

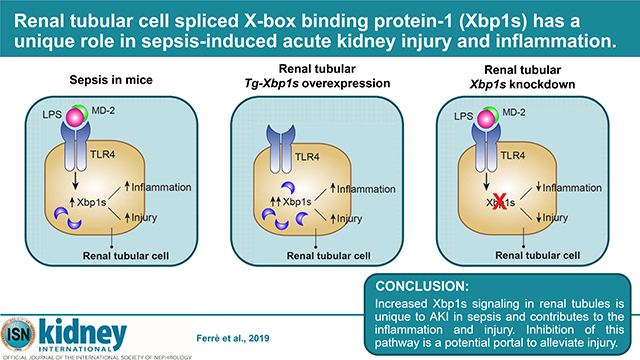
Abstract
Sepsis is a systemic inflammatory state in response to infection, and concomitant acute kidney injury (AKI) increases mortality significantly. Endoplasmic reticulum stress is activated in many cell types upon microbial infection and modulates inflammation. The role of endoplasmic reticulum signaling in the kidney during septic AKI is unknown. Here we tested the role of the spliced X-box binding protein 1 (Xbp1s), a key component of the endoplasmic reticulum stress- activated pathways, in the renal response to sepsis in the lipopolysaccharide (LPS) model. Xbp1s was increased in the kidneys of mice treated with LPS but not in other models of AKI, or several chronic kidney disease models. The functional significance of Xbp1s induction was examined by genetic manipulation in renal tubules. Renal tubule-specific overexpression of Xbp1s caused severe tubule dilation and vacuolation with expression of the injury markers Kim1 and Ngal, the pro-inflammatory molecules interleukin-6 (Il6) and Toll-like receptor 4 (Tlr4), decreased kidney function and 50% mortality in five days. Renal tubule-specific genetic ablation of Xbp1 had no phenotype at baseline. However, after LPS, Xbp1 knockdown mice displayed lower renal NGAL, pro-apoptotic factor CHOP, serum creatinine levels, and a tendency towards lower Tlr4 compared to LPS-treated mice with intact Xbp1s. LPS treatment in Xbp1s-overexpressing mice caused a mild increase in NGAL and CHOP compared to LPS-treated mice without genetic Xbp1s overexpression. Thus, increased Xbp1s signaling in renal tubules is unique to sepsis- induced AKI and contributes to renal inflammation and injury. Inhibition of this pathway may be a potential portal to alleviate injury.
Keywords: ER stress, Xbp1s, AKI, sepsis, inflammation
Graphical Abstract
INTRODUCTION
Sepsis is a systemic inflammatory response elicited by microbial infection. It is frequently encountered in the intensive care unit (ICU) and is associated with significant morbidity and mortality.1,2 Acute kidney injury (AKI) is a frequent complication in critically ill patients occurring in about 45% of septic patients and 60% of those with septic shock.3–5 The combination of sepsis and AKI synergistically increases mortality to beyond 50%.1
The pathophysiology of AKI in sepsis is complex, but results in part from systemic inflammation, nephrotoxins, and hemodynamic alterations.6,7 Systemic endotoxin, or lipopolysaccharide (LPS), released from gram-negative bacteria binds to the Toll-like receptor 4 (TLR4)-myeloid differentiation factor 2 (MD-2) complex on immune cells followed by secretion of various cytokines, such as interleukin (IL)-1, tumor necrosis factor (TNF)-α, and IL-6, resulting in a cytokine storm, hemodynamic instability, and eventually septic shock.6,8,9 LPS can cause direct injury through interactions with epithelial and endothelial TLR4-MD-2 complex in the kidney. Both LPS and TNF-α have direct pro-inflammatory effects on renal epithelial cells,10,11 and can induce renal tubular production of cytokines.12–14
Endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) are induced upon viral, bacterial, or parasitic infection.15,16 UPR is a complex network of intracellular signaling pathways that restores ER homeostasis towards normality, or activates pro-apoptotic towards cellular demise, depending on cell types and severity of the stress.17,18 UPR activation is associated with production of many pro-inflammatory molecules that contribute to the pathogenesis and/or progression of conditions such as diabetes, obesity, inflammatory bowel disease, and cancer.19 However, little is known about the role of the UPR in the renal response to sepsis in vivo.
Among the three UPR signaling cascades, the inositol-requiring enzyme 1α (IRE1α) pathway is the most conserved across species.20 Activated IRE1α has endoribonuclease activity, which cleaves its primary target, X-box-binding protein 1 (Xbp1) mRNA. The specific excision of 26 nucleotides from the Xbp1 mRNA causes a frameshift, generating the spliced Xbp1 form (referred to as Xbp1s), which is translated into a potent transcription factor.17 Xbp1s transcript levels were increased in the lungs of mice treated with LPS and in primary bronchial epithelial cells treated with Pseudomonas aeruginosa virulence factors.21,22 In macrophages, the spliced Xbp1s protein is required downstream of TLR4 for sustained transcription and optimal production of immune mediators such as IL-6.23 Deletion of Xbp1s in macrophages and other hematopoietic cells increases bacterial burden in animals infected with the TLR2-activating human pathogen Francisella tularensis,23 and specific disruption of Xbp1s in the intestinal epithelium results in inability to respond appropriately to inflammatory signals and increased susceptibility to colitis.24 These studies suggest Xbp1s expression may be beneficial in these inflammatory states.
Thus far, studies on the role of Xbp1s in the kidney have mainly focused on podocytes, and Xbp1s response to ER stress secondary to metabolic signals or accumulation of immature proteins. Disruption of Xbp1s in podocytes is not pathogenic,25 but when coupled with persistent hyperglycemia, podocyte Xbp1s deficiency aggravates glucose-induced cellular injury, in part by increased oxidative stress, and exacerbates diabetic nephropathy.26,27 Simultaneous genetic ablation of Xbp1 and Sec63, a heat shock protein-40 chaperone required for protein folding in the ER, in podocytes results in foot process effacement and albuminuria, suggesting that intact Xbp1 is essential for the maintenance of a normal glomerular filtration barrier.25 Concomitant inactivation of both Sec63 and Xbp1 in distal tubules in a polycystic kidney disease model, decreases maturation of polycystin-1 and exacerbated the disease,28 while in the collecting ducts causes chronic tubulointerstitial injury.29 However, the role of Xbp1s in the tubular epithelium in the context of sepsis is unknown.
In the present study, we examined the response of native Xbp1s in response to LPS and used renal tubule-specific Xbp1s-overexpression or deletion to examine the functional role of Xbp1s on kidney injury during LPS-mediated sepsis.
RESULTS
Xbp1s is activated in the mouse kidney upon LPS-induced sepsis
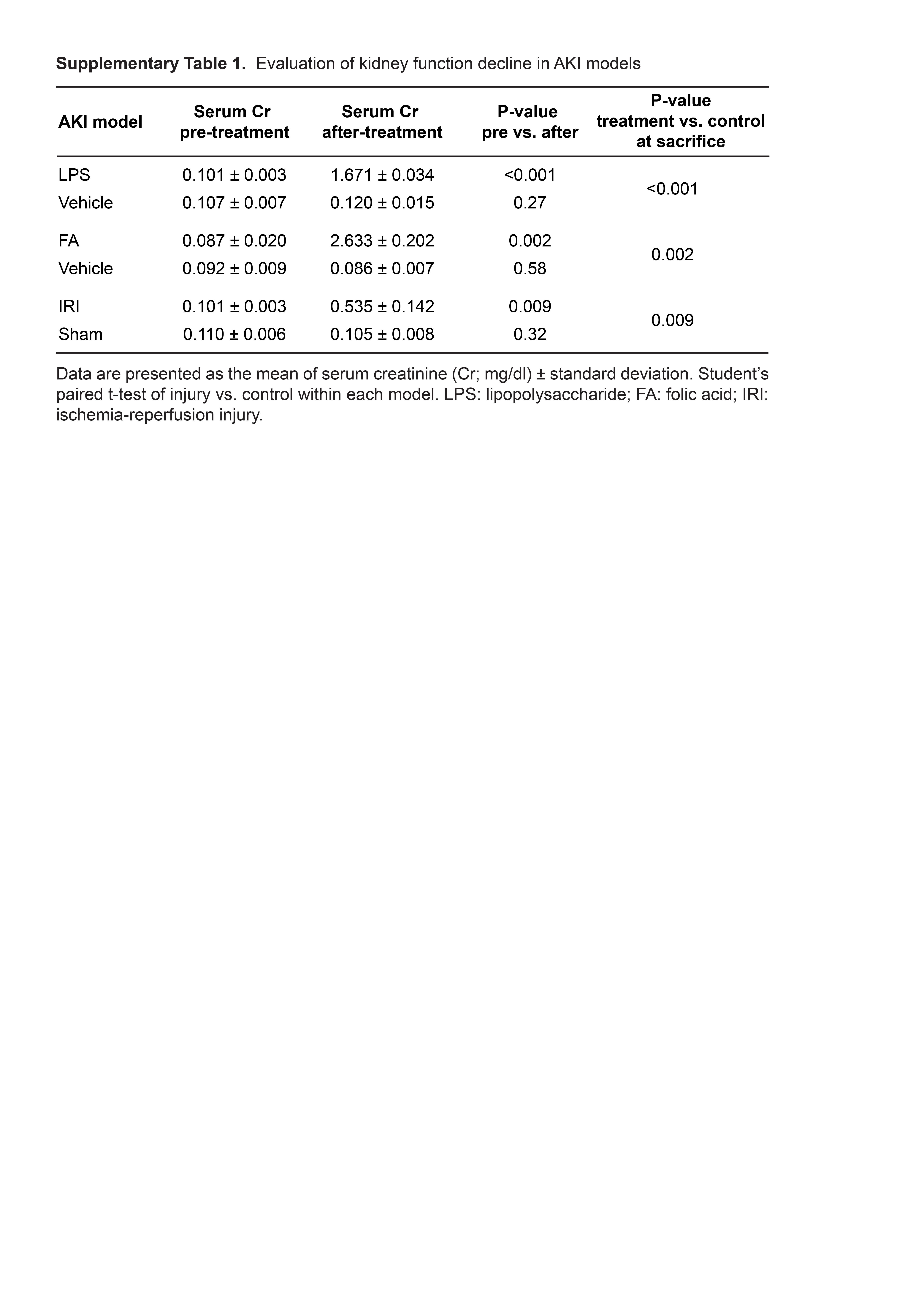
In the kidneys of control mice, Xbp1s expression was detected in the nucleus of LTA (proximal convoluted) and NCC (distal convoluted) positive tubules in the renal cortex, whereas Xbp1s expression in the renal medulla was modest (Figure 1C and Supplementary Figure 1). After acute kidney injury (AKI), kidney Xbp1s mRNA was increased exclusively after LPS injection, but not after folic acid-induced or ischemia-reperfusion AKI (Figure 1A and Supplementary Table 1). The increase of Xbp1s nuclear expression in response to LPS treatment occurred in the nucleus of all renal tubules, both in the renal cortex and medulla (Figure 1C and Supplementary Figure 1). Xbp1s was also increased in the kidneys of mice that developed sepsis after cecal ligation puncture (CLP), a condition that shares pathophysiologic features with LPS (Supplementary Figure 2). Our findings were confirmed by RT-PCR and qPCR using Xbp1s-specific primers that selectively amplify the spliced form of the transcript (Primer set 3, Supplementary Figure 3). Primers that anneal to the Xbp1 mRNA outside the cleavage site showed unchanged levels of unspliced Xbp1 mRNA (Primer set 1, Supplementary Figure 3 and Figure 1A). In order to determine whether the increase in renal Xbp1s expression is mediated by TLR4 activation during bacterial inflammation, we injected Tlr4Lps-d mice, which have an inactivating mutation in Tlr4 and are resistant to LPS, with Ultrapure LPS that only activates the TLR4, but not the TLR2, pathway. Xbp1s response to Ultrapure LPS was blunted in kidneys from Tlr4Lps-d mice, but not in wild-type mice, indicating that the induction of renal Xbp1s mRNA splicing is downstream of TLR4 activation in LPS sepsis (Supplementary Figure 4).
Figure 1. Xbp1s expression in mouse kidney is increased by LPS-induced sepsis.

(A) RT-PCR (left panel) and qPCR (right panel) analysis showing Xbp1s transcript levels in mouse kidney is increased by lipopolysaccharide (LPS; CTR and LPS-injected, n=3 each), but not by other acute kidney injury (AKI) insults such as folic acid (FA; CTR and FA, n=3 each) or ischemia-reperfusion injury (IRI; CTR and IRI, n=4) compared to respective control. (B) RT-PCR (left panel) and qPCR (right panel) showing that Xbp1s transcript levels are unchanged in the kidney of genetic mouse models of chronic kidney disease, i.e. type II diabetes (CTR and ob/ob mice, n=3) and polycystic kidney disease [PKD; Ksp/Cre;Pkd1F/F (Pkd1, n=3) mice at postnatal day 10, P10; Pkhd1/Cre;Pkd2F/F (Pkd2, n=3) mice at postnatal day 21, P21] compared to respective control (n=3 for each condition). Mouse kidney with known Xbp1 splicing was used as positive control. (C) Xbp1s antibody co-staining with nephron segment-specific markers revealed that Xbp1s expression was induced in epithelia cells from all nephron segments in kidneys of mice treated with LPS compared to vehicle-treated controls. LTA: lotus tetragonolobus agglutinin; NKCC2: Na+-K+-Cl- cotransporter; NCC: Na+-Cl- cotransporter; DBA: dolichos biflorus agglutinin. Scale bar, 20 μm. Bars and error bars indicate mean and SEM. Student’s unpaired t-test of injury vs. control within each model. * indicates P<0.05, compared to respective control. CTR: control.
Since ER-stress response is activated in chronic conditions such as diabetes, obesity and cancer,30,31 we tested Xbp1 splice variants in three models of genetically-induced chronic kidney disease (CKD; Figure 1B). We did not detect any changes in Xbp1s levels in ob/ob (obesity and the metabolic syndrome), Ksp/Cre;Pkd1F/F, and Pkhd1/Cre;Pkd2F/F (polycystic kidney disease) mouse kidney samples compared to their corresponding controls (Figure 1B). These results indicate that increased renal Xbp1s mRNA expression is unique to LPS-induced AKI. The next and logical question is whether the increased Xbp1s is a paraphenomenon, protective, or pathogenic for AKI.
Tubular overexpression of Xbp1s induces tubular injury in kidney-inducible Xbp1s (KIXs) mice
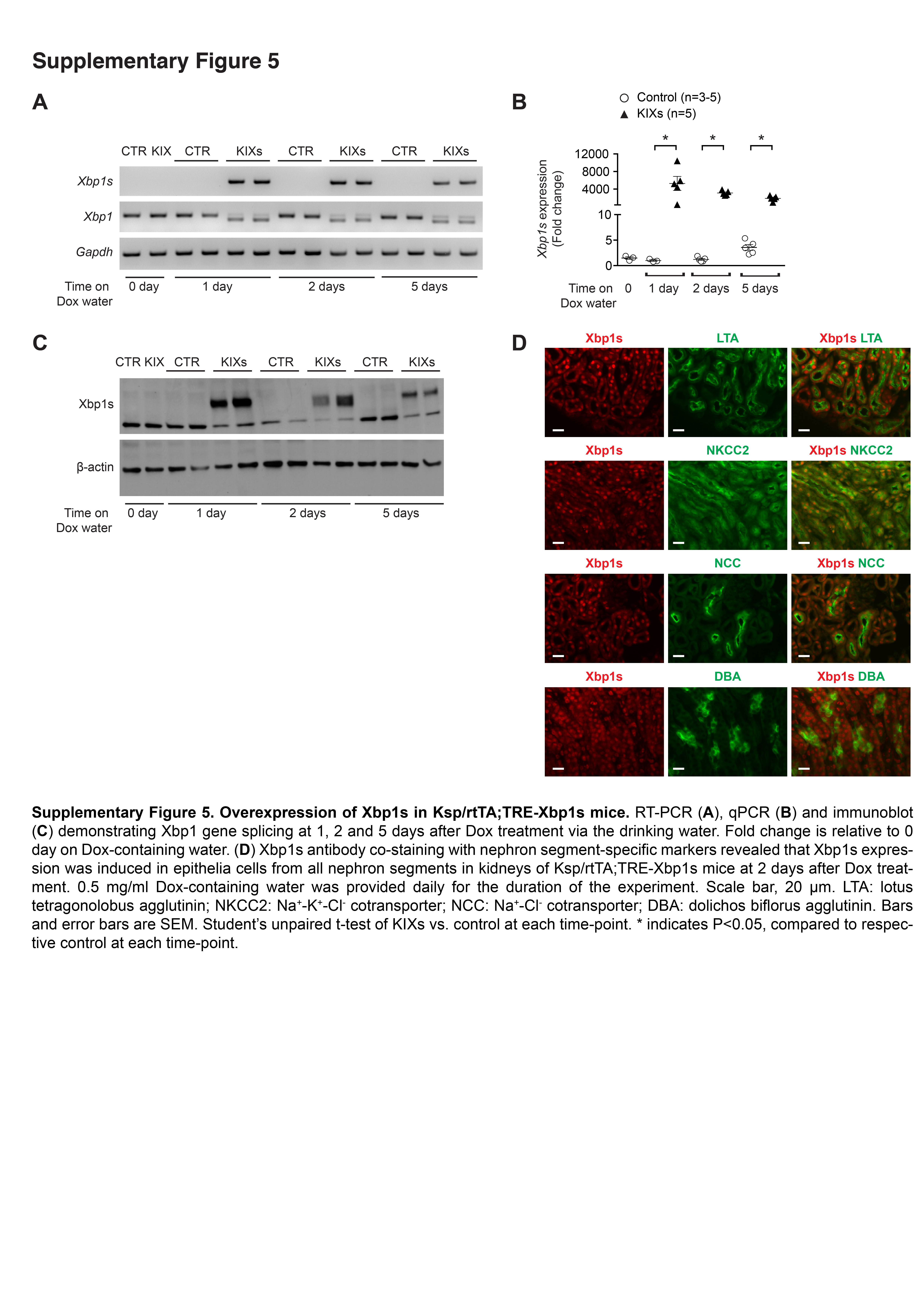
The systemic and multifactorial nature of LPS injection renders it difficult to dissect the role of renal Xbp1s expression in LPS-induced AKI, and disables one from concluding whether the high Xbp1s induced by LPS is a mere biomarker, a protective cellular adaptation, or pathogenic. We made a mouse where the Xbp1s transgene downstream from a tetracycline-responsive element (TRE) allows inducible renal tubule-specific expression of Xbp1s by the tetracycline reverse transcriptional activator (rtTA) driven by a Ksp promoter with doxycycline (Dox), without the need to expose the mice to endotoxin. Crossing TRE/Xbp1s with Ksp/rtTA yielded a mouse with kidney-specific inducible expression of Xbp1s (KIXs; Figure 2A). After Dox, Xbp1s mRNA and protein was readily detected within 24 hours (Figure 2B and Supplementary Figure 5A–C) and persisted 2 and 5 days after induction (Supplementary Figure 5A–C). The functionality of the transgenic Xbp1s was validated by increased expression of known Xbp1s target genes in KIXs mice such as GalE and Dnajb9,32,33 and of genes involved in the unfolded protein response (UPR), namely Bip and CCAAT-enhancer-binding protein homologous protein (CHOP) gene Chop (Figure 3A–B).17 As expected, the expression of Bip, encoding the major chaperone in the ER stress response, rapidly and steadily increased over time, while the expression of Chop, a pro-apoptotic transcription factor involved in the late response to stress, continued to increase up to 5 days. Co-staining of KIXs kidney sections with Xbp1s and nephron segment-specific markers showed that Xbp1s overexpression occurred in both proximal and distal tubules (Supplementary Figure 5D).
Figure 2. Overexpression of Xbp1s in the kidney produces tubular injury.

(A) To generate a kidney-inducible Xbp1s (KIX) mouse model, TRE/Xbp1s mice were crossed with Ksp/rtTA mice. KIXs were Ksp/rtTA;TRE-Xbp1s positive, whereas control animals only expressed either TRE-Xbp1s or Ksp/rtTA (lighter grey color). Doxycycline (Dox) was used to induce Xbp1s expression in renal tubular cells at postnatal day 21 (P21). Control animals also received Dox. (B) qPCR showed that Xbp1s expression increased equally after one day treatment with Dox- containing water at concentrations of either 0.1 mg/ml or 0.5 mg/ml. (C-G) 0.5 mg/ml Dox- containing water was provided daily for the duration of the experiment. (C) Kaplan-Meier survival curves of KIXs and control mice upon treatment with Dox. The experiment was stopped after 5 days. (D) Body weight of KIXs and control mice upon Xbp1s induction. (E) Kidney to body weight ratios for KIXs and control mice at baseline and after Xbp1s induction. (F-G) Kidney function of KIXs and control mice was assessed by serum Cr (F) and blood urea nitrogen (BUN, G) levels. (H) H&E staining revealed histological changes consistent with acute tubular necrosis (e.g. tubule dilation and intracellular vacuoles; arrows) in KIXs mice at 5 days after induction. Kidney histological score is shown (control, n=6; KIXs, n=9). Scale bar, 20 μm. Bars and error bars are mean and SEM. Student’s unpaired t-test of KIXs vs. control at each time-point. * indicates P<0.05, compared to respective control. TRE: tetracycline responsive element; Ksp: Ksp-cadherin promoter; rtTA: reverse tetracycline-controlled transactivator; KIXs: kidney- inducible Xbp1s mice; Cr: creatinine.
Figure 3. Overexpression of Xbp1s in the kidney increases the expression of UPR effectors, kidney injury markers and inflammatory molecules.

qPCR showed that Xbp1s overexpression in KIXs mice induced significant increase of the UPR effectors Bip and Chop (A), and of the Xbp1s target genes GalE and Dnajb9 (B) from one to five days after treatment with Dox. Expression of the kidney injury markers Kim1 and Ngal (C), and of the inflammatory molecules Il6 and Tlr4 (D) were increased starting at two days after treatment. The transcription factor HNF-1β regulates genes important for the homeostatic control of kidney repair during stress-induced proliferation. KIXs mice have significantly reduced Hnf1b expression, increased Socs3, and decreased Pkhd1 expression that are negatively and positively regulated by HNF- 1β, respectively (E). 0.5 mg/ml Dox-containing water was provided daily for the duration of the experiment. Fold change is relative to 0 day on Dox-containing water. Bars and error bars are mean and SEM. Student’s unpaired t-test of control vs. KIXs at each time-point. * indicates P<0.05, compared to respective control at each time-point. KIX: kidney-inducible Xbp1s mice; UPR: unfolded protein response.
KIXs mice showed reduced weight gain over time, 65% increase in kidney-weight-to-body- weight ratio and 50% mortality 5 days after induction (Figure 2C–E). Kidney function was severely impaired in KIXs compared to controls as demonstrated by markedly elevated blood urea nitrogen (BUN) and serum Cr levels at 24 hours after induction, which continued to rise up to 5 days (Figure 2F–G). Kidney histopathology revealed tubular necrosis, tubule dilation, and intracellular vacuoles (Figure 2H). Periodic acid-Schiff (PAS) staining showed loss of brush border in all KIXs mice (Supplementary Figure 6) compatible with proximal tubule injury. Due to the short observational period, fibrosis based on trichrome staining was not observed in KIXs kidneys (Supplementary Figure 6). The expression of the kidney injury marker neutrophil gelatinase-associated lipocalin (Ngal), and inflammatory molecules Il6 and Tlr4 was increased in KIXs compared to control mice at 2 and 5 days after induction (Figure 3C–D). The increase in the injury marker kidney injury molecule-1 (Kim1) was more variable and did not reach statistical significance at 5 days (P=0.14). Recovery of injured tubular cells is achieved by biphasic expression of genes normally expressed during kidney development, and transient activation of cytokine-induced signaling.34,35 We found downregulation of hepatocyte nuclear factor-1beta (Hnf1b), a transcription factor involved in kidney development, and dysregulation of its target genes, suppressor of cytokine signaling 3 (Socs3), and polycystic kidney and hepatic disease 1 (Pkhd1) (Figure 3E). Therefore, overexpression of Xpb1s alone is sufficient to induce renal damage, suggesting that the elevation of Xpb1s in sepsis-induced AKI is not just a biomarker but likely pathogenic for the kidney injury.
Xbp1s deletion is protective against LPS-induced kidney injury
The above studies established that Xbp1s is sufficient to cause kidney injury. We next proceeded to test the necessity of Xbp1s in septic renal injury. In order to determine the role of renal tubular Xbp1s in sepsis-induced AKI, we generated renal tubule-specific Xbp1 deletion using the kidney-specific Six2/Cre (Figure 4A). During kidney development, Six2/Cre is expressed in the nephron progenitors that will form the epithelial tubule cells. Xbp1 expression was decreased 50% in total kidney lysate of Six2/Cre+;Xbp1F/F mice compared to Six2/Cre-; Xbp1F/F mice (Figure 4B). At baseline, Six2/Cre+;Xbp1F/F animals did not exhibit abnormalities in body weight or kidney morphology and function (Figure 4C–D). A similar finding was previously described in Ksp/Cre+;Xbp1F/F mice in which Cre activity resulted in deletion of Xbp1 in distal nephron segments.28
Figure 4. Kidney-specific Xbp1s knockdown mice have normal kidney function.

(A) Floxed Xbp1 mice were bred with Six2/Cre mice to generate kidney-specific deletion of Xbp1. Open triangles indicate loxP sites. (B) Xbp1 expression in kidneys of Six2/Cre+;Xbp1F/F (n=13) mice at P21 was approximately 50% decreased compared to Six2/Cre-;Xbp1F/F (n=10) control mice. Six2/Cre+;Xbp1F/F mice showed similar body weight (C) and kidney function (D) at P21 compared to control animals. Bars and error bars are mean and SEM. Student’s unpaired t-test of Six2/Cre-;Xbp1F/F vs. Six2/Cre+;Xbp1F/F. *** indicates P<0.001, compared to control.
Next, we challenged both the Six2/Cre+;Xbp1F/F and Six2/Cre-;Xbp1F/F control mice with an injection of LPS, and compared them to animals injected with vehicle (Figure 5 and Supplementary Figure 7A). Histology did not show any significant morphologic changes after LPS injection in either the Six2/Cre+;Xbp1F/F or Six2/Cre-;Xbp1F/F control mice (Supplementary Figure 7A), similar to other reports of minimal histological tubular injury in septic AKI.36–38 LPS treatment induced Xbp1s to approximately 20 times in the kidney of Six2/Cre-;Xbp1F/F mice, but failed to increase Xbp1s expression in the kidney of Six2/Cre+;Xbp1F/F mice (Figure 5C). Tlr4 and Kim1 mRNA expression were upregulated in Six2/Cre-;Xbp1F/F LPS-injected mice, but not in Six2/Cre+;Xbp1F/F LPS-injected mice (Figure 5C). The difference in Kim1 and Tlr4 expression between the two LPS-treated genotypes reached statistical significance for Kim1 (P<0.01), but not for Tlr4 (P=0.07). Il6 and Tnfa expression levels were not different across the experimental groups. While Ngal mRNA was equally increased in both genotypes upon LPS treatment (Figure 5C), the increase in kidney protein levels of NGAL was significantly less in kidneys from Six2/Cre+;Xbp1F/F LPS-injected mice compared to those from Six2/Cre-;Xbp1F/F LPS-injected mice (Figure 5B). Within 24 hours after LPS treatment, kidney dysfunction developed as shown by elevated serum Cr in both Six2/Cre-;Xbp1F/F and Six2/Cre+;Xbp1F/F mice (Figure 5A). However, the increase in serum Cr in Six2/Cre+;Xbp1F/F was lower than in controls (Figure 5A). Although IREα-Xbp1s signaling is important in promoting pro-survival UPR gene expression, prolonged or excessive ER stress will lead Xbp1s mediated cell death via pro-apoptotic factor CHOP.39 After LPS challenge, kidneys from Six2/Cre+;Xbp1F/F mice had lower levels of CHOP compared to those from Six2/Cre-;Xbp1F/F mice (Figure 6A–B). In order to examine the association between changes in Xbp1s and renal function, we further analyzed the data obtained from Six2/Cre-;Xbp1F/F mice injected with vehicle or LPS. We found a direct relationship between Xbp1s expression level and serum Cr, Kim1, Ngal, Tlr4 and Chop expression in kidney (Supplementary Figure 8). These data further support the model that Xpb1s has a pathogenic role in LPS-induced AKI.
Figure 5. Kidney-specific Xbp1s knockdown affects kidney function in response to acute LPS-induced AKI.

Mice received an intraperitoneal (IP) injection of LPS (16 mg/kg) or vehicle, and were sacrificed 24 hours later. (A) Both Six2/Cre-;Xbp1F/F and Six2/Cre+;Xbp1F/F animals showed significant increase in serum Cr levels after LPS injection (Six2/Cre-;Xbp1F/F: n=8; Six2/Cre+;Xbp1F/F: n=10) compared to vehicle (Six2/Cre-;Xbp1F/F: n=6; Six2/Cre+;Xbp1F/F: n=10), but the increase was lower in Six2/Cre-;Xbp1F/F mice. (B) After LPS injection, NGAL protein expression levels normalized to β-actin were lower in the kidney of Six2/Cre+;Xbp1F/F mice compared to controls. Fold change is relative to vehicle-treated Six2/Cre-;Xbp1F/F mice. (C) qPCR on mouse kidney samples from mice injected with vehicle (Six2/Cre-;Xbp1F/F: n=6; Six2/Cre+;Xbp1F/F: n=8) or LPS (Six2/Cre-;Xbp1F/F: n=6; Six2/Cre+;Xbp1F/F: n=8). Fold change is relative to vehicle-treated Six2/Cre-;Xbp1F/F mice. Il6 mRNA expression was undetectable in the kidney of mice treated with vehicle. Bars and error bars are mean and SEM. 2-way ANOVA, Sidak’s multiple comparisons test. * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001. Cr: creatinine; LPS: lipopolysaccharide.
Figure 6. Kidney-specific Xbp1s knockdown associates with decreased expression of the pro-apoptotic transcription factor CHOP.

qPCR and immunoblot of the pro-apoptotic marker Chop/CHOP in the kidney of tissue-specific Xbp1s knockdown (A-B) and KIXs (C-D) mice at 24 hours after an intraperitoneal (IP) injection of LPS (16 mg/kg) or vehicle. Fold change is relative to vehicle-treated control mice. KIXs and control mice received 0.1 mg/ml Dox- containing water for 16 hours prior LPS injection. Bars and error bars are mean and SEM. 2-way ANOVA, Sidak’s multiple comparisons test. * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001. LPS: lipopolysaccharide.
Xbp1s overexpression exacerbates LPS-induced kidney injury
To interrogate whether Xbp1s is the sole or major mediator of LPS-induced AKI, we examined whether Xpb1s overexpression and LPS are additive in imparting renal injury. Histology did not show any significant morphologic changes after LPS injection in either the control or KIXs kidneys (Supplementary Figure 7B).36–38 Within the vehicle-treated group, KIXs mice had significantly higher Tlr4 and Chop expression compared to controls (Figure 6C–D and Figure 7C). After LPS injection, control animals showed an increase in NGAL/Ngal levels compared to vehicle-treated controls (Figure 7B–C). Similarly, after LPS injection, KIXs animals showed an increase in NGAL/Ngal levels comparable to vehicle-treated KIXs (Figure 7B–C). The increase in NGAL protein levels were not significantly higher in LPS-treated KIXs than in LPS-treated controls (Figure 7B). Serum Cr levels, Kim1, Tlr4 and Chop/CHOP expression did not differ between KIXs treated with vehicle or LPS (Figure 7A and 7C, Figure 6C–D). Il6 and Tnfa expression levels were not different across the experimental groups. These studies suggest that in the presence of already elevated Xbp1s levels, LPS only modestly increases injury.
Figure 7. Kidney-specific Xbp1s overexpression affects kidney function in response to acute LPS-induced injury.

KIXs and control mice received 0.1 mg/ml Dox-containing water for 16 hours, then an intraperitoneal (IP) injection of LPS 16 mg/kg (control: n=5; KIXs: n=5) or vehicle (control: n=4; KIXs: n=4), and sacrificed 24 hours later. (A) Serum Cr levels from control or KIXs mice injected with vehicle or LPS. (B) NGAL protein expression normalized by β-actin in the kidney of KIXs mice compared to controls. (C) qPCR on kidney samples from mice injected with vehicle or LPS. Fold change is relative to vehicle-treated control mice. Il6 mRNA expression was undetectable in the kidney of mice treated with vehicle. Bars and error bars are mean and SEM. 2-way ANOVA, Sidak’s multiple comparisons test. * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001. Cr: creatinine; LPS: lipopolysaccharide.
DISCUSSION
This study investigated the role of renal tubular UPR transcription factor Xbp1s affects AKI during sepsis. We found that the response of renal tubules to systemic endotoxin involves Xbp1s signaling, and that activation of this pathway contributes to tubular injury, inflammation and renal failure based on the following findings. First, two in vivo models of sepsis increased native Xbp1s in kidney. The increase of Xbp1s in kidney after LPS injection was recently observed also by Hato et al. between 4 hours to 28 hours.40 Second, inducible Xbp1s overexpression in renal tubules per se was sufficient to produce pathology that resembles sepsis-induced AKI including increased serum Cr, injury markers and pro-inflammatory molecules Il6 and Tlr4. Moreover, a frequent observation after Xbp1s overexpression was vacuolation of tubule epithelium which is the most consistent pathology in septic AKI.8 Third, Xbp1s mRNA reduction in renal tubules is associated with lower NGAL and CHOP/Chop levels in the kidney after LPS treatment compared to LPS-treated controls with intact Xbp1s expression. These last two findings support the conclusion that the increased Xpb1s in LPS- induced AKI is pathogenic, necessary, and sufficient. Fourth, concomitant Xbp1s overexpression and LPS injection exhibits additive effects only to a certain extent indicating that LPS utilizes pathways in addition to Xpb1s to induce AKI.
The level of nuclear Xbp1s in renal cortex of wild-type mice concurs with previous studies demonstrating that Xbp1s is required in a tissue-specific manner for constitutive homeostatic UPR.26 However, Xbp1s hyperactivity during infection is detrimental to the kidney. We observed no changes in kidney Xbp1s mRNA expression in AKI models other than LPS, such as FA and IRI experiments, suggesting no significant Xbp1s activation at the whole kidney level. However, it is possible that Xbp1s could be increased in small subsets of cells. Further studies will be needed to determine whether Xbp1s is activated in specific limited cell populations within the kidney in AKI models other than LPS, and are clinically relevant in those disease processes.
Importantly, Xbp1s overexpression per se leads to kidney injury, inflammation, and mortality in the absence of systemic infection and inflammation. KIXs mice had higher renal Chop levels within a day after induction, and higher Ngal, Kim1, Il6 and Tlr4 levels starting at 2 and 5 days after induction. In our LPS-induced sepsis experiments, vehicle-treated KIXs mice showed a significant increase only in Tlr4 and Chop expression compared to vehicle-treated controls, possibly due to the shorter induction time used in this protocol (i.e. 16 hours versus 1 day). The combination of LPS and Xbp1s overexpression did not have additive effect on Tlr4 and Chop expression in KIXs, but it increased renal Chop expression compared to LPS-alone. It is important to consider that the transgenic induction of Xbp1s in KIXs was much higher than the Xbp1s induction by LPS alone (approximately 2,000–4,000 times in KIXs versus 6–20 times after LPS injection, respectively). It is possible that in LPS-treated KIXs the Xbp1s overexpression overwhelmed the cellular UPR response making it difficult to unmask any contribution by other parallel pathways activated by LPS.
Overall, we found that Xbp1s overexpression activates pathways that are common to LPS- induced injury, which contrasts with previous findings showing that pretreatment with ER stress inducers reduces the degree of renal injury.41–44 Nevertheless, Xbp1s in macrophages was shown to be required downstream of TLR4 after LPS recognition for sustained production of innate immune mediators like IL-6 and TNF-α.23 Our data suggest that such a cascade occurs in renal tubular cells. Xbp1s overexpression increased Il6 and Tlr4, and increased Socs3, a major regulator that inhibits the anti-inflammatory response in infection.45,46 The increased Socs3 is compatible with the decrease of its transcriptional repressor Hnf1b. Downregulation of Hnf1b also decreased its target gene Pkhd1 in KIXs.47 The Hnf1b-Socs3-Pkhd1 axis is essential for tubulogenesis, a process that is reactivated during the regeneration of injured tubular cells.47 In KIXs mice, this pathway was silenced meaning that the repair potential of renal tubules is impaired, which in turn may contribute to the burden of kidney failure in these mice.
On the other hand, Six2/Cre+;Xbp1F/F mice had no detectable phenotype at baseline but showed a tendency towards less kidney injury and inflammation upon LPS challenge. Using Six2/Cre, we achieved a 50% decrease in Xbp1 mRNA in total kidney of 21 day old mice (Figure 4). Using Xbp1s-specific primers, we found that Xbp1s levels were similar between Six2/Cre+;Xbp1F/F and Six2/Cre-;Xbp1F/F mice that were 8–12 weeks old and injected with vehicle (Figure 5C). This suggests that in the absence of stress, Xbp1s levels are maintained constant in the kidney of Six2/Cre+;Xbp1F/F mice via increased splicing of newly transcribed or already available Xbp1 mRNA in tubular cells that did not undergo recombination. However, when treated with LPS, Six2/Cre+;Xbp1F/F mice failed to further increase kidney Xbp1s expression compared to LPS-injected Six2/Cre-;Xbp1F/F controls. After LPS injection, Six2/Cre+;Xbp1F/F mice showed lower Kim1 and Chop expression, lower NGAL protein levels, and lower serum Cr values compared to LPS-treated controls. NGAL is also a marker of systemic inflammation as it is produced by both hematopoietic and non-hematopoietic cells as part of the inflammatory response.48 The differences observed in NGAL expression in the KIXs and Xbp1s knockdown kidneys during sepsis may be explained by both changes in the number of infiltrating immune cells and in NGAL production by renal tubular cells.
ER stress signaling controls inflammation in an organ-specific manner.19 While the knockdown of pathways that activate Xbp1s in other systems is associated with increased inflammation,24,49,50 we showed that Xbp1s actually promotes inflammation and injury in LPS-induced AKI, while a decrease in Xbp1s in renal tubular cells attenuates the inflammatory response and injury (Figure 8). Using Tlr4Lps-d mice, which have a whole body expression of mutant Tlr4 and are resistant to LPS, we showed that TLR4 activation by LPS is required for the increase in Xbp1s in kidney. However, due to the global nature of the deletion, this experiment per se is not sufficient to distinguish between the role of tubular TLR4 signaling versus TLR4 activation in infiltrating immune cells; two pathways that can both contribute to the increase in Xbp1s in kidney. Recently, administration of STF-083010, an inhibitor of the endonuclease activity of IRE1α, to rats with ischemic AKI, suppressed Xbp1s, inflammation and apoptosis, and improve renal function and structure.51 In this study, we did not observe any improvement in the kidney histology of Six2/Cre+;Xbp1F/F mice ih the short duration of 24 hours after LPS injection (Supplementary Figure 7), but we cannot exclude that the lower NGAL (and serum creatinine) levels may lead to a better renal outcome in longer follow-up. As sepsis causes multi-organ dysfunction, systemic therapy to suppress Xbp1s activation could have implications for organs other than the kidney. Future studies are needed to determine whether and how LPS sepsis activates Xbp1s in non-renal organs as well as whether renal Xbp1s-mediated AKI affects distant organs.52
Figure 8. The proposed mechanism by which Xbp1s promotes inflammation and injury in renal tubular cells during AKI in sepsis.

LPS is bound to LBP which transfers it to CD14; CD14 then brings LPS to the TLR4-MD-2 complex. The binding of the endotoxin LPS to the TLR4-MD-2 complex activates IRE1 that cleaves the mRNA encoding for Xbp1 by excision of 26 nucleotides (red) from the Xbp1 mRNA, generating the spliced Xbp1 form, Xbp1s, which is translated into a nuclear transcription factor. Xbp1s increases the transcription of its known target gene Il6. Transcriptional pathways not fully characterized lead to an increase in Ngal and a decrease in Hnf1b transcription. HNF-1β is a nuclear transcription factor that inhibits the expression of Socs-3. Altogether these transcriptional networks augment inflammation and injury, while decreasing kidney repair. Other pathways downstream the activation of TLR4, such as the NF-κB pathway, may augment the sepsis-induced AKI. Importantly, both tubular TLR4 signaling and TLR4 activation in infiltrating immune cells can possibly contribute to the increase in Xbp1s in kidney. LPS: lipopolysaccharide; LBP: LPS binding protein; CD14: cluster of differentiation 14; MD-2: myeloid differentiation factor 2; TLR4: toll-like receptor-4; Xbp1s: spliced X-box binding protein-1; ER: endoplasmic reticulum; IRE1: inositol-requiring enzyme 1; Il6: interleukin-6; Ngal: neutrophil gelatinase-associated lipocalin; Hnf1b: hepatocyte nuclear factor 1β; Socs3: suppressor of cytokine signaling 3.
In conclusion, our findings have significant translational potential. Further investigation in septic patients will be required to confirm or refute whether renal Xbp1s signaling drives AKI in sepsis, whether Xbp1s could serve as an early sentinel biomarker to signal impending sepsis- associated AKI and eventually, pharmacologic blockade of Xbp1s signaling can prevent, forestall, or alleviate AKI in sepsis and improve overall survival.
METHODS
Animal studies
Mice were maintained on a 12-hour light/dark cycle (6 am to 6 pm) with unrestricted access to food and water and no more than 4 mice per cage. All protocols were approved by the Institutional Animal Care and Use Committee of University of Texas Southwestern Medical Center. Male and female mice were randomly assigned to experimental groups in all studies.
To study kidney expression of Xbp1s in AKI, several disease models were used. C57BL/6 mice (8–12 weeks old) were injected with intraperitoneal (IP) folic acid (FA, 240 μg/g body weight; F7876, Sigma-Aldrich, St.Louis, MO) or vehicle (NaHCO3, 0.2 ml, 0.3 M; S6014, Sigma-Aldrich, St.Louis, MO), and killed 48 hours later.53 Another set of C57BL/6 mice (8–12 weeks old) received an IP injection of 16 mg/kg lipopolysaccharide (LPS) from Escherichia coli 0111:B4 (LPS-EB; tlrl-eblps, InvivoGen, San Diego, CA) or vehicle, and a single dose of IP injection of 1 mg/kg buprenorphine to control pain. 500 μl of saline was administered subcutaneously as fluid replacement. Animals were sacrificed at 24-hour following LPS injection. Cecal ligation puncture (CLP) was performed in C57BL/6 mice as a second sepsis model in vivo. Briefly, the cecum was ligated 1 cm from the distal end with a silk suture and punctured once with a 23-gauge needle. Sham operated animals underwent laparotomy without ligation or puncture of the cecum. 1 mg/kg sustained release buprenorphine was administered IP at the time of surgery to all animals. Mice were sacrificed 24 hours following surgery. Finally, another set of C57BL/6 mice (8–12 weeks old) underwent right nephrectomy and contralateral ischemia-reperfusion injury (IRI; 20’ ischemia by renal artery cross-clamp, 4-hour reperfusion), and sacrificed 4-hour after surgery. To study the expression of Xbp1s in chronic kidney disease (CKD), three models were tested. First, the ob/ob mouse strain was used as model timed for phases I and II of type II diabetes and obesity. These mice exhibit obesity, hyperphagia, transient hyperglycemia, glucose intolerance, and elevated plasma insulin due to a homozygous mutation in leptin. Next, two previously described mouse models of polycystic kidney disease (PKD) were tested, Ksp/Cre;Pkd1F/F and Pkhd1/Cre;Pkd2F/F.54 Ksp/Cre; Pkd1F/F mice exhibit an aggressive phenotype and die from kidney failure by postnatal day 16 (P16). Pkhd1/Cre;Pkd2F/F mice suffer from a milder form of PKD and die between P7–125. To prove that the Xbp1s increase in kidney after LPS treatment is mediated by TLR4 activation, C3H/HeJ mice, which have a spontaneous mutation in Tlr4 at the LPS response locus (Tlr4Lps-d) that makes C3H/HeJ mice more resistant to endotoxin, were used. C3H/HeJ or wild-type C57BL/6J mice received an IP injection of 16 mg/kg Ultrapure LPS from Escherichia coli 0111 :B4 (Ultrapure LPS-EB: tlrl- 3pelps, InvivoGen, San Diego, CA) that only activates the TLR4, but not TLR2, pathway, or vehicle, and sacrificed after 24 hours.
To generate the kidney-inducible Xbp1s (KIXs) transgenic mice, we crossed the TRE/Xbp1s transgenic mouse32 in C57BL/6 background with the Ksp/rtTA55 mouse model in CD-1 background. Doxycycline (Dox)-containing water (0.1 or 0.5 mg/mL; D9891, Sigma-Aldrich) was used to induce Xbp1s expression at weaning (21 days after birth). Ksp/rtTA transgenic mice given Dox were used as controls to exclude confounding effects of rtTA expression and activation, and Dox exposure. Xbp1s expression was tested at 1, 2 and 5 days after induction. Fresh Dox-containing water was provided 3 days after the initial induction.
Conditional Xbp1 knockdown mice (Xbp1F/F)24 in C57BL/6 background were bred with Six2/Cre56 mice with CD-1 background to achieve renal tubule-specific deletion of Xbp1. KO mice at weaning were compared to age-matched Six2/Cre control animals. KIXs or Six2/Cre+;Xbp1F/F mice (8–12 weeks old) were used for LPS injections. KIXs underwent preconditioning with doxycycline-containing water (0.1 mg/mL) for 16 hours (6 pm to 10 am). At sacrifice, tissues were frozen in liquid nitrogen for subsequent processing.
Tissue harvesting and analysis
Mice were anesthetized and blood was obtained by cardiac puncture, and the right kidney was flash-frozen for molecular assays. Left kidneys were perfused with cold PBS and 4% (w/v) paraformaldehyde (PFA) and then removed. For immunofluorescence, isolated kidneys were fixed in 4% (w/v) PFA overnight, dehydrated in sucrose/PBS 30% (w/v) overnight, embedded in optimal cutting temperature (OCT) compound, and sectioned by cryostat. For immunohistochemistry, kidneys were fixed in 4% (w/v) PFA overnight and embedded in paraffin. Sagittal sections of kidneys were stained with hematoxylin and eosin (H&E), trichrome or Periodic acid-Schiff stain (PAS), and examined by light microscopy using an Axioplan 2 Imaging system (Carl Zeiss Micro-Imaging, Inc. Thornwood, NY).
For histology, kidney sections stained with H&E were examined and photographed by an operator blinded to the experimental protocol. A semi-quantitative pathological scoring system was used as described with minor modifications.57 The severity of renal tubular damage was scored in a blinded fashion with a grading system: 0 = no visible lesions, normal or near normal morphology; 1 = loss of brush border in less than 25% of tubular cells, integrity of basal membrane; 2 = loss of brush border in more than 25% of tubular cells, thickened basal membrane; 3 = as in 2 plus inflammation cast formation, necrosis up to 60% of tubular cells; 4 = as in 3 plus necrosis in more than 60% of tubular cells. Ten fields of cortex and medulla were scored in each kidney sections using ImageJ. The total score for each kidney was calculated by addition of all scores with a numeric maximum of 40.
Blood urea nitrogen (BUN) and creatinine measurements
Blood was collected, kept at room temperature (RT) for 30 minutes, and serum was isolated (3,600 × g for 10 min). Serum creatinine (Cr) was measured using capillary electrophoresis, and BUN using the Vitros 250 Analyzer.
Immunofluorescence
Frozen 8-mm sections were washed with PBS, blocked with 5% (w/v) BSA/PBS (1 h at RT), incubated with a primary antibody anti-Xbp1s (1:50; 619502, BioLegend, San Diego, CA), anti- NKCC2 (1:200; NKCC21-A, Alpha Diagnostic International, San Antonio, TX), or anti-NCC (1:200; SPC-402D, StressMarq, Biosciences Inc., Victoria, Canada) at 4°C overnight, and detected by secondary antibodies conjugated with Alexa Fluor 594 or Alexa Fluor 488 (1:500; Molecular Probes, Eugene, OR). Sections stained with Lotus Tetragonolobus Lectin (LTA) were incubated with fluorescein-labeled LTA (1:500; FL-1321, Vector Laboratories, Burlingame, CA) at RT for 30 min before washing. Sections stained with Dolichos Biflorus Agglutinin (DBA) were incubated with biotinylated DBA (1:400; B-1035, Vector Laboratories, Burlingame, CA) at 4°C overnight, and detected by fluorescein Avidin D (1:500; A-2001, Vector Laboratories, Burlingame, CA).
Immunoblotting
Total protein was extracted from kidneys using T-PER buffer (78510, Thermo Fisher Scientific, Waltham, MA) with proteinase/phosphatase inhibitors (88669, Thermo Fisher Scientific, Waltham, MA). Protein was quantified by the BCA Protein Assay Kit (23227, Thermo Fisher Scientific, Waltham, MA). 20 μg of protein was loaded on a 4–15% SDS-polyacrylamide gel, and the proteins were transferred nitrocellulose membranes. The membrane was blocked in either 5% bovine serum albumin (BSA) or 1% powdered milk, and then incubated with a primary rabbit anti-Xbp1s (1:500; 619502, BioLegend, San Diego, CA), goat anti-Ngal (1:1000; AF1757, R&D Systems, Minneapolis, MN), mouse anti-GADD153 (clone B-3; 1:200; sc-7351, Santa Cruz Biotechnology Inc., Dallas, TX), or mouse anti-β-actin (1:50,000; A3854; Sigma-Aldrich, St.Louis, MO) overnight at 4°C. Goat anti-rabbit, donkey anti-goat or sheep anti-mouse HRP- conjugated IgG were used as secondary antibodies, and blots were developed using the SuperSignal West Dura Extended Duration substrate (Pierce, Thermo Fisher Scientific, Waltham, MA). The protein bands were quantified using Quantity One imaging software from Bio-Rad.
Reverse transcription polymerase chain reaction (RT-PCR) and quantitative RT-PCR (RT-qPCR)
Total RNA from mouse kidney was isolated using Trizol (Gibco, Thermo Fisher Scientific, Waltham, MA). Total RNA (2 μg) was treated with DNase (18068–015, Invitrogen, Thermo Fisher Scientific, Waltham, MA) and reverse transcribed using an iScript cDNA Synthesis Kit (170–8891, Bio-Rad, Hercules, CA). RT-PCR was performed using a Platinum Taq DNA Polymerase High Fidelity (11304–011, Invitrogen, Thermo Fisher Scientific, Waltham, MA). Transcript levels were quantified by RT-qPCR using SybrGreen (172–5121, Bio-Rad, Hercules, CA) on a CFX Connect real-time PCR detection system. Expression amount was normalized to 18S mRNA and was represented as fold change. Data were analyzed using the Bio-Rad CFX software. PCR primers (Supplementary Table 2) were designed with the online NCBI/Primer- BLAST software.
Statistical analyses
All results are depicted as mean ± standard error of the mean (SEM). Unpaired student’s t-test was used for pairwise comparisons, and two-way analysis of variance (ANOVA) with Sidak’s test for multiple comparisons. P<0.05 was considered statistically significant. The correlation between Xbp1s gene expression and serum Cr values, Kim-1, Ngal, Tlr4 and Chop gene expression was assessed with linear regression using Prism 7 (v.7.0a, GraphPad Software Inc, La Jolla, CA).
Supplementary Material
Supplementary Table 1. Evaluation of kidney function decline in AKI models
{kind=link}
Supplementary Table 2. Primer sets used in this study
Supplementary Figure 1. Xbp1s expression is increased in all nephron segments in LPS induced sepsis. Single and merged staining of Xbp1s and nephron segment-specific markers revealed that Xbp1s expression was induced in epithelia cells from all nephron segments in kidneys of mice treated with LPS. LTA: lotus tetragonolobus agglutinin; NKCC2: Na+-K+-Cl cotransporter; NCC: Na+-Cl- cotransporter; DBA: dolichos biflorus agglutinin. Scale bar, 20 μm.
{kind=link}
Supplementary Figure 2. Xbp1s expression in mouse kidney is increased by CLP-induced sepsis. C57BL/6 (8 weeks old) underwent CLP (N=5) or sham (N=5) surgery, and sacrificed 24 hours later. (A) qPCR on kidney samples from mice that underwent CLP or sham surgery. Fold change is relative to sham controls. (B) Serum Cr and (C) BUN levels from mice that underwent CLP or sham surgery. (D) Kidney sections from mice that underwent CLP or sham surgery were stained with hematoxylin and eosin (H&E). Bar represents 40 μm. CLP: cecal ligation puncture; Cr: creatinine; BUN: blood urea nitrogen. Bars and error bars are mean and SEM. Student’s unpaired t-test of CLP vs. sham control. * indicates P<0.05, *** indicates P<0.001, compared to sham control.
{kind=link}
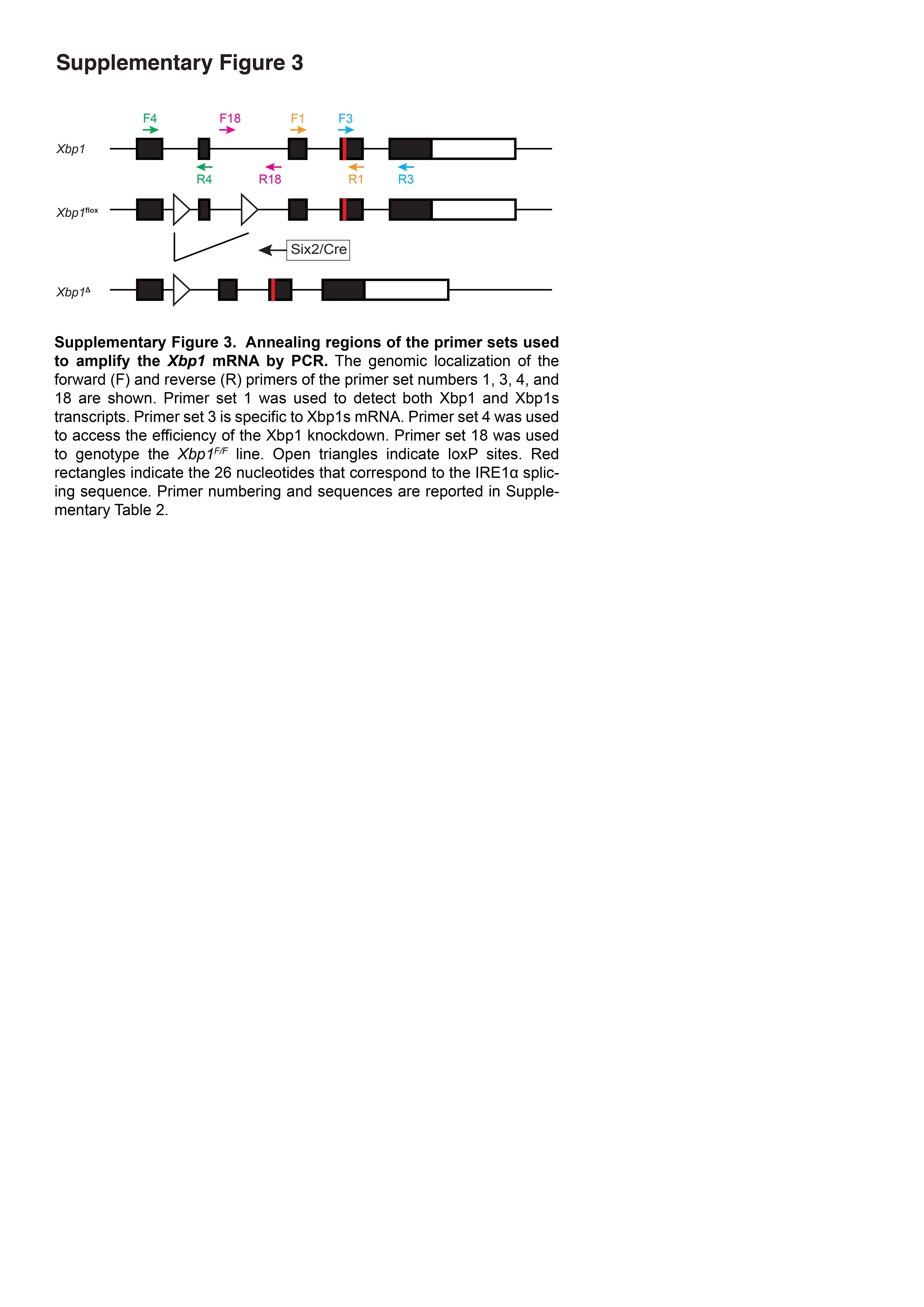
Supplementary Figure 3. Annealing regions of the primer sets used to amplify the Xbp1 mRNA by PCR. The genomic localization of the forward (F) and reverse (R) primers of the primer set numbers 1, 3, 4, and 18 are shown. Primer set 1 was used to detect both Xbp1 and Xbp1s transcripts. Primer set 3 is specific to Xbp1s mRNA. Primer set 4 was used to access the efficiency of the Xbp1 knockdown. Primer set 18 was used to genotype the Xbp1F/F line. Open triangles indicate loxP sites. Red rectangles indicate the 26 nucleotides that correspond to the IRE1α splicing sequence. Primer numbering and sequences are reported in Supplementary Table 2.
{kind=link}
Supplementary Figure 4. Xbp1s increase in kidney after LPS treatment is mediated by TLR4 activation. Wild-type C57BL/6J (WT) or Tlr4Lps-d mice received an IP injection of 16 mg/kg Ultrapure LPS that only activates the TLR4, but not TLR2, pathway, or vehicle, and sacrificed after 24 hours. (A) Xbp1s response was blunted in kidneys from Tlr4Lps-d mice injected with Ultrapure LPS, but not in WT mice. Fold change is relative to vehicle-treated controls within each genotype. (B) Serum creatinine was significantly increased in both strains, but the increase was much lower in Tlr4Lps-d compared to WT mice. (C-D) Body temperature and blood glucose levels at 6 hours after injection reveal that Tlr4Lps-d, but not WT, mice are resistant to Ultrapure LPS. SCr: serum creatinine. Bars and error bars are mean and SEM. Student’s unpaired t-test of Ultrapure LPS vs. vehicle within each genotype for Supplementary Figure 3A–B; 2-way ANOVA, Sidak’s multiple comparisons test for Supplementary Figure 3C–D. * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001.
{kind=link}
Supplementary Figure 5. Overexpression of Xbp1s in Ksp/rtTA;TRE-Xbp1s mice. RT-PCR (A), qPCR (B) and immunoblot (C) demonstrating Xbp1 gene splicing at 1, 2 and 5 days after Dox treatment via the drinking water. Fold change is relative to 0 day on Dox-containing water. (D) Xbp1s antibody co-staining with nephron segment-specific markers revealed that Xbp1s expression was induced in epithelia cells from all nephron segments in kidneys of Ksp/rtTA;TRE-Xbp1s mice at 2 days after Dox treatment. 0.5 mg/ml Dox-containing water was provided daily for the duration of the experiment. Scale bar, 20 μm. LTA: lotus tetragonolobus agglutinin; NKCC2: Na+-K+-Cl- cotransporter; NCC: Na+-Cl- cotransporter; DBA: dolichos biflorus agglutinin. Bars and error bars are SEM. Student’s unpaired t-test of KIXs vs. control at each time-point. * indicates P<0.05, compared to respective control at each time-point.
{kind=link}
Supplementary Figure 6. Overexpression of Xbp1s in the kidney induces tubular injury. Kidney sections from KIXs animals treated for 5 days with Dox-containing water were stained with periodic acid-Schiff (PAS) or trichrome. PAS staining showed loss of brush border in all transgenic mice compared to control animals. No fibrosis was observed as indicated by trichrome staining. 0.5 mg/ml Dox-containing water was provided daily for the duration of the experiment. Bar represents 20 μm. KIXs: kidney-inducible Xbp1s mice.
{kind=link}
Supplementary Figure 7. Kidney histology of tissue-specific Xbp1s knockdown or KIXs mice after LPS-induced sepsis. Hematoxylin and eosin (H&E) staining of kidney sections from Xbp1s knockout (A) or KIXs (B) mice at 24 hours after an intraperitoneal (IP) injection of LPS (16 mg/kg) or vehicle. KIXs and control mice received 0.1 mg/ml Dox-containing water for 16 hours prior LPS injection. Bar represents 40 μm. KIXs: kidney-inducible Xbp1s mice.
{kind=link}
Supplementary Figure 8. Correlation curves of Xbp1s mRNA expression with serum Cr, Kim1, Ngal, Tlr4, or Chop mRNA expression. Data was obtained from Six2/Cre-;Xbp1F/F mice injected with vehicle (n=6) or LPS 16 mg/kg (n=6) as shown in Figure 5A–B. On the X-axis are the Xbp1s mRNA expression levels in kidney determined by qPCR as fold control. On the Y-axis are the serum Cr values (A), or Kim1 (B), Ngal (C), Tlr4 (D) and Chop (E) mRNA levels in the kidney of the same mice. Cr: creatinine; LPS: lipopolysaccharide.
{kind=link}
TRANSLATIONAL STATEMENT.
Increased Xbp1s signaling in renal tubules is unique to sepsis-associated AKI and contributes to inflammation and injury. Inhibition of Xbp1s-mediated pathways is a potential portal to alleviate AKI onset, severity, and progression in sepsis. Xbp1 knockdown mice exposed to endotoxin (LPS) had lower NGAL and serum creatinine levels which may translate to better renal outcomes in longer follow-up. Future studies should determine if inhibition of Xbp1s activity improves renal outcome in septic AKI. Finally, urine Xbp1s may be a potential biomarker of impending AKI onset during sepsis.
ACKNOWLEDGEMENTS
SF was supported by the Ben J. Lipps Research Fellowship Program of American Society of Nephrology Foundation for Kidney Research and the Charles and Jane Pak Center for Mineral Metabolism and Clinical Research Innovative Research Support Award. Work from the authors’ laboratories is supported by the National Institute of Health grants R01-DK091392, and R01- DK091392, (to OM); R01-DK55758, R01-DK099110, P01-DK088761 and P01-AG051459 (to PES); R37DK042921 (to PI); K08-DK110424 and ASN Carl W. Gottschalk Research Scholar Grant (to SH); and R01-DK096251 (to CYL). We thank the UT Southwestern O’Brien Kidney Research Core Center (P30-DK079328 to OM). We thank the UT Southwestern Metabolic phenotyping core for all creatinine and BUN measurements, Laurie H Glimcher (Weill Cornell Medical College) for providing the Xbp1s-Flox mice, Vishal D Patel (UT Southwestern Medical Center) for providing Ksp/Cre;Pkd1F/F and Pkhd1/Cre;Pkd2F/F mouse kidney samples, and Alexandru Bobulescu (UT Southwestern Medical Center) for providing ob/ob mouse kidney samples. We thank Keng-Mean Lin and Matanel Yheskel (UT Southwestern Medical Center) for excellent technical assistance and helpful discussion during the preparation of this manuscript.
Footnotes
DISCLOSURE
None.
Supplementary information is available at Kidney International’s website
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Angus DC, et al. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. [DOI] [PubMed] [Google Scholar]
- 2.Vincent JL, et al. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med. 2006;34:344–353. [DOI] [PubMed] [Google Scholar]
- 3.Bagshaw SM, et al. Septic acute kidney injury in critically ill patients: clinical characteristics and outcomes. Clin J Am Soc Nephrol. 2007;2:431–439. [DOI] [PubMed] [Google Scholar]
- 4.Bagshaw SM, et al. Early acute kidney injury and sepsis: a multicentre evaluation. Crit Care. 2008;12:R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bagshaw SM, et al. Acute kidney injury in septic shock: clinical outcomes and impact of duration of hypotension prior to initiation of antimicrobial therapy. Intensive Care Med. 2009;35:871–881. [DOI] [PubMed] [Google Scholar]
- 6.Zarjou A, Agarwal A. Sepsis and acute kidney injury. J Am Soc Nephrol. 2011;22:999–1006. [DOI] [PubMed] [Google Scholar]
- 7.Honore PM, et al. Renal blood flow and acute kidney injury in septic shock: an arduous conflict that smolders intrarenally? Kidney Int. 2016;90:22–24. [DOI] [PubMed] [Google Scholar]
- 8.Venkatachalam MA, Weinberg JM. The tubule pathology of septic acute kidney injury: a neglected area of research comes of age. Kidney Int. 2012;81:338–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park BS, et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. [DOI] [PubMed] [Google Scholar]
- 10.El-Achkar TM, et al. Pathways of renal injury in systemic gram-negative sepsis. Eur J Clin Invest. 2008;38 Suppl 2:39–44. [DOI] [PubMed] [Google Scholar]
- 11.Sanchez-Nino MD, et al. TNF superfamily: a growing saga of kidney injury modulators. Mediators Inflamm. 2010;2010: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naito M, et al. Endotoxin mediates recruitment of RNA polymerase II to target genes in acute renal failure. J Am Soc Nephrol. 2008;19:1321–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noiri E, et al. Tumor necrosis factor-alpha mRNA expression in lipopolysaccharide- stimulated rat kidney. Chronological analysis of localization. Am J Pathol. 1994;144:1159–1166. [PMC free article] [PubMed] [Google Scholar]
- 14.Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. [DOI] [PubMed] [Google Scholar]
- 15.Watowich SS, et al. Flux of the paramyxovirus hemagglutinin-neuraminidase glycoprotein through the endoplasmic reticulum activates transcription of the GRP78-BiP gene. J Virol. 1991;65:3590–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan CP, et al. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2006;80:9279–9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. [DOI] [PubMed] [Google Scholar]
- 18.Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat Rev Nephrol. 2017;13:681–696. [DOI] [PubMed] [Google Scholar]
- 19.Garg AD, et al. ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol Med. 2012;18:589–598. [DOI] [PubMed] [Google Scholar]
- 20.Mori K Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem. 2009;146:743–750. [DOI] [PubMed] [Google Scholar]
- 21.Endo M, et al. The ER stress pathway involving CHOP is activated in the lungs of LPS- treated mice. J Biochem. 2005;138:501–507. [DOI] [PubMed] [Google Scholar]
- 22.van ‘t Wout EF, et al. Virulence Factors of Pseudomonas aeruginosa Induce Both the Unfolded Protein and Integrated Stress Responses in Airway Epithelial Cells. PLoS Pathog. 2015;11:e1004946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinon F, et al. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaser A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hassan H, et al. Essential Role of X-Box Binding Protein-1 during Endoplasmic Reticulum Stress in Podocytes. J Am Soc Nephrol. 2016;27:1055–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madhusudhan T, et al. Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nat Commun. 2015;6:6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shao D, et al. Suppression of XBP1S mediates high glucose-induced oxidative stress and extracellular matrix synthesis in renal mesangial cell and kidney of diabetic rats. PLoS One. 2013;8:e56124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fedeles SV, et al. Sec63 and Xbp1 regulate IRE1alpha activity and polycystic disease severity. J Clin Invest. 2015;125:1955–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishikawa Y, et al. Spliced XBP1 Rescues Renal Interstitial Inflammation Due to Loss of Sec63 in Collecting Ducts. J Am Soc Nephrol. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ozcan U, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. [DOI] [PubMed] [Google Scholar]
- 31.Garg AD, et al. Targeting the hallmarks of cancer with therapy-induced endoplasmic reticulum (ER) stress. Mol Cell Oncol. 2015;2:e975089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng Y, et al. The Xbp1s/GalE axis links ER stress to postprandial hepatic metabolism. J Clin Invest. 2013;123:455–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee AH, et al. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verdeguer F, et al. A mitotic transcriptional switch in polycystic kidney disease. Nat Med. 2010;16:106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faguer S, et al. Hnf-1beta transcription factor is an early hif-1alpha-independent marker of epithelial hypoxia and controls renal repair. PLoS One. 2013;8:e63585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hotchkiss RS, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230–1251. [DOI] [PubMed] [Google Scholar]
- 37.Takasu O, et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med. 2013;187:509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kosaka J, et al. Histopathology of Septic Acute Kidney Injury: A Systematic Review of Experimental Data. Crit Care Med. 2016;44:e897–903. [DOI] [PubMed] [Google Scholar]
- 39.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hato T, et al. Bacterial sepsis triggers an antiviral response that causes translation shutdown. J Clin Invest. 2019;129:296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chatterjee PK, et al. Lipoteichoic acid from Staphylococcus aureus reduces renal ischemia/reperfusion injury. Kidney Int. 2002;62:1249–1263. [DOI] [PubMed] [Google Scholar]
- 42.He K, et al. Lipopolysaccharide-induced cross-tolerance against renal ischemia- reperfusion injury is mediated by hypoxia-inducible factor-2alpha-regulated nitric oxide production. Kidney Int. 2014;85:276–288. [DOI] [PubMed] [Google Scholar]
- 43.Heemann U, et al. Lipopolysaccharide pretreatment protects from renal ischemia/reperfusion injury : possible connection to an interleukin-6-dependent pathway. Am J Pathol. 2000;156:287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prachasilchai W, et al. A protective role of unfolded protein response in mouse ischemic acute kidney injury. Eur J Pharmacol. 2008;592:138–145. [DOI] [PubMed] [Google Scholar]
- 45.Susnik N, et al. Ablation of proximal tubular suppressor of cytokine signaling 3 enhances tubular cell cycling and modifies macrophage phenotype during acute kidney injury. Kidney Int. 2014;85:1357–1368. [DOI] [PubMed] [Google Scholar]
- 46.Wilson HM. SOCS Proteins in Macrophage Polarization and Function. Front Immunol. 2014;5:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma Z, et al. Mutations of HNF-1beta inhibit epithelial morphogenesis through dysregulation of SOCS-3. Proc Natl Acad Sci U S A. 2007;104:20386–20391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cowland JB, Borregaard N. Molecular characterization and pattern of tissue expression of the gene for neutrophil gelatinase-associated lipocalin from humans. Genomics. 1997;45:17–23. [DOI] [PubMed] [Google Scholar]
- 49.Leonard A, et al. Preconditioning with endoplasmic reticulum stress ameliorates endothelial cell inflammation. PLoS One. 2014;9:e110949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J, et al. Preconditioning with endoplasmic reticulum stress mitigates retinal endothelial inflammation via activation of X-box binding protein 1. J Biol Chem. 2011;286:4912–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu L, et al. STF-083010, an inhibitor of XBP1 splicing, attenuates acute renal failure in rats by suppressing endoplasmic reticulum stress-induced apoptosis and inflammation. Exp Anim. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doi K, Rabb H. Impact of acute kidney injury on distant organ function: recent findings and potential therapeutic targets. Kidney Int. 2016;89:555–564. [DOI] [PubMed] [Google Scholar]
- 53.Kolatsi-Joannou M, et al. Modified citrus pectin reduces galectin-3 expression and disease severity in experimental acute kidney injury. PLoS One. 2011;6:e18683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hajarnis S, et al. microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat Commun. 2017;8:14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pan X, et al. Generation and characterization of KsprtTA and KsptTA transgenic mice. Genesis. 2013;51:430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kobayashi A, et al. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell. 2008;3:169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Di Giusto G, et al. Oat5 and NaDC1 protein abundance in kidney and urine after renal ischemic reperfusion injury. J Histochem Cytochem. 2009;57:17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Evaluation of kidney function decline in AKI models
Supplementary Table 2. Primer sets used in this study
Supplementary Figure 1. Xbp1s expression is increased in all nephron segments in LPS induced sepsis. Single and merged staining of Xbp1s and nephron segment-specific markers revealed that Xbp1s expression was induced in epithelia cells from all nephron segments in kidneys of mice treated with LPS. LTA: lotus tetragonolobus agglutinin; NKCC2: Na+-K+-Cl cotransporter; NCC: Na+-Cl- cotransporter; DBA: dolichos biflorus agglutinin. Scale bar, 20 μm.
Supplementary Figure 2. Xbp1s expression in mouse kidney is increased by CLP-induced sepsis. C57BL/6 (8 weeks old) underwent CLP (N=5) or sham (N=5) surgery, and sacrificed 24 hours later. (A) qPCR on kidney samples from mice that underwent CLP or sham surgery. Fold change is relative to sham controls. (B) Serum Cr and (C) BUN levels from mice that underwent CLP or sham surgery. (D) Kidney sections from mice that underwent CLP or sham surgery were stained with hematoxylin and eosin (H&E). Bar represents 40 μm. CLP: cecal ligation puncture; Cr: creatinine; BUN: blood urea nitrogen. Bars and error bars are mean and SEM. Student’s unpaired t-test of CLP vs. sham control. * indicates P<0.05, *** indicates P<0.001, compared to sham control.
Supplementary Figure 3. Annealing regions of the primer sets used to amplify the Xbp1 mRNA by PCR. The genomic localization of the forward (F) and reverse (R) primers of the primer set numbers 1, 3, 4, and 18 are shown. Primer set 1 was used to detect both Xbp1 and Xbp1s transcripts. Primer set 3 is specific to Xbp1s mRNA. Primer set 4 was used to access the efficiency of the Xbp1 knockdown. Primer set 18 was used to genotype the Xbp1F/F line. Open triangles indicate loxP sites. Red rectangles indicate the 26 nucleotides that correspond to the IRE1α splicing sequence. Primer numbering and sequences are reported in Supplementary Table 2.
Supplementary Figure 4. Xbp1s increase in kidney after LPS treatment is mediated by TLR4 activation. Wild-type C57BL/6J (WT) or Tlr4Lps-d mice received an IP injection of 16 mg/kg Ultrapure LPS that only activates the TLR4, but not TLR2, pathway, or vehicle, and sacrificed after 24 hours. (A) Xbp1s response was blunted in kidneys from Tlr4Lps-d mice injected with Ultrapure LPS, but not in WT mice. Fold change is relative to vehicle-treated controls within each genotype. (B) Serum creatinine was significantly increased in both strains, but the increase was much lower in Tlr4Lps-d compared to WT mice. (C-D) Body temperature and blood glucose levels at 6 hours after injection reveal that Tlr4Lps-d, but not WT, mice are resistant to Ultrapure LPS. SCr: serum creatinine. Bars and error bars are mean and SEM. Student’s unpaired t-test of Ultrapure LPS vs. vehicle within each genotype for Supplementary Figure 3A–B; 2-way ANOVA, Sidak’s multiple comparisons test for Supplementary Figure 3C–D. * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001.
Supplementary Figure 5. Overexpression of Xbp1s in Ksp/rtTA;TRE-Xbp1s mice. RT-PCR (A), qPCR (B) and immunoblot (C) demonstrating Xbp1 gene splicing at 1, 2 and 5 days after Dox treatment via the drinking water. Fold change is relative to 0 day on Dox-containing water. (D) Xbp1s antibody co-staining with nephron segment-specific markers revealed that Xbp1s expression was induced in epithelia cells from all nephron segments in kidneys of Ksp/rtTA;TRE-Xbp1s mice at 2 days after Dox treatment. 0.5 mg/ml Dox-containing water was provided daily for the duration of the experiment. Scale bar, 20 μm. LTA: lotus tetragonolobus agglutinin; NKCC2: Na+-K+-Cl- cotransporter; NCC: Na+-Cl- cotransporter; DBA: dolichos biflorus agglutinin. Bars and error bars are SEM. Student’s unpaired t-test of KIXs vs. control at each time-point. * indicates P<0.05, compared to respective control at each time-point.
Supplementary Figure 6. Overexpression of Xbp1s in the kidney induces tubular injury. Kidney sections from KIXs animals treated for 5 days with Dox-containing water were stained with periodic acid-Schiff (PAS) or trichrome. PAS staining showed loss of brush border in all transgenic mice compared to control animals. No fibrosis was observed as indicated by trichrome staining. 0.5 mg/ml Dox-containing water was provided daily for the duration of the experiment. Bar represents 20 μm. KIXs: kidney-inducible Xbp1s mice.
Supplementary Figure 7. Kidney histology of tissue-specific Xbp1s knockdown or KIXs mice after LPS-induced sepsis. Hematoxylin and eosin (H&E) staining of kidney sections from Xbp1s knockout (A) or KIXs (B) mice at 24 hours after an intraperitoneal (IP) injection of LPS (16 mg/kg) or vehicle. KIXs and control mice received 0.1 mg/ml Dox-containing water for 16 hours prior LPS injection. Bar represents 40 μm. KIXs: kidney-inducible Xbp1s mice.
Supplementary Figure 8. Correlation curves of Xbp1s mRNA expression with serum Cr, Kim1, Ngal, Tlr4, or Chop mRNA expression. Data was obtained from Six2/Cre-;Xbp1F/F mice injected with vehicle (n=6) or LPS 16 mg/kg (n=6) as shown in Figure 5A–B. On the X-axis are the Xbp1s mRNA expression levels in kidney determined by qPCR as fold control. On the Y-axis are the serum Cr values (A), or Kim1 (B), Ngal (C), Tlr4 (D) and Chop (E) mRNA levels in the kidney of the same mice. Cr: creatinine; LPS: lipopolysaccharide.