

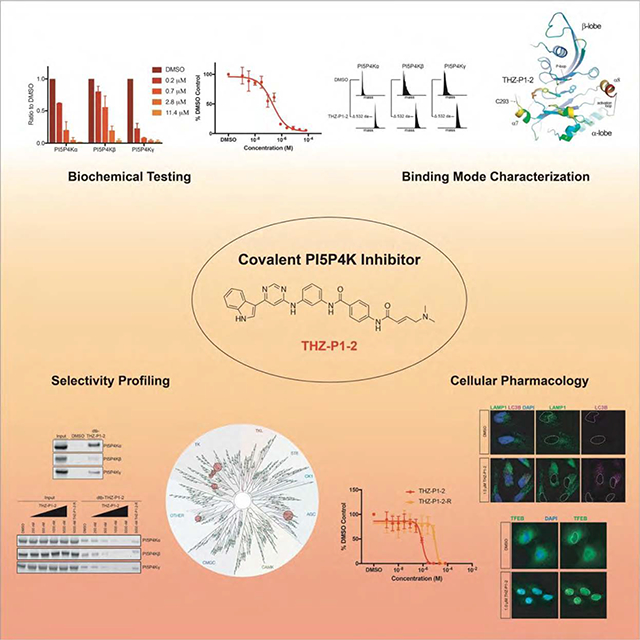
Summary
The PI5P4Ks have been demonstrated to be important for cancer cell proliferation and other diseases. However, the therapeutic potential of targeting these kinases is understudied due to a lack of potent, specific small molecules available. Here we present the discovery and characterization of a pan-PI5P4K inhibitor, THZ-P1-2, that covalently targets cysteines on a disordered loop in PI5P4Kα/β/γ. THZ-P1-2 demonstrates cellular on-target engagement with limited off-targets across the kinome. AML/ALL cell lines were sensitive to THZ-P1-2, consistent with PI5P4K’s reported role in leukemogenesis. THZ-P1-2 causes autophagosome clearance defects and upregulation in TFEB nuclear localization and target genes, disrupting autophagy in a covalent-dependent manner and phenocopying the effects of PI5P4K genetic deletion. Our studies demonstrate that PI5P4Ks are tractable targets, with THZ-P1-2 as a useful tool to further interrogate the therapeutic potential of PI5P4K inhibition and inform drug discovery campaigns for these lipid kinases in cancer metabolism and other autophagy-dependent disorders.
Keywords: PI5P4K, phosphoinositide, autophagy, covalent inhibitor, kinase, cancer, drug discovery
eTOC Blurb
PI5P4K, an understudied kinase family, is essential in various disease contexts. Sivakumaren et al. develop and characterize PI5P4K inhibitor THZ-P1-2 that targets unique cysteines, exhibits effects in biochemical and cellular assays, displays anticancer activity in leukemia cell lines, and causes defects in autophagy similar to PI5P4K gene knockdown or deletion.
Graphical Abstract
Introduction
Phosphatidylinositol 5-phosphate (PI-5-P) is one of the seven phosphosinositides that regulate a wide range of cellular functions (Balla, Szentpetery and Kim, 2009; Bulley et al., 2015). Known to localize in the nucleus, Golgi, endoplasmic reticulum and at the plasma membrane, PI-5-P is an oxidative stress-induced regulator of AKT activation and is also regulated by proline isomerase Pin1 through Pin1’s interactions with the phosphatidylinositol 5-phosphate 4-kinases (PI5P4K) in times of cellular stress (Keune et al., 2012; Keune, Jones and Divecha, 2013). The phosphorylation of the low abundance phosphoinositide PI-5-P at the 4-position, yielding the product phosphatidylinositol-4,5-bisphosphate (PI-4,5-P2), is catalyzed by PI5P4Kα, β and γ (encoded by genes PIP4K2A, PIP4K2B and PIP4K2C) (Rameh et al., 1997; Rameh & Cantley, 1999). PI-4,5-P2 is an important precursor for second messengers inositol-1,4,5-triphosphate (IP3), diacylgycerol (DAG) and phosphatidylinositol-3,4,5-trisphosphate (PI-3,4,5-P3) (Martelli et al., 1992; Divecha et al., 1993; Fiume et al., 2012; Fiume et al., 2015).
While the majority of PI-4,5-P2 is generated by phosphorylation of phosphatidylinositol 4-phosphate (PI-4-P) on the 5-position by the Type I PI4P5K kinases, a PI5P4K-driven alternate route was discovered in 1997, hence the designation Type II (Rameh et al., 1997). The PI5P4Ks were traditionally thought to mainly be crucial direct regulators of PI-5-P levels (Bulley et al., 2015; Stijf-Bultsma et al., 2015; Hasegawa, Strunk and Weisman, 2017). However, PI5P4Kα was found to synthesize a pool of PI-4,5-P2 that is specifically important in mTORC2 regulation (Bulley et al., 2016) and to play a critical role in intracellular cholesterol transport by modulating PI-4,5-P2 homeostasis on peroxisome membranes (Hu et al., 2018). The low-activity isoform PI5P4Kγ was demonstrated to positively regulate Notch1 signaling by facilitating receptor recycling, suggesting that endosome-localized production of PI(4,5)P2 is involved Notch transport (Zheng & Conner, 2018). PI5P4Kα/β were also shown to be required for autophagosome-lysosome fusion during times of metabolic stress, suggesting that they were evolved by multicellular organisms to produce sufficient PI-4,5-P2 in nutrient-deficient conditions (Lundquist et al., 2018). These findings have dispelled the notion of PI5P4K as simply being functionally redundant in PI-4,5-P2 production.
PI5P4K has been suggested to be important in several diseases. PIP4K2B-null mice were found to have increased insulin sensitivity and reduced adiposity (Lamia et al., 2004). PIP4K2A was found to be a dependency in AML and ALL (Jude et al., 2014; Rosales-Rodríguez, et al., 2016; Urayama et al., 2018) and PIP4K2A−/−, PIP4K2B+/−, TP53−/− mice had a dramatic tumor-free life extension compared to TP53−/− mice, uncovering a potential synthetic lethality of PI5P4K with p53 (Emerling et al., 2013). Knockdown of PIP4K2A/B in human retinal pigment epithelial cells and rabbit models abrogated the pathogenesis of proliferative vitreoretinopathy (Ma et al., 2016). Deletion of PIP4K2C in mice resulted in an increase of proinflammatory cytokines and T-helper-cells, as well as a decrease in regulatory T-cells via hyperactivation of mTORC1 signaling (Shim et al., 2016). Pharmacological inhibition or knockdown of PI5P4Kγ reduced mutant huntingtin protein in human patient fibroblasts and aggregates in neurons, and relieved neuronal degeneration in Drosophila models of Huntington’s disease (Al-Ramahi et al., 2017).
The criticai role of the PI5P4Ks in mediating autophagy may explain their induced essentiality in various disease pathologies (Emerling et al., 2013; Vicinanza et al., 2015; Al-Ramahi et al., 2017; Lundquist et al., 2018). Collectively, these studies suggest that the PI5P4Ks represent a lipid kinase family whose underlying biology is important to numerous cellular processes and warrants further investigation of their therapeutic potential across a range of disease states.
The relevance of PI5P4K in a wide range of diseases has motivated efforts to develop PI5P4K inhibitors. Reported pan-PI5P4K inhibitors (Kitagawa et al., 2017) and isoform-specific inhibitors of PI5P4Kα (Davis et al., 2013), PI5P4Kβ (Voss et al., 2014) and PI5P4Kγ (Clarke et al., 2015; Al-Ramahi et al., 2017) have laid the foundation for evidence of PI5P4K druggability and motivated a need for inhibitors with further improved pharmacological properties. Here we present the identification of a potent and selective covalent PI5P4K inhibitor, THZ-P1-2, and characterize its cellular pharmacology in the contexts of autophagy and cancer. Using a multipronged approach combining biochemical and cellular assays, mass spectrometry, and crystallography, we discovered that THZ-P1-2 inhibits the PI5P4K family at sub-micromolar concentrations by reacting covalently with cysteine residues in a flexible loop outside the kinase domain of all three kinase isoforms. We show that THZ-P1-2 exhibits a reasonable selectivity profile across the kinome, with an S-score S(10) of 0.02 (Karaman et al., 2008, Davis et al., 2011) and inhibits cell proliferation at micromolar concentrations in a panel of leukemia cell lines in a manner dependent on covalent targeting. Finally, further investigation of the mechanism of action of THZ-P1-2 led us to the observation that THZ-P1-2 causes defects in the clearance of autophagosomes and lysosomes, halting autophagy and phenocopying the effects of genetic deletion of PI5P4Kα/β/γ (Al-Ramahi et al., 2017; Lundquist et al., 2018). These studies provide evidence that irreversible inhibition of PI5P4K by THZ-P1-2 compromises autophagy, an essential alternative energy source during periods of metabolic stress which cancer cells depend on to maintain cellular homeostasis and prolonged cell viability, further suggesting PI5P4K as a potentially important target in cancer metabolism and other autophagy-dependent diseases.
Results
Chemoproteomic profiling and synthetic chemistry approaches reveal lead PI5P4K inhibitor THZ-P1-2
A previously developed acrylamide-based covalent JNK inhibitor, JNK-IN-7, was serendipitously discovered to also have activity on PI5P4Kγ through KiNativ cellular selectivity profiling (Fig. 1A, Fig. S1A) (Zhang et al., 2012). JNK-IN-7 was found to inhibit the kinase activity of PI5P4Kα/β/γ in a radiometric thin-layer chromatography (TLC) assay (Fig. S1B–C) and mass spectrometry revealed labeling of cysteine residues located on a disordered loop that is not observed in the available PI5P4K crystal structures (Fig. S1D–E). We pursued a focused medicinal chemistry campaign guided by biochemical kinase assays and cellular pulldowns to optimize the potency and selectivity of phenylamino-pyrimidine which resulted in the development of THZ-P1-2 (Fig. 1A) (full structure-activity relationships to be described elsewhere). THZ-P1-2 demonstrated inhibition of PI5P4Kα kinase activity (Fig. 1B), with an IC50 of 190 nM using an ADP-Glo (Promega) bioluminescence assay. THZ-P1-2 exhibited approximately 75% inhibition of PI-4,5-P2 formation by PI5P4Kα and PI5P4K γ and 50% inhibition by PI5P4Kβ at a concentration of 0.7 μM monitored using a thin-layer chromatography (TLC) assay (Fig. 1C–D).
Figure 1. Chemoproteomic profiling and synthetic chemistry approaches reveal PI5P4K inhibitor THZ-P1-2.

(A) Chemical structures of compounds JNK-IN-7 as previously reported in Zhang et al. (2012) and THZ-P1-2 with its reversible and desthiobiotinylated counterparts. See also Fig. S1. (B) THZ-P1-2 potently inhibits PI5P4Kα kinase activity in the ADP-Glo luminescence assay. Data shown are the mean of two biological replicates ± SEM. (C) THZ-P1-2 shows potent inhibition of kinase activity of all three PI5P4K isoforms in a radiometric TLC assay measuring radiolabeled PI-4,5-P2. TLC image shown is representative of two independent experiments. (D) Quantification of (C). Radiolabeled PI-4,5-P2 spots were imaged by autoradiography and quantified by densitometry.
THZ-P1-2 Binds in the Active Site and Covalently Modifies Cysteine Residues of the PI5P4K Kinases
Based on the initial evidence of ATP-competitive inhibition and the covalent binding of JNK-IN-7 to PI5P4Kγ, we sought to confirm covalent binding of THZ-P1-2 to each PI5P4K isoform. Upon incubating THZ-P1-2 with recombinant PI5P4Kα/β/γ protein and analyzing each sample by electrospray mass spectrometry, we discovered that the compound covalently labeled each isoform, as evidenced by the mass shift corresponding to the labeled protein, more efficiently than observed with JNK-IN-7 (Fig. 2A). Notably, THZ-P1-2 achieved 100% labeling of PI5P4Kα/β within 2 h, and PI5P4Kγ in 30 min, corroborating earlier observations in the TLC assay. This observed labeling is dependent on the compound possessing the cysteine-reactive acrylamide moiety, and we confirmed this using a non-cysteine-reactive analog of THZ-P1-2, containing a 4-(dimethylamino)butanamide warhead (Fig. 1A). The reversible counterpart, denoted by THZ-P1-2-R, did not label the recombinant PI5P4K protein (Fig. S2B).
Figure 2. THZ-P1-2 binds covalently to all isoforms of the PI5P4K family on unique cysteine residues located on a disordered loop outside the kinase domain.

(A) Electrospray mass spectrometry of recombinant PI5P4Kα/β/γ incubated with THZ-P1-2 demonstrates covalent labeling of PI5P4K isoforms. See also Fig. S2B. (B) Subsequent protease digestion and tandem mass spectrometry confirms that THZ-P1-2 covalently labels cysteine residues. See also Fig. S2A. (C) Crystal structure of PI5P4Kα in complex with THZ-P1-2, colored according to B factor and shown with covalent warhead extended out towards the covalently-targeted cysteine, C293 (labeled; not resolved in crystal structure). See also Table S1. (D) Ligand interaction map of THZ-P1-2 with residues in the ATP-binding pocket of PI5P4Kα. Related to Fig. S2C–D. (E) Modeled THZ-P1-2 binding showed for all three isoforms based on alignment of published PI5P4Kβ (1BO1; Rao et al., 1998) and γ (2GK9; Uppenberg et al., 2006) structures with obtained PI5P4Kα structure. PI5P4Kα (magenta), PI5P4Kβ (blue), PI5P4Kγ (orange).
To ascertain the specific residues covalently modified by THZ-P1-2, we performed protease digestion and tandem mass spectrometry. Here, we identified peptides containing previously unannotated cysteine residues on a conserved disordered loop residing outside of the PI5P4K isoforms kinase ATP-binding sites – Cys293 on PI5P4Kα, Cys307 and Cys318 on PI5P4Kβ, and Cys313 on PI5P4Kγ – that were covalently modified by THZ-P1-2 (Fig. 2B, Fig. S2A). We hypothesize that this interaction is possible as the disordered loop region is brought into close proximity of the ATP-binding pocket through tertiary structure. These observations add PI5P4K to the list of kinases that possess targetable cysteines distant from the ATP-site in sequence yet accessible proximal to the ATP-site due to the overall kinase conformation (Zhang et al., 2016; Kwiatkowski et al., 2014; Browne et al., 2018).
Elucidation of the crystal structure of THZ-P1-2 bound to PI5P4Kα
To elucidate the detailed molecular interactions involved with the covalent and non-covalent interactions of THZ-P1-2 with the PI5P4Ks, we determined a crystal structure of PI5P4Kα in complex with THZ-P1-2 to 2.21 Å resolution (Fig. 2C, Fig. S2C). The crystal structure showed crucial non-covalent interactions within the ATP-binding pocket. Asn198 forms hydrogen-bonding interactions between the carbonyl of the benzamide and the amine linker connecting the phenylenediamine with the pyrimidine moiety of THZ-P1-2. Furthermore, aromatic π-stacking interactions between the phenylenediamine and Phe200, as well as hydrophobic interactions between the indole head group and linker regions of THZ-P1-2 and several residues in the active site were detected (Fig. 2C–D). Unfortunately, as has been observed in previously published crystal structures, there was insufficient electron density to resolve the disordered loop containing the reactive Cys293 (Fig. S2D). However, it is clear from the co-structure that the cysteine-containing loop could easily traverse the region adjacent to where the acrylamide warhead of THZ-P1-2 is situated. Alignment of crystal structures of PI5P4Kβ (1BO1) and γ (2GK9) deposited in the Protein Data Bank to our resolved co-crystal structure in PyMOL allowed for a more direct comparison between the three isoforms, showing that THZ-P1-2 likely binds to the other two isoforms in a similar manner given the high degree of homology in the active sites and cysteine-containing loops (Fig. 2E).
Cellular target engagement and kinome selectivity of THZ-P1-2
To confirm that the covalent binding observed in vitro is also observed in a cellular context, we synthesized a desthiobiotinylated derivative, dtb-THZ-P1-2, which maintained the biochemical potency of the parental compound (Fig. S3A). A streptavidin pulldown assay in HEK293T cell lysate confirmed that dtb-THZ-P1-2 could capture PI5P4Kα/β/γ at a concentration of 1 μM. We further demonstrated cell permeability and cellular on-target engagement of THZ-P1-2 by performing the streptavidin pulldown assay in a competitive fashion, first pre-treating HEK293T cells with the parent compound, followed by pulldown in cell lysate with the desthiobiotinylated derivative. THZ-P1-2 demonstrated the ability to compete and block pulldown of PI5P4K with dtb-THZ-P1-2 in a dose-dependent manner (Fig. 3B). We observed that engagement measured by the streptavidin pulldown assay also determined covalent binding, as pre-treatment with THZ-P1-2-R was not able to block pulldown by dtb-THZ-P1-2 (Fig. 3B) even though the reversible inactive control compound was still able to inhibit kinase activity to a lesser extent than the covalent compound in the ADP-Glo biochemical assay (Fig. S3A). We validated THZ-P1-2’s continuous on-target engagement typical of covalent inhibitors by performing an additional pulldown with a washout at 2 hours and 4 hours, observing engagement as early as 2 hours with 1 μM compound treatment (Fig. S3B).
Figure 3. Cellular on-target engagement and selectivity profile of THZ-P1-2.

(A) A desthiobiotinylated analog of THZ-P1-2 irreversibly engages all PI5P4K isoforms in a streptavidin pulldown in HEK293T lysate. (B) THZ-P1-2, but not its reversible analog, exhibits dose-dependent on-target engagement of PI5P4K isoforms in a competitive streptavidin pulldown with 1 μM desthiobiotinylated-THZ-P1-2. See also Fig. S3B. (C) Kinome selectivity profile of THZ-P1-2. Complete dataset for off-targets included as Table S2. See also Fig. S4A–C. (D) THZ-P1-2 does not engage the PI4P5Ks or PIKfyve in a streptavidin pulldown. See also Fig. S3C.
To broadly evaluate the kinome-wide selectivity of THZ-P1-2 we used the commercially available DiscoverX KINOMEScan profiling platform which revealed appreciable selectivity across the kinome with a selectivity score at a 10% DMSO control cutoff, S(10), of 0.02 (indicating THZ-P1-2 bound to 2% of queried kinases at the specified cutoff) (Fig. 3C) (Karaman et al., 2008, Davis et al., 2011). We found the related lipid kinase PIKfyve to be an off-target (6.4% DMSO control). Since PIKfyve and PI4P5Kα/β/γ could reasonably be off-targets due to overall similar lipid kinase structure, we first tested THZ-P1-2 inhibitory activity on these kinases. We observed low micromolar IC50s against PI4P5Kα/β/γ and an IC50 of 40 nM on PIKfyve in a fixed time-point ADP-Glo assay (Carna) (Fig. S3C). To further assess if THZ-P1-2 biochemical inhibition of these closely related lipid kinases was maintained in cells, we evaluated dtb-THZ-P1-2 binding of these targets by streptavidin pulldown. We observed no engagement of PI4P5Kα/β/γ and PIKfyve, establishing cellular selectivity of THZ-P1-2 against these related Type 1 and Type 3 lipid kinases (Fig. 3D). Because of THZ-P1-2’s high potency on PIKfyve in the biochemical context, we took a step further to investigate its cellular activity on PIKfyve in a vacuolar enlargement assay. THZ-P1-2 produced the established PIKfyve-inhibitory phenotype in Vero cells only at a high concentration of 10 μM, 1000 × that of positive control apilimod (Cai et al., 2013; Choy & Saffi et al., 2018; Sharma & Guardia et al., 2018) which causes substantial vacuolar enlargement at 10 nM (Fig. S3D), but failed to show inhibition of PIKfyve at 1 μM. THZ-P1-2 was also unable to impair the ability of PIKfyve inhibitor apilimod to induce vacuolar enlargement when cells were either pre-incubated with THZ-P1-2 followed by apilimod, or co-incubated with both compounds (Fig. S3E).
The additional potential off-targets identified through KINOMEScan were further evaluated using the Z’LYTE kinase assay (Invitrogen). All top off-targets except for BRK and ABL1 were found to have IC50s in the micromolar range (Fig. S4A). Engagement of both BRK and ABL1, both of which lack cysteines in or near the vicinity of the ATP-site, were assessed in the cellular pulldown assay with dtb-THZ-P1-2 and found to have little to no pulldown, suggesting that the affinity observed in the Z’LYTE assay may have been due to tight noncovalent binding which was not perpetuated in the cellular context (Fig. S4B). THZ-P1-2 was markedly less potent than the ABL inhibitors imatinib, nilotinib and dasatinib, at killing BCR-ABL translocation containing cell lines (K562 and KU812F) indicating that THZ-P1-2 does not effectively inhibit BCR-ABL in cells (Fig. S4C). Interestingly, even with retention of the indole head group of THZ1 and THZ531, the switch in 2,4- to 4,6-pyrimidine from JNK-IN-7 to THZ-P1-2 was sufficient to alleviate engagement of kinases targeted by related phenylaminopyrimidine acrylamides including JNK, IRAK1, PKN3, CDK7 and CDK12 when probed in a streptavidin pulldown with their respective biotinylated or desthiobiotinylated compounds (Zhang et al., 2012; Kwiatkowski et al., 2014; Browne et al., 2018) (Fig. S4D).
THZ-P1-2 as a probe for potential PI5P4K dependencies in cancer
PIP4K2A was found to be essential for cell survival in AML (Jude et al., 2014) and several variant SNPs in the PIP4K2A locus have been associated with ALL susceptibility and leukemogenesis (Rosales-Rodríguez, et al., 2016; Urayama et al., 2018). We evaluated the sensitivity of a small panel of leukemia cell lines to THZ-P1-2, using a 72 h Cell-Titer Glo assay. We used THZ-P1-2-R as a negative compound to control for effects of THZ-P1-2 that are mediated solely by reversible binding. THZ-P1-2 demonstrated modest anti-proliferative activity in all six AML/ALL cell lines with IC50s in the low micromolar range. We observed approximately 10-fold IC50 shifts between THZ-P1-2 to THZ-P1-2-R (Fig. 4A), indicating that the presence of the PI5P4K cysteine-targeting acrylamide partially contributes to the anti-proliferative activity. ABL1 fusion genes are also heavily involved in hematological malignancies (Braekeleer et al., 2011), but the weak engagement of ABL1 in the cellular context (Fig. S4B–C), together with the covalent dependency, gave confidence that the anti-proliferative activity observed is not solely due to ABL1 inhibition.
Figure 4. Preliminary cancer cell line profiling with THZ-P1-2 to identify potential PI5P4K dependencies.

(A) A panel of AML and ALL cell lines were treated with THZ-P1-2 and THZ-P1-2-R for 72h and cell proliferation was measured with a Cell-Titer Glo luminescence assay. Data shown are the mean of three biological replicates ± SEM.
Inhibition of PI5P4K by THZ-P1-2 results in autophagy defects
Recently, PI5P4K was demonstrated to be necessary for the proper completion of autophagy in mouse models (Lundquist et al., 2018). Loss of PIP4K2A and PIP4K2B in mouse embryonic fibroblasts (MEFs) and mouse liver leads to accumulation of lysosomes and autophagosomes. Lysosomes are enlarged, fused to multiple autophagosomes, and clustered at the nuclear membrane, indicating a defect in the autophagy process. The nuclear localization of the master autophagy transcription factor TFEB was also increased and downstream transcriptional targets were upregulated, indicating an activation of the autophagy gene program (Settembre et al., 2011). However, it has yet to be demonstrated whether these defects were caused by the loss of the full-length protein or the enzymatic activity.
To validate that PI5P4K enzymatic activity is necessary for completion of autophagy, we sought to demonstrate that THZ-P1-2 phenocopies the genetic loss of PI5P4Kα and PI5P4Kβ. We first treated HeLa cells with either DMSO or THZ-P1-2 for 18 hours and stained for the lysosomal and autophagosomal markers LAMP1 and LC3B, respectively. Similar to the genetic knockouts of PI5P4K, treatment with THZ-P1-2 at concentrations as low as 250 nM results in the formation of numerous, enlarged LAMP1 puncta with fusion defects to LC3B-stained autophagosomes (Fig. 5A). This upregulation in autophagosomal/autolysosomal puncta was accompanied by little to no increase in LAMP1 protein levels with THZ-P1-2 treatment for 24 hours, and an increase in LC3B-II and p62 protein levels in serum starvation conditions (HeLa cells cultured in media supplemented with 0.3% FBS) when compared with known autophagy inhibitors bafilomycin A1 and chloroquine (Fig. S5A). Interestingly, we did not observe cumulative effects on autophagy markers by Western blot when THZ-P1-2 treated in combination with these commercially available inhibitors. However, using a RFP-GFP-LC3B tandem-tagged sensor, we were able to further confirm that THZ-P1-2 treatment leads to a marked increase in GFP signal and a moderate increase in RFP signal, suggesting a disruption in autophagy flux, more specifically the induction and upregulation of autophagy without the autolysosome fusion that leads to GFP fluorescence signal loss (Fig. S5B). We also observed an increase in nuclear TFEB accumulation (Fig. 5B–C) and the upregulation of downstream TFEB targets with THZ-P1-2 treatment (Fig. 5F).
Figure 5. Inhibition of PI5P4K with THZ-P1-2 leads to lysosomal-autophagosomal defects and increased TFEB activation.

(A) Similar to genetic loss of PIP4K2A/B, inhibition of PI5P4K activity with THZ-P1-2 increases LAMP1-positive lysosomal size, number and contact with LC3B-positive autophagosomes. HeLa cells were cultured overnight with either DMSO, 0.25 μM, 0.5 μM or 1.0 μM THZ-P1-2 and stained for LC3B (magenta) or LAMP1 (green) with nuclei in blue. Scale bars, 10 μm. See also Fig. S5. (B) Inhibition of PI5P4K activity with THZ-P1-2 increases TFEB nuclear localization. HeLa cells were cultured overnight with either DMSO, 0.25 μM, 0.5 μM or 1.0 μM THZ-P1-2 and stained for TFEB (green) with nuclei in blue. Scale bars, 10 μm. (C) Quantification of results in (B). The intensity of TFEB immunofluorescence was quantified in the nucleus and the cytoplasm, and used to calculate the ratio. Statistical significance determined by ANOVA (***p < 0.0005) with Dunnett multiple comparison post-test. Each group was compared to control control HeLa cells treated with DMSO, (n ≥ 30). (D) Expression of PI5P4K cysteine to serine mutants alleviates lysosomal dysfunction induced by THZ-P1-2 treatment. HeLa cells were infected with viruses expressing GFP, GFP-PI5P4Kα, GFP- PI5P4Kα C293S, GFP-PI5P4Kβ, or GFP-PI5P4Kβ C307S C318S and treated with either DMSO or 250 nM THZ-P1-2 overnight. Expression of both the PI5P4Kα and PI5P4Kβ cysteine to serine mutants alleviated dysfunctional lysosomes (green) with nuclei in blue. Scale bars, 10 μm. (E) Quantification of results in (D). The number of lysosomes was quantified per cell. Statistical significance determined by ANOVA (***p < 0.0005) with Dunnett multiple comparison post-test. Each group was compared to control HeLa cells expressing GFP and treated with DMSO, (n ≥ 30). (F) HeLa cells expressing GFP, GFP-PI5P4Kα, GFP- PI5P4Kα C293S, GFP-PI5P4Kβ, or GFP-PI5P4Kβ C307S C318S were treated with either DMSO or 250 nM THZ-P1-2 overnight for 16 hours and subsequently harvested for qPCR of TFEB targets. Fold change is calculated by comparison to HeLa cells expressing GFP treated with DMSO. *p < 0.05, Student’s t-test, (n ≥ 8).
We next investigated whether the effects of THZ-P1-2 are specific to PI5P4K by rescuing the autophagy defects with the expression of PI5P4Kα/β Cys-Ser mutant isoforms, which are unable to bind the inhibitor covalently. We infected HeLa cell lines to stably express a GFP-only vector, GFP-tagged PI5P4Kα/β, and GFP-tagged PI5P4K Cys-Ser mutants (α: C293S; β: C307,318S;). We treated the cells for 18 hours with either DMSO or THZ-P1-2. As observed with uninfected cells, cell lines expressing GFP alone saw an increase in lysosome/autophagosome dysfunction with THZ-P1-2 treatment (Fig. 5D–E). These effects were partially ameliorated by expression of the PI5P4K constructs and even more so by the cysteine to serine mutants (Fig. 5D–E). The PI5P4K Cys-Ser mutants also partially rescued the nuclear localization of TFEB and the expression of downstream TFEB target genes (Fig. 5D–F).
Discussion
The PI5P4Ks comprise a family of lipid kinases that regulate levels of their substrate and product PI-5-P and PI-4,5-P2, maintaining compartment-specific concentrations of these phosphoinositides at a subcellular level. Initially thought to be mainly a PI-5-P modulator within a noncanonical PI-4,5-P2 generation pathway, the PI5P4Ks have garnered a lot of interest in recent years because of their purported importance in specific disease contexts, spurring efforts to develop PI5P4K inhibitors. In this study, we designed and characterized THZ-P1-2, a first-in-class covalent inhibitor of the PI5P4K family. THZ-P1-2, a small molecule containing an electrophilic acrylamide moiety capable of undergoing a Michael addition, bound in the active site and irreversibly inhibited the enzymatic activity of these kinases. A covalent targeting strategy with THZ-P1-2 resulted in prolonged on-target engagement in cells and modest anti-proliferative activity in a panel of AML and ALL cell lines, while retaining a reasonable selectivity across the kinome. Lastly, we found that THZ-P1-2 treatment phenocopied the effects of genetic deletion of PI5P4K α/β/β in mice by causing autophagy defects in HeLa cells.
Our initial point of compound development was JNK-IN-7, a previously reported covalent JNK inhibitor that was found to be capable of binding to PI5P4Kγ by KiNativ profiling. Originally inspired by the imatinib core structure, this “privileged” kinase scaffold served as a promising starting point for developing a selection of analogs with chemical modifications with which to explore structure-activity relationships. Among this panel of molecules (Manz et al.) we found a critical feature resulting in superior potency to be a switch from a 2,4-pyrimidine to a 4,6-pyrimidine hinge-interacting motif. We established THZ-P1-2’s inhibitory activity with an apparent IC50 of 190 nM on PI5P4Kα in a fixed time-point assay, the isoform with the highest kinase activity and thus easiest to analyze using an ATPase assay (ADP-Glo). We confirmed that THZ-P1-2 specifically prevents phosphate transfer to the PI-5-P substrate at nanomolar concentrations for all isoforms with an orthogonal TLC assay measuring radiolabeled PI-4,5-P2 formation.
THZ-P1-2 covalently labels all three PI5P4K isoforms, with the reactive cysteine residues located on analogous disordered loops of unknown function outside the kinase domain. There has been a resurgence of interest in covalent inhibitors, especially with recent clinical successes of cysteine-directed covalent BTK inhibitor ibrutinib (Pan et al., 2007), EGFR inhibitors neratinib (Rabindran et al., 2004), afatinib (Li et al., 2008), and osimertinib (Cross et al., 2014). Covalent irreversible inhibitors characteristically exhibit an enhanced potency and prolonged pharmacodynamics due to permanent disabling of kinase enzymatic activity, and high selectivity as covalent inhibitors will only bind irreversibly to a kinase with an appropriately placed cysteine, within the trajectory of the electrophilic warhead (Liu et al., 2013). Our approach in developing probes THZ1 against CDK7 (Kwiatkowski et al., 2014), TH531 against CDK12/13 (Zhang et al., 2016) and most recently JZ128 against PKN3 (Browne et al., 2018) showcased to great effect the modification of a reactive cysteine residue outside the active site, widening the scope of “accessible” cysteines. We sought to apply these principles and strategies towards PI5P4K. Our study validates the PI5P4Ks as additional candidate kinases with cysteines that are distant from the active site in sequence but brought close to the ATP-binding site due to kinase tertiary structure, rendering them amenable to covalent targeting. Moreover, our approach in targeting these unique cysteines provides a proof-of-concept in using a combination of inhibitor covalent and noncovalent affinities to achieve selectivity, due to a lack of equivalent off-target cysteines across the kinome.
Structural insights into the binding mode of THZ-P1-2 with PI5P4Kα were obtained through a co-crystal structure. As underscored in the results for Fig. 2C–E, we were unable to resolve the covalent bond between the acrylamide warhead and Cys293 likely due to the degree of disorder in the cysteine-containing loop, as evidenced by the lack of electron density (Fig. S2D). However, we believe the combination of techniques in our study, particularly the MS results (Fig 2A–B) and Cys-Ser rescue in autophagy dysfunction (Fig. 5D–F), lead to observations that taken together support the covalent binding of THZ-P1-2 to the specific PI5P4K cysteine residues, once again emphasizing the value of our multipronged approach. Besides the noncovalent interactions highlighted in the results, hydrogen-bonding between the backbone of Val199 and the 4,6-pyrimidine may account for the increased potency compared to analogs with a 2,4-pyrimidine, especially if they contain substituents on the heterocycle linker. Notably, the Phe205 residue that engages in aromatic p-stacking interactions with the phenylenediamine moiety of THZ-P1-2 is conserved among the PI5P4Ks and has been identified as the residue responsible for the GTP-sensing ability of PI5P4KP (Sumita et al., 2016). This co-crystal structure also serves as an important resource for the scientific community, enabling structure-guided design of PI5P4K inhibitors with improved properties and continuing to bring to attention the challenges of structurally capturing the targeting of regions of disorder. Cell permeability and dose-dependent on-target engagement of THZ-P1-2 was validated in HEK293T cells, chosen for their robust expression of all three PI5P4K isoforms, and the necessity for covalency verified through observing effects after washout and in anti-proliferation assays alongside THZ-P1-2-R.
Our first bona fide look at covalent-dependent on-target engagement was using a streptavidin pulldown assay with a desthiobiotinylated analog of THZ-P1-2 (Fig. 3). In the direct pulldown, THZ-P1-2 engages with PI5P4K preferentially over PI4P5K and PIKfyve. Given that we saw a very mild decrease in activity in the ADP-Glo biochemical assay from covalent to reversible compound (190 nM to 319 nM, Fig. S3A), but observed that THZ-P1-2-R was not able to compete and prevent pulldown by dtb-THZ-P1-2, this indicates that the covalent contribution of the compound’s binding mechanism truly comes into effect and is relevant in the cellular context. Furthermore, this also suggests that our conditions for the ADP-Glo assay may really only be accurate for determining the noncovalent binding potency for this family of kinases, with the covalent aspect needing to be verified in cellular assays.
Our biochemical selectivity profiling of THZ-P1-2 indicates that though the compound exhibits satisfactory selectivity, there are some off-targets observed. The most potent off-targets, such as ABL1, PIKfyve and BRK, were not engaged by dtb-THZ-P1-2 in a cellular pull-down assay. PIKfyve was inhibited by THZ-P1-2 only at concentrations of 10 μM or greater, higher than concentrations relevant for observable PI5P4K targeting. These results indicate that THZ-P1-2 does have some off-target activity on PIKfyve, but maintains a preference for the PI5P4Ks, again possibly due to a combination of covalent and non-covalent binding modes.
A principal part of our study was to further confirm recently discovered functions of the PI5P4Ks beyond their asserted primary role of non-canonical PI-4,5-P2 generation. Treatment with THZ-P1-2 phenocopied genetic deletion of PI5P4K in causing autophagosome and lysosome dysfunction, TFEB nuclear localization and TFEB target gene upregulation, indicating that PI5P4K kinase activity is required for the role of these kinases in autophagy. Cancer cell line profiling demonstrates AML/ALL cancer cell lines to be sensitive to THZ-P1-2 covalent targeting. Consistent with findings that PI5P4K mediates autophagy in times of nutrient stress (Lundquist et al., 2018) our studies using THZ-P1-2 as a tool suggest that the PI5P4Ks display an induced essentiality by facilitating autophagy as an energy source in a metabolically-stressed environment such as cancer (Kimmelman & White, 2017). Additional studies are required to fully dissect the connection between PI5P4K inhibition, autophagy flux disruption and subsequent effects on cell viability, as well as investigate the sensitivities of various cancer types to specific PI5P4K inhibition. THZ-P1-2 may be a useful probe of the pharmacological consequences of covalent inhibition of PI5P4K that can complement genetic approaches (such as CRISPR- or RNAi-based screening) to identify unique vulnerabilities in cancer.
In conclusion, we have characterized a potent and selective covalent PI5P4K inhibitor that exhibits durable cellular pharmacodynamics, causes autophagy dysfunction, and demonstrates modest anti-proliferative activity against leukemia-derived cell lines. THZ-P1-2, THZ-P1-R and the inhibitor resistant PI5P4K Cys to Ser mutants described in this manuscript provide a chemical biology toolbox serving as a valuable resource with which to investigate the pharmacological consequences of covalent inhibition of PI5P4Ks. Our discovery of THZ-P1-2 may be used as a model for targeting other understudied gene families, especially those bearing targetable cysteines in unique sites. Lastly, our small-molecule strategy for disrupting autophagy through covalent PI5P4K inhibition exposes a new Achilles heel and indicates that exploiting this metabolic vulnerability may be a viable therapeutic approach in cancer and potentially other autophagy-addicted disorders.
Significance
Our discovery and characterization of a pan-PI5P4K inhibitor, THZ-P1-2, that covalently targets cysteines on a disordered loop in PI5P4Kα/β/γ presents a new way to target kinases, especially noncanonical lipid kinases, by taking advantage of unique unannotated cysteines outside the kinase domain. THZ-P1-2 demonstrates cellular on-target engagement with limited off-targets across the kinome, displaying requisite potency and selectivity to be a useful tool compound. The sensitivity of leukemia cancer cells to THZ-P1-2 covalent inhibition is evidence of the potential of THZ-P1-2 to be used more universally in larger scale cancer cell line screens to identify PI5P4K dependencies in other cancer types, or other diseases beyond cancer. This can be done in parallel with typical genetic approaches (such as CRISPR- or RNAi-based screening) to identify unique vulnerabilities in a cheaper, faster, less labor-intensive manner. One of the major advantages of THZ-P1-2, as with covalent inhibitors in general, is the ability to verify on-target activity through inhibitor-resistant cysteine-to-serine mutations. In the case of the PI5P4K family of enzymes which has three isoforms, the utility of having both covalent and reversible compounds is extremely valuable, where both compounds can be used in combination to broadly assess PI5P4K dependencies in disease, with follow-up mutant rescue studies to verify the PI5P4K contribution once the scope is narrowed down. Finally, THZ-P1-2-induced autophagy dysfunction phenocopying the effects of PI5P4K genetic deletion lends confidence to the on-target activity of the compound and presents an alternative strategy for targeting autophagy-lysosome homeostasis in disease. Our studies demonstrate that PI5P4Ks are tractable targets, with THZ-P1-2, its reversible and desthiobiotinylated analogs, and wild-type and mutant cell lines as a useful toolbox to further interrogate the therapeutic potential of PI5P4K inhibition, providing a new starting point for drug discovery campaigns for these lipid kinases in cancer metabolism and other autophagy-dependent disorders.
STAR★Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding authors Lewis C. Cantley (LCantley@med.cornell.edu) and Nathanael S. Gray (nathanael_gray@dfci.harvard.edu). All unique/stable reagents generated in this study are available from the corresponding authors with a completed Materials Transfer Agreement.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell lines
Sources of cell lines
All cell lines were sourced from ATCC or DSMZ unless otherwise noted. K562 and KU812F cell lines were obtained from James D. Griffin (Dana-Farber Cancer Institute), and all other leukemia cell lines were obtained from James E. Bradner (formerly Dana-Farber Cancer Institute). HeLa-GFP cell lines were generated using lentiviral or retroviral transduction (see method details). The genders of all cell lines can be found in the Key Resources Table with additional information available on ATCC or DSMZ.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti PI5P4Kα | Cell Signaling | Cat# 5527, RRID:AB_2722636 |
| Rabbit monoclonal anti PI5P4Kβ | Cell Signaling Technologies | Cat# 9694, RRID:AB_2164572 |
| Rabbit monoclonal anti PI5P4Kγ | Sigma-Aldrich | Cat# HPA058551 RRID:AB_2683752 |
| Rabbit monoclonal anti PI4P5Kα | Cell Signaling Technologies | Cat# 9693, RRID:AB_2164698 |
| Rabbit monoclonal anti PI4P5Kβ | Proteintech | Cat# 12541-1-AP |
| Rabbit monoclonal anti PI4P5Kγ | Cell Signaling Technologies | Cat# 3296, RRID:AB_2164719 |
| Mouse monoclonal anti PIKfyve | EMD Millipore | Cat# MABS522 |
| Mouse monoclonal anti c-Abl | Santa Cruz | Cat# sc-56887, RRID:AB_781732 |
| Mouse monoclonal anti Brk | Santa Cruz | Cat# sc-137045, RRID:AB_2174228 |
| Rabbit monoclonal anti IRAK1 | Cell Signaling Technologies | Cat# 4504, RRID:AB_1904032 |
| Mouse polyclonal anti PKN3 | Novus Biologicals | Cat# H00029941-B01P, RRID:AB_2237066 |
| Rabbit monoclonal anti SAP/JNK | Cell Signaling Technologies | Cat# 9252 |
| Mouse monoclonal anti CDK7 | Cell Signaling Technologies | Cat# 2916, RRID:AB_2077142 |
| Rabbit polyclonal anti CDK12 | Cell Signaling Technologies | Cat# 11973, RRID:AB_2715688 |
| Rabbit monoclonal anti LAMP1 | Cell Signaling Technologies | Cat# 9091, RRID:AB_2687579 |
| Rabbit monoclonal anti LC3B | Cell Signaling Technologies | Cat# 2775, RRID:AB_915950 |
| Rabbit monoclonal anti LC3B | Cell Signaling Technologies | Cat# 3868, RRID:AB_2137707 |
| Rabbit polyclonal anti TFEB | Sigma-Aldrich | Cat# SAB4503154, RRID:AB_10761398 |
| Rat monoclonal anti LAMP1 | Abcam | Cat# ab25245, RRID:AB_449893 |
| Mouse monoclonal anti Tubulin | Cell Signaling Technologies | Cat# 3873 |
| Rabbit polyclonal anti Vinculin | Cell Signaling Technologies | Cat# 4650, RRID:AB_10559207 |
| IRDye® 800CW Goat anti-Rabbit IgG (H + L) antibody | LI-COR Biosciences | Cat# 925-32211, RRID:AB_2651127 |
| IRDye 680LT Goat anti-Mouse IgG (H + L) antibody | LI-COR Biosciences | Cat# P/N 925-68020, RRID:AB_2687826 |
| Donkey anti-Rabbit IgG Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-31573, RRID:AB_2536183 |
| Donkey anti-Mouse IgG Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-31571, RRID:AB_162542 |
| Donkey anti-Mouse IgG Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21202, RRID:AB_141607 |
| Donkey anti-Rabbit IgG Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21206, RRID:AB_2535792 |
| Bacterial and Virus Strains | ||
| NEB 10-beta | NEB | Cat# C3019I |
| NEB Stable | NEB | Cat# C3040I |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PI5P diC16 | Echelon Biosciences | Cat# P-5016 |
| DPPS (16:0/16:0 PS) | Echelon Biosciences | Cat# L-3116 |
| Chloroquine | Cell Signaling Technologies | Cat# 14774 |
| Bafilomycin A1 | Cell Signaling Technologies | Cat# 54645 |
| Critical Commercial Assays | ||
| Z’LYTE (TYK2 | Km app) | Invitrogen | Assay ID 864 |
| Z’LYTE (RPS6KA4 (MSK2) | Km app) | Invitrogen | Assay ID 801 |
| Z’LYTE (CSNK2A2 (CK2 alpha 2) | Km app) | Invitrogen | Assay ID 363 |
| Z’LYTE (ABL1 | Km app) | Invitrogen | Assay ID 220 |
| Z’LYTE (KIT | Km app) | Invitrogen | Assay ID 542 |
| Z’LYTE (PDGFRB (PDGFR beta) | Km app) | Invitrogen | Assay ID 687 |
| Z’LYTE (PTK6 (Brk) | Km app) | Invitrogen | Assay ID 771 |
| Adapta (PIP5K1A | 10) | Invitrogen | Assay ID 2340 |
| Adapta (PIP5K1B | 10) | Invitrogen | Assay ID 2341 |
| Adapta (PIP5K1C | 10) | Invitrogen | Assay ID 2342 |
| ADP-Glo (PIKfyve) | Carna Biosciences | N/A |
| ADP-Glo kinase assay | Promega | Cat# V6930 |
| CellTiter Glo | Promega | Cat# G7571 |
| Premo Autophagy Tandem Sensor RFP-GFP-LC3B Kit | Thermo Fisher Scientific | Cat# P36239 |
| CalPhos Mammalian Transfection Kit | Clontech | Cat# 631312 |
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| Superscript VILO cDNA Synthesis Kit | Thermo Fisher Scientific | Cat# 11754250 |
| Quikchange XL Site-Directed Mutagenesis Kit | Agilent | Cat# 200517 |
| Eppendorf MasterCycler RealPlex with SYBR Green | Thermo Fisher Scientific | Cat# K0251 |
| Deposited Data | ||
| PI5P4Kα/THZ-P1-2 co-crystal structure | This paper | PDB: 6OSP |
| PI5P4Kβ crystal structure | Rao et al., 1998 | PDB: 1BO1 |
| PI5P4Kγ crystal structure | Uppenberg et al., 2006 | PDB: 2GK9 |
| Experimental Models: Cell Lines | ||
| HeLa (gender: female) | ATCC | Cat# CCL-2 |
| HEK293T (gender: undetermined) | ATCC | Cat# CRL-3216 |
| THP1 (gender: male) | ATCC | Cat# TIB-202 |
| SEMK2 (gender: female) | DSMZ | RRID:CVCL_S906 |
| HL-60 (gender: female) | ATCC | Cat# CCL-240 |
| SKM1 (gender: male) | DSMZ | Cat# ACC 547 |
| NOMO1 (gender: female) | DSMZ | Cat# ACC 542 |
| K562 (gender: female) | ATCC | Cat# CCL-243 |
| KU812F (gender: male) | ATCC | Cat# CRL-2101 |
| OCI/AML-2 (gender: male) | DSMZ | Cat# ACC 99 |
| HeLa-GFP/GFP-PI5P4K WT and Cys-Ser mutants (α: C293S; β: C307,318S;) (gender: female) | This paper | N/A |
| Oligonucleotides | ||
| qPCR primers | Lundquist et al., 2018 | N/A |
| hPI5P4Kα C293S QuikChange forward primer: catcgttctcctcactctccacttcctcctg | This paper | N/A |
| hPI5P4Kα C293S QuikChange reverse primer: caggaggaagtggagagtgaggagaacgatg | This paper | N/A |
| hPI5P4Kβ C307S QuikChange forward primer: gcagaggacgaggagagtgagaatgatgggg | This paper | N/A |
| hPI5P4Kβ C307S QuikChange reverse primer: ccccatcattctcactctcctcgtcctctgc | This paper | N/A |
| hPI5P4Kβ C318S QuikChange forward primer: gtgtgccataggagctgagtag gttgccacc | This paper | N/A |
| hPI5P4Kβ C318S QuikChange reverse primer: ggtggcaacctactcagctcctatggcacac | This paper | N/A |
| Recombinant DNA | ||
| pCL-Eco | Naviaux et al., 1996 | Addgene plasmid Cat# 12371 |
| pLP1 | Lundquist et al., 2018 | N/A |
| pLP2 | Lundquist et al., 2018 | N/A |
| pVSV-G | Lundquist et al., 2018 | N/A |
| pGEX-2T-PIP4K2A | This paper | N/A |
| pNIC28-Bsa4-PIP4K2A | This paper | N/A |
| pGEX-2T-PIP4K2B | This paper | N/A |
| pNIC28-Bsa4-PIP4K2C | This paper | N/A |
| Software and Algorithms | ||
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/download.html |
| ZEN lite | Carl Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen-lite.html |
| Fiji | NIH | https://imagej.net/Fiji |
| Phenix | Adams et al., 2010 | https://www.phenix-online.org/ |
| XDS | Kabsch, 2010 | http://xds.mpimf-heidelberg.mpg.de/ |
| Phaser | McCoy et al., 2007 | http://www.ccp4.ac.uk/html/phaser.html |
| Coot | Emsley & Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Other | ||
| Streptavidin-agarose beads | Thermo Fisher | Cat# 20349 |
| ProLong Gold DAPI Antifade Mountant | Thermo Fisher | Cat# P36931 |
| Polybrene | Millipore Sigma | Cat# TR-1003-G |
| Puromycin | Invivogen | Cat# ant-pr-1 |
| 4% Affymetrix/Alfa Aesar | Affymetrix/Alfa Aesar | Cat# J19943K2 |
Culture and maintenance of cell lines
All cell culture was performed using standard techniques. HEK293T and HeLa cells were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% FBS and 1% penicillin/streptomycin (Gibco). Leukemia cells were cultured according to ATCC or DSMZ recommendations. All cells were cultured at 370 an d 5% CO2.
METHOD DETAILS
Chemistry
All solvents and reagents were used as obtained. 1H-NMR spectra were recorded with a Varian Inova 600 NMR spectrometer and referenced to DMSO. Chemical shifts are expressed in ppm. Mass spectra were measured with Waters Micromass ZQ using an ESI source coupled to a Waters 2525 HPLC system operating in reverse mode with a Waters Sunfire C18 5 μm, 4.6 × 50 mm column. Purification of compounds was performed with either a Teledyne ISCO CombiFlash Rf system or a Waters Micromass ZQ preparative system. The purity was analyzed on a Waters LC-MS Symmetry (C18 column, 4.6 × 50 mm, 5 μM) using a gradient of 5%−95% methanol in water containing 0.05% trifluoroacetic acid (TFA). Detailed synthetic schemes and characterization data below (see Scheme 1).
ADP-Glo kinase assays
ADP-Glo assay protocol was modified from Davis et al. (2013). DPPS and PI5P (Echelon Biosciences) were dissolved in DMSO (333 μL/mg) and mixed by sonication and vortexing in a ratio of 2:1 DPPS:P15P. First, 63 μL of DMSO was added to 1255 μL of buffer 1 (30 mM Hepes pH 7.4, 1 mM EGTA, 0.1% CHAPS) and 2868 μL of buffer 2 (46 mM Hepes pH 7.4, 0.1% CHAPS). Volumes were multiplied according to number of assays to be run. PI5P4Kα enzyme was added to the buffer mixture at 32 nM concentration (predetermined on a batch-by-batch basis to give maximal signal to background). 10 μL was dispensed into white 384-well plates (Corning #3824). Then, 100 nL of compounds in DMSO were transferred by a pintool (JANUS, PerkinElmer). To initiate the reaction, 5 μL of ATP (Promega) at 15 μM (final assay concentration of 5 μM) and PI5P/DPPS (at concentrations of 0.06 μg/μL and 0.12 μg/μL respectively) in buffer 3 (20 mM Hepes pH 7.4, 60 mM MgCl2, 0.015 mM ATP and 0.1% CHAPS) was added to each well. The final concentration of DMSO in the reaction was less than 5%. The resulting mixture was incubated at room temperature in the dark for one hour, at which time 5 μL of ADP-Glo reagent 1 were added to stop the reaction and remove any remaining ATP. After a 45-minute incubation, 10 μL of the ADP-Glo reagent 2 were added and allowed to incubate for 30 minutes. The luminescence was then read on an EnVision 2104 Multilabel Plate Reader (PerkinElmer). IC50s were determined using the GraphPad Prism nonlinear regression curve fit.
Radiometrie kinase assays
The PI5P4K assay was carried out as described in Rameh et al. (1997). Briefly, the kinase reaction was carried out in a total of 70 μL of kinase buffer (50 mM HEPES pH 7.4, 10 mM MgCl2) with 0.1 μg of purified PI5P4K protein (preincubated with DMSO or compound at various concentrations), followed by addition of 20 μL of the resuspended lipids (sonicated and vortexed), 10 μM non-radiolabeled ATP, and 10 pCi [γ−32P]-ATP for 10 minutes at room temperature. The reaction was terminated by adding 50 μL of 4 N HCl. Phosphoinositides were extracted by adding 100 μL of methanol/chloroform (approximately 1:1, vol:vol) mix and subjected to TLC (thin-layer chromatography) separation using heat-activated 2% oxaloacetate-coated silica gel 60 plates (20 cmx20 cm, EMD Millipore) and a 1-propanol/2 M acetic acid (65:35, vol:vol) solvent system. The radiolabeled product, PI(4,5)P2, was quantified with a Phosphorimager (Molecular Dynamics, STORM840, GE Healthcare).
Immunoblotting
Cells were lysed with Pierce IP Lysis buffer supplemented with a cOmplete™ Mini Protease Inhibitor Cocktail tablet (Roche) on ice for 60 min. Lysates were clarified by centrifugation at 20,000 × g for 15 min at 4°C. Protein concentration was determined by a BCA assay (Pierce), and all samples were run with equal total protein content. Imaging was performed by detection of fluorescently labeled infrared secondary antibodies (IRDye) on the Odyssey CLx Imager (LI-COR).
Pulldown assays
HEK293T cells were lysed with Pierce IP Lysis buffer supplemented with a cOmplete™ Mini Protease Inhibitor Cocktail tablet (Roche) on ice for 60 min. Lysates were clarified by centrifugation at 20,000 × g for 15 min at 4°C. Protein concentration was determined by a BCA assay (Pierce), and all samples were equalized for protein content. DMSO or 1 μM dtb-THZ-P1-2 was added and rotated overnight at 4°C. Samples were rotated for 2h at room temperature to enhance covalent binding and then incubated with streptavidin resin for 2h at 4°C. Beads were washed with lysis buffer five times, resuspended in 1x LDS sample buffer, boiled at 95°C for 10 min and subjected to immunoblotting. For competitive pulldowns, HEK293T cells were grown to 90–95% confluence, pretreated for 6h with DMSO or varying concentrations of THZ-P1-2, washed twice with cold PBS, lysed, and streptavidin pulldown conducted as described above with normalized samples.
Mass Spectrometry
For intact MS analysis, recombinant PI5P4K protein was incubated with DMSO or 5 μM inhibitor (JNK-IN-7, THZ-P1-2 or THZ-P1-2-R) for 2 h at 37°C and analyzed by LC-ESI-MS essentially as described in Zhang et al. (2012). In each analysis, 5 pg protein was injected onto a self-packed reversed phase column (1/32” O.D. * 500 um I.D., 5 cm of POROS 10R2 resin). After desalting for four minutes, protein was eluted with an HPLC gradient (0–100% B in 4 min, A = 0.2M acetic acid in water, B = 0.2 M acetic acid in acetonitrile, flow rate = 10 μL/min) into an LTQ ion trap mass spectrometer (ThermoFisher). Mass spectra were deconvoluted using MagTran1.03b2 software. To determine the site of modification, the samples used for intact analysis were reduced with TCEP (10 mM final concentration), alkylated with iodoacetamide (22.5 mM final concentration) and digested with Asp-N or Glu-C (37 °C, overnight) and analyzed by nanoLC-MS. Digested peptides were injected onto the precolumn (4 cm POROS 10R2, Applied Biosystems) and eluted with an HPLC gradient (NanoAcquity UPLC system, Waters, Milford, MA; 10–70% B in 60 min; A = 0.1 M acetic acid in water, B = 0.1M acetic acid in acetonitrile). Peptides were resolved on a self-packed analytical column (50 cm Monitor C18, Column Engineering, Ontario, CA) and introduced to the mass spectrometer (LTQ Orbitrap XL) at a flow rate of ~30 nL/min (ESI spray voltage = 3.2 kV). The mass spectrometer was programmed to perform data-dependent MS/MS on the five most abundant precursors (35% collision energy) in each MS1 scan (image current detection, 30K resolution, m/z 300–2000). MS/MS spectra were matched to peptide sequences using Mascot (version 2.2.1) after conversion of raw data to .mgf using multiplierz scripts. Precursor and peptide ion mass tolerances were 10 ppm and 0.6 Da, respectively. Supplemental Intact MS Spectra for representative raw MS spectra used for charge deconvolution included separately as Data S1.
Protein expression and purification
Constructs of human PI5P4K in pNIC28-Bsa4/pGEX-2T vectors were overexpressed in E. coli BL21 (DE3) in TB medium in the presence of 50 μg/ml of kanamycin for pNIC28-Bsa4 or 100 μg/ml ampicillin for pGEX-2T. Cells were grown at 37°C to an OD of 0.7, induced overnight at 17°C with 400 μM isopropyl-1-thio-D-galactopyranoside, collected by centrifugation, and stored at −80°C. Cell pellets were resuspended in buffer A (50 mM sodium phosphate, pH 7.4, 500 mM NaCl, 10% glycerol, 20 mM Imidazole, and 14 mM BME), lysed by sonication, and the resulting lysate was centrifuged at 16,000 ×g for 30 min. ~5mL Ni-NTA beads (Qiagen) were mixed with lysate supernatant for 45 min, washed with buffer A, and eluted with buffer B (50 mM sodium phosphate, pH 7.4, 500 mM NaCl, 10% glycerol, 300 mM Imidazole, and 14 mM BME). The eluted sample was gel-filtered through a Superdex-200 16/600 column in buffer C (20 mM HEPES, pH 7.5, 500 mM NaCl, 10% glycerol, 10mM DTT, and 1mM TCEP). Protein fractions were pooled, concentrated, and stored at −80°C. To purify GST-tag proteins, cell pellets were resuspended in buffer G (50 mM sodium phosphate, pH 7.4, 500 mM NaCl, 10% glycerol, 1 mM EDTA, and 14 mM BME), lysed, centrifuged, and purified as described above using ~5mL GSH beads (GE healthcare) and glutathione elution buffer (50 mM sodium phosphate, pH 7.4, 500 mM NaCl, 10% glycerol, 15 mM Glutathione, and 14 mM BME).
Crystallization
A sample of 400 μM protein and 500 μM THZ-P1-2 was co-crystallized in 20% PEG3350 and 200 mM NaMalate by sitting-drop vapor diffusion at 20°C. Crystals were transferred briefly into crystallization buffer containing 25% glycerol prior to flash-freezing in liquid nitrogen.
Data collection and structure determination
Diffraction data from complex crystals were collected at beamline 24ID-E of the NE-CAT at the Advanced Photon Source at the Argonne National Laboratory. The following beamline settings were used: 12662.0 eV (0.9792 Å) beam energy, ADSC QUANTUM 315 detector, 0.2° oscillation, and 0.2s exposure. Multiple crystals were examined, the best of which diffracted to approximately 2.2 Å. XDS was used for integration and scaling (Kabsch, 2010). The structure was solved by molecular replacement using the program Phaser (McCoy et al., 2007) and the search model PDB entry 2YBX, which generated solutions having TFZ scores > 50. Refinement strategy included iterative model building, ligand fitting, and refinement using Phenix (Adams et al., 2010) and Coot (Emsley and Cowtan, 2004). Initial maps had well-defined density (Fo-Fc maps) for the core of the compound, but not for the linker nor electrophile. This was the case for multiple datasets, collected from crystals derived from different crystallization conditions, including primary precipitants Citrate at pH 8 and PEG50000 at pH 6. Despite repeated attempts at including the linker/electrophile segment, with various exit vectors, maps did not improve and did not suggest significant order or occupancy. Also, two residues upstream and 30 downstream of the cysteine in question, Cys293, were disordered. Final refinement led to a model with excellent statistics, shown in Table S1.
Proliferation assays
Cells were plated in 96-well or 384-well format and treated with indicated compounds at varying concentrations for 72h. Anti-proliferative effects of compounds were assessed using the Cell Titer Glo assay kit (Promega) with luminescence measured on an EnVision 2104 Multilabel Plate Reader (PerkinElmer). IC50s were determined using the GraphPad Prism nonlinear regression curve fit.
qRT-PCR
Total RNA was prepared using RNeasy (Qiagen). cDNA was synthesized using Superscript Vilo (Thermo) and qRT-PCR performed utilizing Fast SYBR green (Thermo) and the Realplex Mastercycler (Eppendorf). For a list of primers used see Lundquist et al. (2018). Isolation of mRNA and qPCR was performed as follows. 200,000 cells were plated in 6-well plastic dishes. 24 hours later, the RNA in the lysates was extracted using the RNeasy protocol. The RNA was resuspended in 50 μl H2O at a concentration of 1 μg/μL. cDNA was transcribed using the SuperScript Vilo. The sequences of the oligonucleotides used as primers in the PCR reactions are given in Lundquist et al. (2018). The genes that were quantified here were previously shown to be regulated by TFEB (Perera et al., 2015).
Fluorescence microscopy
HeLa were grown on glass coverslips pre-treated with poly-d-lysine. When indicated cells were treated with 250–1000 nM THZ-P1-2 for 16 hours. Adherent cell lines were rinsed with phosphate-buffered saline, pH 7.4 (PBS) and fixed with 4% paraformaldehyde (PFA) in PBS for 15 minutes at room temperature. After fixation, the cells were permeabilized for 10 minutes with PBS/0.1% Triton X-100, blocked for 30 minutes in blocking buffer (PBS with 3% BSA) and labeled with primary antibodies in blocking buffer for 1 hour at room temperature or overnight at 4°C. Alternatively, for staining, LC3B and LAMP1 (a b25245; Abcam) cells were fixed/permeabilized in −20 MeOH for 20 minutes. Coverslips were washed three times with blocking buffer and incubated with Alexa Fluor-conjugated goat secondary antibodies in blocking buffer for 1 hour at room temperature. After incubation with secondary antibodies, coverslips were washed three times with PBS, once with water, and then mounted on a glass microscope slide with Prolong Gold with DAPI (Thermo). The following primary antibodies were used: TFEB (SAB4503154; Sigma), LC3B (3868; Cell Signaling), LAMP1 (ab25245; Abcam). Alexa Fluor-conjugated secondary antibodies (Thermo) were used at 1:1000. Fluorescent and phase contrast images were acquired on a Nikon Eclipse Ti microscope equipped with an Andor Zyla sCMOS camera. Within each experiment, exposure times were kept constant and in the linear range throughout. When using the 60x and 100x oil immersion objectives, stacks of images were taken and deconvoluted using AutoQuant (Media Cybernetics).
Autophagy flux assays
HeLa cells were plated in 24-well coverslip-bottomed dishes and transduced with 40 particles per cell of the Premo Autophagy Tandem Sensor kit (Thermo Fisher) and cultured for 16–24 hours. Cells were then incubated in either DMSO, 50 μM chloroquine or 1 μM THZ-P1-2 for an additional 16 hours. Cells were mounted in an Okolab Stage Top Incubator warmed to 37°C. DMEM without phenol red (Gibco) was used duri ng image acquisition. All images were collected with a Yokagawa CSU-X1 spinning disk confocal with Spectral Applied Research Aurora Borealis modification on a Nikon Ti motorized inverted microscope equipped with Perfect Focus System. Images were acquired with a Hamamatsu ORCA-ER2 cooled-CCD camera controlled with MetaMorph image acquisition software. Within each experiment, exposure times were kept constant and in the linear range throughout. Images were analyzed and puncta quantified using Fiji software and values plotted using GraphPad Prism.
Generation and transduction of GFP PIP4K2 lentiviral constructs
Replication-deficient lentiviruses were prepared using a third-generation lentiviral system. GFP was inserted by itself or upstream of either human PIP4K2A or PIP4K2B in a lentiviral vector. Cysteine to Serine point mutations were made using the Quikchange XL II site-directed mutagenesis kit (Agilent). Virus was generated by cotransfecting three helper plasmids (pLP1, pLP2, pVSV-G) and the vector containing the gene of interest with c/s-acting sequences for proper packaging were used to generate pseudovirions. A subconfluent culture of HEK293T cells was transfected using the CalPhos Mammalian Transfection Kit (Clontech). The titer of each virus was determined using HEK293T cells. Viral supernatant was then used to express PI5P4Ks in HeLa cells.
Virus production and infection
The 293T packaging cell line was used for retroviral amplification. Cells were transfected at 80–90% confluence with packaging plasmid and viral vectors. The packaging plasmid used for retroviral infection was pCL-Eco (Naviaux et al., 1996). Media was refreshed after 6 hours and the virus-containing media was collected 48 hours later, filtered, and subsequently used for infecting cells in the presence of 8pg/ml polybrene (Sigma). Media was once again refreshed 24 hours post-infection, and cells were subjected to puromycin selection or cell sorting for GFP 48 hours post-infection.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analysis, methods, and details for experiments can be found in the legend of each figure. Figures 1B, 1D, 4, 5C, 5E, 5F, S1C, S3A, S4C, and S5B were generated using GraphPad Prism 7.0.
DATA AND CODE AVAILABILITY
The PI5P4Kα/THZ-P1-2 co-crystal structure generated in this study has been deposited to the PDB under code 6OSP.
Supplementary Material
Table S2, Related to Fig. 3C. Complete dataset for THZ-P1-2 kinome selectivity using the DiscoverX commercial profiling platform.
The kinome selectivity dataset for THZ-P1-2 is 11 pages and provided as a supplemental excel table.
Highlights.
Inhibitor THZ-P1-2 shows PI5P4K enzyme inhibition and target engagement in cells
THZ-P1-2 covalently targets unannotated cysteines outside the PI5P4K active site
AML/ALL cell lines are broadly sensitive to THZ-P1-2’s covalent effects
PI5P4K inhibition causes autophagy disruption and upregulates TFEB signaling
Acknowledgements
The authors acknowledge generous support from NIH grants 1U19AI109740 (to J.M.C.), R21 CA178860 (to J.A.M.), R21CA188881 (to J.A.M.), R35 CA197588 (to L.C.C.), R01 GM041890 (to L.C.C.), U54 U54CA210184 (to L.C.C.), R01 CA197329 (to N.S.G. and S.D.P.), the Dana-Farber Strategic Research Initiative (to J.A.M.) and the Breast Cancer Research Foundation (to L.C.C.). Synchrotron data collection was based upon research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines (NIGMS P41 GM103403). The authors acknowledge the Nikon Imaging Center at Harvard Medical School for valuable training and support with imaging experiments. The authors would like to thank the James D. Griffin and James E. Bradner labs (Dana-Farber Cancer Institute) for generously donating cell lines, and Milka Kostic for advice and guidance with the manuscript submission and revision.
Footnotes
Declaration of interests:
J.A.M. is a member of the scientific advisory board (SAB) of 908 Devices. L.C.C. is a founder and member of the Board of Directors (BOD) of Agios Pharmaceuticals and is a founder and receives research support from Petra Pharmaceuticals. These companies are developing novel therapies for cancer. N.S.G. is a founder, SAB member and equity holder in Gatekeeper, Syros, Petra, C4, B2S and Soltego. The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voronoi, Her2llc, Deerfield and Sanofi. N.S.G., T.Z, and N.P.K. are inventors on a patent application covering chemical matter in this publication owned by Dana Farber Cancer Institute.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, … Zwart PH (2010). PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D: Biological Crystallography, 66(2), 213–221. 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-ramahi I, Srinivas S, Giridharan P, Chen Y, Patnaik S, Safren N, … Marugan JJ (2017). Inhibition of PIP4K g ameliorates the pathological effects of mutant huntingtin protein, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla T, Szentpetery Z, & Kim YJ (2009). Phosphoinositide Signaling: New Tools and Insights. Physiology, 24(4), 231–244. 10.1152/physiol.00014.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braekeleer ED, Douet-Guilbert N, Rowe D, Bown N, Morel F, Berthou C, … Braekeleer MD (2011). ABL1 fusion genes in hematological malignancies: A review. European Journal of Haematology,86(5), 361–371. doi: 10.1111/j.1600-0609.2011.01586.x [DOI] [PubMed] [Google Scholar]
- Browne CM, Jiang B, Ficarro SB, Doctor ZM, Johnson JL, Card JD, … Marto JA (2018). A Chemoproteomic Strategy for Direct and Proteome-Wide Covalent Inhibitor Target-Site Identification. Journal of the American Chemical Society,141(1), 191–203. doi: 10.1021/jacs.8b07911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulley SJ, Clarke JH, Droubi A, Giudici ML, & Irvine RF (2015). Exploring phosphatidylinositol 5-phosphate 4-kinase function. Advances in Biological Regulation, 57, 193–202. 10.1016/j.jbior.2014.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulley SJ, Droubi A, Clarke JH, Anderson KE, Stephens LR, Hawkins PT, & Irvine RF (2016). In B cells, phosphatidylinositol 5-phosphate 4-kinase-α synthesizes PI(4,5)P2 to impact mTORC2 and Akt signaling. Proceedings of the National Academy of Sciences of the United States of America, 113(38), 10571–10576. 10.1073/pnas.1522478113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Xu Y, Cheung AK, Tomlinson RC, Alca A, … Huang Q (2013). Article PIKfyve, a Class III PI Kinase, Is the Target of the Small Molecular IL-12 / IL-23 Inhibitor Apilimod and a Player in Toll-like Receptor Signaling, 912–921. https://doi.org/10.1016d.chembiol.2013.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy CH, Saffi G, Gray MA, Wallace C, Dayam RM, Ou ZA, … Botelho RJ (2018). Lysosome enlargement during inhibition of the lipid kinase PIKfyve proceeds through lysosome coalescence. 10.1242/jcs.213587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke JH, Giudici M, Burke JE, Williams RL, Maloney DJ, Marugan J, & Irvine RF (2015). The function of phosphatidylinositol 5-phosphate 4-kinase γ ( PI5P4K γ ) explored using a specific inhibitor that targets the PI5P-binding site, 367, 359–367. 10.1042/BJ20141333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, … Pao W (2014). AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discovery,4(9), 1046–1061. doi: 10.1158/2159-8290.cd-14-0337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, … Zarrinkar PP (2011). Comprehensive analysis of kinase inhibitor selectivity. Nature Biotechnology,29(11), 1046–1051. doi: 10.1038/nbt.1990 [DOI] [PubMed] [Google Scholar]
- Davis MI, Sasaki AT, Shen M, Emerling BM, Thorne N, Michael S, … Simeonov A (2013). A Homogeneous, High-Throughput Assay for Phosphatidylinositol 5-Phosphate 4-Kinase with a Novel, Rapid Substrate Preparation. PLoS ONE,8(1). doi: 10.1371/journal.pone.0054127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divecha N, Rhee SG, Letcher AJ, Irvine RF, Phosphoinositide signalling enzymes in rat liver nuclei: phosphoinositidase C isoform beta 1 is specifically, but not predominantly, located in the nucleus, Biochem. J. 289 (Pt 3) (1993) 617–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerling BM, Hurov JB, Poulogiannis G, Tsukazawa KS, Choo-Wing R, Wulf GM, … Cantley LC (2013). XDepletion of a putatively druggable class of phosphatidylinositol kinases inhibits growth of p53-Null tumors. Cell, 155(4), 844–857. 10.1016/j.cell.2013.09.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, & Cowtan K (2004). Coot: Model-building tools for molecular graphics. Acta Crystallographica Section D: Biological Crystallography, 60(12 I), 2126–2132. 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- Fiume R, Keune WJ, Faenza I, Bultsma Y, Ramazzotti G, Jones DR, Martelli AM, Somner L, Follo MY, Divecha N, Cocco L, Nuclear phosphoinositides: location, regulation and function, Subcell. Biochem. 59 (2012) 335–361. [DOI] [PubMed] [Google Scholar]
- Fiume R, Stijf-Bultsma Y, Shah ZH, Keune WJ, Jones DR, Jude JG, & Divecha N (2015). PIP4K and the role of nuclear phosphoinositides in tumour suppression. Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids, 1851(6), 898–910. https://doi.org/10.1016Zj.bbalip.2015.02.014 [DOI] [PubMed] [Google Scholar]
- Hasegawa J, Strunk BS, & Weisman LS (2017). PI5P and PI(3,5)P2: Minor, but Essential Phosphoinositides. Cell Structure and Function,42(1), 49–60. doi: 10.1247/csf.17003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu A, Zhao X, Tu H, Xiao T, Fu T, Wang Y, … Song B (2018). PIP4K2A regulates intracellular cholesterol transport through modulating PI(4,5)P2homeostasis. Journal of Lipid Research,59(3), 507–514. doi: 10.1194/jlr.m082149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu A et al. (2018) ‘ PIP4K2A regulates intracellular cholesterol transport through modulating PI(4,5)P 2 homeostasis ‘, Journal of Lipid Research, 59(3), pp. 507–514. doi: 10.1194/jlr.m082149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jude JG, Spencer GJ, Huang X, Somerville TDD, Jones DR, Divecha N, & Somervaille TCP (2014). A targeted knockdown screen of genes coding for phosphoinositide modulators identifies PIP4K2A as required for acute myeloid leukemia cell proliferation and survival. Oncogene, 34(10), 1–10. 10.1038/onc.2014.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010). Integration, scaling, space-group assignment and post-refinement. Acta Crystallographica Section D Biological Crystallography, 66(2), 133–144. 10.1107/s0907444909047374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, … Zarrinkar PP (2008). A quantitative analysis of kinase inhibitor selectivity. Nature Biotechnology,26(1 ), 127–132. doi: 10.1038/nbt1358 [DOI] [PubMed] [Google Scholar]
- Keune WJ, Jones DR, & Divecha N (2013). PtdIns5P and Pin1 in oxidative stress signaling. Advances in Biological Regulation, 53(2), 179–189. 10.1016/j.jbior.2013.02.001 [DOI] [PubMed] [Google Scholar]
- Keune WJ, Jones DR, Bultsma Y, Sommer L, Zhou XZ, Lu KP, & Divecha N (2012). Regulation of phosphatidylinositol-5-phosphate signaling by Pin1 determines sensitivity to oxidative stress. Science Signaling, 5(252), 1–13. 10.1126/scisignal.2003223 [DOI] [PubMed] [Google Scholar]
- Kimmelman AC, & White E (2017). Autophagy and Tumor Metabolism. Cell Metabolism,25(5), 1037–1043. doi: 10.1016/j.cmet.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa M, Liao P, Lee KH, Wong J, Shang SC, Minami N, … Lee SH (n.d.). mitotic pathways leads to cancer-selective lethality. Nature Communications. 10.1038/s41467-017-02287-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, … Gray NS (2014). Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature, 511(7511), 616–20. 10.1038/nature13393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamia KA, Peroni OD, Kim Y, Rameh LE, Kahn BB, & Cantley LC (2004). Increased Insulin Sensitivity and Reduced Adiposity in Phosphatidylinositol 5-Phosphate 4-Kinase −/− Mice. Molecular and Cellular Biology,24(11), 5080–5087. doi: 10.1128/mcb.24.11.5080-5087.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, … Wong K (2008). BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene,27(34), 4702–4711. doi: 10.1038/onc.2008.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, & Gray NS (2013). Developing irreversible inhibitors of the protein kinase cysteinome. Chemistry and Biology, 20(2), 146–159. 10.1016/j.chembiol.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist MR, Goncalves MD, Loughran RM, Possik E, Vijayaraghavan T, Yang A, … Emerling BM (2018). Phosphatidylinositol-5-Phosphate 4-Kinases Regulate Cellular Lipid Metabolism By Facilitating Autophagy. Molecular Cell,70(3). doi: 10.1016/j.molcel.2018.03.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma G, Duan Y, Huang X, Qian CX, Chee Y, Mukai S, … Lei, H. (2016). Prevention of Proliferative Vitreoretinopathy by Suppression of Phosphatidylinositol 5-Phosphate 4-Kinases. Investigative Opthalmology & Visual Science,57(8), 3935. doi: 10.1167/iovs.16-19405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manz TD, Sivakumaren SC, Yasgar A, Hall MD, Davis MI, Seo H-S, Card JD, Ficarro SB, Shim H, Marto JA, Dhe-Paganon S, Sasaki AT, Boxer MB, Simeonov A, Cantley LC, Shen M, Zhang T, Ferguson FM, Gray NS, 2019. Structure-Activity Relationship Study of Covalent Pan-phosphatidylinositol 5-Phosphate 4-Kinase Inhibitors. ACS Medicinal Chemistry Letters. doi: 10.1021/acsmedchemlett.9b00402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli AM, Gilmour RS, Bertagnolo V, Neri LM, Manzoli L, Cocco L, Nuclear localization and signalling activity of phosphoinositidase C beta in Swiss 3T3 cells, Nature 358 (1992) 242–245. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, & Read RJ (2007). Phaser crystallographic software. Journal of Applied Crystallography, 40(4), 658–674. 10.1107/s0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naviaux RK, Costanzi E, Haas M, & Verma IM (1996). The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. Journal of Virology, 70(8), 5701–5. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8764092%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC190538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z, Scheerens H, Li S, Schultz B, Sprengeler P, Burrill L, … Palmer J (2007). Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem,2(1), 58–61. doi: 10.1002/cmdc.200600221 [DOI] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, … Bardeesy N (2015). Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature, 524(7565), 361–365. 10.1038/nature14587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabindran SK, Discafani CM, Rosfjord EC, Baxter M, Floyd MB, Golas J, … Wissner A (2004). Antitumor Activity of HKI-272, an Orally Active, Irreversible Inhibitor of the HER-2 Tyrosine Kinase. CancerResearch,64(11), 3958–3965. doi: 10.1158/0008-5472.can-03-2868 [DOI] [PubMed] [Google Scholar]
- Rameh LE, Tolias KF, Duckworth BC, & Cantley LC (1997). A new pathway for synthesis of phosphatidylinositol-4,5-bisphosphate. Nature,390(6656), 192–196. doi : 10.1038/36621 [DOI] [PubMed] [Google Scholar]
- Rameh LE, and Cantley LC (1999). The role of phosphoinositide 3-kinase lipid products in cell function. J. Biol. Chem. 274, 8347–8350. [DOI] [PubMed] [Google Scholar]
- Rao VD, Misra S, Boronenkov IV, Anderson RA, & Hurley JH (1998). Structure of Type IIP Phosphatidylinositol Phosphate Kinase. Cell, 94(6), 829–839. 10.1016/S0092-8674(00)81741-9 [DOI] [PubMed] [Google Scholar]
- Rosales-Rodríguez B, FernÅndez-Ramírez F, Núñez-Enríquez JC, VelÅzquez-Wong AC, Medina-Sansón A, Jiménez-HernÅndez E, … Rosas-Vargas H (2016). Copy Number Alterations Associated with Acute Lymphoblastic Leukemia in Mexican Children. A report from The Mexican Inter-Institutional Group for the identification of the causes of childhood leukemia. Archives of Medical Research,47(8), 706–711. doi: 10.1016/j.arcmed.2016.12.002 [DOI] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, … Ballabio A (2011). TFEB links autophagy to lysosomal biogenesis. Science (New York, N.Y.), 332(6036), 1429–33. 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Guardia CM, Roy A, Vassilev A, Saric A, Griner LN, … DePamphilis ML (2019). A Family of PIKFYVE Inhibitors with Therapeutic Potential Against Autophagy-Dependent Cancer Cells Disrupt Multiple Events in Lysosome Homeostasis. Autophagy, 0(0), 15548627.2019.1586257. 10.1080/15548627.2019.1586257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim H, Wu C, Ramsamooj S, Bosch KN, Chen Z, & Emerling BM (2016). Deletion of the gene Pip4k2c, a novel phosphatidylinositol kinase, results in hyperactivation of the immune system, 113(27), 7596–7601. 10.1073/pnas.1600934113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stijf-Bultsma Y, Sommer L, Tauber M, Baalbaki M, Giardoglou P, Jones DR, … Divecha N (2015). The basal transcription complex component TAF3 transduces changes in nuclear phosphoinositides into transcriptional output. Molecular Cell, 58(3), 453–467. https://doi.org/10.1016d.molcel.2015.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumita K, Lo YH, Takeuchi K, Senda M, Kofuji S, Ikeda Y, … Sasaki AT (2016). The Lipid Kinase PI5P4KP Is an Intracellular GTP Sensor for Metabolism and Tumorigenesis. Molecular Cell, 61(2), 187–198. https://doi.org/10.1016Zj.molcel.2015.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uppenberg J, Hogbom M, Ogg D, Arrowsmith C, Berglund H, Collins R, Ehn M, Flodin S, Flores A, Graslund S, Holmberg-Schiavone L, Edwards A, Hammarstrom M, Kotenyova T, Nilsson-Ehle P, Nordlund P, Nyman T, Persson C, Sagemark J, Stenmark P, Sundstrom M, Thorsell AG, Van Den Berg S, Weigelt J, Hallberg BM (2006). Human Phosphatidylinositol-4-phosphate 5-kinase, type II, gamma. Structural Genomics Consortium (SGC) doi: 10.2210/pdb2gk9/pdb [DOI] [Google Scholar]
- Urayama KY, Takagi M, Kawaguchi T, Matsuo K, Tanaka Y, Ayukawa Y, … Manabe A (2018). Regional evaluation of childhood acute lymphoblastic leukemia genetic susceptibility loci among Japanese. Scientific Reports,8(1). doi: 10.1038/s41598-017-19127-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicinanza M, Korolchuk VI, Ashkenazi A, Puri C, Menzies FM, Clarke JH, & Rubinsztein DC (2015). PI(5)P regulates autophagosome biogenesis. Molecular Cell, 57(2), 219–234. 10.1016/j.molcel.2014.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss MD, Czechtizky W, Li Z, Rudolph C, Petry S, Brummerhop H, … Schaefer H (2014). Discovery and pharmacological characterization of a novel small molecule inhibitor of phosphatidylinositol-5-phosphate 4-kinase, type II, beta. Biochemical and Biophysical Research Communications,449(3), 327–331. doi: 10.1016/j.bbrc.2014.05.024 [DOI] [PubMed] [Google Scholar]
- Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, MacHleidt T, … Gray NS (2012). Discovery of potent and selective covalent inhibitors of JNK. Chemistry and Biology, 19(1), 140–154. 10.1016/j.chembiol.2011.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Kwiatkowski N, Olson CM, Dixon-clarke SE, Abraham BJ, Greifenberg AK, … Gray NS (2016). Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nature Chemical Biology, 12(10), 876–884. 10.1038/nchembio.2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, & Conner SD (2018). PI5P4Kγ functions in DTX1-mediated Notch signaling. Proceedings of the National Academy of Sciences,115(9). doi: 10.1073/pnas.1712142115 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2, Related to Fig. 3C. Complete dataset for THZ-P1-2 kinome selectivity using the DiscoverX commercial profiling platform.
The kinome selectivity dataset for THZ-P1-2 is 11 pages and provided as a supplemental excel table.
Data Availability Statement
The PI5P4Kα/THZ-P1-2 co-crystal structure generated in this study has been deposited to the PDB under code 6OSP.