

Abstract
Cancer is driven by incremental changes that accumulate, eventually leading to oncogenic transformation. Although genetic alterations dominate the way cancer biologists think about oncogenesis, growing evidence suggests that systemic factors (for example, insulin, oestrogen and inflammatory cytokines) and their intracellular pathways activate oncogenic signals and contribute to targetable phenotypes. Systemic factors can have a critical role in both tumour initiation and therapeutic responses as increasingly targeted and personalized therapeutic regimens are used to treat patients with cancer. The endocrine system controls cell growth and metabolism by providing extracellular cues that integrate systemic nutrient status with cellular activities such as proliferation and survival via the production of metabolites and hormones such as insulin. When insulin binds to its receptor, it initiates a seguence of phosphorylation events that lead to activation of the catalytic activity of phosphoinositide 3-kinase (PI3K), a lipid kinase that coordinates the intake and utilization of glucose, and mTOR, a kinase downstream of PI3K that stimulates transcription and translation. When chronically activated, the PI3K pathway can drive malignant transformation. Here, we discuss the insulin–PI3K signalling cascade and emphasize its roles in normal cells (including coordinating cell metabolism and growth), highlighting the features of this network that make it ideal for co-option by cancer cells. Furthermore, we discuss how this signalling network can affect therapeutic responses and how novel metabolic-based strategies might enhance treatment efficacy for cancer.
The discovery of insulin was initiated when Banting and Best first demonstrated that a pancreatic extract could alleviate diabetic symptoms in canines1. As their work progressed, they confirmed these effects in several mammalian species, including humans; administration of this extract to patients with type 1 diabetes mellitus reduced blood levels of sugar, abolished glycosuria, prevented ketonuria, increased carbohydrate utilization and improved their overall condition2. Since the time of Banting and Best’s initial publications, our knowledge about insulin has expanded considerably. We now know that the insulin signalling pathway is highly conserved through evolution, even pre-dating vertebrates3. In these early species, the precursors of insulin integrated nutrient availability with cell survival and growth4. This function is preserved in later organisms, where insulin acts as a signalling molecule to regulate glucose and amino acid uptake, ultimately controlling systemic metabolic homeostasis. Although these functions are central to our understanding of the activity of insulin at the whole-body level, it is critical to recognize that insulin is also a local signal that connects systemic nutrient status with cellular metabolism, growth and proliferation5,6. These insulin-regulated pathways are frequently mutated in tumour cells, which must transport glucose and amino acids to maintain their hyperproliferative state independent of the nutrient status of the organism5,6. In this regard, tumour cells are able to excise themselves from the normal systemic endocrine regulation that regulates cell growth, proliferation and survival. This Review examines the connections between insulin signalling and oncogenic transformation, highlighting the potential effect of insulin as a pro-tumorigenic factor. Building on this argument, the Review discusses the evolutionarily entrenched feedback mechanisms through which systemic insulin feedback affects the efficacy of therapeutics that target the phosphoinositide 3-kinase (PI3K) signalling cascade and highlights the latest studies examining new approaches to circumvent systemic insulin feedback to increase the antitumour effect of these agents.
Insulin–PI3K signalling axis
Insulin production.
Insulin is secreted by pancreatic β-cells in response to increases in blood levels of glucose. Once secreted, insulin enters the bloodstream and circulates until it binds to a cell surface receptor, the insulin receptor. This extracellular interaction activates the protein–tyrosine kinase domain of the insulin receptor, which allows it to catalyse the phosphorylation of the insulin receptor substrate proteins 1 and 2 (IRS1 and IRS2). This biochemical change propagates the signal to multiple signalling pathways within cells, including both the PI3K and mitogen-activated protein kinase (MAPK) signalling pathways7–11 (FIG. 1). The result of this signalling is a variety of cellular responses, including regulation of hypoxia inducible factor, cyclin D1 and Myc (proteins that inhibit cell death and activate cellular survival and proliferation programmes)12,13 and, most importantly, the shuttling of transporters to the cell membrane to increase glucose uptake into cells.
Fig. 1 |. Insulin regulates glycolysis through both AKT-dependent and AKT-independent mechanisms.
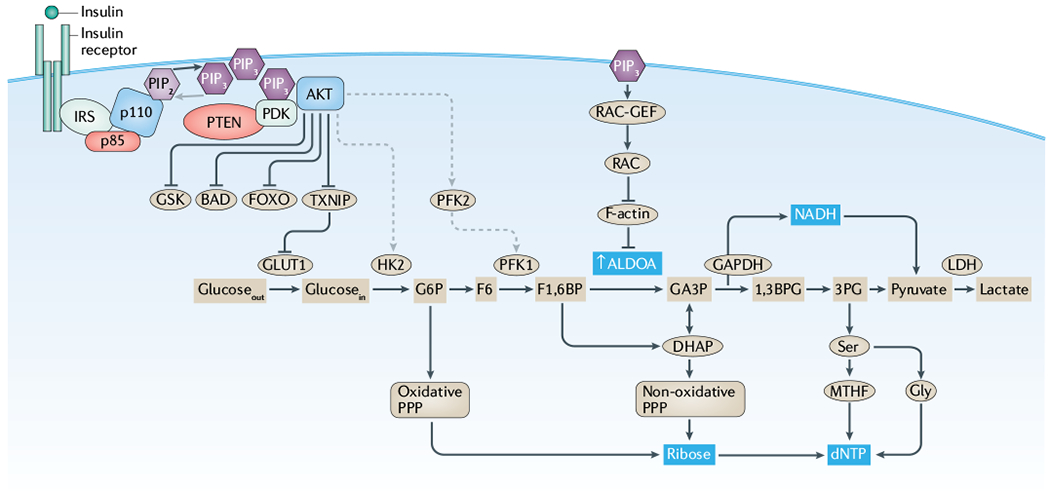
This simplified figure highlights components of the phosphoinositide 3-kinase (PI3K)-regulated cellular signalling events that occur downstream of the interaction between insulin and its receptor. While a myriad of growth factors and their cognate receptors (such as insulin growth factor or epidermal growth factor) activate PI3K signalling, here we highlight the insulin receptor, which is of particular importance to the systemic feedbacks central to this Review. The effects of insulin are mediated through both AKT-dependent (left side) and AKT-independent mechanisms (right side). From an evolutionary perspective, systemic insulin drives the intracellular PI3K signalling pathway to integrate systemic nutrient status and cellular metabolism with cellular functions, including proliferation and survival. This function enables insulin to coordinate cellular activities with systemic features, such as glucose availability, and provides a unified mechanism for the generation and utilization of key metabolites, such as deoxyribonucleotide triphosphates (dNTPs). Under normal conditions, this integration of systemic and cellular activity protects the organism by linking metabolic requirements and cellular growth, thereby constraining cellular activities based on systemic factors. In the context of oncogenic transformation, this integration makes the insulin–PI3K signalling axis a vulnerability, as activating mutations in oncogenes (encoding p110 and AKT) or inactivating mutations in tumour suppressors (encoding p85 and phosphatase and tensin homologue (PTEN)) can decouple this integration. IRS, insulin receptor substrate; LDH, lactate dehydrogenase; MTHF, 5,10-methenyltetrahydrofolate; PDK, phosphoinositide-dependent kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PPP, pentose phosphate pathway.
Function.
Although the insulin–PI3K signalling axis is highly evolutionarily conserved14 and functions in all of the tissues of the human body, it has evolved so that the effects of insulin binding to its receptor elicit responses that are specific to the cell and tissue. For example, in adipose and skeletal muscle cells, activation of the insulin receptor stimulates glucose uptake and promotes glycolysis, whereas in the liver the primary function of activation of the insulin receptor is to retard gluconeogenesis and enhance glycogen storage15,16. Thus, at the level of the human organism, insulin signalling is a critical component of metabolic homeostasis, balancing the storage and production of glucose17 (FIG. 2).
Fig. 2 |. Tissue and tumour response to insulin signalling.
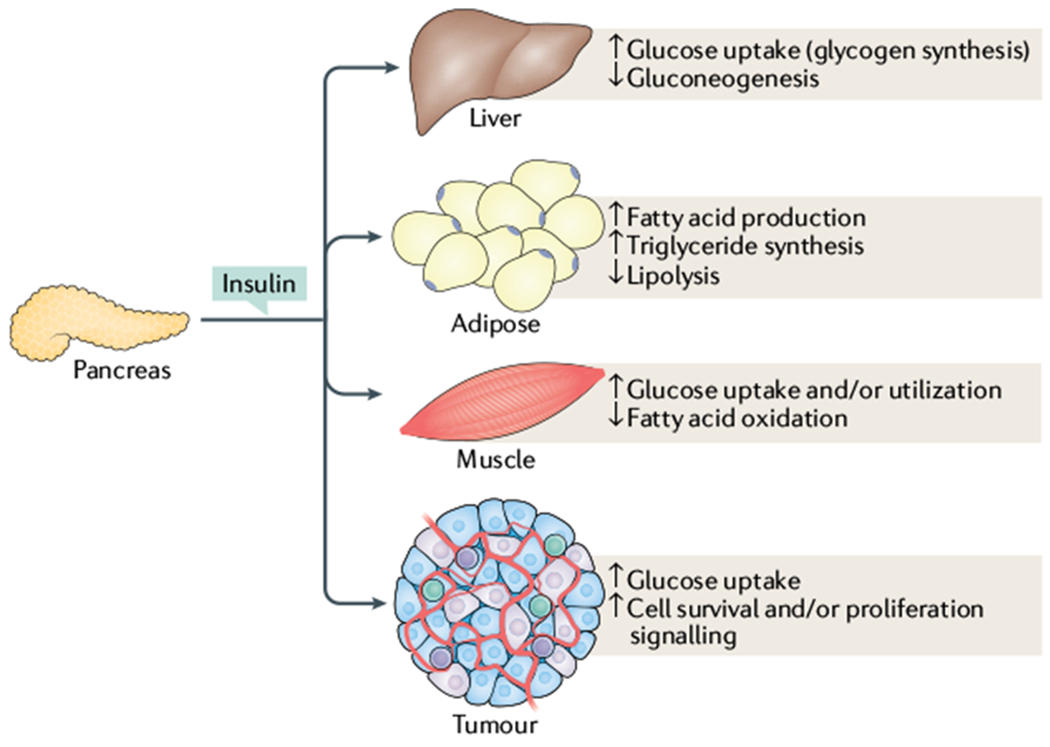
While canonical insulin signalling remains intact across tissue types, activation of the insulin receptor has tissue-specific effects. In muscle, insulin stimulates fatty acid oxidation as well as glucose uptake and utilization. By contrast, in adipose tissues and liver, insulin stimulates a storage phenotype, driving fatty acid production, inhibiting lipolysis in adipose tissue and inducing glucose uptake while suppressing gluconeogenesis in the liver. Similarly, tumours can use insulin signalling to drive glucose uptake as well as inducing cell survival and proliferation.
In 1976, Rinderknecht and Humbel isolated two chemicals from human serum that structurally resembled insulin and exhibited non-suppressible insulin-like activity18. These substances were named insulin-like growth factor 1 and 2 (IGF1 and IGF2). Both IGFs and insulin act as ligands for their respective receptor tyrosine kinases (insulin receptor and IGF1 receptor (IGF1R))11. It is difficult to functionally isolate the effect of the ligands at the cellular level because of the high levels of structural homology between the IGFs and insulin, the ability of these ligands to cross-activate each other’s receptors and the observation that their receptors form the full complement of homodimers and heterodimers with one another12. Although insulin and the IGF ligands share structural homology, the regulation of these ligands is distinct19. Insulin is secreted by the pancreas when blood levels of glucose increase and therefore acts as an acute signal to cells indicating a change in systemic nutrient status. By contrast, IGFs are not subject to such acute regulation. The production of IGF1 is controlled mostly by the action of growth hormone on the liver. It is then further regulated by a series of binding proteins (such as IGF-binding protein 3) that sequester IGF in the blood, thus limiting its ability to activate the IGF1R19,20.
Intracellular signalling.
Once the IGFs or insulin have activated the cell surface receptor tyrosine kinases, the resulting intracellular signalling events are similar. In general, for this family of receptors, a ligand binding to a receptor leads to kinase activation of the receptor and autophosphorylation. This change enhances the binding affinity of the receptor to the IRS proteins, which are then phosphorylated. The phosphorylated IRS proteins can then interact with the regulatory subunit of PI3K (that is, p85), p85 then inhibits the catalytic subunit (p110) of PI3K, which allows it to phosphorylate phosphatidylinositol 4,5-bisphosphate (PIP2), a lipid that is located on the cell surface5. The product of this reaction is a critical second messenger, phosphatidylinositol 3,4,5-trisphosphate (PIP3). The newly formed PIP3 recruits phosphoinositide-dependent kinase 1 (PDK1), AKT and other proteins that contain PH domains to the cell membrane, where they propagate the signal through a series of serine/threonine kinases, tyrosine kinases and G-proteins (FIG. 1). Within cells, the signalling cascades serve to activate vesicle trafficking, cell migration, actin remodelling and the regulation of cell growth5.
In addition to insulin and IGFs, other hormones might alter the function of PI3K. For example, the oestrogen and thyroid hormone receptors might form cytoplasmic complexes with p85, which enhances PI3K signalling following ligand binding21,22. Conversely, catecholamines such as adrenaline (also known as epinephrine) might inhibit PI3K activity through an unclear mechanism23,24. These examples highlight the importance of PI3K as an integrator of the systemic hormonal milieu.
AKT.
The most studied and seemingly critical factor downstream of PI3K is the serine/threonine kinase AKT. AKT phosphorylates a myriad of targets, including GSK3, TXNIP, BAD, AS160 and FOXO. Phosphorylation of FOXO proteins leads to their exclusion from the nucleus, which prevents them from acting as transcriptional regulators (such as for the transcription factor FOXOl)4. Phosphorylation by AKT also leads to the inactivation of GSK3, which in turn regulates a variety of processes, including glycogen storage14. Similarly, AKT regulates the activity of TXNIP, which controls the internalization of glucose transporters, thereby allowing the transporters to remain at the cell surface and regulate the acute influx of glucose downstream of insulin stimulation14. Insulin also has numerous AKT-independent effects (FIG. 1). For example, PDK1 has a number of other cellular targets, such as Polo-like kinase 1 (PLK1), which it phosphorylates to allow cell cycle progression25,26. In addition, insulin stimulates RAC activity to induce a dramatic rearrangement of the cellular cytoskeleton27,28. This structural rearrangement puts the cell in a physically permissive state for migration or cell division, and releases actin-bound aldolase to enhance glycolysis and ATP production27.
mTOR.
The activation of the PI3K and MAPK pathways leads to activation of mTOR, a protein kinase that controls cell growth by regulating protein and nucleotide synthesis. mTOR complex 1 (mTORC1), a complex of mTOR with RAPTOR, regulates S6 kinase and 4EBP1 (which regulate translation) and is inhibited by rapalogues such as everolimus, which was approved by the FDA for the treatment of renal cell carcinoma, subependymal giant cell astrocytoma and progressive neuroendocrine tumours of pancreatic origin. Ultimately, the insulin-induced growth signal needs to be abolished for the successful treatment of some cancers. This ‘off-switch’ occurs mostly through the removal of the 3′-phosphate of PIP3 by phosphatase and tensin homologue (PTEN). PTEN is a bona fide tumour suppressor whose activity is tightly regulated in a number of different ways, including protein–protein interactions, alternative translation initiation, microRNAs and post-translational modifications such as phosphorylation, acetylation, oxidation and ubiquitylation29–33. Numerous other negative feedback loops have evolved at both the systemic and cellular levels that are responsible for bringing PI3K signalling back to homeostasis. For example, hyperactivation of mTORCl in response to chronic insulin stimulation results in stabilization of GRB10, an adaptor protein that can sequester the insulin receptor and dampen PI3K signalling34. Interestingly, the GRB10 gene has been identified as a risk allele for type 2 diabetes mellitus35, which highlights how dysregulation of these feedback loops alters the balance of PI3K activity and leads to clinical disease36.
Regulation of cellular growth and metabolism.
The insulin–PI3K signalling axis has a fundamental role in human cellular growth and metabolism5. The combination of p110 and p85 is an example of a class 1A PI3K; this class is comprised of heterodimers made up of a catalytic subunit (either p110α, p110β or p110δ) and a regulatory subunit (p85α, p85β, p55α, p50α or p55γ). In the context of insulin signalling, the most critical isoform is p110α (encoded by PIK3CA) because it is thought to be the dominant isoform downstream of the insulin receptor13. Activating mutations in PIK3CA are associated with human diseases, including overgrowth syndromes (such as CLOVES syndrome) and malignancies including uterine, breast, head and neck, and colon cancers14. In fact, PIK3CA is the most frequently mutated oncogene in human cancers14. Conversely, a reduction in PI3K activity is found in patients with SHORT syndrome, a rare condition that features mutations in PIK3R1 (which encodes p85); the syndrome is usually diagnosed in early childhood and is characterized by delayed growth and features resembling type 1 diabetes mellitus37. Mouse models that feature knockout of components of PI3K signalling have supported the importance of PI3K in growth and metabolism. For instance, germline and/or tissue-specific manipulation of Pik3ca, Pik3r1 and Pik3r2 (the latter encodes p85β) have confirmed the role of insulin–PI3K signalling in the liver, skeletal muscle and adipose tissue as well as in the control of systemic glucose homeostasis38–44.
Maintaining homeostasis.
Together, the observations from human syndromes and mouse models allow us to support a model in which signalling through the insulin–PI3K axis must be kept in exquisite balance to avoid excessive growth, hypoglycaemia and malignant transformation if too much signalling occurs, and growth restriction with hyperglycaemia if too little signalling is present. As we transition to focus on the role of the insulin–PI3K signalling axis in cancer, the argument will continue to hinge on this need for balance. As we have already suggested, sufficient insulin signalling is crucial for glucose homeostasis and normal cell growth and survival. In the context of oncogenic transformation, a growing body of evidence suggests that this same signalling must be kept in check as its activation can contribute to tumour cell growth and survival, and thus can have a profound effect on tumour initiation and treatment efficacy.
Insulin–PI3K signalling in cancer
Tumour initiation and progression are dependent on tumour cells acquiring the abilities to sustain malignant growth45. One key ability is the acquisition of sufficient nutrients for energy and biomass production. As first described by Otto Warburg, tumours have a propensity to shift away from oxidative phosphorylation in favour of glycolysis46,47. While this shift is in some ways paradoxical as it means that the tumour cells generate far less ATP per molecule of glucose than non-tumour cells, it has been proposed that this change in metabolism enables the necessary flow of metabolites to produce new biomass6. A consequence of this shift away from oxidative phosphorylation is the propensity of cells to take up notably more glucose than normal cells6. This increase in glucose flux is aided by genetic alterations that evolve within tumours (that is, PIK3CA mutations or amplifications) and extracellular signals that promote glucose utilization (that is, insulin). Thus, insulin signalling can be seen as enabling tumour development by providing a mechanism for PI3K activation and enhanced glucose uptake. This idea is supported by studies showing enhanced tumour development in humans and mice with hyperinsulinaemia48,49, and reduced tumour development in states of reduced insulin levels50.
The pro-tumorigenic effects of insulin are dependent on the tissue and genotype. For example, tumours with genetic alterations that sensitize patients to insulin will display a more pronounced effect than tumours that lack the insulin receptor31. In fact, it has been observed that patients who have tumours with activating mutations in the insulin signalling pathway are insensitive to systemic calorie deprivation51, which is a dietary approach to reduce systemic insulin levels. Reciprocally, these mutations might reduce the amount of insulin required to activate PI3K and, therefore, ‘normal’ systemic insulin levels are perceived as hyperinsulinaemia by the tumour cells. This chronic activation of the insulin–PI3K cascade might reduce the barrier to oncogenic transformation. It is interesting to note that certain tissues (such as the endometrium, breast, colon, head and neck) acquire genomic PI3K pathway alterations more frequently than others (FIG. 3). Thus, one could speculate that the local insulin level in the mutation-prone tissues is somehow limiting growth, and an activating mutation overcomes this barrier. Other sites that are chronically exposed to high doses of insulin (such as the pancreas and liver) would not have the need for mutational events that lead to further PI3K signal activation as these signalling events could be driven by the local environment.
Fig. 3 |. Analysis of the frequency of mutations in insulin–PI3K signalling.
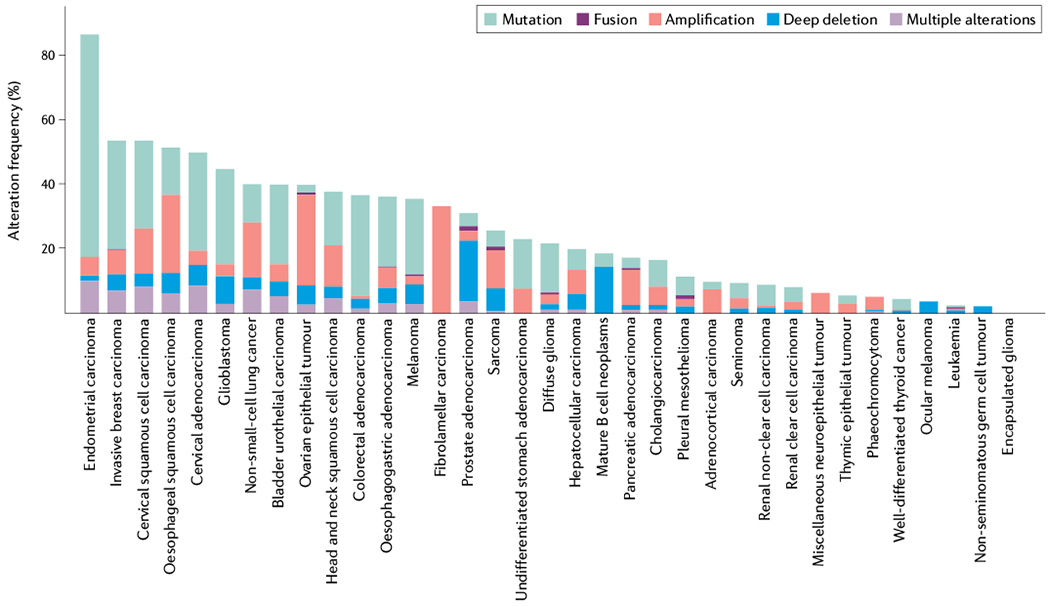
Histogram of the frequency of mutations for the 32 The Cancer Genome Atlas (TCGA) pan-cancer data sets from cBioPortal75,76 showing that the distribution of mutations in genes (PIK3CA, PIK3R1, AKT1, AKT2, AKT3, PTEN and INSR) encoding elements of the insulin–phosphoinositide 3-kinase (PI3K) cascade are not uniform across tumour types. These data highlight the frequency with which this pathway is altered in human cancer and support the idea that different tumour types might be differentially sensitive to genetic modulations of this pathway as the result of the tissues from which they arise or the specific environment of that tissue. Looking across the data set, which covers sequencing from 32 studies, demonstrates the high frequency with which components of this signalling cascade are altered, particularly p110α (encoded by PIK3CA, p110α is a catalytic subunit of PI3K) and phosphatase and tensin homologue (the phosphatase that catalyses the reverse reaction). In total, 38% of the cases in the TCGA pan-cancer studies had mutations in these seven genes.
From these observations, we conclude that insulin can promote or help sustain the growth of certain tumour types. Therefore, hormonal and metabolic approaches that minimize systemic insulin levels might affect tumour initiation and progression. Clinically, this theory can be tested using dietary approaches to manipulate insulin levels, and a general consensus has emerged that individuals with cancer should avoid dietary elements that induce extreme spikes in their glucose and insulin levels, particularly those who have tumours with genomic alterations that enhance the insulin–PI3K pathway14,52. Two phase III combination trials have examined the effect of combining PI3K inhibition with fulvestrant (a selective oestrogen receptor degrader approved for use in some breast cancers). Strikingly, although the compounds that inhibited PI3K showed similar promise, only the trial that accounted for the metabolic state of the patient, excluding those who needed to receive insulin, resulted in FDA approval53,54. Although there were other differences between the compounds, study populations and trial designs, this difference might have been critical to the outcome.
Along these lines, it is worth considering the effect that high glycaemic dietary components have on the metabolism and signalling of tumours, and the potential for these foods to increase tumour cell survival signalling in the context of cytotoxic therapy. In this regard, it might be possible to enhance the efficacy of anticancer therapies by modulating diet; however, no one-size-fits-all approach exists. Different tumours have different capacities for utilizing different fuel sources. For example, patients with endometrial and colon tumours might benefit from low-glycaemic diets to suppress insulin and sugar whereas breast and prostate tumours can thrive on lipid-rich (low-carbohydrate) diets55. Therefore, augmenting a patient’s exposure to insulin through diet requires a precision approach that integrates tumour genetics, tissue of origin and the patient’s hormonal profile. It follows from this line of reasoning that tumours that do not have mutations in the insulin signalling pathway might be alternatively activating this pathway through their systemic metabolism and so could be sensitive to therapeutic strategies that limit a patient’s exposure to insulin, which could be responsible for driving PI3K–mTOR signalling in these patients. It is important to note that not all tumours are dependent on this signalling axis, and while many tumours utilize glycolysis (which is regulated by PI3K signalling) other tumours use alternative metabolic processes such as glutaminolysis56 and, as such, might not be sensitive to approaches that target the insulin–PI3K signalling pathway.
Insulin feedback
The concept of diet augmenting antitumour therapy seems particularly pertinent in the context of targeted inhibitors against the insulin–PI3K pathway. As the activity of this pathway in healthy metabolic tissues (such as liver, skeletal muscle and adipose tissue) coordinates the clearance of glucose from the blood, states of insulin resistance lead to postprandial hyperglycaemia and a consequent enhancement in the release of insulin from the pancreas to restore glucose homeostasis57. This systemic insulin feedback system is the major factor limiting the use of targeted therapies against individual components of the insulin–PI3K signalling cascade. For example, inhibition of the IGF1R as an anticancer therapy results in compensatory activation of PI3K via the closely related insulin receptor58. Furthermore, tumours treated with inhibitors that target both IGF1R and the insulin receptor exhibit extreme hyperglycaemia and extreme hyperinsulinaemia that can then reactivate the insulin receptor52,59,60. These bouts of hyperinsulinaemia can stimulate PI3K and AKT signalling in tumours and promote the growth and survival of tumour cells61–64. The same hyperglycaemic effect is observed when PI3K is directly targeted with small molecules (PI3K inhibitors)65,66. These drugs induce insulin resistance in tumours and normal tissues that is eventually overcome by systemic hyperinsulinaemia. This process limits the clinical efficacy of these drugs, which target the PI3K signalling axis and cause reflex hyperinsulinaemia14,52,67.
If insulin can tip the balance towards oncogenic growth and tumour cell survival, then it might be clinically useful to minimize serum levels of insulin. Fortunately, research on diabetes mellitus has provided multiple avenues to limit hyperglycaemia and hyperinsulinaemia, including diet, exercise and pharmacologic approaches. Before the days of Banting and Best, the very-low-carbohydrate (ketogenic) diet was the primary treatment for patients with diabetes mellitus. Ketogenic diets reduce blood levels of glucose and insulin, and improve insulin sensitivity in humans68 and in mouse models of systemic metabolism69,70. The effects of a ketogenic diet on reducing serum levels of insulin are expected to be beneficial to patients whose tumours express the insulin receptor, and the diet is expected to enhance sensitivity to PI3K and AKT inhibitors by clearing the liver’s glycogen stores and preventing drug-induced hyperglycaemia67. While this diet and drug combination approach is effective in rodent models of cancer52, there is a lack of supportive human data from controlled trials.
In addition to feasibility and efficacy, future studies should also focus on diet composition as it remains unclear how severe the restriction of sugar and rapid-acting carbohydrates needs to be to achieve therapeutic synergy with therapies such as PI3K and AKT inhibitors. In addition to diet, pharmacologic approaches can decrease serum levels of insulin following PI3K inhibition. For example, inhibitors of sodium–glucose co-transporter 2 (SGLT2) in the kidney can also reduce serum levels of glucose by blocking reabsorption of glucose from the renal ultrafiltrate. These drugs essentially clear glucose from the blood by diverting it into the urine. If administered before PI3K inhibitors, the SGLT2 inhibitors can prevent the drug-induced hyperglycaemia and hyperinsulinaemia52,67,71. Another potential pharmacologic approach is to combine PI3K inhibitors with potassium channel inhibitors such as diazoxide to prevent the pancreas from releasing insulin. In this scenario, the drug-induced hyperglycaemia would still occur or be exacerbated at the expense of improved hyperinsulinaemia72,73. Lastly, exercise might be an ideal partner for PI3K inhibitors. If timed properly, the exercise-induced glucose disposal into skeletal muscle could prevent the hyperglycaemia and hyperinsulinaemia induced by the PI3K inhibitor52. By reducing serum levels of glucose and/or increasing insulin sensitivity, these approaches might reduce oncogenic signalling through the insulin–PI3K cascade. According to this theory, these approaches would also be expected to reduce insulin-dependent tumour growth and tumour cell survival signalling (FIG. 4).
Fig. 4 |. Mechanisms to inhibit insulin feedback in tumours.
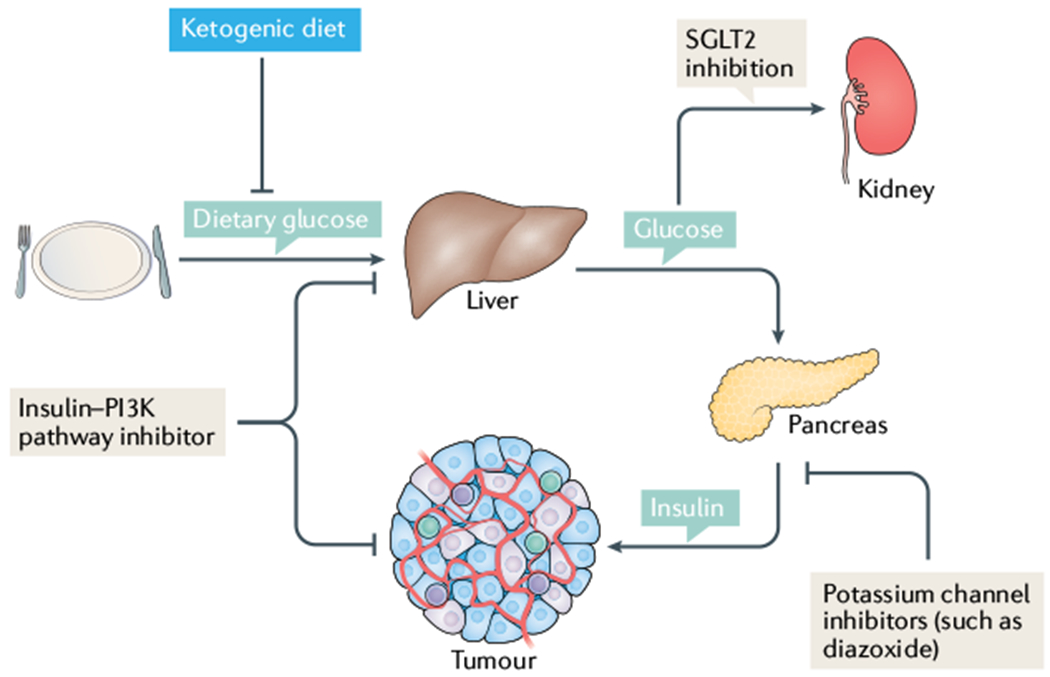
In a patient with cancer who is on a normal carbohydrate-replete diet, normal glucose homeostasis leads to glucose uptake and storage in the liver. When insulin signalling is not active, the liver induces gluconeogenesis to maintain systemic blood levels of glucose. In the clinic, when patients are given inhibitors that target the insulin receptor, phosphoinositide 3-kinase (PI3K) or AKT, their tissues perceive this as a decrease in insulin levels. In the liver, this means increasing the release of glucose, which in turn raises blood levels of glucose and induces increased insulin release from the pancreas. As insulin can act as a mitogen and regulate tumour cell survival signalling, this systemic feedback might be detrimental to treatment efficacy. By administering these agents in combination with sodium–glucose co-transporter 2 (SGLT2) inhibitors that cause the removal of glucose by the kidney, or a ketogenic diet that removes dietary carbohydrate so there are insufficient stores in the liver to cause notable rises in blood levels of glucose, one can prevent therapy-induced spikes in insulin levels, which might limit treatment efficacy. More broadly, limiting insulin exposure in patients might decrease survival and proliferative signalling in tumours where these features are typically induced by systemic insulin.
In a study published in 2018, we demonstrated that co-administration of agents targeting the insulin–PI3K signalling cascade with either an SGLT2 inhibitor or the ketogenic diet limited the on-target hyperglycaemia and hyperinsulinaemia and increased therapeutic efficacy in numerous preclinical models of cancer52. While there are legitimate concerns that these strategies could lead to weight loss, sarcopenia or severe ketosis, which might alter quality of life or increase mortality independently of the tumour, these findings warrant further evaluation in controlled clinical trials. Currently, 178 clinical trials are listed on clinicaltrials.gov evaluating the effect of diet in patients with cancer, including 36 trials examining the effects of the ketogenic diet in cancer, focusing on the effect of the diet on patients from early-stage to advanced cancer and even through surveillance. These trials include 11 combination trials in which the ketogenic diet is being combined with other agents, including standard of care chemotherapies and radiotherapies, to test the feasibility of providing these diets to patients with cancer who might have different nutritional requirements than the patients for whom the ketogenic diet has been provided in the past (such as patients with epilepsy). While drug combinations often result in combined toxicities that limit their clinical utility, diet and exercise regimens are often well tolerated and feasible in patients with cancer74. Clinical trials that evaluate the effect of combining approaches that reduce serum levels of insulin with targeted and/or conventional therapies in a controlled environment need to be conducted to evaluate the efficacy of these approaches so that they can be effectively deployed in the general population; thus, it is imperative that physicians wait to use these approaches until the appropriate trials have been completed.
Clinical cancer care
Moving forward, the limited clinical efficacy of agents that target the insulin–PI3K signalling axis indicates that we need to more rigorously assess the role of systemic factors in treating patients. While this seems apparent in the context of insulin–PI3K inhibitors, it is likely to be more broadly applicable to drugs that target other highly regulated homeostatic pathways in the body. While not all agents will induce such linear feedbacks as targeted inhibitors of insulin signalling, it is clear that circulating factors such as oestrogen, cytokines and growth factors can drive oncogenic pathways in a manner that is independent of tumour mutations, and thus might be critically important for supporting tumour cell survival in the context of therapy. As evidence demonstrating the effect of obesity and systemic metabolism on cancer incidence increases and we come to appreciate the numerous ways in which systemic factors can profoundly affect cancer risk and responses to therapy, it is critical that we begin to implement this knowledge into clinical trials and take note of lifestyle choices made by patients classified as extreme responders, whose tumour responses are either exceptionally good or bad, in order to determine how these choices might affect therapeutic responses. While the example of insulin provided here has direct implications for improving patient responses and clinical deployment of targeted therapies, it also highlights an alternative mechanism by which tumours might be initiated and/or sustained. This thought process has tremendous implications for the way we design clinical trials and the way we evaluate patients. In addition, these ideas might help clinicians to identify effective therapeutic strategies by targeting systemic features, such as increased levels of insulin, instead of or in addition to mutated enzymes.
Conclusions
The integration of systemic and cellular metabolism has evolved to maintain glucose homeostasis by coupling cellular events (for example, replication and survival) with external cues provided by systemic factors (such as insulin), and insulating these networks with a variety of feedback mechanisms that operate at both the cellular and systemic levels to maintain homeostasis. Unfortunately, these pathways are frequently co-opted by tumours and the same feedback mechanisms that evolved to insulate mammals from changes in nutrient availability can serve to promote tumour growth and limit our capacity to effectively target these signals in tumours. As we enter the era of precision medicine, both the effect of the patient’s systemic metabolism and the effect that therapies have on these systemic networks will have a role in determining treatment efficacy. Through the development of therapeutic strategies that contend with these feedback systems, it might be possible to increase treatment efficacy and improve response rates for patients with a wide array of tumour types.
Key points.
Systemic factors such as insulin activate the same signalling pathways as some of the most recurrent mutations in human cancer.
The phosphoinositide 3-kinase (PI3K) signalling cascade, which is activated by insulin, regulates cellular metabolism and cell fate decisions, including cell survival and proliferation.
High insulin levels can promote and sustain tumour growth.
Therapeutic targeting of the PI3K signalling cascade is subject to a variety of cellular and systemic feedback mechanisms, including acute insulin release.
Therapeutic approaches that reduce insulin exposure might increase the efficacy of agents that target the PI3K signalling axis.
Acknowledgements
The authors would like to acknowledge the National Cancer Institute of the National Institutes of Health as research reported in this publication was supported under award numbers R35CA197588 (L.C.C.), R00CA230384 (B.D.H.) and K08CA230318 (M.D.G.). The authors would also like to acknowledge the support of the Grey Foundation’s Basser Initiative (L.C.C. and B.D.H.), a generous gift from The Roger and Susan Hertog Charitable Fund (L.C.C.) and the Lung Cancer Research Foundation (M.D.G.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or these foundations.
Footnotes
Competing interests
B.D.H., M.D.G. and L.C.C. are all founders of and consultants for Faeth, a company developing nutrition for cancer care. L.C.C. is a founder and member of the scientific advisory board and board of directors of Agios and Petra Pharma, which are companies developing drugs to target metabolism.
Peer review information
Nature Reviews Endocrinology thanks F. Janku and the other, anonymous, reviewers for their contribution to the peer review of this work.
References
- 1.Karamitsos DT The story of insulin discovery. Diabetes Res. Clin. Pract 93, S2–S8 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Banting FG & Best CH Pancreatic Extracts (Toronto Univ. Library, 1922). [Google Scholar]
- 3.Pollak M Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 8, 915–928 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Dong MQ et al. Quantitative mass spectrometry identifies insulin signaling targets in C. elegans. Science 317, 660–663 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Engelman JA, Luo J & Cantley LC The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet 7, 606–619 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Vander Heiden MG, Cantley LC & Thompson CB Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White MF & Kahn CR The insulin signaling system. J. Biol. Chem 269, 1–4 (1994). [PubMed] [Google Scholar]
- 8.Ruderman NB, Kapeller R, White MF & Cantley LC Activation of phosphatidylinositol 3-kinase by insulin. Proc. Natl Acad. Sci. USA 87, 1411–1415 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li N et al. Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature 363, 85–88 (1993). [DOI] [PubMed] [Google Scholar]
- 10.Simon MA, Dodson GS & Rubin GM An SH3-SH2-SH3 protein is required for p21Ras1 activation and binds to sevenless and Sos proteins in vitro. Cell 73, 169–177 (1993). [DOI] [PubMed] [Google Scholar]
- 11.Gallagher EJ & LeRoith D Minireview: IGF, insulin, and cancer. Endocrinology 152, 2546–2551 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Pollak M The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat. Rev. Cancer 12, 159–169 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Engelman JA & Cantley LC Chemoprevention meets glucose control. Cancer Prev. Res. 3, 1049–1052 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Fruman DA et al. The PI3K pathway in human disease. Cell 170, 605–635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaham O et al. Metabolic profiling of the human response to a glucose challenge reveals distinct axes of insulin sensitivity. Mol. Syst. Biol 4, 214 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng Z, Tseng Y & White MF Insulin signaling meets mitochondria in metabolism. Trends Endocrinol. Metab. 21, 589–598 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yaribeygi H, Farrokhi FR, Butler AE & Sahebkar A Insulin resistance: review of the underlying molecular mechanisms. J. Cell Physiol. 234, 8152–8161 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Rinderknecht E & Humbel RE Polypeptides with nonsuppressible insulin-like and cell-growth promoting activities in human serum: isolation, chemical characterization, and some biological properties of forms I and II. Proc. Natl Acad. Sci. USA 73, 2365–2369 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pollak M The insulin receptor/insulin-like growth factor receptor family as a therapeutic target in oncology. Clin. Cancer Res. 18, 40–50 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Cignarelli A et al. Insulin and insulin receptors in adipose tissue development. Int. J. Mol. Sci 20, E759 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simoncini T et al. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 407, 538–541 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao X, Kambe F, Moeller LC, Refetoff S & Seo H Thyroid hormone induces rapid activation of Akt/protein kinase B-mammalian target of rapamycin-p70S6K cascade through phosphatidylinositol 3-kinase in human fibroblasts. Mol. Endocrinol 19, 102–112 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Saad MJ et al. Modulation of early steps in insulin action in the liver and muscle of epinephrine treated rats. Endocrine 3, 755–759 (1995). [DOI] [PubMed] [Google Scholar]
- 24.Deibert DC & DeFronzo RA Epinephrine-induced insulin resistance in man. J. Clin. Invest 65, 717–721 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan J et al. PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov. 3, 1156–1171 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Cunningham JT & Ruggero D New connections between old pathways: PDK1 signaling promotes cellular transformation through PLK1-dependent MYC stabilization. Cancer Discov. 3, 1099–1102 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu H et al. Phosphoinositide 3-kinase regulates glycolysis through mobilization of aldolase from the actin cytoskeleton. Cell 164, 433–446 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khayat ZA et al. Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. J. Cell Sci. 113, 279–290 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Hopkins BD, Hodakoski C, Barrows D, Mense SM & Parsons RE PTEN function: the long and the short of it. Trends Biochem. Sci. 39, 183–190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hopkins BD et al. A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 341,399–402 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hodakoski C et al. Regulation of PTEN inhibition by the pleckstrin homology domain of P-REX2 during insulin signaling and glucose homeostasis. Proc. Natl Acad. Sci. USA 111, 155–160 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hodakoski C, Fine B, Hopkins B & Parsons R Analysis of intracellular PTEN signaling and secretion. Methods 77-78, 164–171 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fine B et al. Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-REX2a. Science 325, 1261–1265 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu Y et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 332, 1322–1326 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lyssenko V, Groop L & Prasad RB Genetics of type 2 diabetes: it matters from which parent we inherit the risk. Rev. Diabet. Stud 12, 233–242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hopkins BD, Goncalves MD & Cantley LC Obesity and cancer mechanisms: cancer metabolism. J. Clin. Oncol 34, 4277–4283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martinez-Lopez A et al. CLOVES syndrome: review of a PIK3CA-related overgrowth spectrum (PROS). Clin. Genet 91, 14–21 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Fruman DA et al. Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85 alpha. Nat. Genet 26, 379–382 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Shioi T et al. The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J. 19, 2537–2548 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brachmann SM, Ueki K, Engelman JA, Kahn RC & Cantley LC Phosphoinositide 3-kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Mol. Cell Biol. 25, 1596–1607 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brachmann SM et al. Role of phosphoinositide 3-kinase regulatory isoforms in development and actin rearrangement. Mol. Cell Biol. 25, 2593–2606 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo J et al. Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell Metab. 3, 355–366 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Shaw RJ & Cantley LC Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441, 424–430 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Dibble CC & Cantley LC Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 25, 545–555 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hanahan D & Weinberg RA The hallmarks of cancer. Cell 100, 57–70 (2000). [DOI] [PubMed] [Google Scholar]
- 46.Hsu PP & Sabatini DM Cancer cell metabolism: Warburg and beyond. Cell 134, 703–707 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Warburg O On respiratory impairment in cancer cells. Science 124, 269–270 (1956). [PubMed] [Google Scholar]
- 48.Orgel E & Mittelman SD The links between insulin resistance, diabetes, and cancer. Curr. Diab. Rep 13, 213–222 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Novosyadlyy R et al. Insulin-mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes. Cancer Res. 70, 741–751 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nencioni A, Caffa I, Cortellino S & Longo VD Fasting and cancer: molecular mechanisms and clinical application. Nat. Rev. Cancer 18, 707–719 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kalaany NY & Sabatini DM Tumours with PI3K activation are resistant to dietary restriction. Nature 458, 725–731 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hopkins BD et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560, 499–503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andre F et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med 380, 1929–1940 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Saura C et al. Neoadjuvant letrozole plus taselisib versus letrozole plus placebo in postmenopausal women with oestrogen receptor-positive, HER2-negative, early-stage breast cancer (LORELEI): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 20, 1226–1238 (2019). [DOI] [PubMed] [Google Scholar]
- 55.Goncalves MD, Hopkins BD & Cantley LC Dietary fat and sugar in promoting cancer development and progression. Annu. Rev. Cancer Biol. 3, 255–273 (2019). [Google Scholar]
- 56.Momcilovic M et al. The GSK3 signaling axis regulates adaptive glutamine metabolism in lung squamous cell carcinoma. Cancer Cell 33, 905–921.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rozengurt E, Soares HP & Sinnet-Smith J Suppression of feedback loops mediated by PI3K/ mTOR induces multiple overactivation of compensatory pathways: an unintended consequence leading to drug resistance. Mol. Cancer Ther. 13, 2477–2488 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buck E et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol. Cancer Ther. 9, 2652–2664 (2010). [DOI] [PubMed] [Google Scholar]
- 59.Shirakawa J et al. Effects of the antitumor drug OSI-906, a dual inhibitor of IGF-1 receptor and insulin receptor, on the glycemic control, beta-cell functions, and beta-cell proliferation in male mice. Endocrinology 155, 2102–2111 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Iams WT & Lovly CM Molecular pathways: clinical applications and future direction of insulin-like growth factor-1 receptor pathway blockade. Clin. Cancer Res. 21,4270–4277 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Osborne CK, Bolan G, Monaco ME & Lippman ME Hormone responsive human breast cancer in long-term tissue culture: effect of insulin. Proc. Natl Acad. Sci. USA 73, 4536–4540 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goodwin PJ et al. Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. J. Clin. Oncol 20, 42–51 (2002). [DOI] [PubMed] [Google Scholar]
- 63.Ma J et al. Prediagnostic body mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: a long-term survival analysis. Lancet Oncol. 9, 1039–1047 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dearth RK, Cui X, Kim HJ, Hadsell DL & Lee AV Oncogenic transformation by the signaling adaptor proteins insulin receptor substrate (IRS)-1 and IRS-2. Cell Cycle 6, 705–713 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Gallagher EJ et al. Inhibiting PI3K reduces mammary tumor growth and induces hyperglycemia in a mouse model of insulin resistance and hyperinsulinemia. Oncogene 31, 3213–3222 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bendell JC et al. Phase I, dose-escalation study of BKM120, an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol 30, 282–290 (2012). [DOI] [PubMed] [Google Scholar]
- 67.Goncalves MD, Hopkins BD & Cantley LC Phosphatidylinositol 3-kinase, growth disorders, and cancer. N. Engl. J. Med 379, 2052–2062 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Neal EG et al. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 7, 500–506 (2008). [DOI] [PubMed] [Google Scholar]
- 69.Kennedy AR et al. A high-fat, ketogenic diet induces a unique metabolic state in mice. Am. J. Physiol. Endocrinol. Metab 292, E1724–E1739 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Douris N et al. Adaptive changes in amino acid metabolism permit normal longevity in mice consuming a low-carbohydrate ketogenic diet. Biochim. Biophys. Acta 1852, 2056–2065 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nasiri AR, Rodrigues MR, Li Z, Leitner BP & Perry RJ SGLT2 inhibition slows tumor growth in mice by reversing hyperinsulinemia. Cancer Metab. 7, 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Loves S et al. Effects of diazoxide-mediated insulin suppression on glucose and lipid metabolism in nondiabetic obese men. J. Clin. Endocrinol. Metab 103, 2346–2353 (2018). [DOI] [PubMed] [Google Scholar]
- 73.Loves S et al. High-dose, diazoxide-mediated insulin suppression boosts weight loss induced by lifestyle intervention. J. Clin. Endocrinol. Metab 103, 4014–4022 (2018). [DOI] [PubMed] [Google Scholar]
- 74.Shams-White MM et al. Operationalizing the 2018 World Cancer Research Fund/American Institute for Cancer Research (WCRF/AICR) cancer prevention recommendations: a standardized scoring system. Nutrients 11, 1572 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao J et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cerami E et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]