

Abstract
The Angiotensin II-Angiotensin-(1-7) axis of the Renin Angiotensin System (RAS) encompasses three enzymes that form Angiotensin-(1-7) [Ang(1-7)] directly from Angiotensin II (Ang II): Angiotensin Converting Enzyme 2 (ACE2), Prolyl Carboxypeptidase (PRCP) and Prolyl Oligopeptidase (POP). We investigated their relative contribution to Ang-(1-7) formation in-vivo and also ex-vivo in serum, lungs and kidneys using models of genetic ablation coupled with pharmacological inhibitors. In WT mice infusion of Ang II resulted in a rapid increase of plasma Ang-(1-7). In ACE2−/−/PRCP−/− mice, Ang II infusion resulted in a similar increase in Ang-(1-7) as in WT (563 ± 48 vs 537 ± 70 fmol/ml, respectively), showing that the bulk of Ang-(1-7) formation in circulation is essentially independent of ACE2 and PRCP. By contrast, a POP inhibitor, Z-Pro-Prolinal (ZPP) reduced the rise in plasma Ang-(1-7) after infusing Ang II to control WT mice. In POP −/− mice, the increase in Ang-(1-7) was also blunted as compared to WT mice (309 ± 46 and 472 ± 28 fmol/ml, respectively p=0.01), and moreover, the rate of recovery from acute Ang II-induced hypertension was delayed (p=0.016). In ex vivo studies POP inhibition with ZZP reduced Ang-(1-7) formation from Ang II markedly in serum and in lung lysates. By contrast, in kidney lysates the absence of ACE2, but not POP, obliterated Ang-(1-7) formation from added Ang II. We conclude that POP is the main enzyme responsible for Ang II conversion to Ang-(1-7) in the circulation and in the lungs, whereas Ang-(1-7) formation in the kidney is mainly ACE2-dependent.
Keywords: angiotensin, angiotensin-converting enzyme, angiotensin II, renin angiotensin system
Graphical Abstract
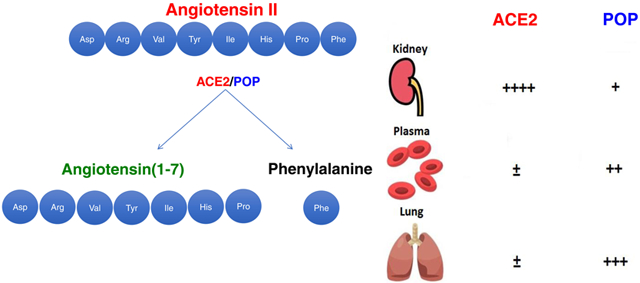
Summary
This study demonstrates the relevance of the POP/Ang-(1-7) axis in the circulation and in the lungs while confirming the importance of ACE2 in kidney Ang-(1-7) formation from Ang II.
Introduction
The knowledge of the Renin Angiotensin System (RAS) has expanded with the identification of enzymes and peptides downstream of Angiotensin (Ang) II, such as Ang-(1-7), Ang III and others1-23. Ang-(1-7) forming enzymes do not only prevent and counteract Ang II overactivity but also foster Ang-(1-7) formation eliciting potentially beneficial actions of the latter peptide7,11,14,15,24,25. There are three known carboxypeptidases that form Ang-(1-7) by cleaving Ang II: Angiotensin Converting Enzyme 2 (ACE2) 1-5,15, Prolyl Carboxypeptidase (PRCP) 19,20,26-28, and Prolyl Oligopeptidase (POP/PEP/PREP/PE)10,23,29-34. The biological importance of conversion of Ang II to Ang-(1-7) is two-fold: by lowering Ang II its potentially detrimental actions may be prevented; in addition, Ang-(1-7) is being formed and this peptide has tissue-protective actions that are generally opposite to the unwanted chronic effects of excessive Ang II. This highlights the importance of enzymes that cleave Ang II to form Ang-(1-7).
It is also known from the work of Yamamoto et al that Ang-(1-7) can be formed directly from Ang I.34 The authors showed that, combined ACE/Neprilysin inhibition decreased Ang-(1-7) formation from Ang I infusion suggesting a critical role of Neprilysin on the formation of Ang-(1-7) directly from Ang I.34 In this study, however, Ang II was not infused. The goal of our study is to examine the relative contribution of enzymes that form Ang-(1-7) directly from Ang II. Of those, ACE2, a homolog of Angiotensin Converting Enzyme (ACE) with carboxypeptidase activity that is able to cleave both Ang I and Ang II is the best studied20,26,35. The enzyme embodies a metalloprotease active site and is widely distributed but principally expressed in the kidney, the heart, the intestine and the testis 36-38.
Two other enzymes are known to form Ang-(1-7) directly from cleavage of Ang II: PRCP and POP. 32-38 PRCP is a serine carboxyprotease that cleaves the C-terminal amino acid of various peptides like Ang II, where the penultimate amino acid is Proline26. The enzyme’s activity is extremely pH dependent with an optimum in the acidic range and essentially no activity at the physiologic pH of plasma20,26,35. POP is a serine protease that is involved in the enzymatic degradation of several biologically active peptides such as Ang I and Ang II36-38. Due to its molecular structure POP hydrolyses peptide hormones and neuropeptides, excluding high molecular weight peptides and proteins 36. POP cuts at the C-side of an internal proline and can cleave Ang I to form Ang-(1-7), and Ang II to form Ang-(1-7) 23,29,37. Ang II contains proline at the 7th amino acid position and Phenylalanine (Phe) at the 8th position. Therefore, the action of POP at the C-terminal end results in the formation of Ang-(1-7) and a release of Phe. POP is an intracellular enzyme localized in the cytoplasm38,39 but a membrane-bound form isolated from bovine brain has also been reported40. The function of POP has mainly been studied in brain tissue, as it could be linked to some neurodegenerative diseases 41, and in neutrophil inflammation42,43 . Enzymatic activity for POP has been detected in lymphocytes, thrombocytes, renal cortex, epithelial cells, fibroblasts and testis38. POP activity also was reported in plasma, but a physiological role of circulatory POP has not been established44,45. In this study we examined formation of Ang-(1-7) following the administration of Ang II and the impact of POP deficiency on acute Ang II-induced hypertension. In addition, we examined the relative contribution to Ang-(1-7) formation from Ang II by POP and ACE2 ex vivo in serum, kidney and lung tissues.
Methods
Methods used will be made available from the corresponding author upon reasonable request.
Ang II injection to evaluate Ang-(1-7) formation
Male WT mice (C57Bl/6 background) at 10-20 weeks of age were intraperitoneally injected with Ang II (0.2 μg/g body weight). In some studies five minutes after the Ang II bolus blood was drawn by cardiac puncture under euthasol anesthesia (see Results) and the mice were euthanized by induction of double pneumothorax. In other studies following intraperitoneal Ang II injection blood was collected from the tail tip snapped at 5 and 30 minutes post infusion (this is specified under results). The same injection protocol was used for POP−/− and ACE2−/−/PRCP−/− mice (males, 20-24 weeks old).
In all the samples, Ang-(1-7) was measured by ELISA (see Data Supplement). In addition, in selected samples the levels of plasma Ang-(1-7) together with Ang II and Ang-(1-5) were measured by LC/MS-MS (Attoquant Diagnostics) because our ELISA and RIA do not distinguish isoforms of those peptides, namely Ang III and Ang-(2-7). A radioimmunoassay (Hypertension Core Lab, Wake Forest University School of Medicine) was also used in the same selected samples. This RIA uses a C-terminal directed antibody, and cross-reacts with Ang-(2-7), and Ang-(3-7) with little or no cross-reactivity with Ang I. 46
ZPP, a POP inhibitor was given IP together with Ang II at a dose (1μg/g BW) calculated to give a circulatory ZPP concentration in the 10 μM range. At this concentration this inhibition is selective for POP whereas at higher concentration (>100 μM) also inhibits PRCP23,47. The dose of ZPP (1μg/g BW) has shown in our preliminary studies a nearly complete inhibition of serum POP activity.
Blood pressure measurement after Ang II injection
The effect of POP deficiency on systolic blood pressure (SBP) after Ang II infusion was studied in male POP−/− and WT mice (C57Bl/6 genetic background) at 20-24 weeks of age. SBP was measured non-invasively in anesthetized mice by determining tail blood volume with a volume-pressure recording (VPR) sensor and an occlusion tail-cuff using a computerized system (CODA System, Kent Scientific, Torrington, CT) as previously described 14-16 (see also Supplement).
Statistical Analyses
For comparison of two independent groups, a two-tailed t-test was used for normally distributed data. For not normally distributed data, a Mann-Whitney test was used. For comparison of more than two independent groups, one-way ANOVA was employed followed by Tukey’s multiple comparisons test. The significance over time between groups was evaluated by GLM model (SPSS, version 23). Results are presented in Mean ± SE and a p-value <0.05 was considered statistically significant.
Results
In vivo formation of Ang-(1-7) in plasma after infusion of Ang II
To examine Ang-(1-7) formation from Ang II, Ang II was injected intraperitoneally to wild-type mice. After an Ang II bolus, blood was collected and plasma levels of Ang-(1-7) 5 minutes later were taken as evidence of rapid formation of this peptide from infused Ang II.
In control animals (n=5) which did not receive an Ang II bolus, plasma Ang II levels measured by ELISA were low (8.8 ± 1.8 fmol/ml), whereas in Ang II-infused animals (n=6) Ang II levels at 5 minutes were about 100-fold higher: (968 ± 213 fmol/ml). The infusion of Ang II resulted in markedly elevated plasma Ang-(1-7) levels (1268 ± 439 fmol/ml), measured by ELISA. To confirm the extremely high levels of plasma Ang-(1-7) after Ang II infusion, additional measurements of Ang-(1-7) were performed by RIA in the same plasma samples from Ang II infused mice. The values of Ang-(1-7) obtained by RIA were also very high and not significantly different from those obtained by ELISA (1702 ± 268 fmol/ml and 1268 ± 439, fmol/ml, respectively).
Plasma Ang II and Ang-(1-7) concentrations were additionally evaluated in the same Ang II post-infusion samples using liquid chromatography tandem mass spectrometry (LC/MS-MS) 16. In addition, a major metabolite, the Ang-(1-5) , formed from Ang-(1-7) was also measured by LC/MS-MS (Figure 1). As previously reported16, non-infused animals had barely detectable levels of Ang II(7.5 ± 1.9 fmol/ml), Ang-(1-7) (3.9 ± 0 fmol/ml) and Ang-(1-5) (3.0±0 fmol/ml) ). In Ang II infused mice, Ang II and Ang-(1-7) were both markedly elevated (233 ± 20 and 854 ± 222 fmol/ml, respectively) (Figure 1). This is consistent with the high levels of these peptides measured by ELISA and RIA following Ang II infusion. A high level of Ang-(1-5), moreover, was also found (233 ± 24 fmol/ml) (Figure 1). This shows that plasma Ang-(1-7) is formed at a high rate when there is a large supply of Ang II and that Ang-(1-7) is further degraded to Ang-(1-5) .
Figure 1: Angiotensin peptide levels in serum measured by LC/MS-MS in Ang II-infused mice.

Plasma showed high levels of Ang II (upper), Ang-(1-7) (middle), and Ang-(1-5) (lower panel) measured by Liquid Chromatography Tandem Mass Spectrometry (LC/MS-MS). Blood was obtained by cardiac puncture from mice (n=6) at 5 minutes after Ang II i.p. infusion.
In vivo formation of Ang-(1-7) after Ang II infusion in a model of genetic ablation of ACE2 and PRCP
We next examined Ang-(1-7) formation in a model of genetic ablation of two known Ang-(1-7) forming enzymes, ACE2 and PRCP. For this, we used a cross of ACE2 KO with PRCP KO that we have generated in our lab (see Methods). Plasma levels of Ang II at 5 minutes post infusion were very high and not significantly different between WT and ACE2−/−/PRCP−/− mice (1229 ± 307 and 783 ± 166 fmol/ml, respectively p=0.3) (Figure 2a). Plasma Ang-(1-7) levels also measured 5 minutes after Ang II infusion were markedly elevated and also not significantly different between WT and ACE2−/−/PRCP−/− (563 ± 51 and 537 ± 70, fmol/ml, respectively p=0.8) (Figure 2b). These in vivo findings show that Ang-(1-7) formation from Ang II in plasma is essentially ACE2- and PRCP-independent.
Figure 2:

Ang II and Ang-(1-7) levels in plasma measured 5 minutes post infusion of Ang II. (A) Plasma Ang II levels in WT and ACE2−/−/PRCP−/− mice were not significantly different from each other in the absence or with administration of ZPP. (B) Ang-(1-7) levels in WT mice with ZPP administration (WT+ZPP) were markedly reduced as compared to WT without ZPP (WT). In the ACE2−/−/PRCP−/− mice, ZPP administration also led to much lower Ang 1-7 levels as compared to ACE2−/−/PRCP−/− mice without ZPP (*p<0.05; **p<0.01). In these studies, blood was obtained from tail vein at 5 minutes and also at 30 minutes (see supplement) after Ang II.
In vivo formation of Ang-(1-7) after Ang II infusion in the face of pharmacological POP inhibition
In these studies, we used ZPP, an inhibitor of POP, given to WT and ACE2−/−/PRCP−/− mice infused with Ang II Plasma levels of Ang II at 5 minutes post infusion were markedly elevated and not significantly different from each other (1651 ± 379 and 1004 ± 169 fmol/ml respectively, p=0.3) (Figure 2a). The administration of ZPP largely prevented the elevated Ang-(1-7) levels seen at 5 minutes as compared to WT not infused with this inhibitor (186 ± 60, fmol/ml and 563 ± 51, respectively p=0.001) (Figure 2b). Similarly, in ACE2−/−/PRCP−/− mice, ZPP administration also resulted in markedly lower Ang-(1-7) plasma levels as compared to mice without ZPP infusion (50 ± 30 and 537 ± 70 fmol/ml respectively, p=0.0002) (Figure 2b). In WT and ACE2−/−/PRCP−/− mice treated with ZPP levels of Ang-(1-7) were reduced and not significantly different from each other (p=0.08).
By 30 minutes post infusion (see supplement Figure S3, panel a) the levels of plasma Ang II had markedly decreased in all four groups studied. Likewise, the levels of plasma Ang-(1-7) had decreased markedly in WT and WT treated with ZPP, as well as in ACE2−/−/PRCP−/−. The latest group had levels of Ang-(1-7) which were barely detectable (see supplement Figure S3, panel b).
Plasma Ang II and Ang-(1-7) levels in WT and POP−/− mice
Baseline Ang II levels in POP−/− mice were higher but not significantly different from WT mice (57.8 ± 14 and 31.1 ± 4.4 fmol/ml, respectively, p=ns) (Figure 3a). Plasma Ang-(1-7) levels in WT mice were significantly higher than in the POP−/− where the levels were not detectable (25.9 ± 11.3 and 0.0 fmol/ml p=0.032) (Figure 3b).
Figure 3: Studies in POPKO mice.

(A) Endogenous Ang II levels in plasma of POP−/− mice were higher but not significantly different from WT controls. (B) Endogenous plasma Ang-(1-7) levels in POP−/− mice were not detectable whereas in WT mice the levels were low, but detectable (p=0.032) (C) Ang II levels in plasma measured 5 minutes after i.p. injection of Ang II were significantly higher in POP−/− mice (n=8) than in WT mice (*p<0.05). (D) Ang-(1-7) levels in plasma also measured 5 minutes after i.p. injection of Ang II were lower in POP−/− mice than in WT mice(**p<0.01). Baseline levels were measured in blood obtained by cardiac puncture while post Ang II infusion blodd was obtained by tail snip.
(E) Systolic blood pressure (SBP) after single i.p. bolus injection of Ang II (time point “0”) under light ketamine anaesthesia WT and POP−/− mice (n=10 in each group). The SBP was recorded every 30 sec. for 15 minutes after Ang II injection. The slope of SBP decline was significantly slower in POP−/− than in WT (3.06±0.48 vs. 4.81±0.45 mmHg/min respectively, *p<0.05.)
In vivo formation of Ang-(1-7) after Ang II infusion in mice with genetic POP ablation
To confirm the effects of ZPP, we next examined Ang-(1-7) formation in POP−/− mice after Ang II infusion using the same protocol as in the sections above. In both WT and POP−/− mice plasma Ang II 5 minutes after its infusion was elevated and significantly higher in the POP−/− than in the WT mice (1120 ± 93 vs. 596 ± 234 fmol/ml, respectively p=0.027) (Figure 3c) . This suggests a major role of POP in the degradation of Ang II since in previous protocols the levels of Ang II after its infusion were not significantly different than those observed in the ACE2−/−/PRCP−/− mice. After Ang II infusion POP−/− mice exhibited lower plasma Ang-(1-7) levels than WT mice (309 ± 46 and 472 ± 28 fmol/ml, respectively, p<0.01) (Figure 3d). Plasma Ang-(1-7) to Ang II ratio after Ang II infusion was also markedly lower in the POP−/− as compared to the WT (0.23±0.02 vs.1.53±0.48 respectively, p=0.017). These findings show that genetic POP deficiency is associated with a significantly reduced ability to convert Ang II to Ang-(1-7) in plasma in vivo. This results in higher levels of Ang II and lower levels of Ang-(1-7) as compared to WT mice.
Blood Pressure Response to Ang II infusion in POP−/− mice
The effect of Ang II bolus injection (0.2 ug/g BW) on systolic blood pressure was examined in POP−/− and WT mice (n=10 in each group) under light anesthesia. Systolic BP peaked in the first minute after Ang II infusion and was not significantly different between POP−/− and WT mice (189±5 vs. 186±8 mmHg) (Figure 3e). The recovery from Ang II-induced acute hypertension was blunted in POP−/− as compared to WT mice (slope 3.06±0.48 vs. 4.81±0.45 mmHg/min respectively, p<0.05). This is consistent with higher levels of Ang II and lower levels of Ang-(1-7) found in POP−/− mice as compared to WT mice shown in (Figures 3c, 3d).
Ex vivo formation of Ang-(1-7) from Ang II in kidney and lung lysates
Since enzymes residing in highly perfused organs, such as kidneys and lungs, might also contribute to circulating Ang-(1-7) levels, ex vivo studies on Ang-(1-7) formation from exogenous Ang II were performed in lysates from those two organs. In WT kidneys (n=10) spiked with Ang II, Ang-(1-7) levels peaked at 15 minutes (Figure 4) As depicted in one example under methods (supplement Figure S2) by 15 minutes there is a rapid decline in Ang II such that by 60 minutes essentially all of the added Ang II has dissipated. ZPP had a minimal and not significant effect on Ang-(1-7) formation at 15 minutes as compared to WT kidneys not incubated with this inhibitor (417 ± 32 vs. 352 ± 75, pmol/ml, respectively p=0.3). To confirm the lack of effect of ZPP in the kidney tissue, we used another inhibitor of POP, S-17092 48,49. In WT kidney lysates, the latter inhibitor also did not reduce Ang-(1-7) formation significantly (peak at 15 min: 417 ± 32 vs. 385 ± 35 pmol/ml, p=ns).
Figure 4: Ex vivo Ang-(1-7) formation from Ang II in kidney and lung lysates as assessed by the peak levels of Ang-(1-7) at 15 minutes (see methods).

Experiments with each lysate were performed on two different occasions in duplicate.
A) In WT kidney lysates, the POP inhibitor, ZPP, did not affect Ang-(1-7) formation from Ang II significantly as compared to WT lysates without ZPP. In ACE2−/−/PRCP−/− kidney lysates Ang-(1-7) was markedly decreased. In ACE2−/−/PRCP−/− kidney lysates ZPP addition decreased Ang-(1-7) to essentially non-detectable levels.
B) In WT lung lysates, ZPP completely prevented Ang-(1-7) formation from Ang II (compare WT and WT+ZPP). In ACE2−/−/PRCP−/− lung lysates Ang-(1-7) levels were not reduced (actually, they were higher) and ZPP resulted in undetectable Ang-(1-7) levels.
C) In POP−/− kidney lysates, Ang-(1-7) formation from Ang II was not different from that of WT lysates. In contrast, in POP−/− kidney lysates an addition of MLN-4760, a specific inhibitor of ACE2, was associated with no detectable Ang-(1-7) formation from Ang II.
D) In POP−/− lung lysates Ang-(1-7) formation was completely prevented as compared to WT lung lysates and addition of MLN-4760 had no further effect.
In kidney lysates from ACE2−/−/PRCP−/− mice the peak Ang-(1-7) levels were much lower than in WT (94 ± 18 vs 417 ± 32 pmol/ml) (Figure 4a). Addition of a POP inhibitor, ZPP (n=7), reduced Ang-(1-7) to undetectable levels at all time points assessed (Figure 4a). In the aggregate, the data show that the formation of Ang-(1-7) in mouse kidneys is highly dependent on ACE2 and minimally dependent on POP.
In lung lysates from WT mice the Ang II decline was slower than with kidney lysates when equivalent amounts of both tissues were exposed to the peptide (See example in supplement, Figure S2). The levels of Ang-(1-7) after 15 minutes of incubation with Ang II, declined at 30 minutes and reached almost undetectable levels at 60 min. of incubation (Figure 4b). Generation of Ang-(1-7) in lung lysates from ACE2−/−/PRCP−/− mice was not significantly different from that of WT lung lysates (peak values at 15 min 313 ± 31 vs. 230 ± 33 pmol/ml, respectively p=0.09) (Figure 4b). Addition of ZPP completely prevented Ang-(1-7) formation from Ang II in lung lysates from both WT and ACE2−/−/PRCP−/− mice (Figure 4b). Using another POP-inhibitor, S-17092, the peak Ang-(1-7) formation at 15 minutes was also prevented entirely (3±3 vs 182.8±23, pmol/ml, respectively) (Supplement figure S4a). These findings show that in mouse lungs Ang-(1-7) formation from Ang II is essentially ACE2 independent and entirely prevented by POP inhibitors. We also measured for comparison the formation of Ang-(1-7) from exogenous Ang I in lung tissue (Supplement Figure S4b). Inhibition of POP with S-17092 significantly, but incompletely reduced the peak formation of Ang-(1-7). An inhibitor of Neprilysin (Thiorphan) also reduced the peak level of Ang-(1-7) from Ang I to an extent similar to S-17092. The combination of both inhibitors markedly reduced Ang-(1-7) formation (Supplement figure S4b).
To further determine the contribution of POP in the formation of Ang-(1-7) from Ang II we used kidney and lung tissues from POP−/− mice and their respective WT counterparts (Figure 4c and 4d). In kidneys from WT and POP−/− mice, Ang-(1-7) formation from Ang II over time was almost identical (Figure 4c). The specific ACE2 inhibitor, MLN-4760 (10−5M) completely obliterated generation of Ang-(1-7) from Ang II (Figure 4c). In contrast to kidney lysates, lung lysates from POP−/−mice showed almost undetectable Ang-(1-7) formation from Ang II (Figure 4d). Addition of MLN had no effect in the POP−/− lungs., In summary, in kidneys lysates Ang-(1-7) formation is predominantly ACE2-dependent whereas in lung lysates the Ang-(1-7) formation from Ang II is essentially ACE2-independent and entirely POP dependent.
Ex vivo formation of Ang-(1-7) from Ang II in mouse serum
To determine the contribution of POP to Ang-(1-7) generation in serum, Ang-(1-7) formation from added Ang II was studied ex vivo with (n=6) and without (n=6) addition of ZPP (10−5M) (Figure 5a). Incubation of the WT sera with Ang II resulted in formation of Ang-(1-7) that peaked at 15 minutes and was no longer detectable at 120 minutes of incubation (Figure 5a). At 15 minutes, ZPP lowered Ang-(1-7) formation by 85,8% (4.5 ± 4.5 vs 31.8±6.3 pmol/ml, p=0.01), and at 30 minutes by 72% (8.9 ± 4.5 vs 31.8 ± 6.7 pmol/ml, p=0.01), respectively.
Figure 5: Ex vivo formation of Ang-(1-7) from Ang II in serum.

Ex vivo Ang-(1-7) formation after incubation with Ang II in sera from A) WT mice, without ZPP and with ZPP and B) serum from from ACE2−/−/PRCP−/− mice and POP−/− mice.
A) In WT sera (n=6) in the presence of the dual POP/PRCP inhibitor, ZPP (10−5M), Ang-(1-7) formation from Ang II is markedly lower than in the absence of ZPP (p<0.01 by GLM statistics over time).
B) In sera from POP−/− mice (n=4), Ang-(1-7) formation from Ang II is markedly lower than in the sera from ACE2−/−/PRCP−/− mice (n=3) (p<0.01 by GLM statistics over time).
We next evaluated the contribution of ACE2, PRCP and POP to ex vivo conversion of Ang II to Ang-(1-7) in sera obtained from mice deficient in those enzymes. For this, Ang II was incubated with sera from the various KO models and the resultant formation of Ang-(1-7) was compared. Incubation of the sera from ACE2−/−/PRCP−/− mice with Ang II resulted in essentially the same formation of Ang-(1-7) as in WT mice (Figure 5b). There was no significant difference between WT mice (n=6) and ACE2−/−/PRCP−/− (n=3) at 30 minutes (32±6 vs 17±7, pmol/ml respectively, p=ns) or at 15 minutes (31.8±6.7 vs 17.8±6, pmol/ml respectively, p=ns), which is the peak of Ang 1-7 formation.
In contrast, the amount of Ang-(1-7) formed from Ang II by the sera from POP−/− mice, was markedly reduced compared to both ACE2−/−/PRCP−/− mice and WT mice (p<0.01) (Figure 5b).
ACE2 and POP proteins in WT sera, lung and kidneys.
By Western blot ACE2 protein was not detectable either in serum or in the lungs but was abundant in kidneys of WT mice. (Figure 6a). In contrast, POP protein was present in serum, lungs and kidney lysates (Figure 6a).
Figure 6: ACE2 and POP proteins in WT sera, lung and kidneys and the effect of rACE2 and rPOP on Ang-(1-7) and Phenylalanine formation.

A) ACE2 and POP proteins in WT sera, lung and kidneys by Western blot. ACE2 protein is abundant in kidneys, but not in serum or lungs. POP protein is present in serum, lungs and kidneys.
B) in vitro Ang II to Ang-(1-7) conversion assessed by equivalent amounts of recombinant (r)ACE2 and rPOP is higher with rACE2 than with rPOP (***p<0.001).
C) generation of free Phenylalanine (Phe) from Ang II as a substrate by equivalent amounts of recombinant (r)ACE2 and rPOP is higher with rACE2 than with rPOP (*** p<0.001).
For Western blot, different amounts of mouse recombinant protein standards were loaded to estimate ACE2 and POP protein expression levels in sera (1ul), lung and kidney lysates (50 ug total protein) from two WT mice.
In vitro Ang-(1-7) and Phenylalanine formation from Ang II
Conversion of Ang II to Ang-(1-7) is associated with a release of its carboxyterminal amino acid, Phenylalanine, that is directly proportional to the amount of the generated Ang-(1-7) 50. We assessed the reaction by both the formation of Ang-(1-7) (Figure 6b) and the Phe formation (Figure 6c) after incubating Ang II with equivalent amounts of murine recombinant (mr) POP and mrACE2 in vitro.
Murine rACE2 formed more Ang-(1-7) than mrPOP (5.3 ± 0.4 vs. 1.3 ± 0.6 pmol/ml, p<0.001) (Figure 6b). In agreement with this finding, Phenylalanine generation by mrACE2 was much higher than that formed by equivalent amount of mrPOP (789±29 RFU/ng mrACE2 vs. 127±12 RFU/ng mrPOP, p<0.001) (Figure 6c). These data show that rACE2 is intrinsically considerably more effective in catalyzing the conversion of Ang II to Ang-(1-7) than rPOP.
Discussion
The ACE2/Ang-(1-7) axis of the RAS has received increased attention and ACE2 is generally considered the main Ang-(1-7) forming enzyme from Ang II. There are two other known angiotensinases, prolyl carboxypeptidase (PRCP) and prolyl endopeptidase (POP), that have not been as extensively studied but could be as important as ACE2 in the processing of Ang II to Ang-(1-7) in body fluids, certain organs and cell types10,19,22,23,29-33. In the current study the conversion of Ang II to Ang-(1-7) by these enzymes was examined in vivo and ex vivo.
We found similarly high levels of Ang-(1-7) in plasma being generated following acute Ang II injections to WT mice as in mice with combined genetic ACE2 and PRCP deficiency. By contrast, reduced plasma Ang-(1-7) levels were found in mice with genetic POP deficiency infused with Ang II. Furthermore, the administration of ZPP, an inhibitor of PEP, markedly decreased plasma Ang-(1-7) formation from infused Ang II in WT and ACE2−/−/PRCP−/− mice. Our study, therefore, has demonstrated that the levels of Ang-(1-7) found in the circulation immediately after Ang II infusion are largely POP dependent and ACE2 and PRCP independent. It should be noted that Ang-(1-7) formation in vivo can originate from Ang I cleavage.34 As demonstrated in a study by Yamamoto et al. the formation of circulatory Ang-(1-7) from infused Ang I was entirely Neprilysin dependent.34 While it is important to note the there is direct formation of Ang-(1-7) from Ang I, in the present study, we are mainly concerned with the relative importance of enzymes that cleave Ang II to form Ang-(1-7).
The physiologic importance of POP in terms of systemic blood pressure was examined in a model of acute hypertension induced by Ang II infusion using the same dosing and timing as in the studies examining the conversion of Ang II to Ang-(1-7). In this model the blood pressure (BP) response was altered in mice with the genetic absence of POP as shown by a blunted recovery from Ang II-induced hypertension This can be attributed to the diminished Ang II degradation and Ang-(1-7) formation in the absence of POP. It is not possible to precisely discern the contribution of Ang II (pro-hypertensive) or Ang-(1-7) (anti-hypertensive) or both on the BP response in this model. Of note, in the POP−/− model the peak level of Ang II after its infusion was higher than in WT . By contrast, it was not significantly different between WT and ACE2−/−/PRCP−/− mice. Since previous studies have shown no effect or only a marginal effect of Ang-(1-7) on BP15,51 we surmise that in this model of acute Ang II-induced hypertension the sluggish recovery observed in POP−/− mice was largely attributable to decreased rate of Ang II degradation as compared to the WT after Ang II infusion . It should be noted that POP is also involved in the metabolism of other vasoactive peptides such as bradykinin. Therefore, in our POP−/− model the changes observed in blood pressure response might be influenced by RAS independent mechanisms. It should also be acknowledged that the i.p. route gives more time for extravascular processing of Ang II to Ang-(1-7) outside the circulation as compared to the i.v. route.
To gain further insight into where in the body the conversion of Ang II to Ang-(1-7) may take place after Ang II infusion we performed ex vivo studies in plasma, lung and kidney lysates. We found that POP protein was present in substantial amounts in mouse serum by western blot. ACE2, by contrast, was scarcely present in serum which is consistent with previous reports14,15,52,53. In sera from WT mice with the POP inhibitor, ZPP, formation of Ang-(1-7) from added Ang II, moreover, was markedly diminished. Consistent with this finding a marked attenuation of Ang-(1-7) formation was also observed in sera from mice with genetic POP deficiency. The presence of POP in plasma and its ability to form Ang-(1-7) demonstrated in these studies supports the role of this enzyme as a contributor to Ang-(1-7) formation in vivo. This does not mean, however, that POP resident in plasma accounts predominantly for the rapid conversion of Ang II to Ang-(1-7). POP could originate from the vascular endothelium and there is evidence that endothelial cells in culture have POP54. In addition, circulating cells such as macrophages that are very rich in this enzyme55 could contribute to the conversion of Ang II to Ang-(1-7) after Ang II infusion.
Another possibility that we think most likely is that POP in plasma comes from the lung circulation, similar to circulating plasma ACE56. This issue was partly addressed by examining Ang-(1-7) formation from exogenous Ang II in ex vivo studies using lung lysates. Our ex vivo studies provide compelling evidence for POP as the main Ang-(1-7) forming enzyme from Ang II in the lungs whereas ACE2 and PRCP have only a marginal if any effect in this organ. According to the Human Protein Atlas ACE2 protein expression was not detectable in human pneumocytes or in alveolar macrophages (https://www.proteinatlas.org/ENSG00000130234-ACE2/tissue). ACE2 mRNA expression was also low and mainly localized in pneumocytes and endothelial cells. Other studies show also low ACE2 mRNA and protein expression in rodent lungs57,58. Our results are therefore consistent with these previous studies showing low lung ACE2 activity.
In sharp contrast to the lungs, in mouse kidneys the conversion of Ang II to Ang-(1-7) was almost entirely ACE2-dependent. Kidney lysates from ACE2−/−/PRCP−/− showed markedly lower Ang-(1-7) generation from Ang II than those of WT kidneys. In POP−/− kidney lysates, which exhibited almost identical generation of Ang-(1-7) from Ang II as the WT kidneys, a very specific ACE2 inhibitor (MLN-4760) completely abrogated the formation of Ang-(1-7). These ex vivo studies overall show that ACE2 is responsible for the formation of Ang-(1-7) from Ang II in mouse kidney which is consistent with findings reported for human kidneys59. In addition, in our study ZPP did not reduce Ang-(1-7) formation from Ang II in kidney lysates from WT mice suggesting minimal, if any, effect of PRCP on Ang-(1-7) formation from Ang II by the kidney at a physiologic pH. One cannot exclude, however, the possibility that PRCP could play an important role in Ang II to Ang-(1-7) conversion in certain regions of the kidney where acidic conditions prevail, such as the lumen of the collecting duct. In fact, PRCP is abundant in the kidney collecting tubules, where the prevailing pH is low20.
The relative potency of each of the Ang-(1-7) forming enzymes and their kinetic properties, need to be taken into consideration together with the relative site abundance of each enzyme. An about 10 times higher catalytic constant for the conversion of Ang II to Ang-(1-7) for ACE2 than POP was previously reported by Chappell et al 60. Earlier studies by Ward et al. documented the kinetic parameters of POP-related Ang II conversion. In these studies, the calculated Km (5.7 ± 0.9 μM) and Vmax (1.70 ± 0.09 μmol*min−1*mg−1) values for POP with Ang II as a substrate61 allowed us an approximation of POP catalytic efficiency (kcat/Km) to be ~5-10 times lower than those reported for human ACE24,5.Using two independent methods: Ang-(1-7) formation and Phenylalanine generation50 (which is a cleavage by-product of Ang II to Ang-(1-7) conversion)50 we found, moreover, that ACE2 is much more efficient than POP which is consistent with Chappell’s et al findings60. We show that both enzymes are present in the kidney by western blot but the higher potency of ACE2 confers much greater capacity for Ang-(1-7) formation from Ang II in this organ. Moreover, one has to take into account the subcellular localization of the enzymes. POP is mainly a cytosolic enzyme but also has been reported to have membrane-bound isoforms62,63. As a limitation of this study, we wish to point out that the doses of Ang II used to demonstrate Ang-(1-7) generation are very high relative to the levels of Ang II normally present in plasma. Peptidases, however, are most efficient at their Km values and the dose of ANG II used in our studies was only twice the Km for POP and ACE2 While the balance between these two peptidases in the metabolism of Ang II that we have demonstrated under conditions of high Ang II levels, both in vivo and ex vivo, may not mimic the physiologic levels of Ang II, the doses of this peptide used are appropriate to demonstrate Ang (1-7) formation. The fact that in the POP KO the levels of Ang II were higher than in the corresponding WT controls supports, moreover, the importance of POP in the metabolism of this peptide in vivo. In contrast, the baseline levels of Ang II were not higher in ACE2 KO mice as compared to wild type64.
Our study did not address the possible function of POP within the kidney or the lungs. Since the conversion of Ang II to Ang-(1-7) within the kidney is largely POP-independent, it is possible that this enzyme exerts kidney actions unrelated to this conversion. For instance, POP has been reported to foster anti-fibrotic processes in kidney tissue by releasing N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) from thymosin-β465. By contrast, POP is the main Ang-(1-7) forming enzyme in the lungs and moreover the lung circulation is possibly a major source of POP protein for conversion of Ang II to Ang-(1-7) in the circulation. Myöhänen et al. reported that POP can be localized in several human peripheral tissues including kidneys and lungs66. Further studies ideally using human tissues and human plasma are needed to elucidate the physiologic function of the POP/Ang-(1-7) axis. Our findings show that conversion of Ang II to Ang-(1-7) in the circulation and lungs is essentially POP-dependent and ACE2-independent. In contrast, this reaction in the kidney is predominantly mediated by ACE2. These findings may have important implications for therapeutic purposes when targeting Ang-(1-7) formation from Ang II in the circulation and specific organs. Future studies are also needed to fully evaluate the kidney and cardiovascular phenotype of POP−/− mice.
Perspectives
There are at least three enzymes known to form Ang-(1-7) directly from AngII but their relative importance in the circulation and various organs is not known .This is the first study to demonstrate that conversion of Ang II to Ang-(1-7) in the circulation and lungs is essentially POP-dependent and ACE2-independent. In contrast, this conversion in the kidney is predominantly mediated by ACE2. Deficiency of POP by slowing down the dissipation of Ang II in the circulation may predispose to hypertension. Our findings therefore may have important implications for therapeutic purposes when targeting Ang-(1-7) formation from Ang II in the circulation and specific organs.
Supplementary Material
Novelty and Significance.
What Is New?
The first study to characterize ACE2 and PRCP independent Ang-(1-7) formation from Ang II in circulation and peripheral tissues using ACE2−/−/PRCP−/− and POP−/− genetic mouse models and pharmacological inhibitors
What Is Relevant?
Circulatory and pulmonary Ang-(1-7) formation from exogenous Ang II is essentially ACE2 independent and POP dependent while in kidneys the conversion is mainly dependent on ACE2
Blood pressure recovery from acute Ang II induced hypertension is delayed in POP deficient mice
Acknowledgments
Sources of funding
This work was supported by National Institute of Diabetes and Digestive Kidney Diseases grants R01DK080089 and R01DK104785 as well as by a gift by the Joseph and Bessie Feinberg Foundation (DB). P. Serfozo and G. Gulua received stipends from BMEP in support of their research in Chicago.
Footnotes
Disclosures
Drs. Batlle and Wysocki are co-inventors of patent 10,443,049
Dr. Batlle is founder/owner of Angiotensin Therapeutics Inc.
References:
- 1.Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 2000;275:33238–43. [DOI] [PubMed] [Google Scholar]
- 2.Donoghue M, Hsieh F, Baronas E, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res 2000;87:E1–9. [DOI] [PubMed] [Google Scholar]
- 3.Crackower MA, Sarao R, Oudit GY, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002;417:822–8. [DOI] [PubMed] [Google Scholar]
- 4.Vickers C, Hales P, Kaushik V, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem 2002;277:14838–43. [DOI] [PubMed] [Google Scholar]
- 5.Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. The Biochemical journal 2004;383:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrario CM, Brosnihan KB, Diz DI, et al. Angiotensin-(1-7): a new hormone of the angiotensin system. Hypertension 1991;18:III126–33. [DOI] [PubMed] [Google Scholar]
- 7.Ferrario CM, Jessup J, Gallagher PE, et al. Effects of renin-angiotensin system blockade on renal angiotensin-(1-7) forming enzymes and receptors. Kidney Int 2005;68:2189–96. [DOI] [PubMed] [Google Scholar]
- 8.Chappell MC, Pirro NT, Sykes A, Ferrario CM. Metabolism of angiotensin-(1-7) by angiotensin-converting enzyme. Hypertension 1998;31:362–7. [DOI] [PubMed] [Google Scholar]
- 9.Yamada K, Iyer SN, Chappell MC, Ganten D, Ferrario CM. Converting enzyme determines plasma clearance of angiotensin-(1-7). Hypertension 1998;32:496–502. [DOI] [PubMed] [Google Scholar]
- 10.Welches WR, Santos RA, Chappell MC, Brosnihan KB, Greene LJ, Ferrario CM. Evidence that prolyl endopeptidase participates in the processing of brain angiotensin. Journal of hypertension 1991;9:631–8. [DOI] [PubMed] [Google Scholar]
- 11.Santos RA, Simoes e Silva AC, Maric C, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proceedings of the National Academy of Sciences of the United States of America 2003;100:8258–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Etelvino GM, Peluso AA, Santos RA. New components of the renin-angiotensin system: alamandine and the MAS-related G protein-coupled receptor D. Current hypertension reports 2014;16:433. [DOI] [PubMed] [Google Scholar]
- 13.Ye M, Wysocki J, William J, Soler MJ, Cokic I, Batlle D. Glomerular localization and expression of Angiotensin-converting enzyme 2 and Angiotensin-converting enzyme: implications for albuminuria in diabetes. J Am Soc Nephrol 2006;17:3067–75. [DOI] [PubMed] [Google Scholar]
- 14.Ye M, Wysocki J, Gonzalez-Pacheco FR, et al. Murine recombinant angiotensin-converting enzyme 2: effect on angiotensin II-dependent hypertension and distinctive angiotensin-converting enzyme 2 inhibitor characteristics on rodent and human angiotensin-converting enzyme 2. Hypertension 2012;60:730–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wysocki J, Ye M, Rodriguez E, et al. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension 2010;55:90–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wysocki J, Ye M, Batlle D. Plasma and Kidney Angiotensin Peptides: Importance of the Aminopeptidase A/Angiotensin III Axis. Am J Hypertens 2015;28:1418–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wysocki J, Ye M, Khattab AM, et al. Angiotensin-converting enzyme 2 amplification limited to the circulation does not protect mice from development of diabetic nephropathy. Kidney Int 2017;91:1336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haber PK, Ye M, Wysocki J, Maier C, Haque SK, Batlle D. Angiotensin-converting enzyme 2-independent action of presumed angiotensin-converting enzyme 2 activators: studies in vivo, ex vivo, and in vitro. Hypertension 2014;63:774–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grobe N, Weir NM, Leiva O, et al. Identification of prolyl carboxypeptidase as an alternative enzyme for processing of renal angiotensin II using mass spectrometry. American journal of physiology Cell physiology 2013;304:C945–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maier C, Schadock I, Haber PK, et al. Prolylcarboxypeptidase deficiency is associated with increased blood pressure, glomerular lesions, and cardiac dysfunction independent of altered circulating and cardiac angiotensin II. J Mol Med (Berl) 2017;95:473–86. [DOI] [PubMed] [Google Scholar]
- 21.Grobe N, Elased KM. Analysis of Angiotensin Metabolism in the Kidney Using Mass Spectrometry. Methods Mol Biol 2017;1614:189–97. [DOI] [PubMed] [Google Scholar]
- 22.Gass J, Khosla C. Prolyl endopeptidases. Cell Mol Life Sci 2007;64:345–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Velez JC, Ierardi JL, Bland AM, et al. Enzymatic processing of angiotensin peptides by human glomerular endothelial cells. Am J Physiol Renal Physiol 2012;302:F1583–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zimmerman D, Burns KD. Angiotensin-(1-7) in kidney disease: a review of the controversies. Clin Sci (Lond) 2012;123:333–46. [DOI] [PubMed] [Google Scholar]
- 25.Li N, Zimpelmann J, Cheng K, Wilkins JA, Burns KD. The role of angiotensin converting enzyme 2 in the generation of angiotensin 1-7 by rat proximal tubules. Am J Physiol Renal Physiol 2005;288:F353–62. [DOI] [PubMed] [Google Scholar]
- 26.Odya CE, Marinkovic DV, Hammon KJ, Stewart TA, Erdos EG. Purification and properties of prolylcarboxypeptidase (angiotensinase C) from human kidney. J Biol Chem 1978;253:5927–31. [PubMed] [Google Scholar]
- 27.Watson B Jr., Nowak NJ, Myracle AD, Shows TB, Warnock DG. The human angiotensinase C gene (HUMPCP) maps to 11q14 within 700 kb of D11S901: a candidate gene for essential hypertension. Genomics 1997;44:365–7. [DOI] [PubMed] [Google Scholar]
- 28.Wallingford N, Perroud B, Gao Q, et al. Prolylcarboxypeptidase regulates food intake by inactivating alpha-MSH in rodents. The Journal of clinical investigation 2009;119:2291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greene LJ, Spadaro AC, Martins AR, Perussi De Jesus WD, Camargo AC. Brain endo-oligopeptidase B: a post-proline cleaving enzyme that inactivates angiotensin I and II. Hypertension 1982;4:178–84. [DOI] [PubMed] [Google Scholar]
- 30.Brandt I, Scharpe S, Lambeir AM. Suggested functions for prolyl oligopeptidase: a puzzling paradox. Clin Chim Acta 2007;377:50–61. [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Horsman JA, Mannisto PT, Venalainen JI. On the role of prolyl oligopeptidase in health and disease. Neuropeptides 2007;41:1–24. [DOI] [PubMed] [Google Scholar]
- 32.Polgar L pH-dependent mechanism in the catalysis of prolyl endopeptidase from pig muscle. Eur J Biochem 1991;197:441–7. [DOI] [PubMed] [Google Scholar]
- 33.Lawandi J, Gerber-Lemaire S, Juillerat-Jeanneret L, Moitessier N. Inhibitors of prolyl oligopeptidases for the therapy of human diseases: defining diseases and inhibitors. J Med Chem 2010;53:3423–38. [DOI] [PubMed] [Google Scholar]
- 34.Yamamoto K, Chappell MC, Brosnihan KB, Ferrario CM. In vivo metabolism of angiotensin I by neutral endopeptidase (EC 3.4.24.11) in spontaneously hypertensive rats. Hypertension 1992;19:692–6. [DOI] [PubMed] [Google Scholar]
- 35.Shariat-Madar Z, Mahdi F, Schmaier AH. Identification and characterization of prolylcarboxypeptidase as an endothelial cell prekallikrein activator. J Biol Chem 2002;277:17962–9. [DOI] [PubMed] [Google Scholar]
- 36.Fulop V, Bocskei Z, Polgar L. Prolyl oligopeptidase: an unusual beta-propeller domain regulates proteolysis. Cell 1998;94:161–70. [DOI] [PubMed] [Google Scholar]
- 37.Welches WR, Brosnihan KB, Ferrario CM. A comparison of the properties and enzymatic activities of three angiotensin processing enzymes: angiotensin converting enzyme, prolyl endopeptidase and neutral endopeptidase 24.11. Life sciences 1993;52:1461–80. [DOI] [PubMed] [Google Scholar]
- 38.Goossens F, De Meester I, Vanhoof G, Scharpe S. Distribution of prolyl oligopeptidase in human peripheral tissues and body fluids. Eur J Clin Chem Clin Biochem 1996;34:17–22. [DOI] [PubMed] [Google Scholar]
- 39.Schulz I, Zeitschel U, Rudolph T, et al. Subcellular localization suggests novel functions for prolyl endopeptidase in protein secretion. Journal of neurochemistry 2005;94:970–9. [DOI] [PubMed] [Google Scholar]
- 40.O’Leary RM, Gallagher SP, O’Connor B. Purification and characterization of a novel membrane-bound form of prolyl endopeptidase from bovine brain. The international journal of biochemistry & cell biology 1996;28:441–9. [DOI] [PubMed] [Google Scholar]
- 41.Brandt I, Gerard M, Sergeant K, et al. Prolyl oligopeptidase stimulates the aggregation of alpha-synuclein. Peptides 2008;29:1472–8. [DOI] [PubMed] [Google Scholar]
- 42.Gaggar A, Jackson PL, Noerager BD, et al. A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. Journal of immunology (Baltimore, Md : 1950) 2008;180:5662–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Reilly PJ, Hardison MT, Jackson PL, et al. Neutrophils contain prolyl endopeptidase and generate the chemotactic peptide, PGP, from collagen. J Neuroimmunol 2009;217:51–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cunningham DF, O’Connor B. A study of prolyl endopeptidase in bovine serum and its relevance to the tissue enzyme. The international journal of biochemistry & cell biology 1998;30:99–114. [DOI] [PubMed] [Google Scholar]
- 45.Tenorio-Laranga J, Montoliu C, Urios A, et al. The expression levels of prolyl oligopeptidase responds not only to neuroinflammation but also to systemic inflammation upon liver failure in rat models and cirrhotic patients. J Neuroinflammation 2015;12:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brosnihan KB, Chappell MC. Measurement of Angiotensin Peptides: HPLC-RIA. Methods Mol Biol 2017;1527:81–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shariat-Madar Z, Mahdi F, Schmaier AH. Recombinant prolylcarboxypeptidase activates plasma prekallikrein. Blood 2004;103:4554. [DOI] [PubMed] [Google Scholar]
- 48.Barelli H, Petit A, Hirsch E, et al. S 17092-1, a Highly Potent, Specific and Cell Permeant Inhibitor of Human Proline Endopeptidase. Biochemical and biophysical research communications 1999;257:657–61. [DOI] [PubMed] [Google Scholar]
- 49.Bellemère G, Morain P, Vaudry H, Jégou S. Effect of S 17092, a novel prolyl endopeptidase inhibitor, on substance P and α-melanocyte-stimulating hormone breakdown in the rat brain. Journal of neurochemistry 2003;84:919–29. [DOI] [PubMed] [Google Scholar]
- 50.Liu P, Wysocki J, Serfozo P, et al. A Fluorometric Method of Measuring Carboxypeptidase Activities for Angiotensin II and Apelin 13. Scientific Reports 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Varagic J, Ahmad S, VonCannon JL, et al. Predominance of AT(1) blockade over mas-mediated angiotensin-(1-7) mechanisms in the regulation of blood pressure and renin-angiotensin system in mRen2.Lewis rats. Am J Hypertens 2013;26:583–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rice GI, Jones AL, Grant PJ, Carter AM, Turner AJ, Hooper NM. Circulating activities of angiotensin-converting enzyme, its homolog, angiotensin-converting enzyme 2, and neprilysin in a family study. Hypertension 2006;48:914–20. [DOI] [PubMed] [Google Scholar]
- 53.Wysocki J, Ye M, Soler MJ, et al. ACE and ACE2 activity in diabetic mice. Diabetes 2006;55:2132–9. [DOI] [PubMed] [Google Scholar]
- 54.Myohanen TT, Tenorio-Laranga J, Jokinen B, et al. Prolyl oligopeptidase induces angiogenesis both in vitro and in vivo in a novel regulatory manner. British journal of pharmacology 2011;163:1666–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Olivo Rdo A, Teixeira Cde F, Silveira PF. Representative aminopeptidases and prolyl endopeptidase from murine macrophages: comparative activity levels in resident and elicited cells. Biochem Pharmacol 2005;69:1441–50. [DOI] [PubMed] [Google Scholar]
- 56.Hodge RL, Ng KK, Vane JR. Disappearance of angiotensin from the circulation of the dog. Nature 1967;215:138–41. [DOI] [PubMed] [Google Scholar]
- 57.Gembardt F, Sterner-Kock A, Imboden H, et al. Organ-specific distribution of ACE2 mRNA and correlating peptidase activity in rodents. Peptides 2005;26:1270–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wiener RS, Cao YX, Hinds A, Ramirez MI, Williams MC. Angiotensin converting enzyme 2 is primarily epithelial and is developmentally regulated in the mouse lung. J Cell Biochem 2007;101:1278–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Domenig O, Manzel A, Grobe N, et al. Neprilysin is a Mediator of Alternative Renin-Angiotensin-System Activation in the Murine and Human Kidney. Sci Rep 2016;6:33678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chappell MC, Marshall AC, Alzayadneh EM, Shaltout HA, Diz DI. Update on the Angiotensin converting enzyme 2-Angiotensin (1-7)-MAS receptor axis: fetal programing, sex differences, and intracellular pathways. Frontiers in endocrinology 2014;4:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ward PE, Bausback HH, Odya CE. Kinin and angiotensin metabolism by purified renal post-proline cleaving enzyme. Biochem Pharmacol 1987;36:3187–93. [DOI] [PubMed] [Google Scholar]
- 62.Tenorio-Laranga J, Venalainen JI, Mannisto PT, Garcia-Horsman JA. Characterization of membrane-bound prolyl endopeptidase from brain. FEBS J 2008;275:4415–27. [DOI] [PubMed] [Google Scholar]
- 63.Myohanen TT, Venalainen JI, Garcia-Horsman JA, Piltonen M, Mannisto PT. Cellular and subcellular distribution of rat brain prolyl oligopeptidase and its association with specific neuronal neurotransmitters. J Comp Neurol 2008;507:1694–708. [DOI] [PubMed] [Google Scholar]
- 64.Wysocki J, Ortiz-Melo DI, Mattocks NK, et al. ACE2 deficiency increases NADPH-mediated oxidative stress in the kidney. Physiological reports 2014;2:e00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zuo Y, Chun B, Potthoff SA, et al. Thymosin beta4 and its degradation product, Ac-SDKP, are novel reparative factors in renal fibrosis. Kidney Int 2013;84:1166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Myohanen TT, Pyykko E, Mannisto PT, Carpen O. Distribution of prolyl oligopeptidase in human peripheral tissues and in ovarian and colorectal tumors. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society 2012;60:706–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.