Scholar]
- [25].Knopman DS, Parisi JE, Boeve BF, Cha RH, Apaydin H, Salviati A, Edland SD, Rocca WA (2003) Vascular dementia in a population-based autopsy study. Arch Neurol 60, 569–575. [DOI] [PubMed] [Google Scholar]
- [26].Gold G, Giannakopoulos P, Herrmann FR, Bouras C, Kovari E (2007) Identification of Alzheimer and vascular lesion thresholds for mixed dementia. Brain 130, 2830–2836. [DOI] [PubMed] [Google Scholar]
- [27].Boise L, Hinton L, Rosen HJ, Ruhl M (2017) Will My Soul Go to Heaven If They Take My Brain? Beliefs and Worries About Brain Donation Among Four Ethnic Groups. Gerontologist 57, 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Boise L, Hinton L, Rosen HJ, Ruhl MC, Dodge H, Mattek N, Albert M, Denny A, Grill JD, Hughes T, Lingler JH, Morhardt D, Parfitt F, Peterson-Hazan S, Pop V, Rose T, Shah RC (2017) Willingness to Be a Brain Donor: A Survey of Research Volunteers From 4 Racial/Ethnic Groups. Alzheimer Dis Assoc Disord 31, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Morris JC, Weintraub S, Chui HC, Cummings J, Decarli C, Ferris S, Foster NL, Galasko D, Graff-Radford N, Peskind ER, Beekly D, Ramos EM, Kukull WA (2006) The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis Assoc Disord 20, 210–216. [DOI] [PubMed] [Google Scholar]
- [30].Weintraub S, Salmon D, Mercaldo N, Ferris S, Graff-Radford NR, Chui H, Cummings J, DeCarli C, Foster NL, Galasko D, Peskind E, Dietrich W, Beekly DL, Kukull WA, Morris JC (2009) The Alzheimer’s Disease Centers’ Uniform Data Set (UDS): the neuropsychologic test battery. Alzheimer Dis Assoc Disord 23, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Blessed G, Tomlinson BE, Roth M (1988) Blessed-Roth Dementia Scale (DS). Psychopharmacol Bull 24, 705–708. [PubMed] [Google Scholar]
- [32].(1997) Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging 18, S1–2. [PubMed] [Google Scholar]
- [33].McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M, Consortium on DLB (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65, 1863–1872. [DOI] [PubMed] [Google Scholar]
- [34].Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chui HC, Zarow C, Mack WJ, Ellis WG, Zheng L, Jagust WJ, Mungas D, Reed BR, Kramer JH, Decarli CC, Weiner MW, Vinters HV (2006) Cognitive impact of subcortical vascular and Alzheimer’s disease pathology. Ann Neurol 60, 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jagust W, Gitcho A, Sun F, Kuczynski B, Mungas D, Haan M (2006) Brain imaging evidence of preclinical Alzheimer’s disease in normal aging. Ann Neurol 59, 673–681. [DOI] [PubMed] [Google Scholar]
- [37].Riedl L, Mackenzie IR, Forstl H, Kurz A, Diehl-Schmid J (2014) Frontotemporal lobar degeneration: current perspectives. Neuropsychiatr Dis Treat 10, 297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rohrer JD, Lashley T, Schott JM, Warren JE, Mead S, Isaacs AM, Beck J, Hardy J, de Silva R, Warrington E, Troakes C, Al-Sarraj S, King A, Borroni B, Clarkson MJ, Ourselin S, Holton JL, Fox NC, Revesz T, Rossor MN, Warren JD (2011) Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain 134, 2565–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM, Lee VM (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122, 111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Besser LM, Kukull WA, Teylan MA, Bigio EH, Cairns NJ, Kofler JK, Montine TJ, Schneider JA, Nelson PT (2018) The Revised National Alzheimer’s Coordinating Center’s Neuropathology Form-Available Data and New Analyses. J Neuropathol Exp Neurol 77, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L (1991) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486. [DOI] [PubMed] [Google Scholar]
- [42].Beach TG, Wilson JR, Sue LI, Newell A, Poston M, Cisneros R, Pandya Y, Esh C, Connor DJ, Sabbagh M, Walker DG, Roher AE (2007) Circle of Willis atherosclerosis: association with Alzheimer’s disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol 113, 13–21. [DOI] [PubMed] [Google Scholar]
- [43].Román GC, Tatemichi TK, Erkinjuntti T, Cummings JL, Masdeu JC, Garcia JH, Amaducci L, Orgogozo JM, Brun A, Hofman A, Moody DM, O’ Brien MD, Yamaguchi T, Grafman J, Drayer BP, Bennett DA, Fisher M, Ogata J, Kokmen E, Bermejo F, Wolf PA, Gorelick PB, Bick KL, Pajeau AK, Bell MA, DeCarli C, Culebras A, Korczyn AD, Bogousslavsky J, Hartmann A, Scheinberg P (1993) Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop [see comments]. Neurology 43, 250–260. [DOI] [PubMed] [Google Scholar]
- [44].Gray MW, Scott LE (1980) A “Statistical” Remedy for Statistically Identified Discrimination. Academe 66, 174–181. [Google Scholar]
- [45].Ferree MM, McQuillan J (1998) Gender-based Pay Gaps. Methodological and Policy Issues in University Studies. Gender & Society 12, 7–39. [Google Scholar]
- [46].Wolf PA, D'Agostino RB, Belanger AJ, Kannel WB (1991) Probability of stroke: a risk profile from the Framingham Study. Stroke; a journal of cerebral circulation 22, 312–318. [DOI] [PubMed] [Google Scholar]
- [47].Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB, American Heart Association Statistics C, Stroke Statistics S (2015) Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation 131, e29–322. [DOI] [PubMed] [Google Scholar]
- [48].Graff-Radford NR, Besser LM, Crook JE, Kukull WA, Dickson DW (2016) Neuropathologic differences by race from the National Alzheimer’s Coordinating Center. Alzheimers Dement 12, 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9, 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Haan MN, Weldon M (1996) The influence of diabetes, hypertension, and stroke on ethnic differences in physical and cognitive functioning in an ethnically diverse older population. Ann Epidemiol 6, 392–398. [DOI] [PubMed] [Google Scholar]
- [51].Haan MN, Shemanski L, Jagust WJ, Manolio TA, Kuller L (1999) The role of APOE epsilon4 in modulating effects of other risk factors for cognitive decline in elderly persons. Jama 282, 40–46. [DOI] [PubMed] [Google Scholar]
- [52].Peila R, Rodriguez BL, Launer LJ, Honolulu-Asia Aging S (2002) Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes 51, 1256–1262. [DOI] [PubMed] [Google Scholar]
- [53].Carmelli D, Swan GE, Reed T, Miller B, Wolf PA, Jarvik GP, Schellenberg GD (1998) Midlife cardiovascular risk factors, ApoE, and cognitive decline in elderly male twins. Neurology 50, 1580–1585. [DOI] [PubMed] [Google Scholar]
- [54].Yin YW, Li JC, Wang JZ, Li BH, Pi Y, Yang QW, Fang CQ, Gao CY, Zhang LL (2012) Association between apolipoprotein E gene polymorphism and the risk of vascular dementia: a meta-analysis. Neurosci Lett 514, 6–11. [DOI] [PubMed] [Google Scholar]
- [55].DeCarli C, Reed T, Miller BL, Wolf PA, Swan GE, Carmelli D (1999) Impact of apolipoprotein E epsilon4 and vascular disease on brain morphology in men from the NHLBI twin study. Stroke 30, 1548–1553. [DOI] [PubMed] [Google Scholar]
- [56].Montine TJ, Koroshetz WJ, Babcock D, Dickson DW, Galpern WR, Glymour MM, Greenberg SM, Hutton ML, Knopman DS, Kuzmichev AN, Manly JJ, Marder KS, Miller BL, Phelps CH, Seeley WW, Sieber BA, Silverberg NB, Sutherland M, Torborg CL, Waddy SP, Zlokovic BV, Corriveau RA, Committee ACO (2014) Recommendations of the Alzheimer’s disease-related dementias conference. Neurology 83, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hampel H, O’Bryant SE, Durrleman S, Younesi E, Rojkova K, Escott-Price V, Corvol JC, Broich K, Dubois B, Lista S, Alzheimer Precision Medicine I (2017) A Precision Medicine Initiative for Alzheimer’s disease: the road ahead to biomarker-guided integrative disease modeling. Climacteric 20, 107–118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.