

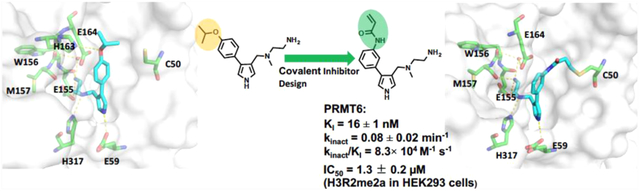
Abstract
Protein arginine methyltransferase 6 (PRMT6) plays important roles in several biological processes associated with multiple cancers. Well-characterized potent, selective, and cell-active PRMT6 inhibitors are invaluable tools for testing biological and therapeutic hypotheses. Although there are several known reversible PRMT6 inhibitors, covalent PRMT6 inhibitors have not been reported. Based on a cocrystal structure of PRMT6-MS023 (a type I PRMT inhibitor), we discovered the first potent and cell-active irreversible PRMT6 inhibitor, 4 (MS117). The covalent binding mode of compound 4 to PRMT6 was confirmed by mass spectrometry and kinetic studies and by a cocrystal structure. Compound 4 did not covalently modify other closely related PRMTs, potently inhibited PRMT6 in cells, and was selective for PRMT6 over other methyltransferases. We also developed two structurally similar control compounds, 5 (MS167) and 7 (MS168). We provide these valuable chemical tools to the scientific community for further studying PRMT6 physiological and pathophysiological functions.
Graphical abstract
INTRODUCTION
Protein arginine methyltransferase 6 (PRMT6) solely resides in the nucleus, and its polypeptide chain consists of a catalytic core sequence that is common to other protein arginine methyltransferases (PRMTs).1 As a member of type I PRMTs, PRMT6 catalyzes the transfer of the methyl group from the cofactor S-5′-adenosyl-l-methionine (SAM) to arginine residues of its substrates to provide post-translationally modified products with monomethylated or asymmetric dimethylated arginine residues.2–5 Besides the well-characterized histone substrate H3R2 (histone H3 arginine 2),6 nonhistone proteins, such as HIV Tat,7 HMGA1a,8 and DNA polymerase β,9 are also PRMT6 substrates. PRMT6 over-expression and hypermethylation of its substrates are associated with multiple cancers, such as prostate,10 bladder,11 melanoma,12 and colorectal13 cancers. Potent, selective, and cell-active reversible or irreversible inhibitors of PRMT6 could be useful chemical tools to investigate cellular processes and/or serve as lead compounds in drug discovery. Several small-molecule noncovalent inhibitors of PRMT6 have been developed,14–16 but covalent, irreversible inhibitors of PRMT6 have not been reported to date. Covalent, irreversible inhibitors have been rapidly gaining momentum in the clinic and more frequently becoming FDA-approved drugs.17 They form inhibitor-target adducts with no dissociation or virtually unlimited residence time. This continuous target engagement could provide extended pharmacodynamic duration without the need to maintain high exposures of the inhibitors.
Based on a cocrystal structure of PRMT6 in complex with MS023, a type I PRMT inhibitor we previously developed,14 we discovered the first potent, selective, and cell-active covalent inhibitor of PRMT6, MS117 (compound 4). We also designed and synthesized two structurally highly similar control compounds, MS167 (compound 5), a potent and cell-active reversible PRMT6 inhibitor, and MS168 (compound 7), a very poor PRMT6 inhibitor but with the same reactive warhead as compound 4. Using mass spectrometry (MS) and kinetic studies, we have demonstrated that compound 4 is a covalent PRMT6 inhibitor, which was further confirmed by the cocrystal structure of PRMT6 conjugated with compound 4. We also found that compound 4 did not covalently modify other closely related PRMTs and was selective for PRMT6 over other methyltransferases. In addition, compound 4 effectively inhibited PRMT6 in cells and was more potent than its close analogue, compound 5, which is a noncovalent inhibitor of PRMT6. Here, we describe structure-based design, chemical synthesis, and biological characterization of these valuable chemical tools in a battery of biochemical, biophysical, and cellular assays.
RESULTS
Structure-Based Inhibitor Design.
We aligned protein sequences of PRMTs and identified that Cys50 is a unique cysteine residue located in the PRMT6 substrate-binding site and other PRMTs lack such a cysteine residue in their substrate-binding sites (Figure S1). Through analysis of the crystal structure of PRMT6 in complex with the type I PRMT inhibitor MS023 we previously developed,14 we noted that the distance between the thiol group of Cys50 and the meta-position of the MS023 phenyl ring is 4.9 Å, which is approximately equal to the length of four to five chemical bonds (Figure 1A). This distance is suitable for introducing an acrylamide group as a Michael addition acceptor to react with the thiol group of Cys50 and form a covalent bond.18 Thus, we designed compound 4 (Figure 1B), which contains an acrylamide moiety at the meta-position of the MS023 phenyl ring. In addition, we designed two structurally highly similar control compounds: 5 and 7 (Figure 1B). Compound 5, which contains the propionamide group instead of the acrylamide moiety, was designed to maintain similar binding affinity to PRMT6 as compound 4, but be incapable of covalently modifying Cys50. We previously reported that the terminal primary amino group of MS023 formed critical hydrogen bonds (H-bonds) with PRMT6 and the replacement of the primary amino group with a primary alcohol group led to significant decreased potency.14 Thus, compound 7, which contains a primary alcohol group, was designed as another control compound to possess the same reactive moiety (warhead) but have much weaker binding affinity to PRMT6.
Figure 1.
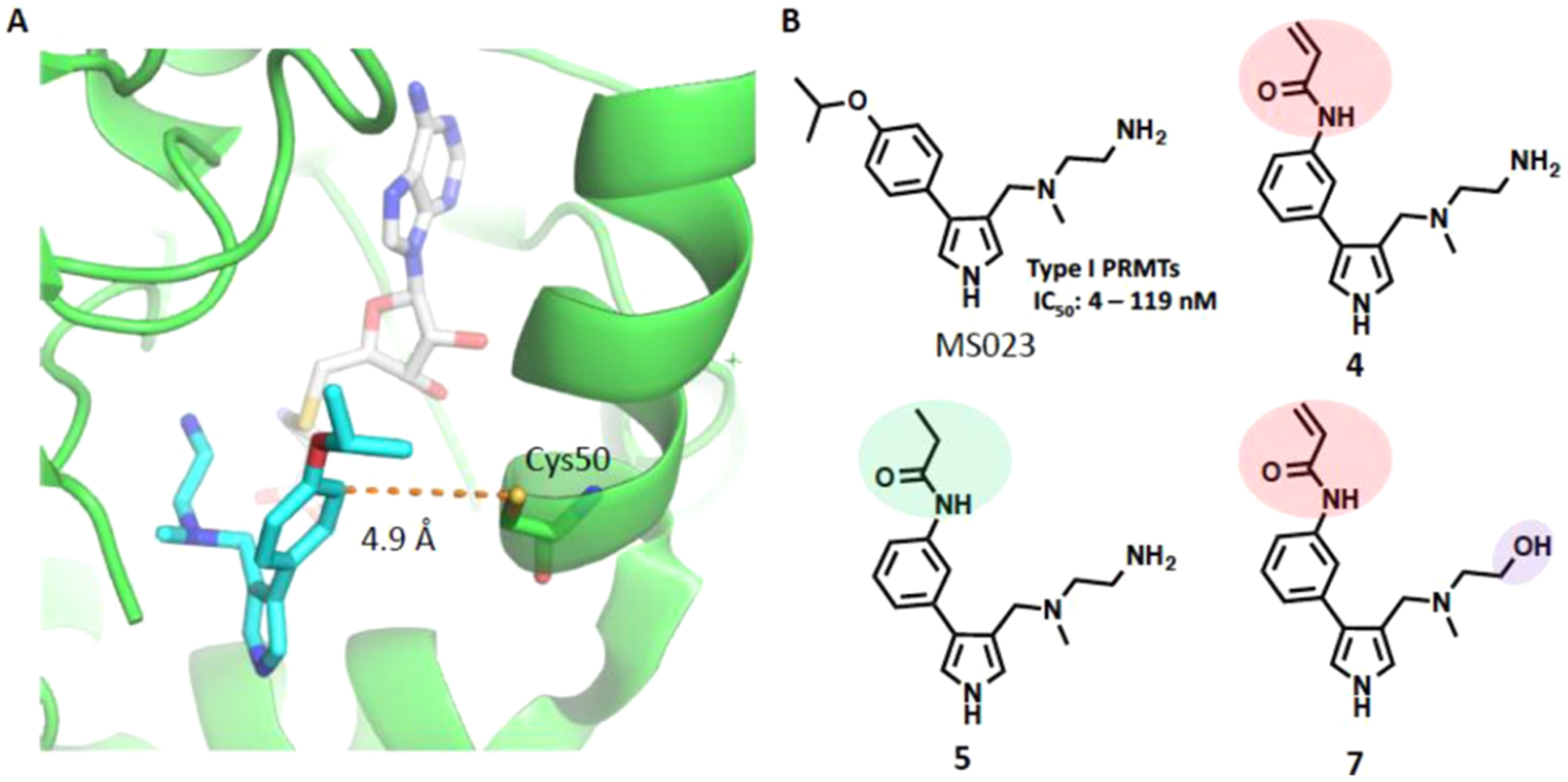
Design of the PRMT6 covalent inhibitor, compound 4, and its two control compounds. (A) Crystal structure (PDB: 5E8R) of PRMT6 (green) in complex with MS023 (cyan) and SAH (gray) reveals that the distance between the Cys50 thiol group and the meta-position of the MS023 phenyl ring (shown by the orange dotted line) is 4.9 Å. (B) Chemical structures of MS023, compound 4 (PRMT6 covalent inhibitor), and two structurally highly similar controls, compounds 5 and 7.
Synthesis.
The synthetic routes for preparing compounds 4, 5, and 7 are depicted in Scheme 1. 1,3-Dipolar cycloaddition between commercially available ethyl (E)-3-(3-nitrophenyl)-acrylate and 1-((isocyanomethyl)sulfonyl)-4-methylbenzene under basic condition, followed by protecting the resulting pyrrole nitrogen with the Boc group, provided compound 1. DIBAL-H-mediated reduction converted the ethyl ester group of compound 1 to an alcohol group, which was subsequently oxidized to yield aldehyde 2 by Dess-Martin periodinane. Reduction amination of the resulting aldehyde intermediate 2 with tert-butyl (2-(methylamino)ethyl)carbamate or 2-(methylamino)ethan-1-ol afforded intermediates 3 and 6, respectively. Under hydrogenation reaction conditions, the nitro group of compound 3 was reduced to an amino group. The subsequent amide coupling reactions between the resulting amine and acryloyl chloride or propionyl chloride, followed by the removal of the Boc-protecting group, provided compounds 4 and 5, respectively. Using a similar reaction sequence, e.g., reductive amination, nitro group reduction, amide coupling, and Boc group deprotection, intermediate 6 was converted to compound 7.
Scheme 1. Synthetic Route of Compounds 4, 5, and 7a.
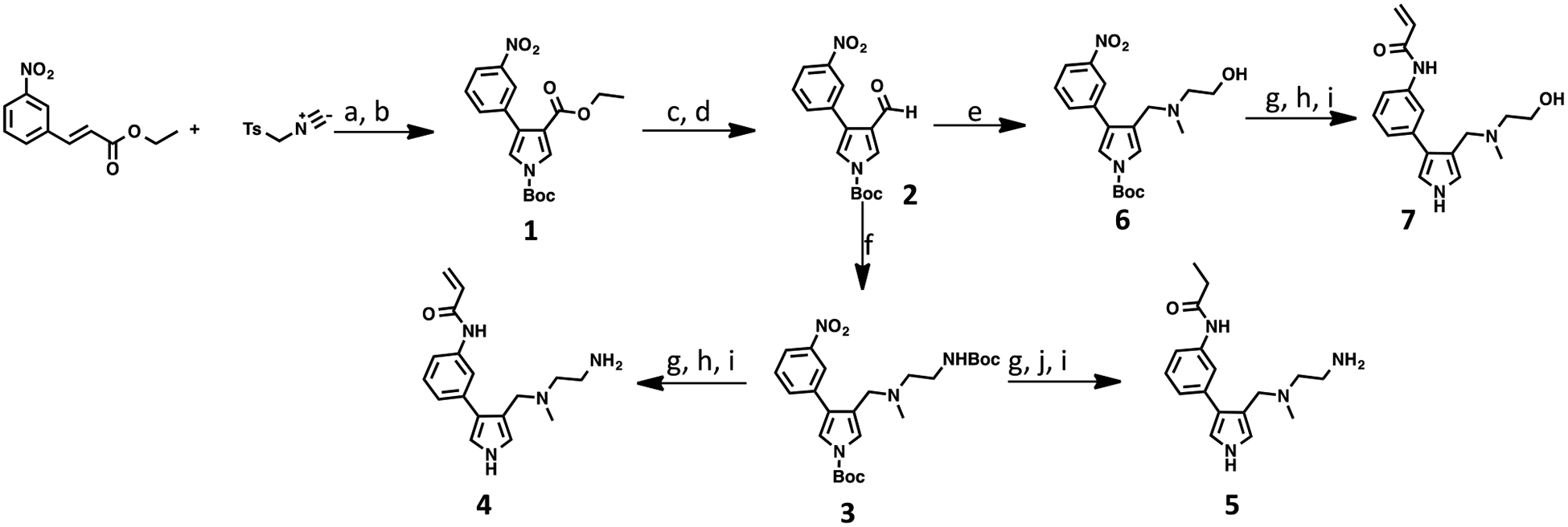
aReagents and conditions: (a) NaH, dimethylformamide/tetrahydrofuran (DMF/THF); (b) Et3N, Boc2O, 4-dimethylaminopyridine (DMAP), dichloromethane (DCM), 37% over two steps. (c) DIBAL-H, THF, −78 °C; (d) Dess-Martin periodinane, DCM, 71%; (e) 2-(methylamino)ethan-1-ol, NaBH(OAc)3, DCM, 74%; (f) tert-butyl (2-(methylamino)ethyl)carbamate, NaBH(OAc)3, DCM, 88%; (g) Pd/C (10%), H2, MeOH; (h) acryloyl chloride, DCM; (i) trifluoroacetic acid (TFA)/DCM, 29–42%; (j) propionyl chloride, Et3N, DCM.
Evaluation in Biochemical and Kinetic Studies.
We first assessed the PRMT6 inhibitory effect of these three compounds using a scintillation proximity assay (SPA), which measures the activity of human PRMT6 in catalyzing the transfer of the tritiated methyl group from 3H-SAM to a peptide substrate in the presence and absence of compounds.19 With 1 h preincubation, compound 4 displayed excellent inhibition potency against PRMT6 (IC50 = 18 ± 2 nM, pIC50 = 7.8 ± 0.01) (Figure 2A). The propionate amide control compound 5 exhibited similar potency to compound 4 (IC50 = 28 ± 1 nM, pIC50 = 7.6 ± 0.1) (Figure 2B). As expected, the alcohol-containing control compound 7 showed poor potency against PRMT6 (IC50 = 9800 ± 980 nM, pIC50 = 5.0 ± 0.04) (Figure 2C).
Figure 2.
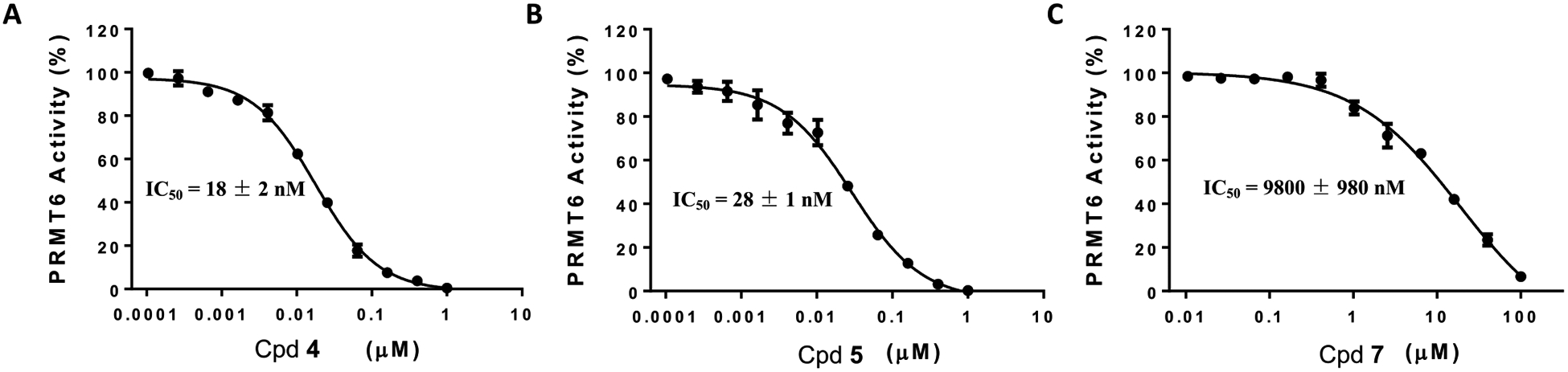
Compounds 4 and 5, but not compound 7, potently inhibited PRMT6. Concentration-response curves of compounds 4 (A), 5 (B), and 7 (C) in inhibiting PRMT6 in a scintillation proximity assay. IC50 determination experiments were performed in triplicate, and the values are presented as mean ± standard deviation (SD).
While IC50 values are appropriate for quantifying the potency of reversible inhibitors, the IC50 values of irreversible inhibitors are time-dependent. Thus, irreversible inhibitors are commonly quantified using the kinact/KI rate constants, derived from the inhibition constant (KI) and the rate of enzyme inactivation (kinact).20 To further characterize the PRMT6 kinetic inhibition nature of compounds 4 and 5, a series of IC50 values were measured using the SPA assay by preincubating the compounds with PRMT6 at different time points ranging from 1 to 30 min. Compound 4 displayed time-dependent potencies against PRMT6 with a kinact/KI value of 8.3 × 104 M−1 s−1, indicating the irreversible inhibition mechanism (Figure 3A).20 On the other hand, no significant change in IC50 values was observed for compound 5 at various time points ranging from 1 to 30 min, indicating that compound 5 is a reversible inhibitor (Figure 3B).
Figure 3.
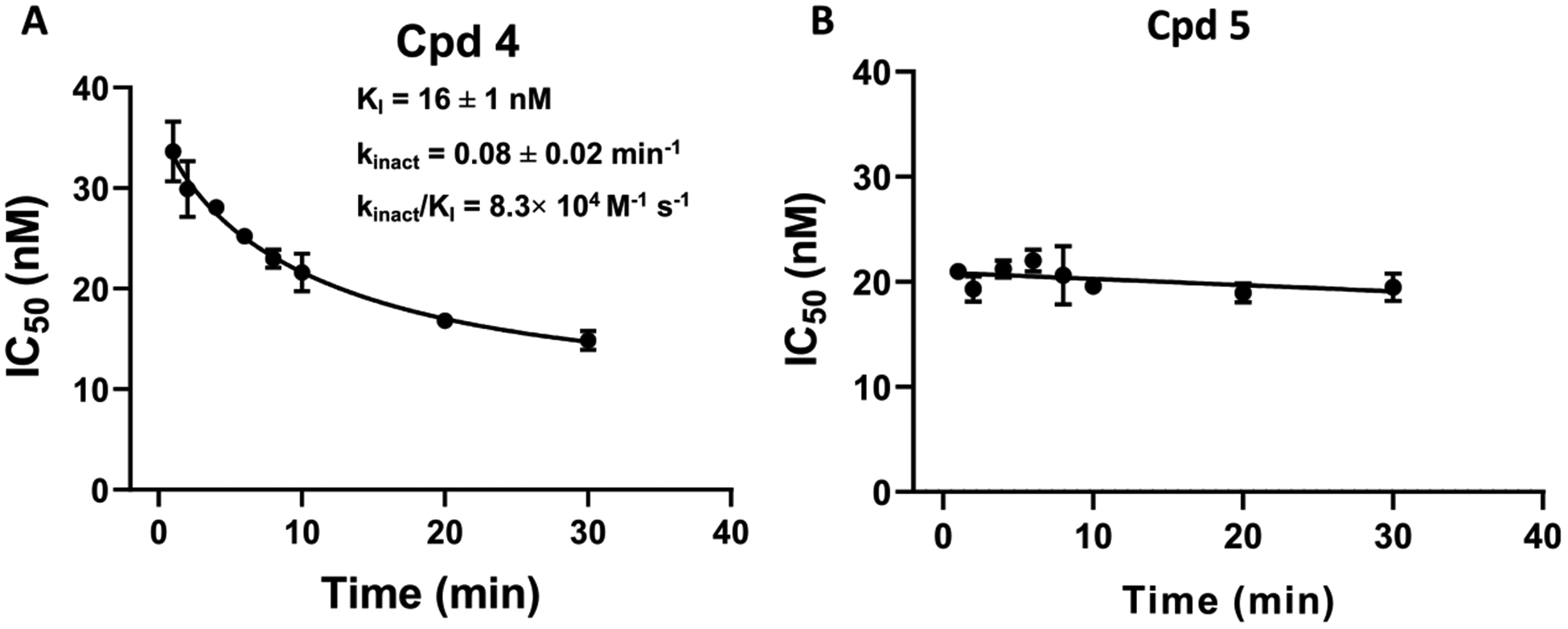
Kinetics of PRMT6 inhibition by compounds 4 and 5. (A) Compound 4 inhibited the PRMT6 methyltransferase activity in a time-dependent manner. (B) Compound 5 inhibited PRMT6 in a time-independent manner. The indicated compound was preincubated with PRMT6 at different time points ranging from 1 to 30 min, and IC50 values were determined using the SPA assay. The experiments were performed in triplicate. The IC50 values are presented as mean ± SD.
Characterization in a Mass Spectrometry-Based Assay.
To confirm that compound 4 covalently modifies PRMT6, an excess amount of compound 4 was preincubated with PRMT6 for 1 h and potential protein-ligand adducts were analyzed by mass spectrometry (MS). Our MS analyses revealed an adduct with mass equal to PRMT6 plus compound 4, with no detectable unmodified PRMT6 (Figure 4A,B), indicating that compound 4 efficiently forms a single modified covalent adduct. Under the same assay conditions, neither compound 5 nor compound 7 could modify PRMT6 in a detected level (Figure 4C,D). In addition, compound 4 was screened for modification of other type I PRMTs (PRMT1, 3, 4, and 8) and no adducts with other type I PRMTs could be detected (Figures S2–S5). This is in agreement with the PRMT protein sequence alignment results that PRMT6, but not other PRMTs, contains a cysteine residue (Cys50) in its substrate-binding site, which can be selectively targeted to form a covalent bond (Figure S1).
Figure 4.
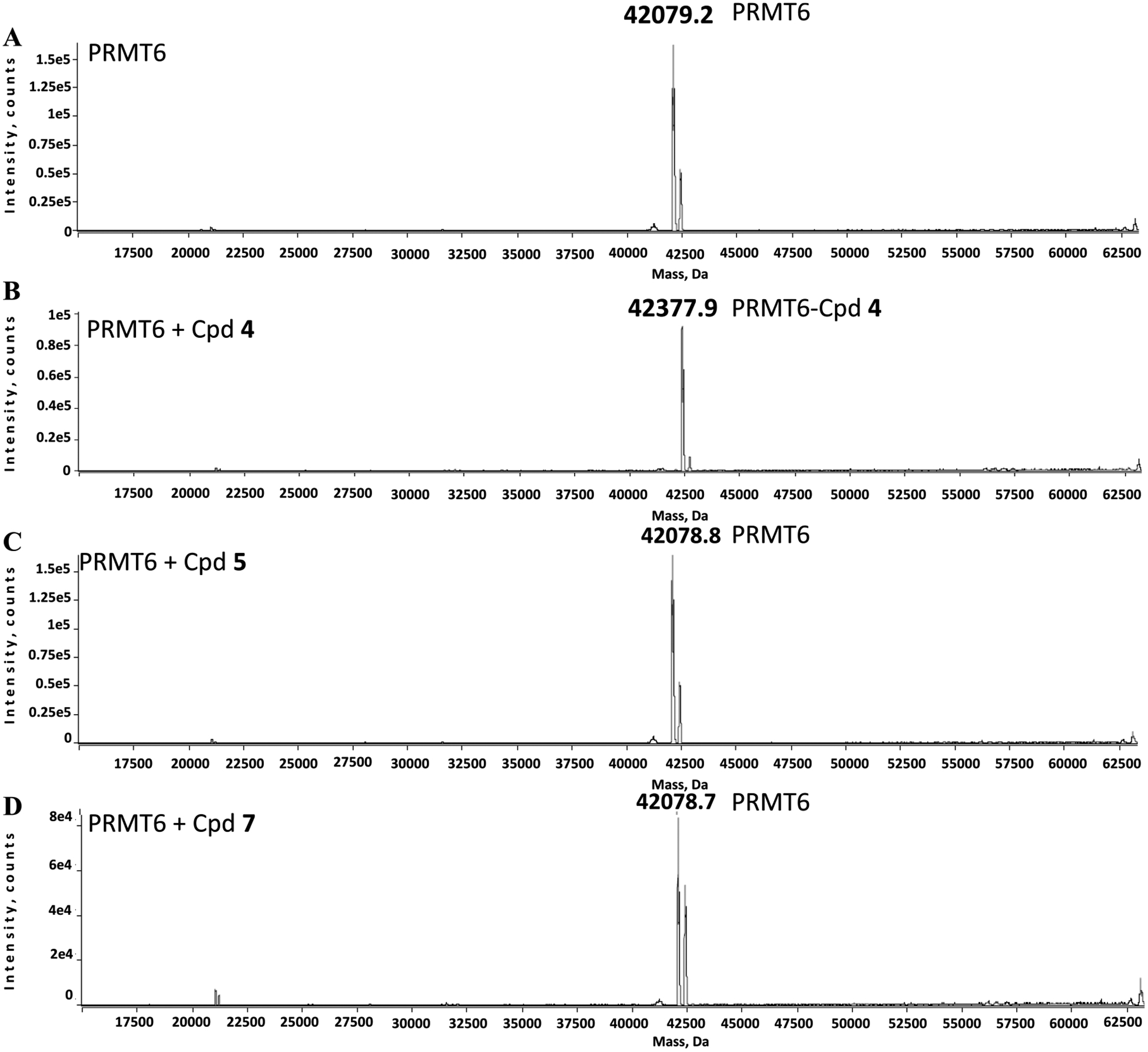
Compound 4, but not compounds 5 and 7, covalently modified PRMT6 in an MS assay. Mass spectra of PRMT6 alone (A), PRMT6 preincubated with compound 4 (MW: 298) (B), PRMT6 preincubated with compound 5 (C), and PRMT6 preincubated with compound 7 (D).
Cocrystal Structure of the PRMT6-Compound 4 Complex.
To confirm that Cys50 was covalently modified, we co-crystallized PRMT6 with compound 4 in the presence of the cofactor product S-5′-adenosyl-l-homocysteine (SAH) and solved a high-resolution (2 Å) cocrystal structure (PDB: 6P7I, Figure 5A). The asymmetric cocrystal structure contains four conformational similar copies of PRMT6, with both compound 4 and SAH in all of the four active sites (Figure 5A). Electron density map at the active sites indicates that the acrylamide group is close to the Cys50 residue. Although there is not enough electron density to confirm the covalent C-S bond in chains a, b, and c, the C-S bond formation is unambiguously confirmed by the electron density in chain d (Figure 5A). The alignment of MS023-PRMT6 and compound 4-PRMT6 cocrystal structures reveals that compound 4 occupies the substrate pocket of PRMT6 with a similar binding mode to MS023 (Figure 5B).14 Compared to MS023, the phenyl ring of compound 4 slightly flipped about 0.9 Å away from Cys50 to favor the Michael addition between acrylamide and Cys50 (Figure 5B). As the isopropoxy group of MS023 was removed in compound 4, the H-bond between the isopropoxy group and residue His163 in the MS023-PRMT6 complex cannot be observed anymore in the PRMT6-compound 4 adduct. Otherwise, all other H-bonds can be found in the PRMT6-compound 4 adduct, not only including direct H-bonds between the primary amino group and Glu155 or Met157, between the ternary amino group and His317, and between the pyrrole nitrogen and Glu59, but also including the water-mediated H-bonds between the primary amino group and Trp156 and Glu164 (Figure 5A).
Figure 5.
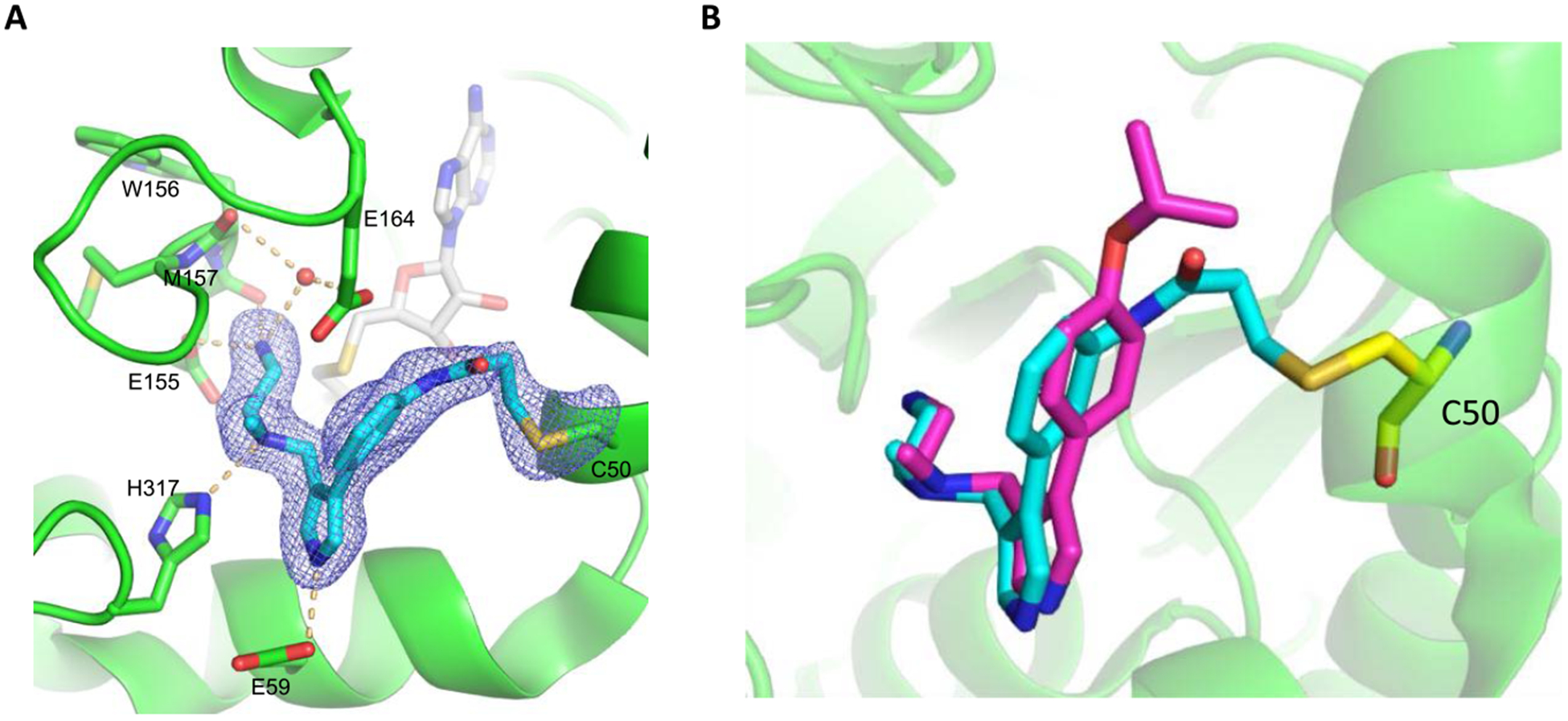
(A) X-ray cocrystal structure of PRMT6 (green, chain d) conjugated with compound 4 (cyan) in complex with SAH (gray) (PDB: 6P7I). Compound 4 covalently bound to the PRMT6 Cys50 residue. Key H-bond interactions between compound 4 and PRMT6 are shown in orange dotted lines. (B) Superimposition of the PRMT6-MS023 (magenta) (PDB: 5E8R) and PRMT6-compound 4 cocrystal structures.
Selectivity Assessment.
To assess the selectivity of compound 4, we tested it against 22 protein lysine methyltransferases (PKMTs), 3 DNA methyltransferases (DNMTs), and 8 PRMTs at 10 and 50 μM (Figure 6A). At 10 μM, compound 4 did not significantly inhibit any PKMTs, DNMTs, and type II, III PRMTs tested. However, compound 4 significantly inhibited type I PRMTs with >90% inhibition. At 50 μM, in addition to type I PRMTs, compound 4 inhibited SUV39H1, SETD7, PRMT7, and DNMT3A/3L (>50% inhibition). We also tested control compounds 5 and 7 against these 33 methyltransferases (Figure 6B,C). Similar to compound 4, compound 5 was very selective for type I PRMTs over PKMTs, DNMTs, and type II, III PRMTs but inhibited all type I PRMTs (>40% inhibition at 10 μM; >80% inhibition at 50 μM) (Figure 6B). On the other hand, compound 7 showed modest inhibition activity only for PRMT1 (~60% inhibition at 10 μM; ~70% inhibition at 50 μM) and PRMT6 (~50% inhibition at 10 μM; ~80% inhibition at 50 μM) (Figure 6C).
Figure 6.
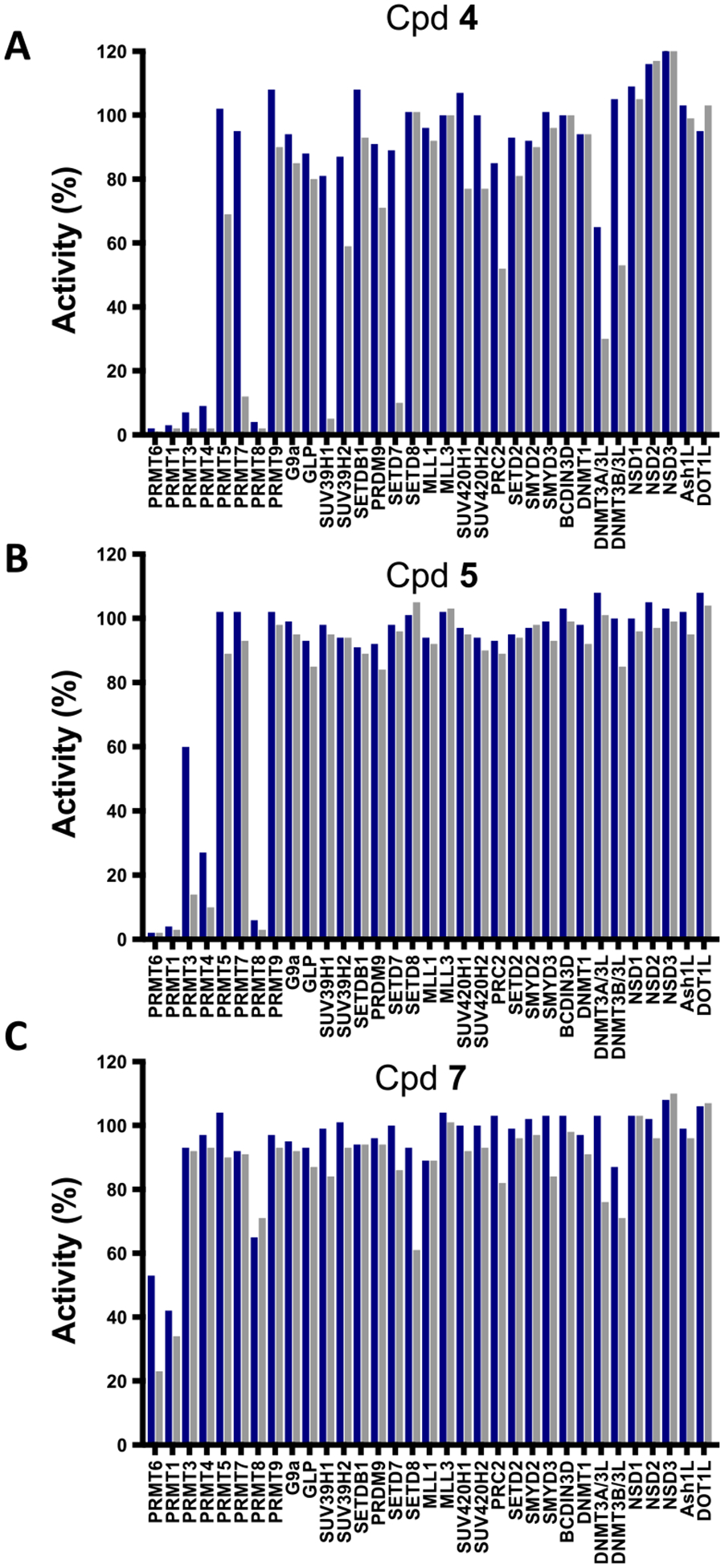
Selectivity assessment of compounds 4 (A), 5 (B), and 7 (C) against a variety of methyltransferases at 10 μM (blue) and 50 μM (gray).
To further assess the selectivity profiles of compounds 4, 5, and 7 within the type I PRMT subfamily, we determined potencies of these compounds against PRMT1, PRMT3, PRMT4, and PRMT8 using the SPA assay (Table 1). Compound 4 showed good selectivity for PRMT6 (IC50 = 0.018 ± 0.002 μM) over PRMT3 (IC50 = 3 ± 0.2 μM) and PRMT4 (IC50 = 0.48 ± 0.1 μM), with 167- and 27-fold selectivities, respectively. In addition, compound 4 showed moderate selectivity (6-fold) for PRMT6 over PRMT1 (IC50 = 0.1 ± 0.025 μM) and PRMT8 (IC50 = 0.11 ± 0.01 μM). Similar to compound 4, compound 5 showed good selectivity for PRMT6 (IC50 = 0.028 ± 0.001 μM) over PRMT3 (IC50 = 15 ± 5 μM) and PRMT4 (IC50 = 2 ± 0.2 μM) and moderate selectivity over PRMT1 (IC50 = 0.17 ± 0.03 μM) and PRMT8 (IC50 = 0.13 ± 0.01 μM). As expected, compared to compounds 4 and 5, compound 7 was much weaker at inhibiting the methyltransferase activity of all type I PRMTs.
Table 1.
Potencies of Compounds 4, 5, and 7 at Inhibiting Type I PRMTsa
| IC50 (μM) | |||||
|---|---|---|---|---|---|
| compound | PRMT1 | PRMT3 | PRMT4 | PRMT6 | PRMT8 |
| 4 | 0.1 ± 0.025 | 3 ± 0.2 | 0.48 ± 0.1 | 0.018 ± 0.002 | 0.11 ± 0.01 |
| 5 | 0.17 ± 0.03 | 15 ± 5 | 2 ± 0.2 | 0.028 ± 0.001 | 0.13 ± 0.01 |
| 7 | 15 ± 3 | >200 | 51 ± 1.6 | 9.8 ± 0.098 | 23 ± 1.3 |
IC50 determination experiments were performed at substrate and cofactor concentrations equal to the respective Km values for each enzyme. IC50 determination experiments were performed in triplicate, and the values are presented as mean ± SD.
Taken together, these results indicate that while compound 7 is a very weak inhibitor against methyltransferases in general, compounds 4 and 5 are highly selective for PRMT6 over PKMTs, DNMTs, and type II and III PRMTs. Within the type I PRMT subfamily, compounds 4 and 5 displayed good selectivity for PRMT6 over PRMT3 and PRMT4 and were moderately selective for PRMT6 over PRMT1 and PRMT8.
Evaluation in Cellular Assays.
Encouraged by the good potency and selectivity data of compound 4 in biochemical assays, we next evaluated the effects of pharmacological inhibition of PRMT6 and other type I PRMT off-targets, including PRMT1, PRMT3, and PRMT4, by compound 4 and its two control compounds 5 and 7 in cells (Figure 7). Because PRMT8 is mainly expressed in the brain,21 we did not investigate the cellular effect of compound 4 against this target. First, the cellular potency of compound 4 against ectopically expressed PRMT6 in HEK293 cells was evaluated (Figure 7A–C). Compound 4 potently and concentration-dependently reduced cellular levels of H3R2 asymmetric dimethylation (H3R2me2a, IC50 = 1.3 ± 0.2 μM), the known PRMT6 asymmetric dimethylation product (Figure 7A). Similarly, both the PRMT6 catalytically inactive V86K/D88A mutation and the type I PRMT inhibitor MS02314 at 2 μM effectively inhibited H3R2me2a. As expected, compound 5 also inhibited H3R2me2a, but was less effective than compound 4, and compound 7 did not significantly inhibit H3R2me2a at concentrations up to 10 μM (Figure 7A,B).
Figure 7.
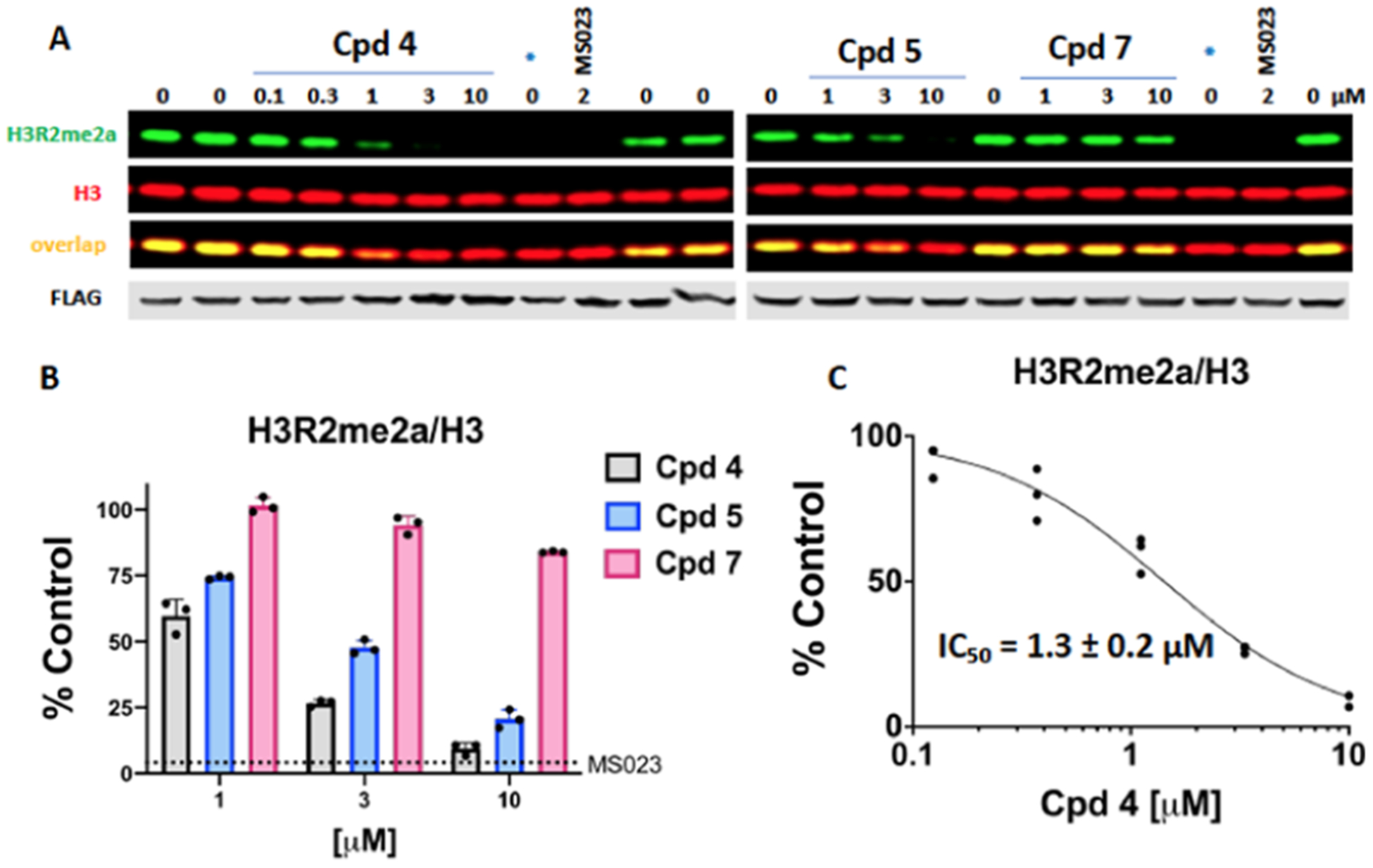
Compound 4 inhibits PRMT6-dependent H3R2 asymmetric dimethylation in cells with the highest potency. HEK293T cells were transfected with Flag-tagged wild-type PRMT6 or PRMT6-V86K/D88A catalytically inactive mutant (*) and treated with indicated compounds for 20 h. Type I PRMT inhibitor MS023 was used as a positive control. (A) Western blot representation of the effect of compounds 4, 5, and 7 on PRMT6 activity. (B) Quantification of the effects of compounds 4, 5, and 7 on PRMT6 activity. The graph shows H3R2me2a fluorescence intensities normalized to intensities of H3. The dotted line indicates the methylation level after MS023 treatment, which represents the background signal. The results are presented as the mean ± SD of three replicates. (C) IC50 determination of compound 4. The graph represents the nonlinear fit of H3R2me2a fluorescence intensities normalized to intensities of H3. The individual data points are indicated.
Next, we used MCF7 cells to assess the cellular activity of compound 4 against endogenous PRMT1-catalyzed asymmetric dimethylation of H4R3 (H4R3me2a) using MS023 (1 μM) as a positive control (Figure 8A,B). Compound 4 treatment resulted in a concentration-dependent inhibition of H4R3me2a levels with an IC50 value of 5.6 ± 0.3 μM, indicating that compound 4 is less potent at inhibiting PRMT1 than PRMT6 in cells. Because compound 4 is not a covalent inhibitor of PRMT1, we expected that compounds 4 and 5 would have similar potencies in this cellular assay. Indeed, compound 5 displayed similar cellular potency (IC50 = 6.7 ± 0.6 μM) to compound 4 (Figure 8A,B). In addition, as predicted, compound 7 was inactive in this cellular assay at concentrations up to 20 μM.
Figure 8.
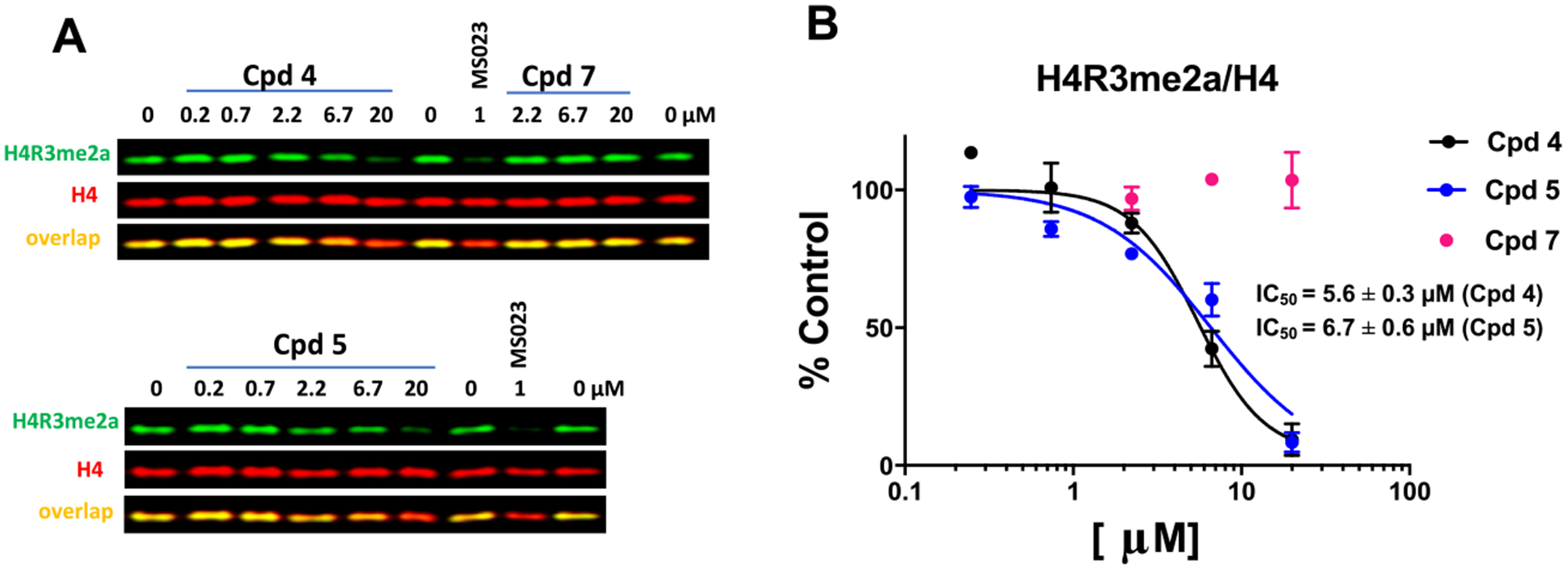
Inhibition of PRMT1-dependent H4R3 asymmetric dimethylation in cells. (A) Compounds 4 and 5, but not compound 7, inhibited H4R3me2a. MCF7 cells were treated with compounds for 2 days. MS023 was used as a positive control. (B) Graph representing the nonlinear fit of H4R3me2a fluorescence intensities normalized to intensities of H4. The results are presented as the mean ± SD of three replicates.
We next investigated the cellular effects of compounds 4, 5, and 7 on the overexpressed PRMT3-catalyzed methylation of exogenously introduced H4 (Figure 9).22,23 While the catalytically inactive PRMT3 mutant (E335Q) and the PRMT3 selective inhibitor SGC70722 (1 μM) effectively inhibited H4R3me2a, compounds 4, 5, and 7 did not significantly reduce this mark at concentrations up to 20 μM.
Figure 9.
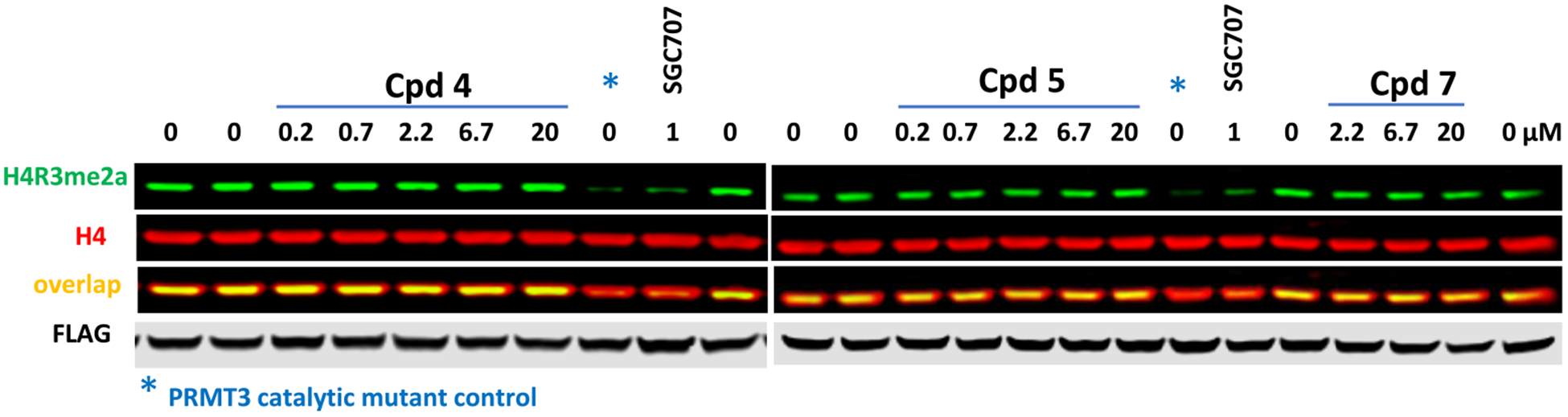
Inhibition of PRMT3-dependent H4R3 arginine asymmetric dimethylation in cells. Compounds 4, 5, and 7 did not inhibit the PRMT3 activity at concentrations up to 20 μM. HEK293T cells were co-transfected with GFP-tagged histone H4 peptide (1–50) and PRMT3 WT or catalytically inactive mutant (E338Q) and treated with compounds for 20 h. PRMT3 selective inhibitor SGC707 was used as a positive control. The WB results are representative of two independent experiments.
We also assessed the effects of compounds 4, 5, and 7 on inhibiting the endogenously expressed PRMT4 in MCF7 cells (Figure 10). In contrast to the PRMT4 selective inhibitor TP064 (2 μM),24 compounds 4, 5, and 7 did not reduce BAF155 arginine asymmetric dimethylation (BAF155-Rme2a) levels at concentrations up to 20 μM.
Figure 10.
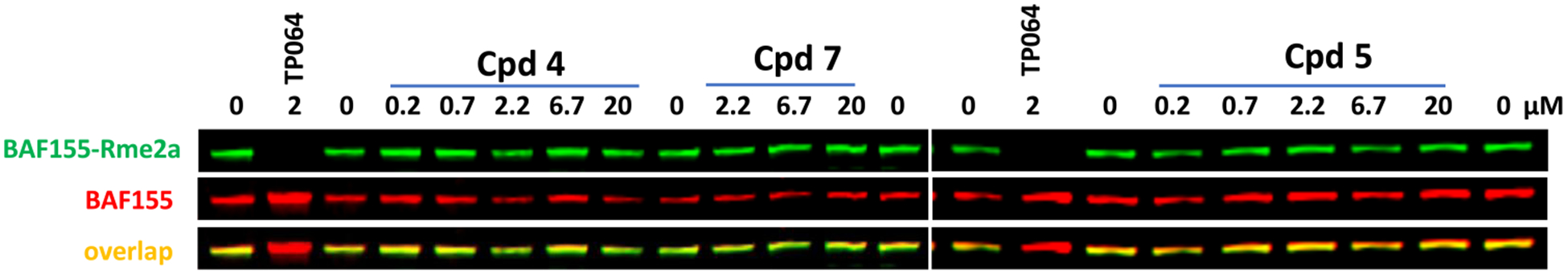
Inhibition of PRMT4-dependent BAF155 asymmetric dimethylation in cells. Compounds 4, 5, and 7 did not inhibit PRMT4 activity at concentrations up to 20 μM. MCF7 cells were treated with compounds for 2 days. PRMT4 selective inhibitor TP064 was used as a positive control. The WB results are representative of two independent experiments.
Finally, to assess the cellular toxicity of compounds 4, 5, and 7, we investigated the effect of these compounds on inhibiting the growth of MCF-7, PNT2 (a nontumorigenic prostate cell line), and HEK293T cells. Each of the three cell lines was treated with compound 4, 5, or 7 at concentrations up to 20 μM for 3 days, and we found that none of these compounds was cytotoxic in MCF-7, PNT2, and HEK293T cells (Figure 11). Thus, compounds 4, 5, and 7 are valuable chemical tools without significant cell toxicity.
Figure 11.
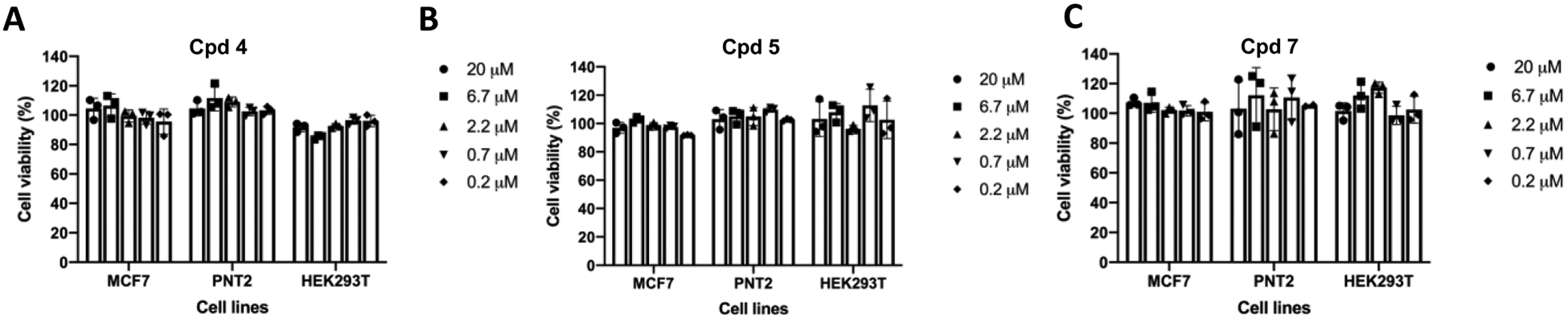
Compounds 4, 5, and 7 are not cytotoxic in MCF-7, PNT2, and HEK293T cells. MCF-7, PNT2, and HEK293T cells were treated with compound 4 (A), 5 (B), or 7 (C) at indicated concentrations for 3 days. Results are presented as mean ± SD from three replicate experiments.
Taken together, these results indicate that compound 4 can potently inhibit PRMT6 and is selective for PRMT6 over PRMT3 (>20-fold), PRMT4 (>20-fold), and PRMT1 (~4-fold) in cells without significant cell toxicity, and compounds 5 and 7 are excellent control compounds for compound 4.
CONCLUSIONS
We discovered a first-in-class PRMT6 covalent inhibitor, compound 4. The covalent inhibition nature of compound 4 was confirmed by kinetic and MS studies. We also obtained a cocrystal structure of PRMT6 conjugated with compound 4, which confirmed that Cys50, a unique cysteine residue in the PRMT6 active site, was modified by compound 4. Compound 4 is a potent PRMT6 inhibitor and remarkably selective for PRMT6 over 28 PKMTs, DNMTs, and type II and III PRMTs. Within the type I PRMT subfamily, compound 4 displayed excellent selectivity over PRMT3 and PRMT4 and moderate selectivity over PRMT1 and PRMT8 and did not covalently modify these closely related type I PRMTs. In cellular assays, compound 4 potently inhibited PRMT6, was less potent against PRMT1 and inactive against PRMT3 and PRMT4, and was not cytotoxic in general. In addition, we developed two structurally highly similar control compounds: compound 5, a potent and cell-active reversible PRMT6 inhibitor, and compound 7, a very poor inhibitor but with the same reactive acrylamide warhead. Collectively, compounds 4, 5, and 7 are a set of valuable tool compounds for the biomedical community to investigate PRMT6 biological functions and disease associations. Furthermore, our discovery has paved the way for the research community to develop the next generation of improved PRMT6 covalent inhibitors.
EXPERIMENTAL SECTION
Chemistry General Procedure.
All commercially available chemical reagents were directly used without further purification. A Teledyne ISCO CombiFlash Rf+ instrument equipped with a variable wavelength UV detector and a fraction collector was used to conduct flash column chromatography. RediSepRf HP C18 and flash silica columns were used for purification. High-performance liquid chromatography (HPLC) spectra for all compounds were acquired using an Agilent 1200 Series system with a DAD detector. Chromatography was performed on a 2.1 mm × 150 mm Zorbax 300SB-C18 5 μm column with water containing 0.1% formic acid as solvent A and acetonitrile containing 0.1% formic acid as solvent B at a flow rate of 0.4 mL/min. The gradient program was as follows: 1% B (0–1 min), 1–99% B (1–4 min), and 99% B (4–8 min). High-resolution mass spectra (HRMS) data were acquired in positive-ion mode using an Agilent G1969A API-TOF with an electrospray ionization (ESI) source. Nuclear magnetic resonance (NMR) spectra were acquired on a Bruker Avance-III 800 MHz spectrometer (800 MHz 1H NMR, 201 MHz 13C NMR). Chemical shifts are reported in ppm (δ). Preparative HPLC was performed on Agilent Prep 1200 series with UV detector set to 254 nm or 220 nM. Samples were injected into a Phenomenex Luna 250 mm × 30 mm, 5 μM C18 column at room temperature (rt). The flow rate was 40 mL/min. A linear gradient was used with 10% of MeOH or CH3CN (A) in H2O (with 0.1% TFA) (B) to 100% of MeOH or CH3CN (A). HPLC was used to establish the purity of target compounds. All final compounds had >95% purity using the HPLC methods described above. All final compounds are characterized as trifluoroacetic acid salt form.
1-(tert-Butyl) 3-Ethyl 4-(3-nitrophenyl)-1H-pyrrole-1,3-dicarboxylate (1).
To a suspension of sodium hydride (60% in mineral oil, 1.44 g, 36.0 mmol) in DMF/THF (50 mL, v/v, 1:1) at 0 °C was slowly added a solution of 1-((isocyanomethyl)sulfonyl)-4-methylbenzene (2.3 g, 12.0 mmol) and ethyl (E)-3-(3-nitrophenyl)acrylate (2.6 g, 12.0 mmol) in DMF/THF (50 mL, v/v, 1:1) over 30 min. After the completion of addition, the reaction was warmed to room temperature and stirred for 1 h. The reaction was then quenched with water and diluted with ethyl acetate. The organic layer was washed twice with water, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting crude intermediate was used in the next step without further purification. To a solution of the above intermediate, triethylamine (1.8 g, 18.0 mmol), and 4-dimethylaminopyridine (110 mg, 0.9 mmol) in dichloromethane (100 mL) was added a solution of di-tert-butyl dicarbonate (2.6 g, 12.0 mmol) in dichloromethane (20 mL). After being stirred overnight, the reaction was quenched with water, separated, dried over anhydrous sodium sulfate, concentrated, and purified by flash chromatography on silica gel column (EtOAc/hexane, 0–25%) to afford the title compound as a white solid (1.6 g, yield 37%). 1H NMR (800 MHz, CDCl3) δ 8.31 (s, 1H), 8.11 (dd, J = 8.3, 2.2 Hz, 1H), 7.92 (d, J = 2.5 Hz, 1H), 7.79 (d, J = 7.6 Hz, 1H), 7.48 (t, J = 7.9 Hz, 1H), 7.33–7.27 (m, 1H), 4.22 (q, J = 7.0 Hz, 2H), 1.63 (s, 9H), 1.24 (t, J = 7.1 Hz, 3H). 13C NMR (201 MHz, CDCl3) δ 163.42, 147.83, 147.59, 135.39, 135.29, 128.53, 126.86, 126.06, 124.07, 121.83, 119.99, 116.86, 85.68, 60.26, 27.80, 14.12.
tert-Butyl 3-Formyl-4-(3-nitrophenyl)-1H-pyrrole-1-carboxylate (2).
To a solution of 1-(tert-butyl) 3-ethyl 4-(3-nitrophenyl)-1H-pyrrole-1,3-dicarboxylate (1.6 g, 4.4 mmol) in THF (100 mL) at −78 °C was slowly added diisobutylaluminium hydride (13.2 mL, 1 M in hexane, 13.2 mmol) over 30 min. The resulting solution was stirred at −78 °C for 3 h before the reaction was quenched with 2 mL of methanol. The solution was poured to saturated aqueous solution of potassium sodium tartrate (150 mL). The resulting mixture was vigorously stirred until two phases can be separated. The organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude intermediate was used in the next step without further purification. To a solution of the above intermediate in dichloromethane (50 mL) was added Dess-Martin periodinane (1.87 g, 4.4 mmol) in portions. The resulting reaction mixture was stirred for 30 min and filtered. The filtrate was concentrated and purified by silica gel flash chromatography (EtOAc/hexane, 0–25%) to afford the title compound as an oil (1 g, yield 71%). 1H NMR (800 MHz, CDCl3) δ 9.92 (s, 1H), 8.39 (s, 1H), 8.19–8.14 (m, 1H), 8.01–7.97 (m, 1H), 7.91 (d, J = 7.6 Hz, 1H), 7.56 (t, J = 7.9 Hz, 1H), 7.45 (d, J = 2.3 Hz, 1H), 1.67 (s, 9H). 13C NMR (201 MHz, CDCl3) δ 185.21, 148.21, 147.36, 134.95, 134.39, 130.97, 129.14, 125.54, 125.04, 123.40, 122.22, 120.93, 86.41, 27.86.
tert-Butyl 3-(((2-((tert-Butoxycarbonyl)amino)ethyl)(methyl)-amino)methyl)-4-(3-nitrophenyl)-1H-pyrrole-1-carboxylate (3).
To a solution of tert-butyl 3-formyl-4-(3-nitrophenyl)-1H-pyrrole-1-carboxylate (760 mg, 2.41 mmol) and tert-butyl (2-(methylamino)-ethyl)carbamate (630 mg, 3.62 mmol) in dichloromethane (30 mL) was added sodium triacetoxylborohydride (1.02 g, 4.82 mmol) in portions. The reaction mixture was stirred overnight at room temperature before it was quenched with 20 mL of saturated aqueous solution of sodium bicarbonate. The aqueous layer was extracted with dichloromethane (3 × 20 mL). The combined organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by silica gel flash chromatography (EtOAc/hexane: 0–50%) to afford the title compound 3 as an oil (1.0 g, yield 88%). 1H NMR (800 MHz, CD3OD) δ 8.67 (s, 1H), 8.17 (d, J = 8.2 Hz, 1H), 8.03 (d, J = 7.8 Hz, 1H), 7.67 (t, J = 7.9 Hz, 1H), 7.55 (d, J = 2.3 Hz, 1H), 7.43 (s, 1H), 3.78–3.65 (m, 2H), 3.25 (t, J = 6.3 Hz, 2H), 2.76–2.64 (m, 2H), 2.42 (s, 3H), 1.69 (s, 9H), 1.42 (s, 9H). 13C NMR (201 MHz, CD3OD) δ 155.47, 147.10, 146.78, 134.69, 132.43, 127.97, 124.82, 121.26, 120.43, 119.62, 117.39, 82.99, 58.69, 53.82, 50.98, 38.71, 35.66, 25.89, 25.38. MS (ESI) m/z 475.2 [M + H]+.
N-(3-(4-(((2-Aminoethyl)(methyl)amino)methyl)-1H-pyrrol-3-yl)-phenyl)acrylamide (4).
To a solution of tert-butyl 3-(((2-((tert-butoxycarbonyl)amino)ethyl)(methyl)amino)methyl)-4-(3-nitrophenyl)-1H-pyrrole-1-carboxylate (200 mg, 0.42 mmol) in methanol (8 mL) was added Pd/C (30 mg, 10 wt % Pd basis). After the resulting mixture was stirred under a hydrogen atmosphere for 2 h, the mixture was filtered through a Celite cup and the filtrate was concentrated under reduced pressure. The crude intermediate was used in the next step without further purification. To a solution of the above intermediate in dichloromethane (10 mL) at 0 °C was added a solution of acryloyl chloride (38 mg, 0.42 mmol) in dichloromethane (2 mL). The resulting solution was stirred for 10 min before half of the solvent was removed under reduced pressure. The reaction was treated with trifluoroacetic acid (4 mL) for 1 h before the volatile was removed under reduced pressure. The resulting residue was purified by reverse prep-HPLC to afford compound 4 as TFA salt (82 mg, yield 37%) after lyophilization. 1H NMR (800 MHz, CD3OD) δ 7.84 (s, 1H), 7.43–7.36 (m, 2H), 7.19 (d, J = 8.0 Hz, 2H), 6.99 (d, J = 2.1 Hz, 1H), 6.50 (dd, J = 17.0, 10.2 Hz, 1H), 6.41 (d, J = 16.9 Hz, 1H), 5.82 (d, J = 10.2 Hz, 1H), 4.53 (s, 2H), 3.44–3.13 (m, 4H), 2.73 (s, 3H). 13C NMR (201 MHz, CD3OD) δ 165.14, 138.56, 135.86, 131.06, 129.26, 126.65, 125.05, 124.56, 122.18, 120.68, 118.33, 117.86, 107.60, 52.06, 50.83, 38.61, 33.94. MS (ESI) m/z 299.3 [M + H]+. HRMS (ESI): calcd for C17H22N4O + H: 299.1866; found: 299.1868 [M + H]+.
N-(3-(4-(((2-Aminoethyl)(methyl)amino)methyl)-1H-pyrrol-3-yl)-phenyl)propionamide (5).
Compound 5 was synthesized according to the procedures for the preparation of compound 4. tert-Butyl 3-(((2-((tert-butoxycarbonyl)amino)ethyl)(methyl)amino)methyl)-4-(3-nitrophenyl)-1H-pyrrole-1-carboxylate (100 mg, 0.21 mmol) and propionyl chloride (27 mg, 0.30 mmol) were used to afford compound 5 (32 mg, yield 29%) after lyophilization. 1H NMR (800 MHz, CD3OD) δ 7.73 (s, 1H), 7.38 (t, J = 7.8 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.25–7.06 (m, 2H), 6.97 (d, J = 2.1 Hz, 1H), 4.51 (s, 2H), 3.60–3.04 (m, 4H), 2.72 (s, 3H), 2.45 (q, J = 7.6 Hz, 2H), 1.24 (t, J = 7.5 Hz, 3H). 13C NMR (201 MHz, CD3OD) δ 174.46, 138.76, 135.75, 129.18, 125.13, 124.23, 122.15, 120.68, 118.26, 117.80, 107.55, 52.02, 50.84, 38.60, 33.95, 29.62, 8.82. MS (ESI) m/z 301.2 [M + H]+. HRMS (ESI): calcd for C17H25N4O + H: 301.2023; found: 301.2008 [M + H]+.
tert-Butyl 3-(((2-Hydroxyethyl)(methyl)amino)methyl)-4-(3-nitrophenyl)-1H-pyrrole-1-carboxylate (6).
To a solution of tert-butyl 3-formyl-4-(3-nitrophenyl)-1H-pyrrole-1-carboxylate (240 mg, 0.76 mmol) and 2-(methylamino)ethan-1-ol (85.5 mg, 1.14 mmol) in dichloromethane (10 mL) was added sodium triacetoxylborohydride (322 mg, 1.52 mmol) in portions. The mixture was stirred overnight at room temperature before the reaction was quenched with 5 mL of saturated aqueous solution of sodium bicarbonate. The aqueous layer was extracted with dichloromethane (3 × 10 mL). The combined organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by silica gel flash chromatography (EtOAc/hexane: 0–50%) to afford the title compound as an oil (210 mg, yield 74%). 1H NMR (800 MHz, CD3OD) δ 8.63 (s, 1H), 8.17–8.07 (m, 1H), 8.02 (d, J = 7.7 Hz, 1H), 7.60 (t, J = 7.9 Hz, 1H), 7.50 (d, J = 2.4 Hz, 1H), 7.31 (d, J = 2.3 Hz, 1H), 3.65 (t, J = 6.3 Hz, 2H), 3.48 (s, 2H), 2.60 (t, J = 6.3 Hz, 2H), 2.28 (s, 3H), 1.65 (s, 9H). 13C NMR (201 MHz, CD3OD) δ 148.51, 148.44, 136.45, 133.92, 129.20, 126.45, 122.61, 122.05, 121.11, 120.85, 118.42, 84.19, 59.18, 58.24, 53.06, 40.93, 26.79. MS (ESI) m/z 376.2 [M + H]+.
N-(3-(4-(((2-Hydroxyethyl)(methyl)amino)methyl)-1H-pyrrol-3-yl)phenyl)acrylamide (7).
To a solution of tert-butyl 3-(((2-hydroxyethyl)(methyl)amino)methyl)-4-(3-nitrophenyl)-1H-pyrrole-1-carboxylate (210 mg, 0.56 mmol) in methanol (8 mL) was added Pd/C (30 mg, 10 wt % Pd basis). After the resulting mixture was stirred under a hydrogen atmosphere for 2 h, it was filtered through a Celite cup, and the filtrate was concentrated under reduced pressure. The crude intermediate was used in the next step without further purification. To a solution of the above intermediate in dichloromethane (10 mL) at 0 °C was added a solution of acryloyl chloride (50 mg, 0.56 mmol) in dichloromethane (2 mL). The resulting solution was stirred for 10 min before half of the solvent was removed under reduced pressure. The reaction was treated with trifluoroacetic acid (4 mL) for 1 h before the volatile was removed under reduced pressure. The resulting residue was purified by reverse prep-HPLC to afford compound 7 as TFA salt (98 mg, yield 42%) after lyophilization. 1H NMR (800 MHz, CD3OD) δ 7.83 (s, 1H), 7.41 (dt, J = 15.3, 8.1 Hz, 2H), 7.18 (d, J = 7.2 Hz, 1H), 7.14 (d, J = 2.3 Hz, 1H), 6.98 (d, J = 2.1 Hz, 1H), 6.51–6.45 (m, 1H), 6.40 (d, J = 16.9 Hz, 1H), 5.81 (d, J = 10.2 Hz, 1H), 4.53 (d, J = 14.0 Hz, 1H), 4.43 (d, J = 14.0 Hz, 1H), 3.71 (t, J = 5.2 Hz, 2H), 3.18 (dt, J = 12.2, 5.6 Hz, 1H), 2.92 (dt, J = 13.3, 4.8 Hz, 1H), 2.66 (s, 3H). 13C NMR (201 MHz, CD3OD) δ 164.99, 138.67, 136.13, 131.08, 129.15, 126.56, 125.14, 124.36, 122.05, 120.37, 118.01, 117.73, 107.86, 55.86, 55.00, 51.13, 38.60. MS (ESI) m/z 300.1 [M + H]+. HRMS (ESI): calcd for C17H21N3O2 + H: 300.1707; found: 300.1699 [M + H]+.
Protein Expression, Purification, Co-crystallization, and Structural Determination.
Human PRMT6 protein was expressed and purified according to the previously published protocol.25 PRMT6 at 5.6 mg/mL was mixed with a 5-fold molar excess of S-adenosyl-l-homocysteine (SAH) and a 3-fold molar excess of compound 4 (dissolved from a previously prepared 100 mM dimethyl sulfoxide (DMSO) stock solution). Diffraction-quality crystals were obtained by setting a 96-well vapor-diffusion sitting drop at room temperature, in a precipitant solution containing 0.1 M sodium malonate pH 7.0, 12% (w/v) PEG 3350. The PRMT6-compound 4 crystal was cryo-protected by immersing it into a precipitant solution supplemented with 10% glycerol and then into paratone, and cryo-cooled in liquid nitrogen. The PRMT6-compound 4 dataset was collected with an in-house Rigaku FR-E SuperBright and processed with HKL3000.26 Initial phases were obtained by Fourier transform in refmac527 using PRMT6 (PDB ID: 5EGS) as the initial model. Model building was performed in COOT,28 and the structure was validated with MolProbity.29 Restraints for compound 4 was generated using Grade Web Server (http://grade.globalphasing.org).
Biochemical Assays.
The inhibitory effects of compound 4, compound 5, and compound 7 against PRMT1, PRMT3, PRMT4, PRMT6, and PRMT8 were tested using the published scintillation proximity assay (SPA).19 The tritiated S-adenosyl-l-methionine (3H-SAM, PerkinElmer Life Sciences) was used as the donor of the methyl group. The 3H-methylated biotin-labeled peptide (subsrate) was captured in a streptavidin/scintillant-coated microplate (FlashPlate PLUS; PerkinElmer Life Sciences), which brings the incorporated 3H-methyl and the scintillant to close proximity resulting in light emission that is quantified by tracing the radioactivity signal (counts per minute), as measured by a TopCount NXT Microplate Scintillation and Luminescence Counter (PerkinElmer Life Sciences). The IC50 values were determined under balanced conditions at Km concentrations of both substrate and cofactor by titration of test compounds in the reaction mixture.
Mass Spectrometry Analysis for Assessing the Covalent Binding.
PRMT proteins (15 μM) were incubated with 20 molar excess of the PRMT inhibitors (300 μM) for 1 h at rt before being quenched by adding 0.1% trifluoroacetic acid (aq.). The resulting samples were separated over an HPLC column with 5–95% acetonitrile in water as an eluent. And the MS data were analyzed using an Agilent LC/MSD Time-of-Flight Mass Spectrometer equipped with an electrospray ionization source.
Methyltransferase Selectivity Assays.
The inhibitory effects of compound 4, compound 5, and compound 7 on 33 methyltransferase (MT) activities were assessed at two concentrations of 10 and 50 μM, as previously described.30
KI and kinact Determinations.
The values of KI and kinact were estimated by time-dependent IC50 values using XLfit, as described previously.20 A series of IC50 values were measured using the SPA by preincubating compound 4 and compound 5 with PRMT6 at different time points ranging from 1 to 30 min.
Cellular Assay.
The effects of compound 4, compound 5, and compound 7 inhibiting PRMT1 in MCF7 cells,14 PRMT3 in HEK293T cells,22 PRMT4 in MCF7 cells,24 or PRMT6 in HEK293T cells14 were assessed as previously reported.
Cell Viability Assay.
MCF7, PNT2, and HEK293T were seeded (1 × 104) into 96-well plates in triplicate. After 24 h, the cells were treated with serially diluted compounds (compound 4, compound 5, and compound 7) from 20 μM for 3 days. Cell viability was assessed using CCK-8 (cell counting kit-8, WST-8). Briefly, a 1× solution of CCK-8 (Dojindo, CK04) was warmed up at room temperature. Then, CCK-8 was added to each of the wells (10 μL per 100 μL medium) and then incubated for 4 h at 37 °C. The absorbance was read at 450 nm using Infinite F PLEX plate reader (TECAN, Morrisville, NC). The data were analyzed using GraphPad Prism 8. Results are shown as mean ± SD from three replicate experiments.
Supplementary Material
ACKNOWLEDGMENTS
The research described here was supported in part by the grant R01GM122749 (to J.J.) from the U.S. National Institutes of Health. The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada through Ontario Genomics Institute [OGI-055], Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA, Darmstadt, Germany, MSD, Novartis Pharma AG, Ontario Ministry of Research, Innovation and Science (MRIS), Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome [106169/ZZ14/Z]. The authors also thank Drs. Jithesh Kottur and Prashasti Kumar for helpful suggestions.
ABBREVIATIONS USED
- PRMT
protein arginine methyltransferase
- SAM
S-adenosyl-methionine
- TFA
trifluoroacetic acid
- DMSO
dimethyl sulfoxide
- ESI
electrospray ionization
- SPA
scintillation proximity assay
- H3R2me2a
H3R2 asymmetric dimethylation
- H4R3me2a
H4R3 asymmetric dimethylation
- BAF155-Rme2a
BAF155 arginine asymmetric demethylation
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00406.
Crystallography data and refinement statistics; selectivity data of compound 4 against 33 methyltransferases; sequence alignment of type I PRMTs; MS assay results for PRMT1, PRMT3, PRMT4, and PRMT8 preincubated with compound 4; and 1H and 13C NMR spectra of compounds 1–7 (PDF)
Molecular formula strings for all compounds (CSV)
The authors declare no competing financial interest.
Accession Codes
The structure presented in this article has been deposited in the Protein Data Bank under accession code 6P7I for the PRMT6-MS117 complex. The authors will release the atomic coordinates upon article publication.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c00406
Contributor Information
Yudao Shen, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Fengling Li, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
Magdalena M. Szewczyk, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
Levon Halabelian, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
Kwang-su Park, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Irene Chau, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
Aiping Dong, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
Hong Zeng, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
He Chen, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Fanye Meng, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Dalia Barsyte-Lovejoy, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
Cheryl H. Arrowsmith, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada; Princess Margaret Cancer Centre and Department of Medical Biophysics, University of Toronto, Toronto, Ontario M5G 2M9, Canada;.
Peter J. Brown, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada
Jing Liu, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States;.
Masoud Vedadi, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada; Department of Pharmacology and Toxicology, University of Toronto, Toronto, Ontario M5S 1A8, Canada;.
Jian Jin, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States;.
■ REFERENCES
- (1).Frankel A; Yadav N; Lee JH; Branscombe TL; Clarke S; Bedford MT The novel human protein arginine N-methyltransferase PRMT6 is a nuclear enzyme displaying unique substrate specificity. J. Biol. Chem 2002, 277, 3537–3543. [DOI] [PubMed] [Google Scholar]
- (2).Bedford MT; Clarke SG Protein arginine methylation in mammals: who, what, and why. Mol. Cell 2009, 33, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kaniskan HÜ; Martini ML; Jin J Inhibitors of protein methyltransferases and dimethylases. Chem. Rev 2018, 118, 989–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Yang Y; Bedford MT Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [DOI] [PubMed] [Google Scholar]
- (5).Wei H; Mundade R; Lange KC; Lu T Protein arginine methylation of non-histone proteins and its role in diseases. Cell Cycle 2014, 13, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Guccione E; Bassi C; Casadio F; Martinato F; Cesaroni M; Schuchlautz H; Luscher B; Amati B Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature 2007, 449, 933–937. [DOI] [PubMed] [Google Scholar]
- (7).Boulanger MC; Liang C; Russell RS; Lin R; Bedford MT; Wainberg MA; Richard S Methylation of Tat by PRMT6 regulates human immunodeficiency virus type 1 gene expression. J. Virol 2005, 79, 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sgarra R; Lee J; Tessari MA; Altamura S; Spolaore B; Giancotti V; Bedford MT; Manfioletti G The AT-hook of the chromatin architectural transcription factor high mobility group A1a is arginine-methylated by protein arginine methyltransferase 6. J. Biol. Chem 2006, 281, 3764–3772. [DOI] [PubMed] [Google Scholar]
- (9).El-Andaloussi N; Valovka T; Toueille M; Steinacher R; Focke F; Gehrig P; Covic M; Hassa PO; Schar P; Hubscher U; Hottiger MO Arginine methylation regulates DNA polymerase beta. Mol. Cell 2006, 22, 51–62. [DOI] [PubMed] [Google Scholar]
- (10).Vieira FQ; Costa-Pinheiro P; Ramalho-Carvalho J; Pereira A; Menezes FD; Antunes L; Carneiro I; Oliveira J; Henrique R; Jeronimo C Deregulated expression of selected histone methylases and dimethylases in prostate carcinoma. Endocr. Relat. Cancer 2014, 21, 51–61. [DOI] [PubMed] [Google Scholar]
- (11).Yoshimatsu M; Toyokawa G; Hayami S; Unoki M; Tsunoda T; Field HI; Kelly JD; Neal DE; Maehara Y; Ponder BAJ; Nakamura Y; Hamamoto R Dysregulation of PRMT1 and PRMT6, type I arginine methyltransferases, is involved in various types of human cancers. Int. J. Cancer 2011, 128, 562–573. [DOI] [PubMed] [Google Scholar]
- (12).Limm K; Ott C; Wallner S; Mueller DW; Oefner P; Hellerbrand C; Bosserhoff AK Deregulation of protein methylation in melanoma. Eur. J. Cancer 2013, 49, 1305–1313. [DOI] [PubMed] [Google Scholar]
- (13).Pan R; Yu H; Dai J; Zhou C; Ying X; Zhong J; Zhao J; Zhang Y; Wu B; Mao Y; Wu D; Ying J; Duan S Significant association of PRMT6 hypomethylation with colorectal cancer. J. Clin. Lab. Anal 2018, 32, No e22590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Eram MS; Shen YD; Szewczyk MM; Wu H; Senisterra G; Li FL; Butler KV; Kaniskan HÜ; Speed BA; dela Sena C; Dong AP; Zeng H; Schapira M; Brown PJ; Arrowsmith CH; Barsyte-Lovejoy D; Liu J; Vedadi M; Jin J A potent, selective, and cell-active inhibitor of human type I protein arginine methyltransferases. ACS Chem. Biol 2016, 11, 772–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Mitchell LH; Drew AE; Ribich SA; Rioux N; Swinger KK; Jacques SL; Lingaraj T; Boriack-Sjodin PA; Waters NJ; Wigle TJ; Moradei O; Jin L; Riera T; Porter-Scott M; Moyer MP; Smith JJ; Chesworth R; Copeland RA Aryl pyrazoles as potent inhibitors of arginine methyltransferases: identification of the first PRMT6 tool compound. ACS Med. Chem. Lett 2015, 6, 655–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shen Y; Szewczyk MM; Eram MS; Smil D; Kaniskan HÜ; de Freitas RF; Senisterra G; Li F; Schapira M; Brown PJ; Arrowsmith CH; Barsyte-Lovejoy D; Liu J; Vedadi M; Jin J Discovery of a potent, selective, and cell-active dual inhibitor of protein arginine methyltransferase 4 and protein arginine methyltransferase 6. J. Med. Chem 2016, 59, 9124–9139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ghosh AK; Samanta I; Mondal A; Liu WR Covalent inhibition in drug discovery. ChemMedChem 2019, 14, 889–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Flanagan ME; Abramite JA; Anderson DP; Aulabaugh A; Dahal UP; Gilbert AM; Li C; Montgomery J; Oppenheimer SR; Ryder T; Schuff BP; Uccello DP; Walker GS; Wu Y; Brown MF; Chen JM; Hayward MM; Noe MC; Obach RS; Philippe L; Shanmugasundaram V; Shapiro MJ; Starr J; Stroh J; Che Y Chemical and computational methods for the characterization of covalent reactive groups for the prospective design of irreversible inhibitors. J. Med. Chem 2014, 57, 10072–10079. [DOI] [PubMed] [Google Scholar]
- (19).Liu F; Li F; Ma A; Dobrovetsky E; Dong A; Gao C; Korboukh I; Liu J; Smil D; Brown PJ; Frye SV; Arrowsmith CH; Schapira M; Vedadi M; Jin J Exploiting an allosteric binding site of PRMT3 yields potent and selective inhibitors. J. Med. Chem 2013, 56, 2110–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Krippendorff BF; Neuhaus R; Lienau P; Reichel A; Huisinga W Mechanism-based inhibition: deriving K(I) and k(inact) directly from time-dependent IC(50) values. J. Biomol. Screening 2009, 14, 913–923. [DOI] [PubMed] [Google Scholar]
- (21).Lee J; Sayegh J; Daniel J; Clarke S; Bedford MT PRMT8, a new membrane-bound tissue-specific member of the protein arginine methyltransferase family. J. Biol. Chem 2005, 280, 32890–32896. [DOI] [PubMed] [Google Scholar]
- (22).Kaniskan HÜ; Szewczyk MM; Yu Z; Eram MS; Yang X; Schmidt K; Luo X; Dai M; He F; Zang I; Lin Y; Kennedy S; Li F; Dobrovetsky E; Dong A; Smil D; Min SJ; Landon M; Lin-Jones J; Huang XP; Roth BL; Schapira M; Atadja P; Barsyte-Lovejoy D; Arrowsmith CH; Brown PJ; Zhao K; Jin J; Vedadi M A potent, selective and cell-active allosteric inhibitor of protein arginine methyltransferase 3 (PRMT3). Angew. Chem., Int. Ed 2015, 54, 5166–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kaniskan HÜ; Eram MS; Zhao K; Szewczyk MM; Yang X; Schmidt K; Luo X; Xiao S; Dai M; He F; Zang I; Lin Y; Li F; Dobrovetsky E; Smil D; Min SJ; Lin-Jones J; Schapira M; Atadja P; Li E; Barsyte-Lovejoy D; Arrowsmith CH; Brown PJ; Liu F; Yu Z; Vedadi M; Jin J Discovery of potent and selective allosteric inhibitors of protein arginine methyltransferase 3 (PRMT3). J. Med. Chem 2018, 61, 1204–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Nakayama K; Szewczyk MM; Dela Sena C; Wu H; Dong A; Zeng H; Li F; de Freitas RF; Eram MS; Schapira M; Baba Y; Kunitomo M; Cary DR; Tawada M; Ohashi A; Imaeda Y; Saikatendu KS; Grimshaw CE; Vedadi M; Arrowsmith CH; Barsyte-Lovejoy D; Kiba A; Tomita D; Brown PJ TP-064, a potent and selective small molecule inhibitor of PRMT4 for multiple myeloma. Oncotarget 2018, 9, 18480–18493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ferreira de Freitas R; Eram MS; Szewczyk MM; Steuber H; Smil D; Wu H; Li F; Senisterra G; Dong A; Brown PJ; Hitchcock M; Moosmayer D; Stegmann CM; Egner U; Arrowsmith C; Barsyte-Lovejoy D; Vedadi M; Schapira M Discovery of a potent class I protein arginine methyltransferase fragment inhibitor. J. Med. Chem 2016, 59, 1176–1183. [DOI] [PubMed] [Google Scholar]
- (26).Minor W; Cymborowski M; Otwinowski Z; Chruszcz M HKL-3000: the integration of data reduction and structure solution-from diffraction images to an initial model in minutes. Acta Crystallogr., Sect. D: Biol. Crystallogr 2006, 62, 859–866. [DOI] [PubMed] [Google Scholar]
- (27).Steiner RA; Lebedev AA; Murshudov GN Fisher’s information in maximum-likelihood macromolecular crystallographic refinement. Acta Crystallogr., Sect. D: Biol. Crystallogr 2003, 59, 2114–2124. [DOI] [PubMed] [Google Scholar]
- (28).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Williams CJ; Headd JJ; Moriarty NW; Prisant MG; Videau LL; Deis LN; Verma V; Keedy DA; Hintze BJ; Chen VB; Jain S; Lewis SM; Arendall WB III; Snoeyink J; Adams PD; Lovell SC; Richardson JS; Richardson DC MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Scheer S; Ackloo S; Medina TS; Schapira M; Li F; Ward JA; Lewis AM; Northrop JP; Richardson PL; Kaniskan HÜ; Shen Y; Liu J; Smil D; McLeod D; Zepeda-Velazquez CA; Luo M; Jin J; Barsyte-Lovejoy D; Huber KVM; De Carvalho DD; Vedadi M; Zaph C; Brown PJ; Arrowsmith CH A chemical biology toolbox to study protein methyltransferases and epigenetic signaling. Nat. Commun 2019, 10, No 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.