

Abstract
Introduction
Sulodexide (SDX) is used for the treatment of many vascular disorders due to its anticoagulant, anti-inflammatory and anti-atherosclerotic properties. However, the detailed molecular mechanism of its endothelioprotective action is still not completely understood. There is increasing evidence suggesting that antioxidant enzymes play an important role in anti-ischemic properties of SDX. We postulate that up-regulation of glutathione-S-transferase P1 (GSTP1) mediated by the transcription factor Nrf2 could be associated with the antioxidant effect of SDX on vascular endothelial cells.
Material and methods
In the present study, we investigated whether SDX affects GSTP1 and Nrf2 in oxygen glucose deprivation (OGD) treated human umbilical vein endothelial cells (HUVECs). The cells treated with/without SDX (0.5 LRU/ml) were subjected to OGD for 1–6 h. To study the influence of SDX on the Nrf2 nucleus accumulation, the cells were incubated with 0.5 LRU/ml SDX in OGD for 1 h.
Results
We found that after short-term OGD (1–3 h), the drug increased the expression of both GSTP1 and Nrf2 mRNA/protein in HUVECs (p < 0.05), as determined by real-time PCR and enzyme-linked immunosorbent assay (ELISA). SDX treatment also enhanced the nuclear accumulation of Nrf2 in HUVECs after 1 h of OGD (p < 0.05).
Conclusions
SDX induces a rapid onset of the antioxidant response by up-regulating the expression of GSTP1 and Nrf2 in endothelial cells subjected to in vitro simulated ischemia.
Keywords: sulodexide, endothelium, ischemia, glutathione S-transferase P1, Nrf2
Introduction
Ischemia plays a crucial role in the pathogenesis of many human diseases, including peripheral artery disease (PAD), myocardial infarction and stroke [1]. Vascular endothelial cell damage is an initial factor of ischemic injury. Depletion of oxygen and glucose in endothelium causes disturbances in energy metabolism and protein synthesis, intracellular calcium overload, overproduction of inflammatory cytokines and induction of adhesion molecules. These conditions lead to oxidative damage through protein oxidation, inactivation of enzymes, lipid peroxidation and DNA base modifications [2]. An altered oxidant-antioxidant balance contributes to disruption of structural and functional endothelial integrity and the development of vascular injury [3]. Reactive oxygen species (ROS), a major mediator of endothelial injury, are generated at the early stage of ischemia through the mitochondrial respiratory chain [2]. Therefore, a promising therapeutic strategy for protecting endothelium during ischemia is enhancement of the endogenous antioxidant system.
Glutathione S-transferases (GSTs) are antioxidant and phase II detoxifying enzymes. Glutathione S-transferase π1 (GSTP1), the major extra-hepatic GST isoform, appears to play a key role in protecting against ischemic stress. GSTP1 can protect cells against ischemia via several potential mechanisms, including facilitation of the metabolism and detoxification of small-chain aldehydes (e.g. acrolein) generated during peroxidation of membrane lipids or direct inhibition of the proapoptotic c-Jun N-terminal kinase (JNK) [4, 5]. The release of cytosolic GSTs into the extracellular space is a valuable marker of ischemia/reperfusion injury [6]. Normally, cell membranes are impermeable to enzymes; for this reason, the levels of GSTP1 released in the culture medium of human umbilical vein endothelial cells (HUVECs) under OGD may indicate a loss of membrane integrity and cellular damage.
During ischemia and the accompanying enhanced oxidative stress, phase II detoxifying and antioxidant genes become active, and this activation leads to the detoxification of ROS or the prevention of ROS generation. Antioxidant response element (ARE) is a cis-acting sequence located in promoter regions of genes encoding phase II enzymes. The activation of gene transcription via the ARE is regulated by nuclear factor-erythroid 2-related factor (Nrf2) signaling [7]. In an inactive state, Nrf2 is retained in the cytoplasm by binding to Kelch-like ECH-associated protein (Keap1). Keap1 limits the basal activity of Nrf2 by directing its ubiquitination and degradation via the Keap1/Cul3/Rbx1 E3–ubiquitin ligase complex [8]. Upon activation, Nrf2 dissociates from Keap1, translocates into the nucleus and dimerizes with the Maf protein. The Nrf2-Maf heterodimer recognizes the ARE sequence and initiates transcription of target genes, including GSTs, heme oxygenase-1 (HO-1), NAD(P)H:quinine oxidoreductase (NQO1) and thioredoxin reductase (TR) [7, 9]. Furthermore, recent studies have proposed that both the activation and increase in transcriptional activity of Nrf2, as well as its antioxidant genes, are involved in protecting vascular endothelial cells against ischemic damage. Among phase II enzymes, GSTP1 is one of the most important targets in Nrf2-linked vasoprotective regulation [10].
Sulodexide (SDX) is a mixed glycosaminoglycan formulation composed of fast-moving heparin and dermatan sulfate. SDX exhibits antithrombotic, profibrinolytic, and anti-inflammatory effects [11]. Furthermore, SDX restores endothelial glycocalyx and inhibits proteolytic activity of metalloproteinases [12]. SDX is capable of penetrating into endothelial cells. Thus, SDX exerts many endothelial-protective effects such as anti-inflammatory, antioxidant, antiapoptotic and anti-atherosclerotic impacts [11, 13]. Recently, it has become more widely used in the treatment and secondary prevention of deep venous thrombosis, venous leg ulcers, ischemic cardiovascular events and diabetic nephropathy [11]. However, the effects of SDX on injury of endothelial cells induced by ischemia remain unclear. We hypothesize that the anti-ischemic activity of SDX in endothelium is associated with the induction of GSTP1 via the Nrf2/ARE pathway. We evaluated the putative mechanism of SDX using HUVECs as a cellular model and oxygen-glucose deprivation (OGD) to mimic ischemic injury in vitro.
Material and methods
Cell culture
HUVECs were purchased from Lonza (USA) and cultured in endothelial basal medium (EGM-2) with EGM-2 BulletKit in standard conditions (humidified atmosphere, 5% (v/v) CO2, 37°C). The cells were seeded at a density of 1 × 103 into 75 cm2 cell culture flasks, 100 mm dishes or 24-well plates (CytoOne, USA). All experiments using HUVECs were conducted on confluent cultures between passages 3 and 4.
OGD model and cell treatment
HUVECs were washed twice with pre-warmed glucose-free Dulbecco’s modified Eagle’s medium (DMEM, no glucose; Thermo Fisher Scientific, USA). To initiate OGD, the cells maintained in DMEM without glucose were immediately transferred to a hypoxic chamber (incubator Galaxy 48 R, Eppendorf Inc., USA) with an atmosphere of 92% N2, 5% CO2 and 3% O2 at 37°C for 1–6 h.
To evaluate the effect of 0.5 LRU/ml SDX treatment (Vessel due F, Alfa Wasserman, Italy), this drug was added directly to the medium at the start of OGD, and cells were harvested after different OGD exposure times (1, 2, 3, 4, 5 and 6 h). The SDX concentration was selected based on literature data [14, 15]. To study the influence of SDX on the Nrf2 nucleus accumulation, the cells were incubated with 0.5 LRU/ml SDX in OGD for 1 h. The cells of the external control group in standard EGM-2 supplemented with EGM-2 BulletKit were exposed to normoxia for 6 h. Cell cultures that were exposed to OGD conditions but were not treated with SDX served as internal controls.
Real-time PCR
The relative gene expression of GSTP1 and Nrf2 was measured by two-step quantitative real-time PCR (qRT-PCR). Extraction of total RNA from cells was performed using the Chomczynski method [16]. One microgram of total RNA was reverse transcribed to cDNA with a high-capacity cDNA reverse transcription kit (PE Applied Biosystems, USA) according to the manufacturer’s protocol. Primers used for qRT-PCR were retrieved from the database RTPrimerDB (http://www.rtprimerdb.org/). The following sequences were synthesized by Genomed S.A. Company (Poland): GSTP1, 5′-TGTCGGGTGGGTAAGGAGATAG-3′ and 5′-TTGCCCTTAGGAGACTCCAAAC-3′; Nrf2, 5′-GACTCCCGTCCCAGCAG-3′ and 5′ GCTCATACTCTTTCCGTCGC 3′; GAPDH, 5′-GAAGGTGAAGGTCGGAGTC-3′ and 5′-GAAGATGGTGATGGGATTTC3′. Real-time PCR reactions were conducted using SYBR select Master Mix (PE Applied Biosystems) on a Roche LightCycler 480 (Germany). The protocol started with 2 min enzyme activation at 95°C, followed by 45 cycles of 95°C for 15 s and final elongation at 60°C for 1 min. Melting curve analysis for each sample was performed to confirm the specificity of the reaction. GAPDH expression from the same sample was used for data normalization. The 2–ΔΔCt relative method was employed to assess fold changes in mRNA expression [17].
ELISA
The GSTP1 protein concentrations were detected in cell lysates and supernatants using an ELISA kit (Wuhan Fine Biological Technology Co., Ltd., China) following the manufacturer’s protocol. Total, cytosolic and nuclear Nrf2 protein levels were determined using the commercially available human NFE2L2 (Nuclear factor, Erythroid derived 2 like 2) ELISA kit (Wuhan Fine Biological Technology Co., China) according to the manufacturer’s instructions. Absorbance was measured at the wavelength of 450 nm using a Multiskan RC microplate reader.
Preparation of nuclear and cytosolic fractions
Nuclear and cytosolic fractions were collected according to the manufacturer’s description of the Nuclear Extraction Kit (Abcam, USA).
Statistical analysis
The values are presented as the mean ± SD. Data were analyzed by one-way ANOVA followed by Bonferroni’s test, using GraphPad Prism 7 Software (USA). Differences were considered significant at p < 0.05.
Results
Effects of SDX on GSTP1 mRNA and protein expression
To evaluate the effects of SDX (0.5 LRU/ml) on GSTP1, HUVECs were incubated in OGD conditions for 1–6 h, and the relative mRNA and protein levels were measured by qRT-PCR and ELISA. As shown in Figure 1 A, in the untreated cultures, a marked decrease in the GSTP1 mRNA expression level was observed in the first 3 h of OGD. Incubation with SDX for 1–3 h strikingly increased the GSTP1 transcripts in ischemic HUVECs. Then, after 4–6 h of OGD, the expression of GSTP1 mRNA after SDX was similar to the relative mRNA level observed after the treatment with OGD alone. Similarly, the results of ELISA showed that treatment with SDX for 1–3 h strongly induced GSTP1 protein expression in ischemic HUVECs (Figure 1 B). The administration of SDX did not affect the GSTP1 protein levels in HUVECs exposed to 4–6 h of OGD.
Figure 1.

Effects of SDX on expression of GSTP1 detected by qRT-PCR and ELISA. HUVECs were exposed to normoxia or 1–6 h of OGD in the absence or presence of SDX (0.5 LRU/ml). A – Relative levels of GSTP1 mRNA in HUVECs of each group measured by qRT-PCR. Data are mean ± SD (n = 4). B – Intracellular GSTP1 protein levels in HUVECs of each group measured by ELISA. Data are mean ± SD (n = 4–6)
*p < 0.05 vs. normoxia; #p < 0.05 vs. 1 h OGD; ^p < 0.05 vs. 2 h OGD. $p < 0.05 vs. 3 h OGD.
Effect of SDX on GSTP1 release into culture media
To determine whether activation of GSTP1 was also associated with attenuation of OGD-induced endothelial cell damage, GSTP1 release into the cell culture medium was assessed at different times of SDX treatment (Figure 2). GSTP1 is a cytosolic protein that belongs to the pi class of the GST superfamily. We assumed that the increased release of GSTP1 into the media may reflect the dynamics of the cell damage process. The concentration of GSTP1 in HUVEC supernatants was increased significantly after 1–6 h of OGD compared with normoxic conditions. At all these time points, treating ischemic HUVECs with SDX caused a significant decrease in the amount of GSTP1 released into the cell culture medium.
Figure 2.
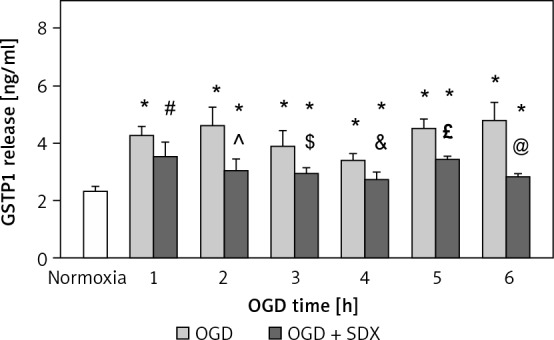
Effects of SDX on the release of GSTP1. HUVECs were exposed to normoxia or 1–6 h of OGD in the absence or presence of SDX (0.5 LRU/ ml). GSTP1 protein concentrations in cell culture supernatants were determined by ELISA
Data are mean ± SD (n = 6). *p < 0.05 vs. normoxia; #p < 0.05 vs. 1 h OGD; ^p < 0.05 vs. 2 h OGD; $p < 0.05 vs. 3 h OGD; &p < 0.05 vs. 4 h OGD; £p < 0.05 vs. 5 h OGD; @p < 0.05 vs. 6 h OGD.
SDX-induced nuclear translocation of Nrf2
Normally, Nrf2 is retained in the cytoplasm by Keap1, which disables Nrf2 translocation to the nucleus [18]. Once released from Keap1 repression, activated Nrf2 translocates to the nucleus, where it binds to ARE regulatory DNA sequences and regulates transcription of target genes, such as GSTP1 [10, 19]. Therefore, we wondered whether SDX-induced activation of GSTP1 in HUVECs exposed to OGD was regulated via Nrf2. To answer this question, we determined the Nrf2 protein concentrations in the nuclear and cytosolic fractions after 1 h of OGD. As shown in Figure 3, when endothelial cells were exposed to 1 h of OGD, the nuclear Nrf2 protein level was lower than that in normoxic cells. The ELISA results show how 1 h of SDX administration dramatically increased the Nrf2 concentration in the nuclear fraction of OGD-treated HUVECs, suggesting involvement of the Nrf2 pathway in the activation of GSTP1.
Figure 3.
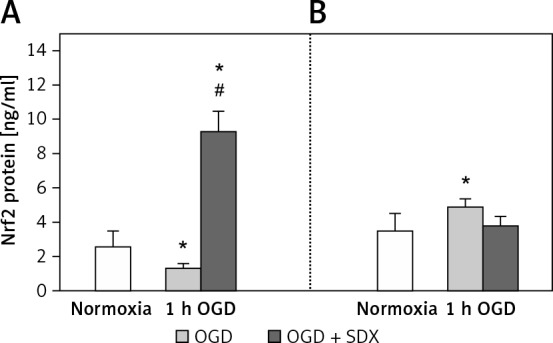
Effects of SDX on nuclear translocation of Nrf2. HUVECs were subjected to normoxia or 1 h of OGD in the absence or presence of SDX (0.5 LRU/ ml). Nrf2 protein concentrations in the nuclear (A) and cytosolic (B) fractions were determined by ELISA
Data are mean ± SD (n = 4). *p < 0.05 vs. normoxia; #p < 0.05 vs. 1 h OGD.
Effect of SDX on Nrf2 mRNA and protein expression
Quantitative analysis revealed a significant decrease in the amount of Nrf2 transcript in HUVECs following 3, 4 and 6 h of OGD (Figure 4 A). Additionally, after incubation of the cultures with SDX, an increase in Nrf2 mRNA expression occurred between the 2nd and 4th h of OGD and then it returned to the basal normoxic level.
Figure 4.

Effects of SDX on expression of Nrf2 detected by qRT-PCR and ELISA. HUVECs were exposed to normoxia or 1–6 h of OGD in the absence or presence of SDX (0.5 LRU/ml). A – Relative levels of Nrf2 mRNA in HUVECs of each group measured by qRT-PCR. Data are mean ± SD (n = 4). B – Total Nrf2 protein levels in HUVECs of each group determined by ELISA. Data are mean ± SD (n = 4–6).
*p < 0.05 vs. normoxia; #p < 0.05 vs. 1 h OGD; $p < 0.05 vs. 3 h OGD; &p < 0.05 vs. 4 h OGD.
Evaluation of total Nrf2 protein by ELISA showed that exposure of HUVECs to 1, 2 and 4 h of OGD significantly reduced the Nrf2 concentration compared to normoxia. As shown in Figure 4 B, treating OGD-stimulated HUVECs with SDX for 1–4 h increased the total Nrf2 protein level.
Discussion
Cumulative evidence has suggested that induction of GSTP1 might be beneficial in protecting the endothelium against ischemic injury [4]. GSTP1, a major GST isoform in murine cardiovascular tissues, is a highly inducible multifunctional enzyme that catalyzes the glutathione conjugation with reactive aldehydes derived from lipid peroxidation (e.g. 4-hydroxynonenal) and ROS. Furthermore, GSTP1 regulates the intracellular protein glutathionylation under oxidative and/or nitrosative stress [4, 20]. Therefore, the pharmacological up-regulation of GSTP1 may be an important therapeutic strategy for limiting ischemic endothelial damage.
Our study demonstrated that SDX treatment significantly induced GSTP1 in the first 3 h of OGD, as evidenced by a dramatic increase in both the GSTP1 mRNA and protein expression in HUVECs (Figure 1). There is much evidence that ischemia may induce the generation of free radicals and ROS, and the production of these species is increased further upon reperfusion. One of the earliest events in the ischemic area may be oxidative injury and apoptosis of vascular endothelium, but the molecular and biochemical mechanisms regulating ischemia-induced dysfunction of the endothelium are incompletely understood [21].
GSTP1 is described as an endogenous inhibitor of proapoptotic JNK kinase that plays a key role in the ischemia-induced apoptosis of vascular endothelial cells [6, 22]. In the normal state, catalytic activity of JNK is maintained at a low level via the GSTP1-JNK complex [21]. Dissociation of this protein complex under ischemic stress releases and activates JNK. The liberated JNK regains its activity by phosphorylation and further phosphorylates c-Jun, resulting in induction of the intrinsic apoptotic pathway [23]. It was shown that pharmacological downregulation of GSTP1 destabilizes the GSTP1-JNK complex concomitantly with JNK activation and inhibition of protein S-glutathionylation [21].
The results of the present study indicate that GSTP1 is strongly induced in HUVECs by SDX treatment after 1–3 h of OGD and suggest that GSTP1 activation could aggravate the endothelial protective effects of SDX. In addition, SDX inhibited the release of GSTP1 into the cell culture medium after 1–6 h of OGD (Figure 2). Measurement of GSTP1 leakage into the culture medium was performed to evaluate the cellular integrity damage induced by OGD and cytoprotection of SDX. The intracellular localization of GSTP1 explains its utility as a marker of endothelial cell membrane ischemic injury [24]. Our findings suggest that SDX at a clinically relevant concentration (0.5 LRU/ml) [14] may prevent GSTP1 release under cell-damaging ischemic conditions.
The present study also attempted to investigate whether SDX induces the fast accumulation of Nrf2 in the nuclei of OGD-stimulated HUVECs. Nrf2 is a transcription factor that regulates a battery cytoprotective and antioxidant genes, including GSTP1 [25]. It was proved that the rapid accumulation of Nrf2 in the nuclear compartment, which results in the stimulation of transcription by binding to ARE regulatory DNA sequences in target genes, effectively protects endothelial cells against ischemia-induced insult [10]. However, the effects of SDX on Nrf2 activation in OGD-treated endothelial cells have not previously been studied. As shown in Figure 3, when HUVECs were treated with SDX for 1 h, the Nrf2 protein level in the nuclear fraction was significantly greater than that in cells subjected to normoxia or OGD alone. Thus, SDX markedly stimulated the transport of Nrf2 from the cytosolic compartment to the nucleus after 1 h of OGD and at the same time significantly increased the mRNA level of GSTP1 (Figures 1 and 3). This evidence implies that SDX may protect against endothelial injury induced by OGD via the Nrf2/ARE signaling pathway. However, such a large SDX-induced increase in the nuclear Nrf2 level in HUVECs exposed to 1 h of OGD also suggests Keap1-independent alternative mechanisms involving enhanced expression of Nrf2 at the transcriptional or translational level [26]. In our study, the endothelial Nrf2 mRNA level was significantly increased by SDX treatment between 2 and 3 h of OGD, and the amounts of total protein were significantly increased after 1–4 h of OGD (Figure 4). It suggests that both transcriptional modulation and de novo synthesis of Nrf2 could also be regulatory mechanisms of SDX action on Nrf2 in ischemic HUVECs. In resting cells, Nrf2 is rapidly degraded in the cytosol through the ubiquitin-proteasome system [27]. When the endothelial cells are exposed to various antioxidants, the Nrf2-Keap1 interaction is disturbed, thus preventing proteasome-dependent degradation, allowing the nuclear accumulation of Nrf2 and activating the expression of cytoprotective genes [27–29]. It should be stressed that, even though the most described mechanism for pharmacological activation of Nrf2 is focused on proteasome inhibition, the tight control of Nrf2 level and activity involves not only proteasomal degradation but also transcription, translation and post-translational modifications [27]. We demonstrated that SDX (0.5 LRU/ml) could increase both the Nrf2 mRNA and total Nrf2 protein in OGD-injured HUVECs (Figure 4). An increase in the level of total Nrf2 protein after SDX administration implies possibilities such as increased transcription, protein stabilization or an increase in Nrf2 protein translation. It was shown that the translational control is one of the Keap-independent mechanisms involved in Nrf2 modulation and de novo synthesized Nrf2 is required for detoxification of cytotoxic electrophiles [30]. Furthermore, a recent study revealed that well-known antioxidants, such as apigenin and resveratrol, activated Nrf2 translation and that the upregulated Nrf2 overcomes the Nrf2-Keap1 pathway, translocates into the nucleus and stimulates ARE. In turn, the presence of the ARE sequence in the Nrf2 promoter region suggests that Nrf2 is a positive regulator of its own expression [31]. It was proposed that Nrf2 transcription is activated by an autoregulation mechanism through the ARE region of its own promoter, leading to enhanced cell defense [32]. So, although we cannot exclude a possible direct effect of SDX on Nrf2-Keap1 interaction, indirect mechanisms, such as enhancement of Nrf2 translation leading to its nuclear accumulation and induction of GSTP1, can explain the protective effects of SDX on OGD-injured HUVECs.
In conclusion, our results indicate that SDX induces a rapid onset of the antioxidant response by up-regulating the expression of GSTP1 and Nrf2 in HUVECs subjected to OGD (Figure 5). Therefore, we believe that the present study may provide a pharmacological basis for the clinical application of SDX in oxidative-stress related vascular diseases. However, further experimental and clinical studies need to be carried out in order to confirm these mechanisms of SDX action on ischemic vascular endothelium.
Figure 5.

Schematic illustration of potential targets of beneficial SDX action in HUVECs injured by simulated ischemia
SDX – sulodexide, Nrf2 – nuclear factor-erythroid 2-related factor, Keap1 – Kelch-like ECH-associated protein, Ub – ubiquitin, ARE – antioxidant response element, Maf – musculoaponeurotic fibrosarcoma protein.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 2000; 87: 840-4. [DOI] [PubMed] [Google Scholar]
- 2.Yang Q, He GW, Underwood MJ, Yu CM. Cellular and molecular mechanisms of endothelial ischemia/reperfusion injury: perspectives and implications for postischemic myocardial protection. Am J Transl Res 2016; 8: 765-77. [PMC free article] [PubMed] [Google Scholar]
- 3.Higashi Y, Maruhashi T, Noma K, Kihara Y. Oxidative stress and endothelial dysfunction: clinical evidence and therapeutic implications. Trends Cardiovasc Med 2014; 24: 165-9. [DOI] [PubMed] [Google Scholar]
- 4.Conklin DJ, Guo Y, Jagatheesan G, et al. Genetic deficiency of glutathione S-transferase P increases myocardial sensitivity to ischemia-reperfusion injury. Circ Res 2015; 117: 437-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang J, Grek C, Ye ZW, Manevich Y, Tew KD. Pleiotropic functions of glutathione S-transferase P. Adv Cancer Res 2014; 122: 143-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagy T, Kovács V, Hardi P, et al. Inhibition of glutathione S-transferase by ethacrynic acid augments ischemia-reperfusion damage and apoptosis and attenuates the positive effect of ischemic postconditioning in a bilateral acute hindlimb ischemia rat model. J Vasc Res 2015; 52: 53-61. [DOI] [PubMed] [Google Scholar]
- 7.Gu J, Sun X, Wang G, Li M, Chi M. Icariside II enhances Nrf2 nuclear translocation to upregulate phase II detoxifying enzyme expression coupled with the ERK, Akt and JNK signaling pathways. Molecules 2011; 16: 9234-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin AH, Chen HW, Liu CT, Tsai CW, Lii CK. Activation of Nrf2 is required for up-regulation of the pi class of glutathione S-transferase in rat primary hepatocytes with L-methionine starvation. J Agric Food Chem 2012; 60: 6537-45. [DOI] [PubMed] [Google Scholar]
- 9.Imai T, Takagi T, Kitashoji A, Yamauchi K, Shimazawa M, Hara H. Nrf2 activator ameliorates hemorrhagic transformation in focal cerebral ischemia under warfarin anticoagulation. Neurobiol Dis 2016; 89: 136-46. [DOI] [PubMed] [Google Scholar]
- 10.Chen B, Lu Y, Chen Y, Cheng J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J Endocrinol 2015; 225: R83-99. [DOI] [PubMed] [Google Scholar]
- 11.Coccheri S, Mannello F. Development and use of sulodexide in vascular diseases: implications for treatment. Drug Des Devel Ther 2018; 8: 49-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mannello F, Ligi D, Canale M, Raffetto JD. Sulodexide down-regulates the release of cytokines, chemokines, and leukocyte colony stimulating factors from human macrophages: role of glycosaminoglycans in inflammatory pathways of chronic venous disease. Curr Vasc Pharmacol 2014; 12: 173-85. [DOI] [PubMed] [Google Scholar]
- 13.Jarząbek K, Gabryel B, Urbanek T. Sulodexide in the treatment of vascular disease: its therapeutic action on endothelium. Phlebol Rev 2016; 24: 51-9. [Google Scholar]
- 14.Borawski J, Dubowski M, Pawlak K, Mysliwiec M. Sulodexide induces hepatocyte growth factor release in humans. Eur J Pharmacol 2007; 558: 167-71. [DOI] [PubMed] [Google Scholar]
- 15.Sosińska P, Baum E, Maćkowiak B, et al. Sulodexide reduces the proinflammatory effect of serum from patients with peripheral artery disease in human arterial endothelial cells. Cell Physiol Biochem 2016; 40: 1005-12. [DOI] [PubMed] [Google Scholar]
- 16.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987; 62: 156-9. [DOI] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and 2(-Delta Delta C(T)). Method Methods 2001; 25: 402-8. [DOI] [PubMed] [Google Scholar]
- 18.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol 2003; 23: 8137-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol 2003; 43: 233-60. [DOI] [PubMed] [Google Scholar]
- 20.Wetzelberger K, Baba SP, Thirunavukkarasu M, et al. Postischemic deactivation of cardiac aldose reductase: role of glutathione S-transferase P and glutaredoxin in regeneration of reduced thiols from sulfenic acids. J Biol Chem 2010; 285: 26135-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Röth E, Marczin N, Balatonyi B, et al. Effect of a glutathione S-transferase inhibitor on oxidative stress and ischemia-reperfusion-induced apoptotic signalling of cultured cardiomyocytes. Exp Clin Cardiol 2011; 16: 92-6. [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, Jiang S, Yang PY, Zhang YF, Li TJ, Rui YC. EF1A1/HSC70 cooperatively suppress brain endothelial cell apoptosis via regulating JNK activity. CNS Neurosci Ther 2016; 22: 836-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis W Jr, Ronai Z, Tew KD. Cellular thiols and reactive oxygen species in drug-induced apoptosis. J Pharmacol Exp Ther 2001; 296: 1-6. [PubMed] [Google Scholar]
- 24.Khurana S, Corbally MT, Manning F, Armenise T, Kierce B, Kilty C. Glutathione S-transferase: a potential new marker of intestinal ischemia. J Pediatr Surg 2002; 37: 1543-8. [DOI] [PubMed] [Google Scholar]
- 25.Duan Q, Sun W, Yuan H, Mu X. MicroRNA-135b-5p prevents oxygen-glucose deprivation and reoxygenation-induced neuronal injury through regulation of the GSK-3β/Nrf2/ARE signaling pathway. Arch Med Sci 2018; 14: 735-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purdom-Dickinson SE, Sheveleva EV, Sun H, Chen QM. Translational control of nrf2 protein in activation of antioxidant response by oxidants. Mol Pharmacol 2007; 72: 1074-81. [DOI] [PubMed] [Google Scholar]
- 27.Bryan HK, Olayanju A, Goldring CE, Park BK. The Nrf2 cell defence pathway: Keap1-dependent and-independent mechanisms of regulation. Biochem Pharmacol 2013; 85: 705-17. [DOI] [PubMed] [Google Scholar]
- 28.Zhu Y, Zhang YJ, Liu WW, Shi AW, Gu N. Salidroside suppresses HUVECs cell injury induced by oxidative stress through activating the Nrf2 signaling pathway. Molecules 2016; 21: pii: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen M, Zhang M, Zhang X, et al. Limb ischemic preconditioning protects endothelium from oxidative stress by enhancing Nrf2 translocation and upregulating expression of antioxidases. PLoS One 2015; 10: e0128455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perez-Leal O, Barrero CA, Merali S. Translational control of Nrf2 within the open reading frame. Biochem Biophys Res Commun 2013; 437: 134-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwak MK, Itoh K, Yamamoto M, Sutter TR, Kensler TW. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dimethiole-3-thione. Mol Med 2001; 7: 135-45. [PMC free article] [PubMed] [Google Scholar]
- 32.Perez-Leal O, Barrero CA, Merali S. Pharmacological stimulation of nuclear factor (erythroid-derived 2)-like 2 translation activates antioxidant responses. J Biol Chem 2017; 292: 14108-21. [DOI] [PMC free article] [PubMed] [Google Scholar]