

Abstract
Nutrition deprivation (ND) is a common feature of the tumor microenvironment. Tumor cells, therefore, frequently develop resistance mechanisms against ND. One of these mechanisms is the activation of the AMP-activated protein kinase (AMPK), which promotes cell survival under ND. AMPK activation promotes the activity of eukaryotic elongation factor 2 kinase (eEF2K), thereby blocking protein synthesis. The results of the present study indicated the inhibiting effect of AMPK activation on mitogen-activated protein kinase (ERK1/2) activity, which in turn downregulates G1/S transition and promotes cell survival by mediating eEF2K under ND. The knockdown of ERK1/2 enhances cancer cell survival under ND. In the presence of nutrients, eEF2k interacts with dual-specificity mitogen-activated protein kinase kinase (MEK)1/2, conferring a positive feedback loop via MEK1/2-ERK1/2-ribosomal protein S6 kinase α-1-eEF2K signaling, leading to the constitutive activation of ERK1/2. By contrast, under acute ND, AMPK activation blocked the interaction between eEF2K and MEK1/2, contributing to the increased resistance of cancer cells to ND. The present findings reveal a previously undiscovered mechanism that uses AMPK activation to mediate ERK1/2-regulated protein synthesis and cell survival by inhibiting eEF2K-MEK1/2 interaction under ND conditions.
Keywords: eukaryotic elongation factor 2 kinase, nutrition deprivation, AMP-activated protein kinase, positive feedback loop
Introduction
Nutrition deprivation (ND) is a common feature of the microenvironment of tumor cells. Under the stress of ND, biosynthesis is suppressed, preserving energy to enable the survival of the cancer cells (1). AMP-activated protein kinase (AMPK) is a key energy sensor, which functions by regulating the intracellular metabolism for the maintenance of energy homeostasis, and serves an important role in the survival of cancer cells under ND (2). AMPK activation maintains energy by inhibiting biosynthesis via the suppression of mammalian target of rapamycin (mTOR) complex 1 (3). It has been reported that AMPK activation promotes cell survival by phosphorylating eukaryotic elongation factor 2 kinase (eEF2K) and blocking protein translation elongation under chronic ND conditions (4,5).
Mitogen-activated protein kinase (ERK1/2) serves a key role in mediating cell growth and the G1/S transition during the cell cycle (6,7). ERK1/2 signaling is closely associated with the induction of cyclin D1, which regulates the functions of cyclin D-dependent kinases (CDKs) and the phosphorylation of the retinoblastoma (Rb) protein. Rb phosphorylation disrupts its association with transcription factor E2F, allowing the coordinated transcription of genes required for DNA replication (8,9). Furthermore, ERK1/2 drives the development of some types of cancer by activating CDKs and the mTOR pathway, leading to cell cycle progression and protein synthesis (10,11). Conversely, overactivation of ERK1/2 may lead to proliferation arrest, apoptosis, autophagy and senescence (12). Therefore, it is conceivable that ERK1/2-induced G1/S transition, and the ensuing protein synthesis may be suppressed in order to support the survival of cancer cells under ND. However, the underlying mechanisms for ND-induced suppression of G1/S transition and protein synthesis remain unclear.
The results of the present study indicated the blocking effect of ND-induced AMPK activation on the interaction between eEF2K and dual-specificity mitogen-activated protein kinase kinase (MEK)1/2, leading to the disruption of the MEK1/2-ERK1/2-ribosomal protein S6 kinase α-1 (p90RSK)-eEF2K-MEK1/2 signaling loop, and thus to the deactivation of ERK1/2. The findings uncover a mechanism that uses AMPK activation to deactivate ERK1/2 by suppressing the interaction between eEF2K and MEK1/2, thus promoting the survival of cancer cells under ND conditions.
Materials and methods
Materials
Cell lines MKN45 and MG-63 were purchased from the Shanghai Institute of Biochemistry and Cell Biology. Gibco RPMI-1640 and Dulbecco's modified Eagle's medium (DMEM)were from Thermo Fisher Scientific, Inc., and fetal bovine serum (FBS) was from Hangzhou Sijiqin Biological Engineering Materials Co., Ltd. Radioimmunoprecipitation assay and NP-40 lysis buffers, PD98059 [2-(2-amino-3-methoxyphenyl)chromen-4-one, a specific inhibitor of MEK1/2 for downregulating ERK1/2 activity], MTT, phenylmethylsulfonylfluoride (PMSF), and BeyoECL Star detection reagents were from Beyotime Institute of Biotechnology. Monoclonal antibodies against p-ERK1/2 Thr202/Tyr204 (cat. no. 4370S), p-eEF2K Ser366 (cat. no. 3691S), and AlexaFluor® 555-(cat. no. 4413S) and 488-(cat. no. 4408S) conjugated antibodies were purchased from Cell Signaling Technology Inc., and polyclonal antibodies against ERK1/2 (cat. no. 16443-1-AP), MEK1/2 (cat. no. 20348-1-AP), eEF2K (cat. no. 13510-1-AP) and α-actin (cat. no. 23660-1-AP) were obtained from Wuhan Sanying Biotechnology. All primary antibodies were diluted in PBS at 1:500. Horseradish peroxidase-conjugated mouse anti-rabbit secondary antibodies (cat. no. D110059; 1:1,000 dilution) were purchased from Shanghai Sangon Biotech Co., Ltd.).
Cell culture
Cells were cultured at 37°C in RPMI-1640, or DMEM supplemented with 10% FBS. Experiments were performed on 70–80% confluent cells. The cells were treated with acadesine (AICAR; Beyotime Institute of Biotechnology) or ND. ND was established by incubating cells in Hank's balanced salt solution (HBSS)-4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (0.185 g/l CaCl2•2H2O, 0.2 g/l MgSO4•7H2O, 0.4 g/l KCl, 0.06 g/l KH2PO4, 0.35 g/l NaHCO3, 8 g/l NaCl, 0.09 g/l Na2HPO4•7H2O, 20 mM HEPES, pH 7.4) (11) containing neither glucose nor amino acids.
Cell death assay
Cells were separated into 4 groups, CTRL (control), PD98059 (20 µg/ml, the inhibitor of MEK1/2), siCTRL (siRNA control), and sieEF2K (siRNA interference of eEF2K). All the groups were cultured under ND. In order to assess cell death, cells were diluted 1:10 in 0.4% Trypan blue (Sigma-Aldrich; Merck KGaA) and counted in a hemocytometer. The mean percentage of dead (Trypan blue-positive) cells was calculated from three independent samples of each cell suspension.
Western blotting
Protein samples were collected by lysing cells with radioimmunoprecipitation buffer (Beyotime Institute of Biotechnology) following the various treatments. Protein concentrations were determined using the Bradford assay (Beyotime Institute of Biotechnology). Proteins (20–30 µg) were separated by SDS-PAGE (10% gels). The separated proteins were then transferred to a nitrocellulose membrane. Membranes were then blocked with 5% skimmed milk in PBS at room temperature for 50 min. Primary antibodies (as aforementioned) were used in a 1:500 dilution and secondary antibodies in 1:2,000. The protein bands on the membrane were visualized using BeyoECL Star detection reagents (Beyotime Institute of Biotechnology) and quantified with ImageJ v.1.48 (National Institutes of Health).
RNA interference of eEF2K and ERK1/2
Short RNA sequences for RNA interference were designed and synthesized by Shanghai GenePharma Co., Ltd. MKN45 and MG-63 cells were placed in 96-well plates (for measurement of cell growth), 24-well plates (for cell death assay) or six-well plates (for western blotting or reverse transcription-quantitative polymerase chain reaction) with a density of 1.5×104, 1×105 or 4×105 cells per well, respectively. The cells were then transfected with 25 nM control siRNA (5′-UUCUCCGAACGUGUCACGUTT-3′), a human eEF2K-targeting siRNA (5′-GCUGGCCAUGAUGGUGAUUTT-3′) or a human ERK1/2-targeting siRNA (5′-GCAAUGACCAUAUCUGCUATT-3′), using the siRNA transfection reagent (Santa Cruz Biotechnology, Inc.) according to the manufacturer's protocols. The transfection efficiency was evaluated by western blotting.
Confocal fluorescence microscopy
Cells grown on glass coverslips were fixed with 4% formalin for 20 min at room temperature, permeabilized with 0.5% Triton X-100/PBS for 15 min and blocked with 5% bovine serum albumin (Beyotime Institute of Biotechnology) at room temperature for 1 h. The cells were incubated with primary antibodies(anti-MEK1/2 and anti-p-eEF2k Ser366) at room temperature for 2 h, washed and then incubated at room temperature with AlexaFluor-conjugated secondary antibodies for 1 h, prior to washing and mounting on glass slides. Finally, the cells were observed using a confocal fluorescence microscope (Olympus I xSLA; Olympus Corporation, Tokyo, Japan) and images were captured (magnification, ×200).
Co-immunoprecipitation (co-IP) assay
The native protein was prepared in NP-40 lysis buffer or in PBS solution (100 mM PMSF). Antibodies against MEK1/2 (1:50 dilution) and ERK1/2 (1:50 dilution) were incubated with the total protein lysate with rotation at 4°C for 6 h. Protein A/G-agarose beads (cat. no. sc-2003; Santa Cruz Biotechnology, Inc.) were added and rotated for another 2 h at 4°C. The agarose beads were then washed 3 times with ice-cold PBS. Finally, the proteins were eluted from the agarose beads using SDS and analyzed against p-eEF2K Ser366 (1:500 dilution, cat. no. 3691S; Cell Signaling Technology, Inc.) by western blotting as aforementioned.
Statistical analysis
Cell death data are expressed as the mean ± standard deviation and analyzed with one-way analysis of variance and Bonferroni's post-hoc test. Statistical calculations were performed using SPSS (v.16.0; SPSS Inc.). P<0.05 was considered to indicate a statistically significant difference.
Results
ERK1/2 activation promotes cell death induced by ND
The signaling pathways for ND-induced cell death remain unclear. It is known that the activation of ERK1/2 can either promote cell survival and proliferation or induce apoptotic cell death (13). To investigate the role of ERK1/2 in ND-induced cell death, its activation was inhibited by siRNA knockdown of ERK1/2 or the upstream signaling of ERK1/2 was inhibited by the MEK1/2 inhibitor PD98059 in the gastric cancer MKN45 cell line. The cell death assay revealed that MKN45 cell death significantly increased following ND treatment from 12 to 18%, at 4 and 8 h, respectively (Fig. 1), while ERK1/2 siRNA or PD98059 significantly attenuated cell death comparing siRNA control or ND treatment only, respectively. This result indicates that downregulating ERK1/2 activity promotes the resistance of cells under ND.
Figure 1.
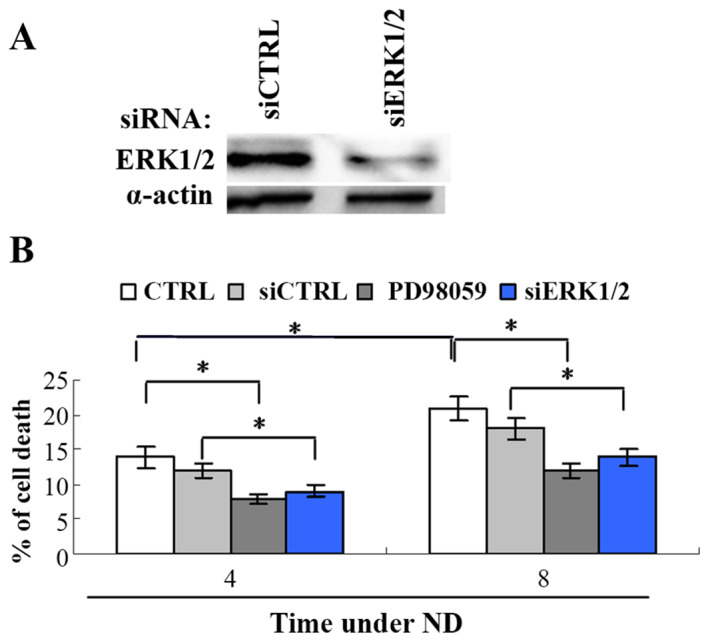
Downregulation of the activity of MEK1/2 or ERK1/2 inhibitedND-induced cell death. MEK1/2 activity was inhibited by its inhibitor PD98059 and ERK1/2 activity was inhibited by siRNA knockdown of ERK1/2 in MKN45 cells. Cells were transiently transfected with 25 nM siCTRL or siERK1/2. (A) Western blot demonstrating the knockdown of ERK1/2 by siRNA. (B) Following transfection (24 h), cells were treated with ND with or without PD98059 (20 µg/ml) for 4 or 8 h, and cell death was analyzed (n=3). *P<0.05. ND, nutrition deprivation; MEK1/2, dual-specificity mitogen-activated protein kinase kinase 1/2; ERK1/2, mitogen-activated protein kinase; siCTRL, siRNA control; siERK1/2, ERK1/2 siRNA.
ERK1/2 activation is regulated by eEF2K through a feedback mechanism
It is known that eEF2K is a downstream mediator in the MEK-ERK-p90RSK-eEF2K signaling pathway (14). Notably, when the knockdown of eEF2K was performed, the phosphorylation of ERK1/2 was significantly reduced (Fig. 2A). Furthermore, ND treatment led to an increase in ERK1/2 phosphorylation, which was then decreased following eEF2K knockdown (Fig. 2B). These data indicate that ERK1/2 is regulated by eEF2K in a feedback manner. To support this conclusion, a possible interaction between eEF2K and ERK1/2, or its upstream factor MEK1/2, was investigated. The co-IP results indicated that p-eEF2K Ser366 interacted with MEK1/2, but not ERK1/2 (Fig. 3A). Furthermore, the immunofluorescence results revealed that the colocalization of MEK1/2 and eEF2K was enhanced when cells were treated with ND for 3 h (Fig. 3B). These data support the conclusion that eEF2K interacts with MEK1/2 to form a feedback loop regulating ERK1/2 activity. The ability of non-phosphorylated eEF2K to physically bind to MEK1/2 was not investigated, however, it is possible that EGF signaling enhances p-eEF2K Ser366 and its binding to MEK1/2 based on the above results.
Figure 2.

Knockdown of eEF2K reduced the activity of ERK1/2. Cells were transiently transfected with 25 nM siCTRL or sieEF2K, cultured for 48 h, treated and analyzed by western blotting. (A) The effect of eEF2K knockdown on ERK1/2 activity in complete medium. Representative immunoblot (left) and quantification of the relative gray intensities of p-ERK1/2 T202/Y204 normalized to ERK1/2 (right). *P<0.05 vs. siCTRL. (B) The effect of ND treatment and eEF2K knockdown on ERK1/2 activity. The cells were treated under ND for 3 h and analyzed by western blotting. Representative immunoblot (left) and quantification of the relative gray intensities of p-ERK1/2 T202/T204 normalized to ERK1/2 (right). *P<0.05. vs. ND + siCTRL. eEF2K, eukaryotic elongation factor 2 kinase; ERK1/2, mitogen-activated protein kinase; P-ERK1/2, phosphorylated ERK1/2; siCTRL, siRNA control; sieEF2K, eEF2K siRNA; ND, nutrition deprivation.
Figure 3.

p-eEF2K Ser366 physically interacts with MEK1/2. (A) Co-IP assay of proteins MEK1/2 and p-eEF2K Ser366 in MKN45 cells. Representative co-IP immunoblot (upper) and quantification of the relative gray intensities (lower). IgG was used as a control. The relative gray intensities represent the ratios of p-eEF2K Ser366 between the co-IP of anti-MEK1/2/ERK1/2 or IgG and the co-IP of CK (control condition) were calculated and normalized to the IP in CK (n=3). *P<0.05 vs. IgG. NS, not significant. (B) Immunofluorescence confocal microscopy of eEF2K and MEK1/2 in MKN45 cells grown in complete medium or under nutrition deprivation for 3 h. The cells were incubated with anti-eEF2K or anti-MEK1/2 and with fluorescent secondary antibodies conjugated with AlexaFluor 555 (red) and 488 (green) for eEF2K and MEK1/2, respectively. Scale bar, 25 µm. eEF2K, eukaryotic elongation factor 2 kinase; MEK1/2, dual-specificity mitogen-activated protein kinase kinase 1/2; IP, immunoprecipitation; CK, cell lysate; IgG, immunoglobulin G.
AMPK activation inhibits the interaction between eEF2K and MEK1/2
It is known that AMPK is activated under ND to promote cell survival (15). In order to investigate the effect of AMPK activation on the feedback regulation of eEF2K on MEK1/2-ERK1/2, the AMPK activator AICAR was used on ND-treated cells. The immunofluorescence data indicated that AICAR inhibited the interaction between eEF2K and MEK1/2 (Fig. 4). This supported the conclusion that AMPK activation may inhibit the eEF2K-MEK1/2 interaction, leading to the deactivation of downstream ERK1/2, which inhibits cell death under ND.
Figure 4.

AICAR downregulates the physical interaction between eEF2K and MEK1/2. Immunofluorescence confocal microscopy of eEF2K and MEK1/2 in MKN45 cells treated under ND with and without AICAR (400 µM) for 3 h. The cells were incubated with primary antibodies against eEF2K or MEK1/2 and then with fluorescent secondary antibodies conjugated with AlexaFluor 555 (red) and 488 (green) for eEF2K and MEK1/2, respectively. Scale bar, 25 µm. AICAR, acadesine; eEF2K, eukaryotic elongation factor 2 kinase; MEK1/2, dual-specificity mitogen-activated protein kinase kinase 1/2; ND, nutrition deprivation.
Discussion
AMPK is critical for cell survival under ND, as it inhibits protein elongation to conserve energy, via phosphorylating eEF2K. Although AMPK can physically interact with ERK1/2 (16,17), the results of the present study indicated that AMPK can also inhibit the interaction between eEF2K and MEK1/2 in the MEK1/2-ERK1/2-p90RSK-eEF2K-MEK1/2 feedback loop. The present study reveals a previously unknown signaling pathway in AMPK-induced cell survival under the stress of ND.
ERK1/2 activity is another critical regulation point for cell survival under ND, as siRNA ERK1/2 blocks the cell cycle at the G1/S transition. However, ERK1/2 activity is mediated not only by exogenous positive signals, but also by endogenous negative signals, such as that of AMPK. The sensitivities of AMPK to energy stresses are diverse among cell lines. In certain cancer cell lines, such as HeLa and NIH 3T3 Ras (15), one hypothesis could be that the consistent AMPK activity under ND may be due to the immediate suspension of cell proliferation and the alternative pathways stimulated for ATP production. Therefore, the effect of anti-ND via mediating ERK1/2 activity depends on the cell line.
eEF2K serves a positive role in regulating the MEK1/2-ERK1/2 axis, as eEF2K siRNA led to a decrease in the MEK1/2-ERK1/2 activity in MKN45 cells in the present study. However, eEF2K activity can be regulated by a variety of upstream factors, including calcium, AMPK, protein kinase A and ND (18–20). Therefore, ND may cause diverse eEF2K responses in various cell lines or cell culture conditions. Activated AMPK may directly phosphorylate eEF2K (at Ser491 or Ser492) (5,21) and negatively affect the phosphorylation of eEF2K at Ser366. The positive regulation loop of ERK1/2-p90RSK-eEF2K-MEK1/2 is critical for the continuous proliferation of cancer cells, even following the removal of growth factor stimulation.
The present study presents evidence that eEF2K physically interacts with MEK1/2 and forms a positive feedback loop consisting of MEK1/2-ERK1/2-p90RSK-eEF2K, and that the potential positive loop is mediated by activated AMPK. However, the way in which AMPK-modified eEF2K affects MEK1/2 activity is unclear. Any other signaling input, other than AMPK, that may modify eEF2K and affect its binding to MEK1/2 remains unknown. Furthermore, it is unclear whether MEK1/2 undergoes direct phosphorylation by eEF2K and whether the binding of both proteins is necessary for MEK1/2 activation by upstream stimuli. In order to provide answers to these questions and explain the relevant mechanisms of the regulation of MEK1/2, specific modifications of eEF2K should be identified, and the occurrence of the potential phosphorylation events must be examined.
Acknowledgements
Not applicable.
Funding
The present study was supported by a fund from the Priority Academic Program for the Development of Jiangsu Higher Education Institutions (Integration of Chinese and Western Medicine).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
ST and TZ conceived and designed the experiments. ST, TZ, JC, YM and DX performed the experiments. JC analyzed the data and wrote the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Caro-Maldonado A, Muñoz-Pinedo C. Dying for something to eat: How cells respond to starvation. Open Cell Signal J. 2011;3:42–51. doi: 10.2174/1876390101103010042. [DOI] [Google Scholar]
- 2.Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hardie DG. Molecular pathways: Is AMPK a friend or a foe in cancer? Clin Cancer Res. 2015;21:3836–3840. doi: 10.1158/1078-0432.CCR-14-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu LL, Xie T, Zhang SY, Liu B. Eukaryotic elongation factor-2 kinase (eEF2K): A potential therapeutic target in cancer. Apoptosis. 2014;19:1527–1531. doi: 10.1007/s10495-014-1019-7. [DOI] [PubMed] [Google Scholar]
- 5.Johanns M, PyrDitRuys S, Houddane A, Vertommen D, Herinckx G, Hue L, Proud CG, Rider MH. Direct and indirect activation of eukaryotic elongation factor 2 kinase by AMP-activated protein kinase. Cell Signal. 2017;36:212–221. doi: 10.1016/j.cellsig.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 6.Meloche S, Pouysségur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26:3227–3239. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- 7.Lavoie JN, L'Allemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem. 1996;271:20608–20616. doi: 10.1074/jbc.271.34.20608. [DOI] [PubMed] [Google Scholar]
- 8.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 9.Bracken AP, Ciro M, Cocito A, Helin K. E2F target genes: Unraveling the biology. Trends Biochem Sci. 2004;29:409–417. doi: 10.1016/j.tibs.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 10.Zi X, Agarwal R. Modulation of mitogen-activated protein kinase activation and cell cycle regulators by the potent skin cancer preventive agent silymarin. Biochem Biophys Res Commun. 1999;263:528–536. doi: 10.1006/bbrc.1999.1398. [DOI] [PubMed] [Google Scholar]
- 11.Yuan HX, Xiong Y, Guan KL. Nutrient sensing, metabolism, and cell growth control. Mol Cell. 2013;49:379–387. doi: 10.1016/j.molcel.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cagnol S, Chambard JC. ERK and cell death: Mechanisms of ERK-induced cell death-apoptosis, autophagy and senescence. FEBS J. 2010;277:2–21. doi: 10.1111/j.1742-4658.2009.07366.x. [DOI] [PubMed] [Google Scholar]
- 13.Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death is subcellular localization the answer? Cell Cycle. 2009;8:1168–1175. doi: 10.4161/cc.8.8.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001;20:4370–4379. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leprivier G, Remke M, Rotblat B, Dubuc A, Mateo AR, Kool M, Agnihotri S, El-Naggar A, Yu B, Somasekharan SP, et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell. 2013;153:1064–1079. doi: 10.1016/j.cell.2013.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang Q, Zhao S, Wu J, Zheng F, Yang L, Hu J, Hann SS. Inhibition of integrin-linked kinase expression by emodin through crosstalk of AMPKα and ERK1/2 signaling and reciprocal interplay of Sp1 and c-Jun. Cell Signal. 2015;27:1469–1477. doi: 10.1016/j.cellsig.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Zheng F, Wu J, Zhao S, Luo Q, Tang Q, Yang L, Li L, Wu W, Hann SS. Baicalein increases the expression and reciprocal interplay of RUNX3 and FOXO3a through crosstalk of AMPKα and MEK/ERK1/2 signaling pathways in human non-small cell lung cancer cells. J Exp Clin Cancer Res. 2015;34:41. doi: 10.1186/s13046-015-0160-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Browne GJ, Proud CG. A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol Cell Biol. 2004;24:2986–2997. doi: 10.1128/MCB.24.7.2986-2997.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie J, Mikolajek H, Pigott CR, Hooper KJ, Mellows T, Moore CE, Mohammed H, Werner JM, Thomas GJ, Proud CG. Molecular mechanism for the control of eukaryotic elongation factor 2 kinase by pH: Role in cancer cell survival. Mol Cell Biol. 2015;35:1805–1824. doi: 10.1128/MCB.00012-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tavares CD, Ferguson SB, Giles DH, Wang Q, Wellmann RM, O'Brien JP, Warthaka M, Brodbelt JS, Ren P, Dalby KN. The molecular mechanism of eukaryotic elongation factor 2 kinase activation. J Biol Chem. 2014;289:23901–23916. doi: 10.1074/jbc.M114.577148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Browne GJ, Finn SG, Proud CG. Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, Serine 398. J Biol Chem. 2004;279:12220–12231. doi: 10.1074/jbc.M309773200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.