

Abstract
BACKGROUND
Successful partner notification can improve community-level outcomes by increasing the proportion of persons living with HIV who are linked to HIV care and virally suppressed, but it is resource intensive. Understanding where HIV transmission pathways may be undetected by routine partner notification may help improve case finding strategies.
METHODS
We combined partner notification interview and HIV sequence data for persons diagnosed with HIV in Wake County, NC in 2012-2013 to evaluate partner contact networks among persons with HIV pol gene sequences ≤ 2% pairwise genetic distance. We applied a set of multivariable generalized estimating equations to identify correlates of disparate membership in genetic vs. partner contact networks.
RESULTS
In the multivariable model, being in a male-male pair (adjusted odds ratio (AOR)=16.7, p=0.01), chronic HIV infection status (AOR=4.5, p<0.01), and increasing percent genetic distance between each dyad member’s HIV pol gene sequence (AOR=8.3 per each 1% increase, p<0.01) were all associated with persons with HIV clustering but not being identified in the partner notification network component. Having anonymous partners or other factors typically associated with risk behavior were not associated.
CONCLUSIONS
Based on genetic networks, partnerships which may be stigmatized, may have occurred farther back in time, or may have an intervening partner were more likely to be unobserved in the partner contact network. HIV genetic cluster information contributes to public health understanding of HIV transmission networks in these settings where partner identifying information is not available.
Keywords: HIV, social network analysis, phylogenetics, contact tracing, public health
Short summary:
Comparing partnerships disclosed during public health HIV investigations to HIV genetic clusters around Raleigh, North Carolina, identified factors associated with failing to observe partnerships during contact tracing.
INTRODUCTION
The Seek, Test, Treat, and Retain strategy aims to identify persons living with HIV infection who do not know their status and remove barriers to retaining them in clinical care and on therapy.1,2 “Seeking” and “testing” rely on several public health actions, including partner notification. Soon after an HIV-infected person is newly diagnosed, partner notification often leads to identifying additional undiagnosed HIV-infected persons.3,4 But, partner notification is limited by interview refusal and inability to locate some partners;3 both result in fewer cases found. As diagnosis represents the HIV care continuum entry point, missed diagnosis opportunities during partner notification lead to suboptimal continuum outcomes and perpetuates potential onward transmission.2,5
Partner notification is name-based, allowing partnerships elicited during interviews to be combined into a contact network of index cases and their located partners. Analysis of networks constructed from partner notification data in North Carolina (NC) from multiple syphilis outbreaks revealed high rates of anonymous, transactional sex among heterosexuals6 and a substantial proportion of partners who could not be located.7,8 Consequently, the observable sociosexual network based on disclosed and located partners had many “missing” partnerships. Identifying where and why partnerships are unobserved can provide clues for finding persons who are active in the network but not identified during the index case’s partner notification interview. To identify gaps in sexual networks where active transmission occurs, HIV gene sequencing offers a complementary methodology which is increasingly available with technological advances in pathogen genetic testing.9
For newly diagnosed HIV cases in NC, we compared the partner notification network to phylogenetic clusters inferred from HIV pol sequences. We observed that many persons with newly diagnosed HIV had no located partners in the partner notification network; but, more than half of these singletons in the partner notification network were identified in a genetic cluster if they had a HIV sequence available.10 In recently diagnosed persons, having an HIV sequence similar enough to cluster with a sequence from at least one other person, yet having no locatable partners indicates gaps where notification of recent partners could be improved. This study presents a comparison of partner notification network and genetic clusters to identify where the partner notification network did not approximate the clusters, with the goal of refining direct efforts for improved partner notification case finding.
MATERIALS AND METHODS
Parent Study Design and Population
We sought to evaluate whether HIV-infected persons identified in the same genetic cluster (“cluster”) were directly or indirectly connected within the same sociosexual network component, as observed through partner notification data (“network”). We identified 116 genetic clusters (defined as ≤3.5% genetic distance between all sequences) using standard techniques of molecular phylogeny11 to analyze a dataset of 15,246 pol gene sequences collected during routine clinical care among HIV patients across the state of NC, as described previously.12 We then matched the clusters to a sociosexual network derived from disclosed partnerships elicited during routine partner notification investigation of cases with newly diagnosed HIV in Wake County from 2012-2013. The partner notification method most closely resembles exponential discriminative snowball sampling,13 as the number of partners named is not limited (unlike respondent driven sampling); the element of discrimination is introduced as only partners who are newly diagnosed with HIV are then asked to disclose their partners. Among all 116 clusters previously identified, 34 (29%) included 2 or more partner notification network members.12 These 34 clusters included 287 total persons; 87 were in the partner notification network and 200 stemmed from the reference sequences collected across NC. We further restricted analysis pairs to those within 2% pairwise genetic distance to capture those partners who were recent enough to have been identified during partner notification, while also allowing clusters to encompass two people who may have intervening partners who were unidentified or unsampled for sequencing. Among the 87 cluster members who were also part of the partner notification network, 84 (97%) were ≤ 2% pairwise genetic distance between another partner notification network member in their cluster. These 84 persons formed the basis for the pairs analyzed here (Figure 1).
Figure 1.
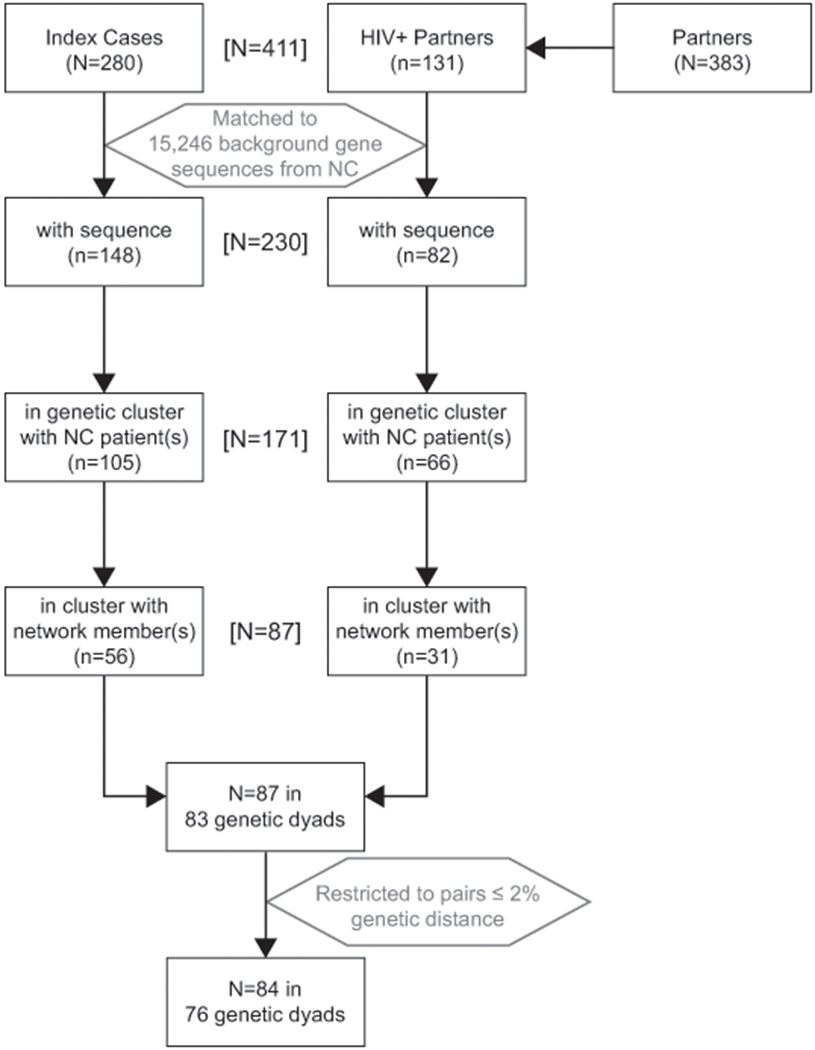
Flow chart of inclusion into the analysis dataset (N=84 people in 76 genetic dyads), starting from the parent study Wake County sociosexual network (N=663, 62% HIV-positive (n=411)). We further analyzed genetic clusters which included at least 2 persons from the partner notification network.
Measures
Genetic Dyads for Observation
We evaluated pairwise genetic distance between all 84 partner notification members. Each pair examined with genetic distance difference ≤ 2% was considered a “genetic dyad” which could represent linked transmission, either directly or indirectly, including through an unsampled partner. These dyads were the unit of observation for this analysis.
Comparison of Genetic Dyads to Partner Notification Components
We compared partner notification network component membership for each member of the genetic dyad. Among persons in the same genetic dyad, membership in different network components suggests that partnerships or other infected persons were not observed, thereby disaggregating the true network into multiple components, as depicted in Figure 2a.
Figure 2.
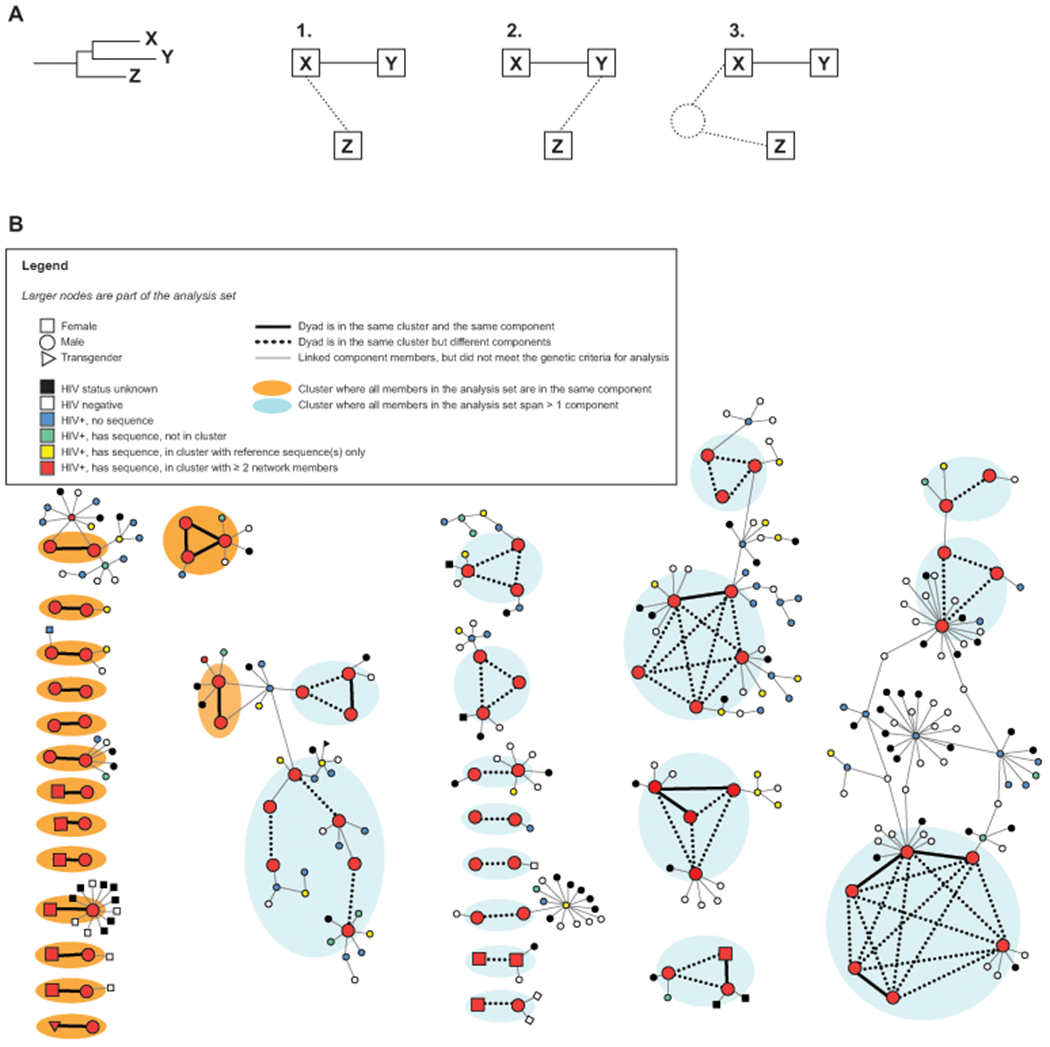
A, Illustration of minimum possible missing relationships (dotted lines) if the HIV genetic cluster includes persons who are not linked in the partner notification network. The person, Z, who is disconnected in the partner notification-based sociosexual network could have (1) been a partner to X, (2) been a partner to Y, or (3) been connected to an unsampled person who is in turn connected to X or Y. B, Partner notification network showing the observed differences between the genetic clusters and sociosexual network based upon partner elicitation interviews. Node color depicts HIV and genetic cluster status: red nodes were in one of the local genetic clusters, defined as ≤ 3.5% genetic distance among all cluster members. Line type depicts relationship and analysis status: thicker lines represent genetic dyads analyzed as they are in the same genetic cluster and ≤ 2% genetic distance, where thick solid lines also represent a first-degree partnership elicited during partner elicitation interview and thick dotted lines connect pair members who met the genetic dyad definition but were not observed to be in the same partner notification network component, and thinner solid lines were partnerships elicited during interview but among people who did not meet the cluster analysis criteria. Clusters grouped in orange only include genetic dyads where all dyad members were in the same cluster and network component. Clusters grouped in blue indicate dyads which span multiple network components.
We used the partner notification network component as the comparator, rather than known first-degree partners, because the component serves somewhat analogously for a genetic cluster: the cluster represents people who are estimated to be close together in the transmission chain, though not necessarily first-degree partners (which cannot be inferred from a genetic cluster). Similarly, a network component is a group of kth-degree partners who are directly or indirectly linked. We considered the genetic dyads as a proxy for transmission linkages because we would expect more partnership links in the partner notification network than what is represented in the genetic cluster because not every sexual contact results in transmission. Therefore, partner notification network components may contain HIV-positive persons from several genetic clusters.
Individual and Partnership Characteristics
We used person-level traits collected in the parent study12 for this analysis, including race, age, gender, Wake County residence, total number of sex partners disclosed, anonymous partners, and HIV stage at diagnosis (acute, recent, or chronic infection). Individual characteristics of each dyad member were compared and then ascribed to the dyad as independent variables. Dyad-level traits for each pair in the cluster included age difference, race/ethnicity difference, gender and sexual preference, days between HIV diagnoses, and similarity among sexual risk behaviors. Risk factor assortativity, the tendency of partners to group by risk behaviors such as engaging in anonymous sex or having a high number of recent sexual partners, occurs in sexual networks14 and HIV genetic clusters.15
Statistical Analyses
The unit of analysis was the dyads within the same genetic cluster. They did not have to be named as each other’s sex partner; because they shared similar genetic sequences, they were inferred to be either directly or indirectly connected in the transmission network. Our outcome was a dichotomous variable indicating whether the persons in each genetic dyad were also in the same or different partner notification network component. We used χ2 tests for categorical comparisons.
Generalized Estimating Equations to Compare Linkages
We examined genetic dyad characteristics that were associated with the outcome of being in different network components using a set of generalized estimating equations (GEE) with a binomial distribution, logit link function, and robust variance. Huber-White robust standard errors accounted for potential autocorrelation among network members. We selected an exchangeable correlation matrix because it permitted us to treat each pair of persons (dyad) in the cluster equally since we could not infer first-degree partnerships. Model predictors selected a priori included whether genetic dyad members shared the same race and whether they were within 5 years of age, as these factors influence partner selection and network formation,16,17 and whether either person was diagnosed during acute HIV infection (AHI). In NC, persons diagnosed with AHI receive priority for interviews because they are highly infectious and the transmitting relationship is in the very recent past, so their links to the transmitting cluster may be more complete. Other predictors tested (Table 2) were selected based on the bivariate relationship between the covariate and the outcome using the odds ratio (OR) and confidence interval, at an alpha level of 0.20. The multivariable GEE was then refined with backward selection of predictors using the quasilikelihood independence model criterion (QIC);18,19 the final model was the one with the lowest QIC. Predictors were considered significant in the final multivariable model if the adjusted OR (AOR) p-value was ≤0.05. Stata 1220 was used for all modeling.
Table 2.
Bivariate and multivariable relationship in the GEE between explanatory variables and the outcome of being in the same genetic dyad but not in the same partner notification interview-based sexual network component, by odds ratio (OR) and confidence intervals (CI) [N=76 pairs].
| Network Component |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Characteristics of Genetic Dyads | Total (N=76) | Different (N=49) | Same (N=27) | Bivariable | Multivariable | |||||||
| N | (col %) | N | (row %) | N | (row %) | OR | (95%CI) | P-value | Adjusted OR | (95%CI) | P-value | |
| Race Assortativity | ||||||||||||
| Different race persons in pair | 18 | (24) | 13 | (72) | 5 | (28) | 1.6 | (0.5-5.1) | 0.43 | |||
| Both persons identify as the same race | 53 | (76) | 36 | (62) | 22 | (38) | ref | |||||
| Black Race in Dyad | ||||||||||||
| Neither person African-American race | 17 | (22) | 13 | (76) | 4 | (24) | 2.1 | (0.6-7.2) | 0.25 | |||
| At least one person identifies as Black | 59 | (78) | 36 | (61) | 23 | (39) | ref | |||||
| Gender in Dyad | ||||||||||||
| Male-male pair | 66 | (87) | 46 | (70) | 20 | (30) | 5.3 | (1.3-22.9) | 0.02* | 16.7** | (1.8-159.2) | 0.01 |
| Male/female or female/female pair | 10 | (13) | 3 | (30) | 7 | (70) | ref | ref | ||||
| Age at HIV Diagnosis | ||||||||||||
| Both age 30 or younger at diagnosis | 44 | (58) | 31 | (70) | 13 | (30) | 1.9 | (0.7-4.8) | 0.20 | |||
| At least one person >30 years of age | 32 | (42) | 18 | (56) | 14 | (44) | ref | |||||
| Age Difference | ||||||||||||
| > 5 years | 36 | (47) | 25 | (69) | 11 | (31) | 1.5 | (0.6-3.9) | 0.39 | |||
| ≤ 5 years of age | 40 | (53) | 24 | (60) | 16 | (40) | ref | |||||
| Wake County Residence in Dyad | ||||||||||||
| Both resided in Wake County at diagnosis | 49 | (64) | 32 | (65) | 17 | (35) | 1.1 | (0.4-2.9) | 0.84 | |||
| At least one person not a Wake resident | 27 | (36) | 17 | (63) | 10 | (37) | ref | |||||
| Time Between HIV Diagnoses in Dyad | ||||||||||||
| < 5 years | 62 | (82) | 42 | (68) | 20 | (32) | 2.1 | (0.6-6.8) | 0.22 | |||
| ≥ 5 years between HIV diagnoses | 14 | (18) | 7 | (50) | 7 | (50) | ref | |||||
| Interview of Person Diagnosed Later in Time | ||||||||||||
| Partners not disclosed | 17 | (22) | 14 | (82) | 3 | (18) | 3.2 | (0.8-12.4) | 0.09* | 4.9 | (0.5-44.6) | 0.16 |
| Partner elicitation completed | 59 | (78) | 35 | (59) | 24 | (41) | ref | ref | ||||
| Acute HIV Infection at Diagnosis in Dyad | ||||||||||||
| Neither acutely infected at diagnosis | 67 | (88) | 45 | (67) | 22 | (33) | 2.6 | (0.6-10.5) | 0.19* | 4.5** | (1.8-11.0) | <0.01 |
| At least one person acutely infected | 9 | (12) | 4 | (44) | 5 | (56) | ref | ref | ||||
| Genetic Distance (per 1% increase) | 0.76 +/− 0.52 | 0.89 +/− 0.48 | 0.52 +/− 0.50 | 4.9 | (1.6-14.8) | <0.01* | 8.3** | (2.4-28.5) | <0.01 | |||
| Anonymous Partners among Dyad | ||||||||||||
| Either member had ≥1 anonymous partner | 35 | (73) | 27 | (77) | 8 | (23) | 1.5 | (0.4-6.2) | 0.56 | |||
| Neither had anonymous partners | 13 | (27) | 9 | (69) | 4 | (31) | ref | |||||
| Behavioral Assortativity (total sex partners) | ||||||||||||
| Fewer than 10 sex partners difference | 66 | (87) | 44 | (67) | 22 | (33) | 2.0 | (0.5-7.6) | 0.31 | |||
| ≥ 10 sex partners difference among pair | 10 | (13) | 5 | (50) | 5 | (50) | ref | |||||
Tested in the multivariable GEE based upon bivariate relationship with outcome
Significant in the multivariable GEE at alpha=0.05
RESULTS
Study Population
From the complete sociosexual network derived from partner notification of the entire county (N=663 index cases and HIV-infected partners), we identified 84 unique persons who could be classified into genetic clusters that included at least one other network member, including 54 index cases and 30 HIV-infected partners (Table 1). Most were male (n=73; 87%) and Black race (n=63; 75%). When compared to HIV-positive persons in the parent study partner notification network who were excluded from this analysis, due to not sharing a genetic cluster with another network member, dyad members were more likely to be Black (75% v. 66%, p=0.11) and younger (mean 32 v. 36 years, p=0.01), but less likely to have zero connections in the partner notification network (“network singleton”: 8% v 30%, p<0.01); 7% refused partner notification interview in this set compared to 20% in the parent dataset. In total, we identified 76 genetic dyads among these 84 persons; 43% (36/84) were included in >1 dyad since their HIV sequence was ≤ 2% pairwise distance from multiple partner notification members.
Table 1.
Characteristics of index cases and HIV-infected partners in the complete sexual network (N=663) and the subset of persons with genetic clusters within ≤ 2% pairwise genetic distance of another network member (N=84), 2012–2013 in Wake County, NC.
| Parent Study Sexual Network (N=663) |
Persons in Genetic Dyads (N=84) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Index (n=280) | Partner (n=383) | Index (n=54) | Partner (n=30) | ||||||
| n | (%) | n | (%) | n | (%) | n | (%) | ||
| Gender | |||||||||
| Male | 232 | (83) | 327 | (85) | 47 | (87) | 26 | (87) | |
| Female | 44 | (16) | 53 | (14) | 6 | (11) | 4 | (13) | |
| Transgender (M to F) | 4 | (1) | 3 | (1) | 1 | (2) | 0 | ||
| Race/ethnicity | |||||||||
| non-Hispanic White | 69 | (25) | 120 | (31) | 13 | (24) | 7 | (23) | |
| non-Hispanic Black | 183 | (65) | 238 | (62) | 40 | (75) | 23 | (77) | |
| Hispanic | 23 | (8) | 12 | (3) | 1 | (1) | 0 | ||
| Other | 5 | (2) | 8 | (2) | 0 | 0 | |||
| unknown | 0 | 5 | (1) | 0 | 0 | ||||
| Age | |||||||||
| mean (SD) | 36 | (12) | 31 | (11) | 32 | (12) | 31 | (12) | |
| HIV status | |||||||||
| Positive, with HIV sequence | 148 | (53) | 82 | (21) | |||||
| Positive, no HIV sequence | 132 | (47) | 49 | (13) | |||||
| Negative | 0 | 148 | (39) | ||||||
| unknown | 0 | 104 | (27) | ||||||
Person-Level Genetic Cluster and Partner Notification Network Overlap
Just under half of persons (39/84, 46%) were in a different partner notification network component than all other network members from their cluster; the other 54% (n=45) were identified in the same partner notification component as at least one other network member in their genetic cluster (Figure 2b). Persons who were not in the same component as any other cluster members were significantly more likely to be a singleton in the partner notification network (18% v. 0%, p<0.01). Cluster members disconnected in different components were less likely to be Black (69% Black) and/or women (8% women) compared to cluster members who shared components with another cluster member (80% Black; 18% women) but these differences were not statistically significant.
Dyad-Level Network Overlap
As shown in Figure 2b, 36% of dyads (27/76) were in the same network component (solid lines) and 64% (49/76) spanned two components (dotted lines). The mean genetic distance for genetic dyads was 0.8% (95%CI: 0.6-0.9%). Among genetic dyads in different network components, genetic distance was significantly greater overall compared to those in the same component (0.9% v. 0.5%, p<0.01), where genetic distance was smaller and positively skewed (Figure 3).
Figure 3.
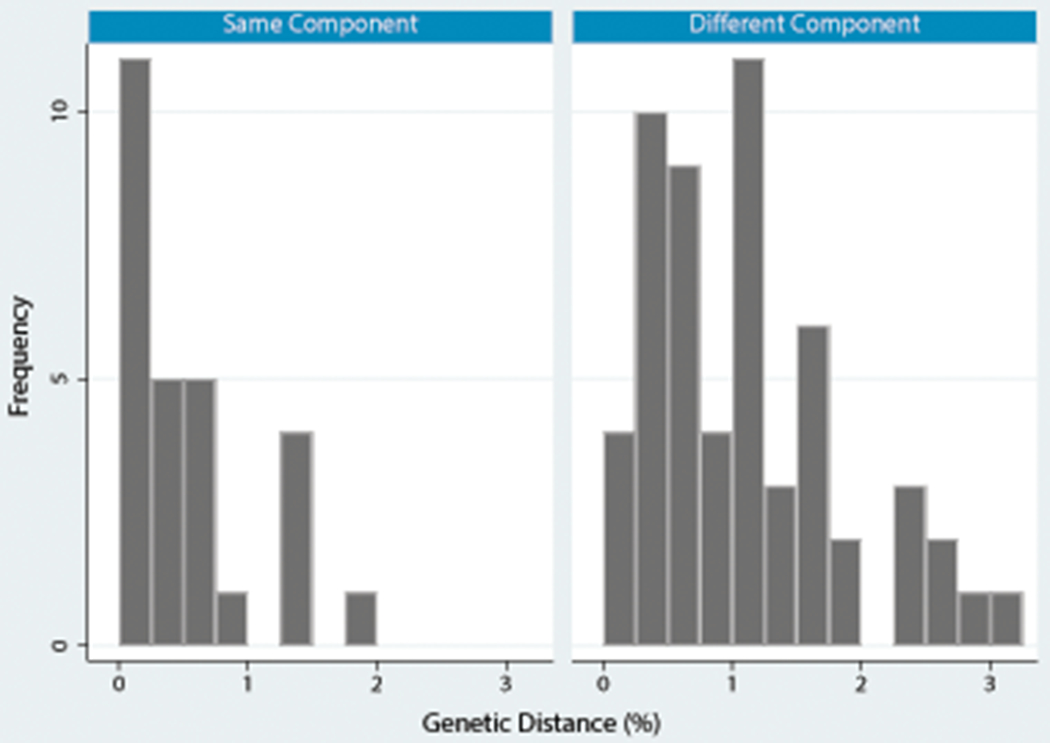
HIV genetic distance by outcome (pairwise network component location) among pairs of members of the same genetic cluster. Clusters were defined as ≤ 3.5% pairwise genetic distance across all possible pairs of cluster members (N=83 pairs), but this analysis was restricted to the analysis set of pairs ≤ 2.0% pairwise genetic distance (N=76 pairs). All 83 possible pairs among cluster members (≤ 3.5%) are included in this graph so that the full distribution of genetic distance can be seen.
Correlates of Membership in the Same Dyad but Different Components
We examined factors associated with membership in different partner notification components compared to persons in the same component among genetic dyads (Table 2). In bivariable analyses, factors associated with membership in different components included being in a male-male dyad (OR=5.4, 95%CI=1.3-22.9) and linear percent genetic distance (OR=4.9 for each 1% increase, 95%CI=1.6-14.8). All male-female dyads were in the same component. Being in a dyad with a person with a subsequent HIV diagnosis who refused partner notification interview (OR=3.2, 95%CI=0.8-12.4) and diagnosis during AHI (OR=2.6, 95%CI=0.6-10.5) were less strongly associated. Neither age, nor race, nor reporting anonymous partners was associated with being in different partner notification components.
The final multivariable GEE included AHI status at diagnosis, pair gender, genetic distance, and whether the person diagnosed later was interviewed (Table 2). Both partners being male (AOR=16.7, 95%CI=1.8-159.2), genetic distance (AOR=8.3 per 1% distance increase, 95%CI=2.4-28.5), and neither person being acutely infected at diagnosis (AOR=4.5, 95%CI=1.8-11.0) were all highly significant in the multivariable GEE as correlates of membership in the same genetic dyad but different components. The person diagnosed later not being interviewed was also retained (AOR=4.9, 95%CI=0.5-44.6) based on the QIC.
DISCUSSION
In this study, we examined persons with new HIV diagnoses and their HIV-infected partners to compare HIV genetic linkages to connections disclosed during partner notification. Nearly half of persons who were members of the same genetic cluster were not connected to anyone else in their cluster by partner notification contact tracing, an indication that potential transmission pathways, including some which may be ongoing, were not identified during partner notification. Strategically, public health resources will ideally be focused on support of people linked to ongoing transmission and not virally suppressed. In this way, HIV gene sequence analysis can supplement partner notification. In support of this mission, we characterized traits which were more common among persons with genetically similar HIV infections who were not recognized as partners or having partners in common during elicitation interviews.
Correlates of being disconnected in contact tracing among pairs of persons with close genetic linkages included pairs where both people were male, neither person was diagnosed with acute HIV infection, and greater genetic distance between each dyad member’s pol sequence. Partner elicitation appeared to be less successful among men; men with male partners were more likely to be in partner notification network components that were more disjointed than the phylogenetic analysis suggested. Conversely, while most clusters were composed of MSM, we identified multiple male-female genetic clusters–all of these clusters were part of the same partner notification component, indicating partner notification success in identifying possible heterosexual transmission sources. This observation has potential public health implications: men with male partners are not captured as well during partner elicitation, as has been shown previously in NC and beyond.21 We previously reported that male gender was associated with disagreement between the partner notification network and genetic clusters in this cohort.12 This multivariable analysis supports those findings, as male-male dyads strongly predicted disagreement between network types. But it also shows that even when holding dyad gender constant, HIV stage at diagnosis and increasing genetic distance provide opportunities to change work flow to better observe the network, potentially through finding new ways to incentivize partner disclosure (if intervening contacts are missing) or by including contacts from more than one year ago (if genetic distance represents time).
Diagnosis with chronic HIV, rather than with AHI, for both dyad members was predictive of being in different components. Chronic HIV indicates a longer time elapsed from infection until diagnosis which affects both partner notification interview completeness and the genetic distance divergence. Persons with AHI at diagnosis are emphasized in partner tracing, which can lead to more partners elicited and notified than testing of chronically-infected22 due to less recall bias and more resources directed toward interview completion. Any persons newly identified as being HIV-positive during partner notification are in turn interviewed to elicit partnerships, which can increase the size, density, and completeness of the partner notification network component. This increased activity leads to more opportunities for the transmitting partner of the acutely infected person to be included in the network and have an available sequence for the phylogeny. Persons with AHI are also more likely to cluster in the phylogeny23 due to less genetic divergence.
As genetic distance increased between dyad members, so did the likelihood of failing to approximate the partner notification components. Overall, genetic distance was larger among persons in different network components. First-degree transmissions cannot be implied in the phylogeny due to potential unsampled persons involved in the transmission chain, nor can we derive transmission direction. Among dyads in different components, smaller genetic distance may represent fairly recent and/or first-degree partnerships that occurred between the first dyad member’s diagnosis and the later dyad member’s diagnosis and then were not captured if the later person to be diagnosed refused the opportunity for disclosure. This hypothesis cannot be verified without partner notification data from the later person; but, an indicator of not having an interview for the person with the later diagnosis improved the fit of our multivariable model.
The greater genetic distances observed among dyads in different components represent a few possibilities. First, higher genetic distance may indicate higher likelihood of intervening partners, and each intervening relationship adds another opportunity for partnerships to be unobserved. Greater distance could indicate unsampled, unobserved intervening persons in the genetic cluster due to the high number of untraced partners and we are seeing second-degree partners clustered together instead of first-degree partners. If that is true then having unreported and/or anonymous partnerships would be substantively significant even if not informative to the model. Second, elicited partnerships are restricted to a time period based upon stage at diagnosis (2 months, 6 months, and 12 months prior to diagnosis for persons diagnosed with AHI, recent infection, and established infection, respectively). Larger distances could represent partnerships which occurred longer ago and were not captured due to recall bias or having occurred prior to the partner notification period of interest.
In support of our findings, many MSM with AHI in NC had no locatable HIV-positive partners24 and disjoint partner notification components were frequently connected by genetically similar viruses.25 These observations could be due to unwillingness or inability of cases to disclose partners to the disease intervention specialists (DIS) performing the interviews or to inability of the DIS to locate partners. Public health efforts directed at cultural competency and support for stigmatized persons may elicit more partners for testing, thereby filling in some of the partner notification network gaps and keeping NC on track to reduce the burden of HIV. Efforts to identify persons who are missed during partner notification can maintain or improve the proportion of people living with HIV infection who are diagnosed, especially persons of color26 and MSM. This analysis quantifies the effect of missing interviews on the observed sociosexual network of people who are diagnosed HIV who have accessed at least one HIV care visit.
This analysis provides a methodological approach to fill partner notification gaps. Using GEE, we identified factors associated with partner notification network gaps, as measured by having genetic clusters which spanned several partner notification-based sociosexual network components. These factors may be amenable to intervention and are worth exploring despite imprecision due to the small sample size. In future studies, the proportion of cases with available sequences should be increased; also, the number of missing interviews and untraced partners could be decreased. With a more complete sample, we might have been able to refine the dyads to more representative pairs rather than assessing all possible pairs within the same genetic cluster. Instead, we used GEE to account for clustering in the data, but the nature of the clustering could not be specified.
Genetic cluster analysis is a promising area to guide HIV reduction interventions.27,28 The quantitative comparison of the partner notification network and genetic clusters in this analysis provide one such possibility. In ongoing work, NC has intensified partner notification towards members of genetic clusters that are growing over time, a possible indication of ongoing HIV transmission. Clusters that demonstrate ongoing growth and span larger genetic distances, yet have no apparent ties linking cluster members in the partner notification network might indicate where partner notification efforts can be intensified. This analysis provides information to fill partner notification gaps by prioritizing efforts toward people with these traits (two men in the same cluster, neither diagnosed during AHI, with increasing genetic distance difference, but where there was interview refusal). Given that increasing genetic distance was predictive in the multivariable GEE, dyad members would be less likely to be recent, first-degree partners. Therefore, re-interviewing both dyad members would be recommended to identify partners of partners (second-degree or higher partners) who are involved in the same putative transmission cluster and who “link” the identified dyad members together. During these interviews, persons who are infected and either not diagnosed or not virally suppressed may be discovered. Our work provides a novel way to identify characteristics associated with potentially unobserved network partnerships, and thereby HIV-positive persons whose partners would benefit from testing but who are not being located during routine partner notification services.
ACKNOWLEDGMENTS
The project described was supported by the National Center for Advancing Translational Sciences (NCATS), NIH, through Grant Award Number UL1TR001111; the National Institute of Allergy and Infectious Diseases (NIAID), NIH, through Grant Award Numbers K08AI112432-02 and 5T32AI070114-10; and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), NIH, Grant Award 1R25HD079352-01A1. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the NC Department of Health and Human Services (NC DHHS). We would like to thank NC DHHS and the Disease Intervention Specialists serving Wake County.
Sources of support: The project described was supported by the National Center for Advancing Translational Sciences (NCATS), NIH, through Grant Award Number UL1TR001111 (AMD and IAD) and the National Institute of Allergy and Infectious Diseases (NIAID), NIH, through Grant Award Number K08AI112432-02 (AMD). DKP was supported by the NIH T32 Grant Award 5T32AI070114-10 to UNC-Chapel Hill and NIH / NICHD Grant Award R25HD079352 to Duke University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. For the remaining authors none were declared.
Footnotes
Conflicts of interest: None were declared.
Presented in part at SER 2016 Epidemiology Congress of the Americas (Miami, FL, USA, June 2016) as “Phylogenetic analysis of HIV enhances contact tracing and sexual network ascertainment”
REFERENCES
- 1.Chandler RK, Kahana SY, Fletcher B, et al. Data collection and harmonization in HIV research: The seek, test, treat, and retain initiative at the national institute on drug abuse. Am J Public Health, 2015. 105(12): p. 2416–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.UNAIDS. 90-90-90: An ambitious treatment target to help end the AIDS epidemic. 2014, Joint United Nations Programme on HIV/AIDS. p. 1–40. [Google Scholar]
- 3.Hogben M, McNally T, McPheeters M, et al. The effectiveness of HIV partner counseling and referral services in increasing identification of HIV-positive individuals a systematic review. Am J Prev Med, 2007. 33(2 Suppl): p. S89–100. [DOI] [PubMed] [Google Scholar]
- 4.Cope AB, Powers KA, Kuruc JD, et al. Ongoing HIV transmission and the HIV care continuum in North Carolina. PLoS One, 2015. 10(6): p. e0127950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gardner EM, McLees MP, Steiner JF, et al. The spectrum of engagement in HIV care and its relevance to test-and-treat strategies for prevention of HIV infection. Clin Infect Dis, 2011. 52(6): p. 793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doherty IA, Serre ML, Gesink D, et al. Sexual networks, surveillance, and geographical space during syphilis outbreaks in rural North Carolina. Epidemiology, 2012. 23(6): p. 845–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoots BE, MacDonald PD, Hightow-Weidman LB, et al. Developing a predictive model to prioritize human immunodeficiency virus partner notification in North Carolina. Sex Transm Dis, 2012. 39(1): p. 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hurt CB, Beagle S, Leone PA, et al. Investigating a sexual network of black men who have sex with men: Implications for transmission and prevention of HIV infection in the United States. J Acquir Immune Defic Syndr, 2012. 61(4): p. 515–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delva W, Leventhal GE and Helleringer S. Connecting the dots: Network data and models in HIV epidemiology. AIDS, 2016. 30(13): p. 2009–20. [DOI] [PubMed] [Google Scholar]
- 10.Pasquale DK, Doherty IA, Sampson LA, et al. HIV transmission hotspot detection combining sexual and phylogenetic network analysis. in 2016 Conference on Retroviruses and Opportunistic Infections 2016. Boston, MA. [Google Scholar]
- 11.Hassan AS, Pybus OG, Sanders EJ, et al. Defining HIV-1 transmission clusters based on sequence data. AIDS, 2017. 31(9): p. 1211–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pasquale DK, Doherty IA, Sampson LA, et al. Leveraging phylogenetics to understand HIV transmission and partner notification networks. J Acquir Immune Defic Syndr, 2018. 78(4): p. 367–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Etikan I, Alkassim R and Abubakar S. Comparision of snowball sampling and sequential sampling technique. Biometrics & Biostatistics International Journal, 2016. 3(1). [Google Scholar]
- 14.Doherty IA, Schoenbach VJ and Adimora AA. Sexual mixing patterns and heterosexual HIV transmission among african americans in the southeastern United States. J Acquir Immune Defic Syndr, 2009. 52(1): p. 114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan PA, Hogan JW, Huang A, et al. Phylogenetic investigation of a statewide HIV-1 epidemic reveals ongoing and active transmission networks among men who have sex with men. J Acquir Immune Defic Syndr, 2015. 70(4): p. 428–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doherty IA, Adimora AA, Muth SQ, et al. Comparison of sexual mixing patterns for syphilis in endemic and outbreak settings. Sex Transm Dis, 2011. 38(5): p. 378–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dennis AM, Hue S, Hurt CB, et al. Phylogenetic insights into regional HIV transmission. AIDS, 2012. 26(14): p. 1813–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cui J QIC program and model selection in GEE analyses. The Stata Journal, 2007. 7(2): p. 209–20. [Google Scholar]
- 19.Pan W Akaike’s information criterion in generalized estimating equations. Biometrics, 2001. 57(1): p. 120–25. [DOI] [PubMed] [Google Scholar]
- 20.StataCorp. Stata statistical software: Release 12. 2011, StataCorp LP: College Station, TX. [Google Scholar]
- 21.Wertheim JO, Kosakovsky Pond SL, Forgione LA, et al. Social and genetic networks of HIV-1 transmission in New York city. PLoS Pathog, 2017. 13(1): p. e1006000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore ZS, McCoy S, Kuruc J, et al. Number of named partners and number of partners newly diagnosed with HIV infection identified by persons with acute versus established HIV infection. J Acquir Immune Defic Syndr, 2009. 52(4): p. 509–13. [DOI] [PubMed] [Google Scholar]
- 23.Volz EM, Koopman JS, Ward MJ, et al. Simple epidemiological dynamics explain phylogenetic clustering of HIV from patients with recent infection. PLoS Comput Biol, 2012. 8(6): p. e1002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dennis AM, Pasquale DK, Billock R, et al. Integration of contact tracing and phylogenetics in an investigation of acute HIV infection. Sex Transm Dis, 2018. 45(4): p. 222–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peters PJ, Gay CL, Beagle S, et al. HIV infection among partners of HIV-infected black men who have sex with men — North Carolina, 2011–2013. MMWR, 2014. 63(5): p. 90–94. [PMC free article] [PubMed] [Google Scholar]
- 26.Hall HI, Frazier EL, Rhodes P, et al. Frlbx05 - oral abstract: Continuum of HIV care: Differences in care and treatment by sex and race/ethnicity in the United States. in International AIDS Conference 2012 . July 22-27, 2012 Washington, D.C. [Google Scholar]
- 27.Dennis AM, Herbeck JT, Brown AL, et al. Phylogenetic studies of transmission dynamics in generalized HIV epidemics: An essential tool where the burden is greatest? J Acquir Immune Defic Syndr, 2014. 67(2): p. 181–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Billock RM, Powers KA, Pasquale DK, et al. Prediction of HIV transmission cluster growth with statewide surveillance data. J Acquir Immune Defic Syndr, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]