

Abstract
Previous glioma classification based primarily on tumour histology resulted in considerable inter-oberver variability and substantial variation in patient survival within grades. Furthermore, there were few known risk factors for glioma. Discoveries over the past decade have deepened our understanding of the molecular alterations underlying glioma and have led to the identification of numerous heritable genetic risk factors. The advances in glioma molecular characterization reframed our understanding of glioma biology and led the World Health Organization (WHO) to develop a new classification system for glioma. The WHO 2016 classification system comprises five glioma subtypes categorized by both tumour morphology and molecular genetic information, which led to reduced misclassification and more-uniform outcomes within glioma subtypes. To date, 25 risk loci for glioma have been identified and several rare inherited mutations that might cause glioma in some families have been discovered. This Review focuses on the two dominant trends in glioma science: the characterization of diagnostic and prognostic tumour markers and the identification of genetic and non-genetic risk factors. An overview of the many challenges still facing glioma researchers is also included.
Introduction
Every year, approximately 100,000 people worldwide are diagnosed as having diffuse gliomas1. Although they comprise less than 2% of all newly diagnosed cancers, diffuse glioma is associated with substantial mortality and morbidity2. Glioblastoma, the most lethal glioma, accounts for 70–75% of all diffuse glioma diagnoses and has a median overall survival of 14–17 months. Globally, there are vast differences in glioma incidence between European and Asian populations3, as discussed more below. In addition to geographic differences, glioma incidence varies by age, sex, ethnicity and tumour histology while glioma survival varies by age and sex.
Historically, gliomas were considered to originate from differentiated astrocytic and oligodendrocytic components of the central nervous system (CNS)4. As such, categorization of diffuse gliomas previously relied only on tumour histology based on the World Health Organization (WHO) 2007 glioma classification criteria5. Over the past decade, molecular studies in human tumours have provided important new insights into the complex genetic, chromosomal, and epigenetic changes within gliomas that accompany glioma formation and maintenance6–11. As a result, a new WHO integrated classification system was introduced in 2016 based on tumour morphology and molecular alterations12. Most recently, stem-like cells within the CNS are thought to be the cells of origin of several primary brain tumour types, including glioblastoma13. Data obtained in experimental animal models support the notion that neural stem cells14, particularly in the subventricular zone, can give rise to at least a subset of glioblastomas15. Also, during the past 10 years, studies discovered many new genetic risk factors for gliomas including rare mutations confined to specific families and more common inherited variants in 25 independent genetic loci16–19.
In this Review of adult diffuse gliomas, we focus on research advances that led from the previous WHO 2007 classification criteria to the WHO 2016 integrated classification system12,20. We present the latest incidence and survival data for adult diffuse glioma from several population based reports including that of the 2018 Central Brain Tumor Registry of the United States (CBTRUS)21, which rely on the WHO 2007 classification system as the WHO 2016 system has not yet been used widely enough in population data sources. We also provide an update on clinical research and genetic and other risk factors for glioma and, where possible, relate these to the WHO 2016 glioma subgroups.
Classification of glioma
The 2007 WHO guidelines for classifying malignant gliomas were based on histologic criteria, in which several morphologic subtypes corresponded to the general appearance of the tissue of origin: astrocytoma, oligodendroglioma and mixed oligoastroctyoma. Tumour malignancy was graded II–IV according to morphological criteria. High-grade tumours generally had a poor prognosis. However, this classification system had high inter-observer variability22–24 and survival varied substantially within grades. Thus, for many decades, neuropathologists and glioma biologists have been investigating markers to improve the characterization of clinically relevant subgroups25.
In 2015, two research groups simultaneously published papers describing molecular and chromosomal subtypes that clarified glioma classification26,27. The first study included over 1,000 patients with WHO grade II–IV glioma from from three large studies in the United States27. These researchers focused on the presence or absence of three tumour markers: promoter mutations in TERT (which encodes telomerase reverse transcriptase); mutations in IDH1 (encoding isocitrate dehydrogenase [NADP] cytoplasmic) or IDH2 (encoding isocitrate dehydrogenase [NADP], mitochondrial)8,28–32, collectively referred to as IDH mutations, which had been shown to be common in some types of glioma and to be associated with survival25; and co-deletion of chromosome arms 1p and 19q, as observed in oligodendrogliomas33. These three markers classified patients with glioma into five groups that were distinguished by differences in survival (especially between patients with grade II or III tumours), age at diagnosis, germline risk alleles, and other tumour mutations and chromosome copy number changes. Notably, grade II or III astrocytomas with only TERT promoter mutations had outcomes very similar to glioblastoma. The second study included almost 300 patients with WHO grade II and III gliomas from the TCGA and found three distinct subgroups based on IDH mutation, TP53 mutation, and 1p19q co-deletion status26.
In 2016, building on data from these and other seminal studies6, the WHO integrated tumour morphology, IDH mutation and 1p19q co-deletion status into a new classification system for adult diffuse glioma12 (Figure 1). This classification system includes five primary designations of adult diffuse glioma: glioblastoma, IDH-wild type; glioblastoma, IDH-mutant; diffuse or anaplastic astrocytomas, IDH-wild type; diffuse or anaplastic astrocytomas, IDH-mutant; and oligodendroglioma or anaplastic oligodendroglioma, IDH-mutant and 1p19q co-deletion (Figure 1). Characterization of the third group was subsequently updated20, as substantial evidence showed that many such tumours had WHO grade II or III histology but were associated with a clinical outcome similar to that of WHO grade IV tumours8,27,34,35 (discussed below). A sixth group, based on histologic appearance in the absence of molecular determination, is designated malignant glioma not otherwise specified. A seventh group, diffuse midline glioma, H3 K27M-mutant (that is, with a histone 3 Lys27>Met amino acid substitution), is also discussed below. Two aspects of the new WHO integrated diagnosis are particularly important: oligoastrocytomas are no longer recognized as a separate entity and instead, via IDH mutation and 1p19q co-deletion status, reflect the genetic profile of either astrocytoma (1p19q intact) or oligodendroglioma (1p19q co-deletion)36; and when histology and molecular features are discordant, molecular features often become the primary determinant of classification.
Figure 1. Overview of key molecular subtypes in the WHO 2016 classification of newly diagnosed adult diffuse glioma.
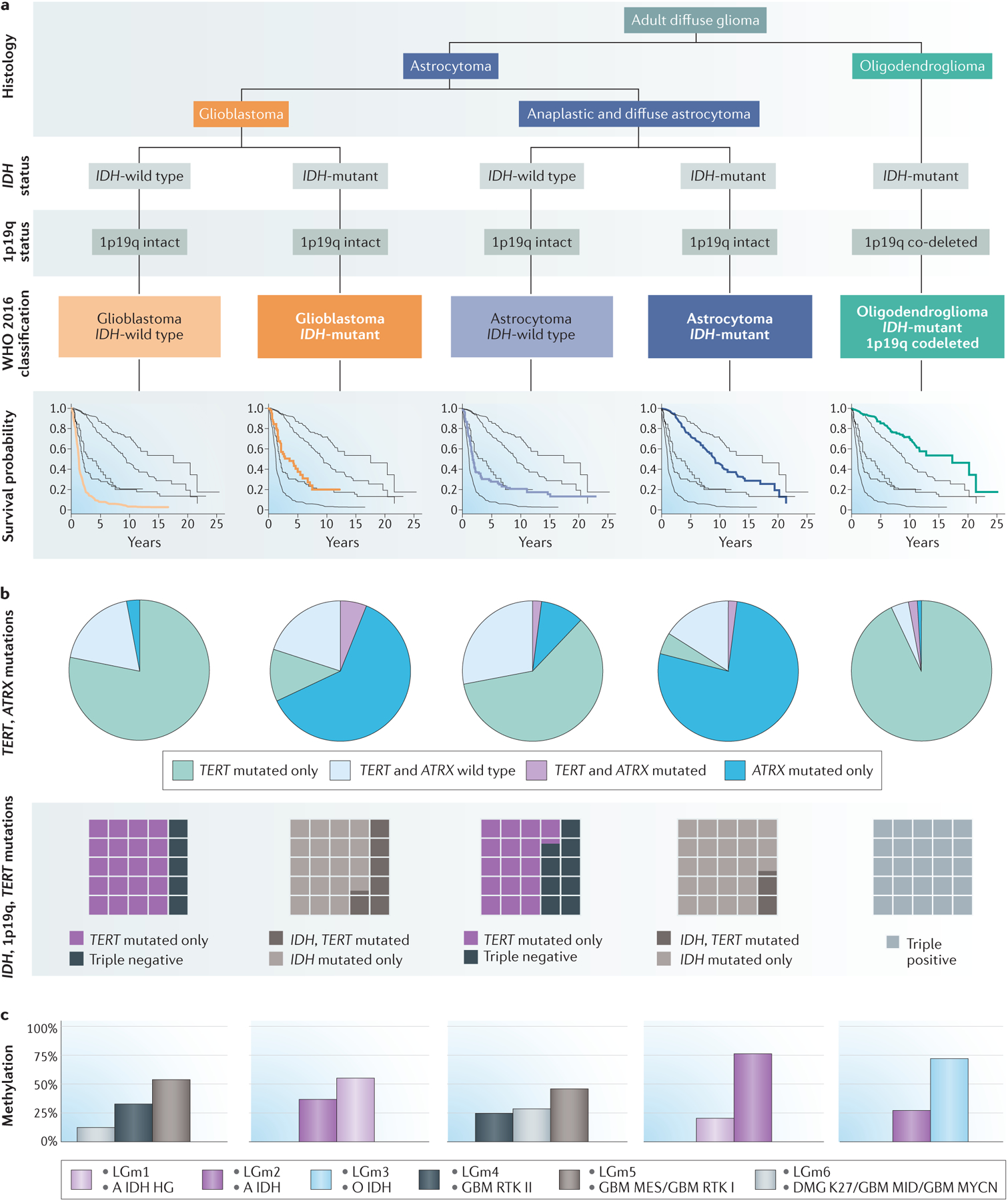
a | Histological assessment integrated with molecular diagnosis based on IDH mutation and 1p19q co-deletion status defines the five subtypes of adult diffuse glioma included in the WHO 2016 classification12,26. Kaplan–Meier survival curves for each type are based on data from 1989–201237. b | For each WHO 2016 disease subtype, telomerase-related mutation frequencies are depicted in pie charts showing the proportion of tumours with mutations in TERT and/or ATRX (green, TERT mutations only; blue, ATRX mutations only; purple, mutations in both TERT and ATRX; light blue, wild type TERT and wild type ATRX)37. Waffle plots show the relative proportions of IDH-mutant, TERT-mutant and 1p19q co-deleted tumours in each WHO 2016 disease subtype27. c | Graphs showing methylation classifications for each WHO 2016 subtype. The methylation categories depicted49 overlap to a great extent with those identified in a subsequent study46 (pink, LGm1 and A IDH HG; purple, LGm2 and A IDH; light blue, LGm3 and O IDH; black, LGm4 and GBM RTK II; gray, LGm5 and GBM MES/GBM RTK I; brown-grey, LGm6 and DMG K27/GBM MID/GBM MYCN). WHO 2016 classification from REFS.12,26. Survival data and telomerase mutation data from REF.37 with additional mutation data from REF.27. Methylation profile data from REFS.46,49.
The five new principal groups have accurately delineated ages at diagnosis and prognosis (Fig 1 and Table 1)8,11,37–39. Patients with glioblastoma, IDH-wild type have, on average, the highest age at diagnosis (median 59 years) and the worst prognosis (median overall survival 1.2 years). Patients with glioblastoma, IDH-mutant tend to be younger (median age at diagnosis 38 years) and have a better prognosis (median overall survival 3.6 years) than those with glioblastoma, IDH-wild type. Patients with astrocytoma, IDH-wild type, regardless of grade II or grade III histology, have a median age at diagnosis of 52 years; their median overall survival (only 1.9 years) is more similar to that of patients with glioblastoma, IDH-wild type than to that of patients with glioblastoma, IDH-mutant. By contrast, patients with astrocytoma, IDH-mutant have a median age at diagnosis of 36 years and median overall survival of 9.3 years37,38. Patients with oligodendroglioma, IDH-mutant and 1p19q co-deletion are also relatively young at diagnosis (median age 44 years) and have the longest median overall survival (17.5 years).
Table 1 –
Additional frequent tumor alterations in and information about the five WHO 2016 subtypes of adult diffuse glioma.
| WHO 2016 classification | Frequent molecular aberrationsa | MGMT promoter methylation (%)b | Median age at diagnosis (years)c | Median overall survival (years)c | Proportion of 2019 diagnoses (%)d |
|---|---|---|---|---|---|
| Glioblastoma, IDH-wild type |
|
~40 | 59 | 1.2 | 71 |
| Glioblastoma, IDH-mutant |
|
~90 | 38 | 3.6 | 7 |
| Astrocytoma, IDH-wild type |
|
55 | 52 | 1.9 | 5 |
| Astrocytoma, IDH-mutant |
|
~85 | 36 | 9.3 | 12 |
| Oligodendroglioma, IDH-mutant and 1p19q co-deletion |
|
~65–100 | 44 | 17.5 | 5 |
As the WHO 2016 integrated diagnosis incorporates both IDH mutation and 1p19q co-deletion status, population-based incidence and survival data for the five WHO classes is not yet available. On the basis of calculations relating to the molecular subtypes (IDH and 1p19q status)27,40 and the most recent CBTRUS data21, we estimate that in 2019, approximately 71% of newly diagnosed diffuse gliomas will be classified as glioblastoma, IDH-wild type; 7% as glioblastoma, IDH-mutant; 5% as astrocytoma, IDH-wild type; 12% as astrocytoma, IDH-mutant; and 5% as oligodendroglioma, IDH-mutant and 1p19q co-deletion (Table 1).
Diffuse midline glioma, H3 K27M-mutant
The seventh group of the WHO 2016 integrated diagnosis (referenced above) is the diffuse midline gliomas, H3 K27M-mutant. According to the 2007 WHO classification system, these tumours could have met the histological criteria for any grade of astrocytoma, although their poor clinical outcomes more closely resemble those of patients with glioblastoma. First identified in paediatric intrinsic pontine gliomas, this subgroup is defined by gain-of-function mutations in genes encoding histone H3 (H3F3A, HIST1H3B, HIST1H3C or HIST1H3I) that result in a Lys>Met amino acid substitution at position 27.41,42 This alteration, which appears early and homogeneously, was also identified in adults with diffuse glioma, predominantly those with young ages of onset and tumours in midline locations such as the spinal cord, thalamus, brainstem, and cerebellum. Although the histological classification of these tumours is typically low-grade, their clinical outcome is poor42. Consequently, the WHO 2016 classification identified diffusely infiltrating gliomas that arise in the midline and harbour the H3 K27M mutation as a separate entity regardless of the presence of anaplastic histologic features.12,43
Diffuse astrocytoma, IDH-wild type
Particular efforts have been made to further characterize tumours in the diffuse astrocytoma, IDH-wild type group according to the WHO 2016 classification, because patient outcomes in this group vary widely20. This group includes patients with very low grade tumours, such as glioneuronal tumours and pilocytic astrocytomas (not discussed in detail in this Review) that have a very favorable course, as well as patients who have a very poor prognosis similar to that of patients with glioblastoma, IDH-wild type. The inclusion of the latter group probably results from sampling of the tumour during resection being insufficient to show histological features of glioblastoma.
Other markers
The Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy (cIMPACT-NOW) is a working group formed to incorporate advances in defining further subtypes of the WHO 2016 classification system into clinical practice20,43,44. For example, cIMPACT-NOW has recommended that diffuse astrocytoma, IDH-wild type tumours should undergo further characterization: testing for EGFR amplification; gain of chromosome 7 and loss of chromosome 10; and the presence of TERT promoter mutation. Regardless of tumour histology, if any of these molecular alterations are found, the patient is now considered to have diffuse astrocytic glioma, IDH-wild type, with molecular features of glioblastoma WHO grade IV20.
Similarly, several other tumour markers that are not incorporated into the formal WHO 2016 classification system are being used to fine-tune the prognosis of some categories of glioma. Below, we summarize the most promising additional markers. Additional genetic alternations and pathway abnormalities are reviewed elsewhere6,45 and summarized in Table 1.
Telomerase alterations.
Numerous studies have shown the classification and prognostic importance of mutations in telomere maintenance genes, including TERT and ATRX7,27,46–48. Typically, TERT and ATRX alterations are mutually exclusive, probably because of functional redundancy7,27,46. A study of over 1,200 adult patients with diffuse glioma aimed to characterize the distribution and additional prognostic value of TERT and ATRX alterations within the five WHO 2016 groups37 (Fig. 1). In the glioblastoma, IDH-wild type subgroup, over 75% of tumours had a TERT mutation. Approximately 3% of glioblastoma, IDH-wild type tumours had alterations in ATRX, which were associated with improved survival (adjusted HR 0.36; 95% CI 0.17–0.81). In the astrocytoma, IDH-wild type subgroup, over 60% of tumours had a TERT mutation whereas 12% had alterations in ATRX; TERT-wild type tumours were associated with improved survival (adjusted HR 0.48; 95% CI 0.27–0.87). Both the astrocytoma, IDH-mutant and glioblastoma, IDH-mutant subgroups showed high proportions of ATRX mutations (63% and 78%, respectively) and low proportions of TERT mutations (5% and 12%, respectively), although neither TERT nor ATRX alterations were associated with survival in these two groups. Among patients with oligodendroglioma, IDH-mutant and 1p19q co-deletion, TERT mutations were seen in 94% of tumours and ATRX alterations in 3%; TERT-wild type tumours were associated with markedly poor survival (adjusted HR 2.46; 95% CI 0.94–6.42). The authors concluded that in some subgroups, additional testing for ATRX and/or TERT mutations might be warranted.
Methylation signatures.
A tumour’s methylome includes both somatically acquired DNA methylation changes and characteristics derived from the cell of origin. Distinct DNA-methylation signatures are associated with prognosis in glioma9,11,46,49,50. For example, mutations in IDH cause aberrant methylation of DNA and histones. Hypermethylation of CpG islands is referred to as the glioma CpG-island methylator phenotype (G-CIMP), which is associated with a favourable prognosis11,25,51. A study that used both methylation and gene expression analyses to examine over 1,100 newly diagnosed diffuse gliomas included in TCGA found six distinct pan-glioma DNA methylation and transcriptome subtypes, which they termed LGm1–LGm6 (Fig. 1)46.
The IDH-mutant gliomas were separated into three groups: LGm1 with 1p19q co-deletion (18%); LGm2 (G-CIMP-high) with 1p19q-intact and highly methylated (25%); and LGm3 (G-CIMP-low) with 1p19q intact and low methylation levels (3%). Interestingly, the LGm1 and LGm2 groups had a similar prognosis; only the LGm3 group had distinctly better prognosis. The IDH-wild type gliomas were separated into four subgroups: LGm4 glioblastomas with classic gene expression profiles (18%); LGm5 glioblastomas with mesenchymal gene expression profiles (27%); and LGm6 glioblastomas with a distinct methylation pattern and stable telomeres (9%). A subset of low-grade gliomas in LGm6 that were similar to pilocytic astrocytoma (that is, with a young age at onset and favourable survival) were referred to as ‘pilocytic astrocytoma -like’46. Of note, only the pilocytic astrocytoma-like subset showed a survival advantage. All other IDH-wild type tumours had indistinguishable survival.
DNA methylation profiling has repeatedly been shown to be a robust method of classifying tumours23,52. Accordingly, DNA methylation-based classification has been used to circumvent the interobserver variability highlighted in the methylation and gene expression study mentioned above and thereby to improve diagnostic precision49. The reference dataset comprised 2,800 CNS tumours including ~100 tumour types, which were divided into 82 CNS tumour methylation classes. Eight of these classes were subgroups of glioblastoma, which suggested a possible future role of DNA methylation profiling in differentiation of the two glioblastoma subgroups included in the WHO 2016 classification. The six TCGA pan-glioma methylation classes shown in Fig. 1 (LGm1–LGm6) overlap with nine of the 82 CNS tumour methylation classes (A IDH HG, A IDH, O IDH, GBM TRK II, GBM MES, GBM RTK I, DMG K27, GBM MID, and GBM MYCN). In the validation dataset, which included over 1,000 prospectively collected CNS tumours, the DNA methylation-based classifier was concordant with histopathology in 76% of tumours, and another 12% (129 tumours) were reclassified based on their methylation profile. In 70% of these 129 tumours, the WHO 2016 classification was either upgraded or downgraded, and several astrocytoma, IDH-wild type tumours were reclassified as glioblastoma, IDH-wild type. Thus, this DNA methylation classifier could substantially reduce tumour misclassification and decrease the subset of tumours for which diagnosis is difficult because the histology and molecular features are discordant.
MGMT:
The DNA damage induced by temozolomide can be repaired by the enzyme methylated-DNA–protein-cysteine methyltransferase (MGMT, also known as 6-O-methylguanine-DNA methyltransferase). Accordingly, high levels of MGMT mRNA and MGMT protein have been linked to DNA alkylating agent resistance53, whereas methylation of CpG islands in the promoter region of MGMT (which suppresses its transcription) increases chemosensitivity to these agents54. The importance of MGMT promoter methylation as both a prognostic and a predictive biomarker has been shown in multiple clinical trials and research studies55–59. A conclusion drawn from several of those studies is that patients with glioblastoma over 65–70 years of age whose tumour lacks MGMT promoter methylation clearly derive a reduced benefit from temozolomide.
Several studies have examined the interaction between MGMT promoter methylation and IDH1 mutation status because IDH1 mutation is associated with the globally hypermethylated G-CIMP phenotype11,51. Among patients with glioblastoma, MGMT promoter methylation rates range from ~40% in IDH-wild type tumours to ~90% in IDH-mutant tumours (a similar pattern is seen in the limited available data on non-glioblastoma astrocytomas60,61; Table 1). In grade III and IV gliomas, MGMT promoter methylation is a prognostic indicator of improved survival and decreased progression in patients with IDH-mutant glioma62, and predicts a favourable therapeutic response to alkylating agents in IDH-wild type patients63,64. In a study that further explored the survival benefit associated with MGMT promoter methylation in patients with glioblastoma, IDH1-wild type, this benefit was only seen in the 75% of patients whose tumour also harboured a TERT promoter mutation (Figure 1)65, which suggested that both markers should be included in future risk stratification. Thus, the research to date supports testing for MGMT promoter methylation in clinical practice, in particular for two groups: elderly patients with glioblastoma and those with a diagnosis of anaplastic (IDH-wild type) glioma61. Several studies in older patients (variably defined as age >60 or >65 years) have confirmed MGMT promoter methylation as a favourable prognostic factor and predictive of response to temozolomide58,66,67. Given the increased toxicity of combined radiotherapy and chemotherapy in elderly patients, individuals with MGMT promoter methylation might be treated with chemotherapy alone67 or patients without MGMT promoter methylation might be treated with radiotherapy alone58,66,67.
Historically, MGMT promoter methylation testing was not widespread owing to difficulties with the assays and their standardization as well as the existence of few therapeutic alternatives to the current standard of care54. However, a data set including over 4,000 patients with glioblastoma multiforme (GBM) pooled from the control arms of four large clinical trials, all of whom received radiation and temozolomide and underwent centralized MGMT promoter methylation testing using quantitative methylation-specific PCR, confirmed that patients without MGMT promoter methylation had a worse prognosis than patients with MGMT promoter methylation. This study also identified that 10% of patients had low MGMT promoter methylation. The outcome of these ‘grey zone’ patients was aligned more closely with the methylated than the nonmethylated group. These data support those from ongoing studies showing that both patients with MGMT promoter methylation and those with indeterminate MGMT promoter methylation status should receive treatment with temozolomide68. This agent should, therefore, only be withheld in patients who truly lack MGMT promoter methylation.
CDKN2A and/or CDKN2B deletion.
Aberrations in signalling pathways affecting the cell cycle are a common mechanism of uncontrolled cell growth in glioma. In glioblastoma, this dysregulation most frequently manifests as homozygous deletions in CDKN2A and/or CDKN2B (50–60%), copy number alterations in other cyclin-dependent pathways (~20%), or mutations in the retinoblastoma-associated protein pathway (7–10%).6,10
Although 50–60% of patients with glioblastoma have loss of CDKN2A and/or CDKN2B, this mutation were not initially associated with prognosis6–8. However, homozygous deletions in CDKN2A and/or CDKN2B have now been identified as a histology-independent negative prognostic marker in IDH-mutant astrocytomas69, including in glioblastoma, IDH-mutant70,71. The loss of CDKN2A and/or CDKN2B, regardless of WHO 2016 classification, was seen in 11–18% of all tested astrocytomas, IDH-mutant, and these patients clinical outcomes’ were similar to those of patients with glioblastoma, IDH-mutant. Although homozygous deletion of CDKN2A and/or CDKN2B was the strongest predictor of worse survival, the effect was diminished when combined with other aberrations in the cell cycle pathway, such as amplification of CDK4 or CDK6 or deficiency of RB1. The adverse effect of CDKN2A and/or CDKN2B deletion on prognosis might be unique to IDH-mutant astrocytomas with TP53 mutations, and might not be seen in oligodendrogliomas or IDH-wild type astrocytomas, including glioblastoma38,69.
Incidence, Risk and Survival
Incidence and survival
Globally, the incidence of adult diffuse gliomas differs substantially between countries72–78. In older adults over 40, the age adjusted annual incidence rate of astrocytic tumors is 6.8/100,000 people79. Countries with predominantly Northern European populations have a higher rate (ranging from 7.8 in the USA to 9.6 in Australia/New Zealand) than countries with predominantly Asian or African populations (ranging from 1.9 in Southeast Asia to 3.3 in India)79. Similar patterns in incidence are seen in oligodendroglial tumors. The wide range of incidence values is confounded by differences in access to medical imaging, case ascertainment and surveillance. Nonetheless, the large difference in incidence between European and Asian populations is observed both when comparing different countries and within the USA and UK79,80. The fourfold higher incidence in Western Europe (8.5/100,000) compared to prosperous East Asian countries (such as Japan and Singapore) (1.9/100,000) suggests that ethnic, cultural and/or environmental differences might be more likely to explain the observed differences in incidence than reporting bias3,78,79. Future studies comparing genetic, environmental and regional lifestyle risk factors within and between countries will be important to understand these differences78.
The latest incidence data in the USA for adult diffuse glioma are derived from the 2018 Central Brain Tumor Registry of the United States (CBTRUS) report21, which relies on the WHO 2007 classification system as the WHO 2016 system has not yet been used widely enough in population data sources. As the CBTRUS definition of glioma includes pilocytic astrocytoma, unique astrocytoma variants, ependymal tumours, and malignant glioma not otherwise specified, when citing CBTRUS data, we have only included data on diffuse glioma in adults. However, other references cited contain variable definitions of glioma, which sometimes include paediatric patients. In this article, therefore, we differentiate between adult diffuse glioma and glioma. As noted above, the WHO 2016 classification does not include the prior histologic category of oligoastrocytoma. However, CBTRUS and other reporting registries retain that category and have not yet published population data based on the new classification. Therefore, in this section we use the older WHO 2007 classification used by registries for population statistics, and when available, limited data from studies that used the WHO 2016 subtypes.
In the United States, diffuse gliomas account for 72% of malignant brain tumours in adults and 21% of all primary brain and other CNS tumours in adults21,81. In adults, glioblastoma, the most rapidly fatal form, accounts for 73% of diffuse gliomas, 52% of malignant non-metastatic brain tumours, and 15% of all primary brain and other CNS tumours21. Although diffuse gliomas account for less than 1% of all new cancers21,82, they are associated with considerable morbidity and mortality. Since 2000, the incidence of glioma has been relatively stable, with a small but statistically significant increase from 2000 to 2008 followed by a statistically significant decrease from 2008 to 201421,72,83,84. In 2019, new cases of adult diffuse glioma in the USA are estimated to approach 17,000 whereas during 1990–1994 <10,000 cases annually were reported85. The large increase observed after 1994 is likely to be due to improved detection through advances in diagnostic imaging.
Most population-based studies show that, in addition to the geographical differences, glioma incidence varies by age, sex, ethnicity and tumour histology (or WHO 2016 subtype), and that survival for patients with glioma also consistently varies by age, sex and tumour subtype.
Age and sex
Within diffuse glioma subtypes, population incidence varies according to age and sex (Figure 2). Astrocytoma (including glioblastoma and diffuse or anaplastic astrocytoma) incidence increases with age, and peaks between the ages of 75 years and 84 years. Men have a 40–50% higher incidence than women at all ages (Figure 2)21,86. In 2000–2014, 1-year and 5-year relative survival for adults with glioblastoma was 41% and 5%, respectively. Corresponding 1-year and 5-year relative survival for those with non-glioblastoma astrocytomas was 72% and 44%, respectively (Figure 2)81. In a large study using the WHO 2016 classification, the median age at diagnosis is lower for patients with IDH-mutant astrocytomas (36 years) and glioblastomas (38 years) than for those with IDH-wild type astrocytomas (52 years) and glioblastomas (59 years)37 (Table 1). Median overall survival is better in patients with IDH-mutant astrocytomas (9.3 years) and IDH-mutant glioblastomas (3.6 years) than for those with IDH-wild type glioblastoma (1.9 years) and IDH-wild type astrocytoma (1.2 years)37 (Table 1).
Figure 2. Annual average age-adjusted incidence and relative survival data for non-glioblastoma and glioblastoma CNS tumours.
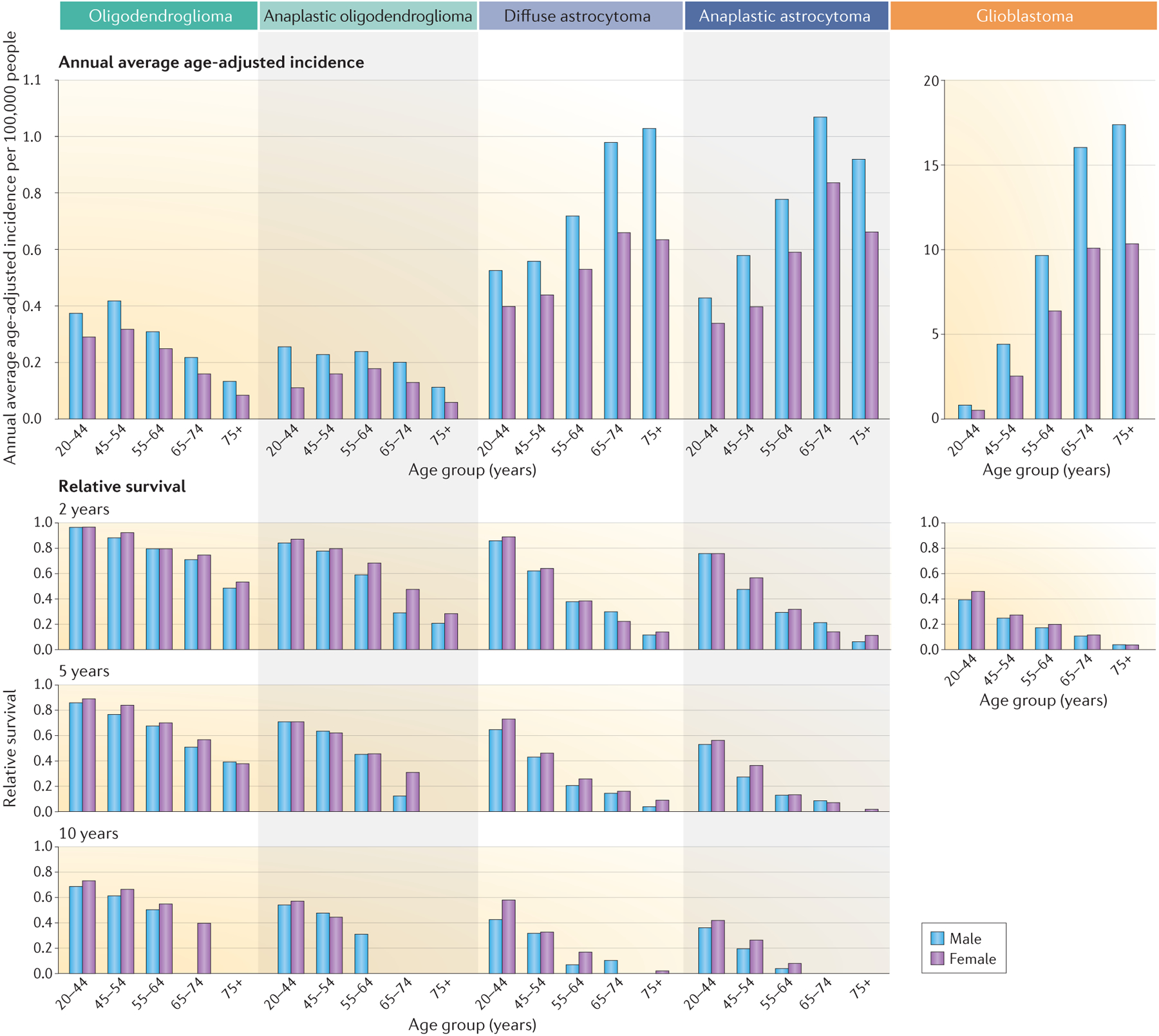
The Central Brain Tumor Registry of the United States (CBTRUS) and other global reporting registries have not yet published glioma statistics based on the WHO 2016 classification system. Accordingly, this Figure presents the latest available published CBTRUS statistics from 2011–2015 as well as data accessed directly from CBTRUS, both based on the WHO 2007 classification system. Data from REF.21 Note that different axes are used for incidences of glioblastoma versus the other types of glioma.
The population incidence of grade II oligodendrogliomas and oligoastrocytomas peaks at age 35–44 years (Figure 2). Men have a ~25–60% higher incidence than women depending on age. In 2000–2014, 1-year and 5-year relative survival for oligodendroglioma was 90% and 70%, respectively. The incidence of anaplastic oligoastrocytic tumours peaks between the ages of 55 years and 64 years; in this age group, men show a 20% higher incidence than women (range 20–40% across all age groups). The median age at diagnosis is 44 years for those with IDH-mutant and 1p19q co-deletion oligodendrogliomas and median overall survival is 17.5 years (Table 1)37.
Similar age and sex patterns for astrocytomas (including glioblastoma) are also seen in Europe87,88. These age patterns may be related to the time needed to acquire the multiple genetic alterations required for malignant transformation. As the world population continues to grow and age, the incidence of diffuse glioma is expected to increase. As yet, no comprehensive explanations account for the peaks in glioma incidence observed in particular age groups nor for the increased incidence of glioma in men. The lack of definitive associations between hormone exposure and glioma risk in multiple large studies,86,89–92 as well as the fact that most cancers show increased incidence in men, suggests the existence of unidentified risk factors.
Ethnicity
In the USA, the incidence of adult diffuse glioma is highest in non-Hispanic white individuals (versus Hispanic white, black, American Indian or Alaska Native, and Asian or Pacific Islander people). Non-Hispanic white people have over a twofold higher rate of glioblastoma than do black, Asian or Pacific Islander and American Indian or Alaska Native individuals, and a 30% higher rate than Hispanic white individuals81. This pattern is also consistently seen in other diffuse glioma subtypes, for which incidence rates are similarly highest in non-Hispanic white individuals. Data from the USA also suggest that the lifetime risk of developing a malignant brain tumour is 25% higher in non-Hispanic white than in Hispanic white individuals, and is twice as high as the risk in black individuals81. In glioblastoma, 5-year relative survival is lowest (5%) in non-Hispanic white individuals and highest (9%) in Asian or Pacific Islander individuals. For non-glioblastoma astrocytoma, the 5-year relative survival is lowest in non-Hispanic white people (43.2%) and higher in all other ethnic groups (44.1% to 50.8%), whereas for oligodendroglioma 5-year relative survival is lowest in black individuals (63.8%)81.
Additional survival factors
Additional factors associated with survival are the extent of tumour resection, Karnofsky performance score and treatment. Multiple studies have indicated that complete resection is beneficial93–95, even in low-grade tumours (for which early resection is preferable to watchful waiting or biopsy alone96). Although difficult to prove, the influence of extent of tumour resection on survival could be confounded by tumour location, whether it is resectable or non-resectable and/or by clinical judgement97. After 2005, when clinical trial results showing an increase in median overall survival from 12.1 months to 14.6 months were published, the standard of care for patients with newly diagnosed glioblastoma has been treatment with the DNA-alkylating agent temozolomide, radiotherapy and surgery (termed the Stupp protocol)98. Among patients with glioblastoma treated according to this protocol, median survival in contemporary clinical trials is 14–17 months57,72,99–104. In one trial, for which preliminary data were published in 201597 and the complete report in 201798, the addition of tumour-treating fields (an antimitotic treatment modality) to maintenance temozolomide resulted in median overall survival of 21 months, versus 16 months in the temozolomide-alone group, and 5-year overall survival of 13%. Although survival in glioblastoma remains poor, the conditional probability of surviving more than one year past 2 years has repeatedly been shown to be much more favourable than surviving more than one year past initial diagnosis100,105,106.
Inherited genetic risk factors
Single-gene hereditary cancer syndromes such as Li–Fraumeni syndrome, neurofibromatosis, Lynch syndrome, melanoma–neural system tumour syndrome, Ollier disease, and tuberous sclerosis cause a small percentage of diffuse gliomas in adults40,107. Gliomagenesis in these familial cancer syndromes might differ from that in gliomas presumed to be spontaneous108. Gliomas arising in patients with neurofibromatosis type 1 lack mutations in IDH1, IDH2 and the H3 K27M mutation. In addition, high-grade tumours in patients with neurofibromatosis type 1 often harbour loss of ATRX whereas low-grade tumours in these patients often show copy number gain of TERT. The methylation signature of these tumours falls into the LGm6 cluster and this subgroup can probably be subdivided further on the basis of ATRX status109. Gliomas in patients with germline TP53 mutations and Li–Fraumeni syndrome show enrichment of the IDH1 R132C mutation, which accounts for <5% of all IDH mutations, and suggests a pathologic relationship with the TP53 mutation110–112.
In the large majority of patients with glioma who do not have these familial cancer syndromes, 5–10% have a family history of glioma113–115. In fact, first-degree relatives of patients with glioma have a twofold increased risk of developing a primary brain tumour compared to first-degree relatives of people who do not have glioma114–116. Possible reasons for the observed familial clustering of glioma are shared genetic and/or environmental factors. Given the paucity of identified environmental risk factors (described below) and the likelihood that glioma familial aggregation is a polygenic effect117,118, linkage analysis, whole-exome sequencing, and genome-wide association studies (GWAS) are ongoing to improve our understanding of how germline variants contribute to the risk of glioma. A potential risk locus for familial glioma was detected on chromosome 15 in an early, small linkage analysis study of a high-risk family16. A larger Gliogene Consortium study later identified a single locus on chromosome 17 and three other loci on chromosomes 6, 12, and 1817,18. Whole-exome sequencing of POT1, which modulates telomerase activity, identified germline DNA mutations linked to oligodendroglioma in multiply affected families119.
A large GWAS including over 12,000 patients with glioma and 18,000 controls19 identified or validated a total of 25 single nucleotide polymorphisms (SNPs) that were strongly associated with glioma risk in adults17,19,120–122. Of these 25 SNPs, 11 were associated with glioblastoma risk, 19 were associated with the risk of non-glioblastoma glioma, and five of these SNPs were associated with risk of both glioblastoma and non-glioblastoma gliomas. The strongest relative risk is for variants at 8q24.21, which confer more than a sixfold relative risk of developing astrocytoma, IDH-mutant or oligodendroglioma40,123,124.
These 25 risk loci were subsequently included in a model to assess the relative risk and lifetime risk of developing glioma125. In another study, these 25 risk loci were studied in over 1,600 tumours for associations with the IDH mutation, TERT promoter mutation, and 1p19q co-deletion molecular subgroups126. The association of 8q24.21 variants with oligodendrogliomas was validated126. These same researchers also examined univariate and multivariate associations between these risk loci and the WHO 2016 glioma subtypes. The majority of the SNPs are in or near genes known to be involved in specific pathways, summarized in Fig. 3. For example, a SNP at 17p13.1 lies in the 3ʹ untranslated region of TP53 (this SNP alters the polyadenylation signal of TP53, thereby impairing processing of TP53 mRNA) and is associated with the risk of several types of cancer.
Figure 3. Heritable germline risk factors for the WHO 2016 subtypes of adult glioma.
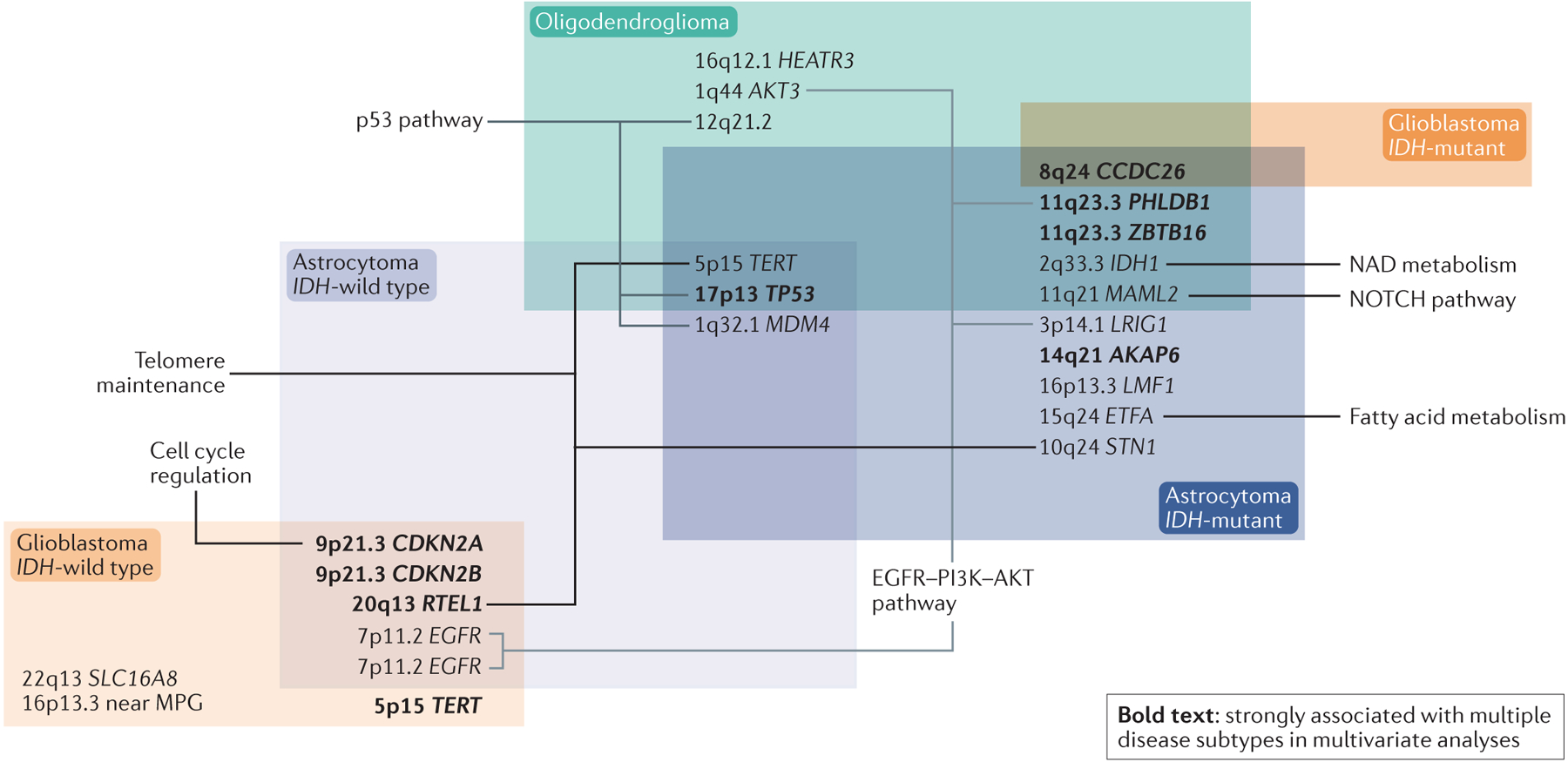
Summary of the relationship between heritable germline risk factors and WHO 2016 classification. Overlapping boxes contain alterations shared by different diffuse glioma tumour subtypes. Involved genes are known to be relevant to the indicated biological pathways, but with a few exceptions the functional consequences of individual single-nucleotide polymorphisms (SNPs) are unknown. SNPs and gene names in bold were strongly associated with more than one WHO 2016 glioma subgroup in multivariant analyses. Data from REFS.19,126.
Telomere maintenance-related genes are important in glioma prognosis and classification. SNPs near TERT and RTEL1 (encoding regulator of telomere elongation helicase 1), both of which genes encode proteins involved in telomere maintenance, were associated with gliomas that had TERT promoter mutations, reinforcing the relationship between SNP genotype and biology126. SNPs near TERT, STN1 and RTEL1 are associated with one to four of the five WHO 2016 groups of diffuse glioma (Fig. 3). Only glioblastoma, IDH-mutant is not associated with these SNPs, probably owing to small sample size (n=68) and, thus, lack of power126. One of these SNPs, in TERT, results in alternate splicing, which results in lack of telomerase activity127.
Given the results of these GWAS as well as a Mendelian randomization study showing that genetically increased telomere length is associated with a fivefold increase in the risk of glioma128, interference with telomere maintenance is clearly a hallmark of gliomagenesis. However, with the exceptions of the TP53 SNP and the telomere-associated SNPs mentioned above, the reader should note that neither the actual functional effects of the glioma risk-associated SNPs nor the developmental window within which these SNPs act to increase glioma risk is known.
Non-genetic risk factors
Very few non-genetic risk factors for development of adult diffuse gliomas have been established despite decades of research129. Only ionizing radiation exposures are considered causal non-genetic risk factors for glioma, but they account for an overall small number of cases. Thus, the specific etiology of most diffuse gliomas remains unknown. Now that more is known about genetic risk factors, it eventually may be possible to understand the interplay between environmental exposures, developmental factors and genetic susceptibilities that may contribute to tumor formation130. Box 1 summarizes the evidence for and against several non-genetic risk factors, as discussed in more detail below.
Box 1.
Non-genetic risk factors for glioma
Association with increased risk
Ionizing radiation from atomic bombs
Therapeutic radiation exposure to the brain during childhood
Radiation in childhood for treatment of tinea capitis
Radiation for childhood leukaemia
CT scans in childhood to doses >50 mGy
Consistently reported association with decreased risk
Allergy and atopic conditions – asthma, hay fever, eczema
Possible but unconfirmed association with increased risk
Non-ionizing radiation such as radiofrequency electromagnetic fields
No clear association
Pesticide exposure
Obesity
Head injury
Cigarette use
Alcohol consumption
Non-genetic risk factors for adult glioma have been thoroughly reviewed in Ostrom et al. (2014)72
Radiation
Exposure to ionizing radiation, particularly in childhood, remains the strongest environmental risk factor for developing diffuse glioma. Ionizing radiation is a known carcinogen that damages DNA which may lead to oncogenesis. This may occur as early as 7 – 9 years after radiation exposure130 and is dose dependent. This is true for both lower dosage ionizing radiation exposures, such as in atomic bomb survivors131, but also for higher therapeutic radiation used to treat childhood infections and cancers. For example, therapeutic radiation has been shown to increase glioma risk ranging from a 3- to 7-fold increase129,132–134.
The roles of various types of non-ionizing radiation exposures for glioma risk including extremely low frequency exposures, microwaves and radiofrequencies used in cell phones have been widely studied, but results are not conclusive because of difficulties in establishing lifetime exposures to such radiation and lack of convincing biologic plausibility for the carcinogenic effects of these exposures129,135–138.
Immune, allergic and atopic conditions
Numerous studies have suggested that history of allergies or other atopic conditions – defined as hay fever, eczema, and asthma – are associated with decreased glioma risk139,140.
The largest study on this topic, from the Glioma International Case-Control (GICC) study confirmed a significant reduced risk of glioma with history of allergy139. Moreover, a recent Mendelian randomization study of 12,488 glioma cases and 18,169 controls reported SNPs associated with atopic dermatitis to be associated with a reduced risk of glioma141. However, the same study did not find correlations between glioma risk and SNPs which have been shown to be associated with other atopic conditions such as asthma, hay fever, and IgE levels141. These conflicting results highlight the complexity of elucidating the relationship between atopy and allergy with glioma risk.
One hypothesis put forth to explain this inverse association is that the atopic state translates into a heightened immunosurveillance, allowing the early detection and suppression of tumor growth140,142,143. Another hypothesis, supported by the inverse relationship seen between elevated immunoglobulin E (IgE) and glioma risk, is the ability of the overactive immune system to eradicate potential environmental toxins, particularly respiratory allergens that may lead to tumor formation144. However, since certain medications that brain tumor patients receive such as steroids and temozolomide affect IgE levels, the potential effects of these exposures on the relationships of IgE and glioma risk warrant further investigation141,145.
In 1977, it was first noted that glioma patients have altered peripheral blood immune cell profiles146. Deficiencies in CD4 T cells, and other lymphocytes147–149, as well as accumulation of myeloid cells150–154 are observed in patients with glioma and is especially dramatic in patients with glioblastoma. Although glioma therapies including steroid, radiation and alkylating agents may adversely impact peripheral blood counts, glioma related immune cell defects can precede and be independent of therapies155. The prognostic importance of these changes is under study and is of obvious importance in the development of immunotherapies. Moreover, the etiologic significance of systemic immune profiles with respect to glioma risk is essentially unknown. New approaches for characterizing peripheral immune profiles based on archival DNA156,157 may be useful to promote large scale epidemiologic exploration that address the etiologic role of systemic immune alterations in gliomagenesis.
Other non-genetic risk factors:
As shown in Box 1, a number of other non-genetic risk factors have not been linked to or not consistently linked to adult glioma risk including, pesticides or chlorinated solvent exposures158–160, obesity/high BMI, cigarette smoking161–165, alcohol consumption164 traumatic brain injury166,167,168,169.
Conclusions
Discoveries over the past 15 years have provided tremendous insight into the molecular pathways and inherited genetic risks associated with gliomagenesis, which have provided a solid foundation for future research. The integration of molecular data into the WHO 2016 classification system for diffuse glioma is critical for both improving prediction of prognosis and tailoring treatment. However, more work is needed to understand the interactions between genetic and non-genetic risk factors in gliomagenesis and to identify risk variants within specific molecular subtypes of adult diffuse glioma (as shown in Fig. 4). Furthermore, the role of patients’ immune status at various stages of the disease process also remains to be elucidated. To address these critical efforts, future studies will need increased international collaboration and large databases of well-annotated biospecimens that include clinical outcome and imaging data as well as detailed exposure histories.
Figure 4. Hypothesized pathways of glioma development.
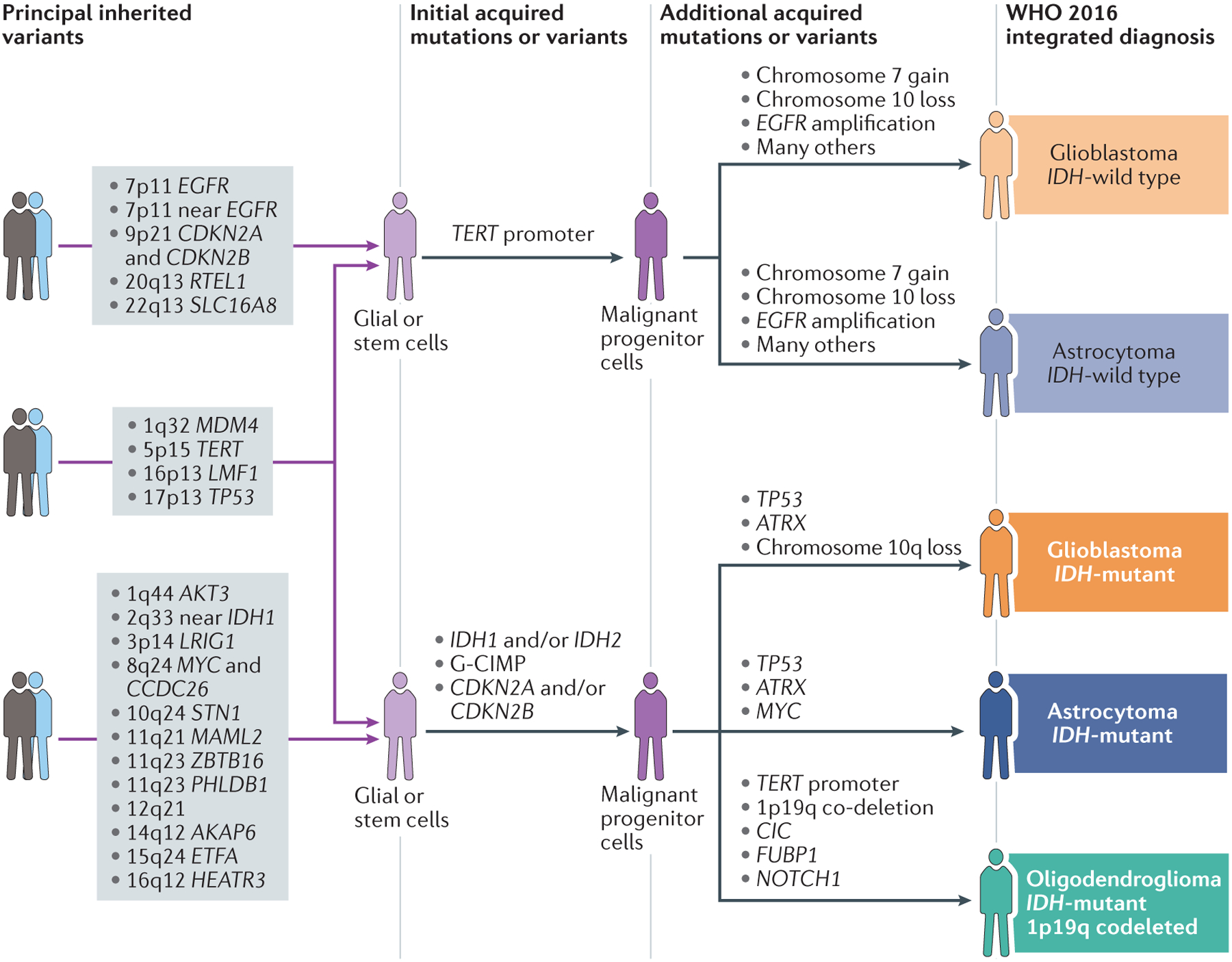
Our current understanding of gliomagenesis involves an interplay of inherited risk variants and acquired alterations. Additional SNPs associated with glioma not included in the figure are: 1p31 (RAVER2); 10q25 (VTI1A); 11q14 (11p14); 16p13 (near MPG); and 16p13 (LMF1). Data from REFS.17,19,120,122,126.
Key points.
Glioma incidence differs by age, sex, ethnicity, and geography. Survival varies by tumour subtype, age, and sex.
In the past decade, multiple discoveries have expanded our understanding of glioma biology and led to a new classification system (WHO 2016) that incorporates molecular alterations with histology for an integrated diagnosis.
The WHO 2016 classification system defines five glioma subtypes that are more homogenous in their clinical outcomes.
Twenty-five risk loci for glioma have been identified as well as several rare inherited mutations that may cause glioma in some families. However, only ionizing radiation is a confirmed environmental risk factor.
Acknowledgements
The authors would like to thank Kenneth Probst for his artistic support and Terri Rice, Sabrina Lin, Gayathri Warrier, Pranathi Chunduru, Yalan Zhang for their analytical support. The authors also thank Joanna Phillips, Jennifer Clarke, Paige Bracci, Quinn Ostrom, Jill Barnholtz-Sloan, Carol Kruchko, Robert Jenkins, Jeanette Eckel-Passow, Arie Perry, Melike Pekmezci, Susan Chang, and Mitchel Berger for help with data and their ongoing insights and intellectual support. The authors’ research work is supported by NIH grant numbers P50CA09725 and the loglio collective (to all four authors), R01 CA207360 (to A.M.M., J.K.W. and M.W.), the Lewis Chair in Brain Tumor Research (held by M.W.) and the Robert Magnin Newman Chair in Neurooncology (held by J.K.W.).
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bray F et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68, 394–424, doi: 10.3322/caac.21492 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer 144, 1941–1953, doi: 10.1002/ijc.31937 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Miranda-Filho A, Pineros M, Soerjomataram I, Deltour I & Bray F Cancers of the brain and CNS: global patterns and trends in incidence. Neuro Oncol 19, 270–280, doi: 10.1093/neuonc/now166 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanai N, Alvarez-Buylla A & Berger MS Neural stem cells and the origin of gliomas. N Engl J Med 353, 811–822, doi: 10.1056/NEJMra043666 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Louis DN et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114, 97–109, doi: 10.1007/s00401-007-0243-4 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The Cancer Genome Atlas Research, N. et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068, doi: 10.1038/nature07385 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Killela PJ et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A 110, 6021–6026, doi: 10.1073/pnas.1303607110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan H et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 360, 765–773, doi: 10.1056/NEJMoa0808710 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sturm D et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22, 425–437, doi: 10.1016/j.ccr.2012.08.024 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Brennan CW et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477, doi: 10.1016/j.cell.2013.09.034 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noushmehr H et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17, 510–522, doi: 10.1016/j.ccr.2010.03.017 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Louis DN et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. 131, 803–820, doi: 10.1007/s00401-016-1545-1 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Alcantara Llaguno S et al. Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nature Neuroscience, doi: 10.1038/s41593-018-0333-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alcantara Llaguno SR & Parada LF Cell of origin of glioma: biological and clinical implications. Br J Cancer 115, 1445–1450, doi: 10.1038/bjc.2016.354 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JH et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 560, 243–247, doi: 10.1038/s41586-018-0389-3 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Paunu N et al. A novel low-penetrance locus for familial glioma at 15q23-q26.3. Cancer Res 62, 3798–3802 (2002). [PubMed] [Google Scholar]
- 17.Shete S et al. Genome-wide high-density SNP linkage search for glioma susceptibility loci: results from the Gliogene Consortium. Cancer Res 71, 7568–7575, doi: 10.1158/0008-5472.CAN-11-0013 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jalali A et al. Targeted sequencing in chromosome 17q linkage region identifies familial glioma candidates in the Gliogene Consortium. Sci Rep 5, 8278, doi: 10.1038/srep08278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melin BS et al. Genome-wide association study of glioma subtypes identifies specific differences in genetic susceptibility to glioblastoma and non-glioblastoma tumors. Nat Genet 49, 789–794, doi: 10.1038/ng.3823 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brat DJ et al. cIMPACT-NOW update 3: recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol, doi: 10.1007/s00401-018-1913-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ostrom QT et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro-Oncology 20, iv1–iv86, doi: 10.1093/neuonc/noy131 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van den Bent MJ Interobserver variation of the histopathological diagnosis in clinical trials on glioma: a clinician’s perspective. Acta Neuropathol 120, 297–304, doi: 10.1007/s00401-010-0725-7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sturm D et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164, 1060–1072, doi: 10.1016/j.cell.2016.01.015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ellison DW et al. Histopathological grading of pediatric ependymoma: reproducibility and clinical relevance in European trial cohorts. J Negat Results Biomed 10, 7, doi: 10.1186/1477-5751-10-7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiestler B et al. Integrated DNA methylation and copy-number profiling identify three clinically and biologically relevant groups of anaplastic glioma. Acta Neuropathol 128, 561–571, doi: 10.1007/s00401-014-1315-x (2014). [DOI] [PubMed] [Google Scholar]
- 26.Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. 372, 2481–2498, doi: 10.1056/NEJMoa1402121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eckel-Passow JE et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med 372, 2499–2508, doi: 10.1056/NEJMoa1407279 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parsons DW et al. An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812, doi: 10.1126/science.1164382 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balss J et al. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116, 597–602, doi: 10.1007/s00401-008-0455-2 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Cohen A, Holmen S & Colman H IDH1 and IDH2 Mutations in Gliomas. Current neurology and neuroscience reports 13, 345–345, doi: 10.1007/s11910-013-0345-4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai A et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol 29, 4482–4490, doi: 10.1200/JCO.2010.33.8715 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki H et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet 47, 458–468, doi: 10.1038/ng.3273 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Jenkins RB et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 66, 9852–9861, doi: 10.1158/0008-5472.CAN-06-1796 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Hartmann C et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol 120, 707–718, doi: 10.1007/s00401-010-0781-z (2010). [DOI] [PubMed] [Google Scholar]
- 35.Weller M et al. Molecular classification of diffuse cerebral WHO grade II/III gliomas using genome- and transcriptome-wide profiling improves stratification of prognostically distinct patient groups. Acta Neuropathol 129, 679–693, doi: 10.1007/s00401-015-1409-0 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Sahm F et al. Farewell to oligoastrocytoma: in situ molecular genetics favor classification as either oligodendroglioma or astrocytoma. Acta Neuropathol 128, 551–559, doi: 10.1007/s00401-014-1326-7 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Pekmezci M et al. Adult infiltrating gliomas with WHO 2016 integrated diagnosis: additional prognostic roles of ATRX and TERT. Acta Neuropathol 133, 1001–1016, doi: 10.1007/s00401-017-1690-1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aoki K et al. Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro Oncol 20, 66–77, doi: 10.1093/neuonc/nox132 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reuss DE et al. IDH mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: a grading problem for WHO. Acta Neuropathol 129, 867–873, doi: 10.1007/s00401-015-1438-8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rice T et al. Understanding inherited genetic risk of adult glioma - a review. Neurooncol Pract 3, 10–16, doi: 10.1093/nop/npv026 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu G et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44, 251–253, doi: 10.1038/ng.1102 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwartzentruber J et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231, doi: 10.1038/nature10833 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Louis DN et al. cIMPACT-NOW update 2: diagnostic clarifications for diffuse midline glioma, H3 K27M-mutant and diffuse astrocytoma/anaplastic astrocytoma, IDH-mutant. Acta Neuropathol 135, 639–642, doi: 10.1007/s00401-018-1826-y (2018). [DOI] [PubMed] [Google Scholar]
- 44.Louis DN et al. cIMPACT-NOW update 1: Not Otherwise Specified (NOS) and Not Elsewhere Classified (NEC). Acta Neuropathol 135, 481–484, doi: 10.1007/s00401-018-1808-0 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Reifenberger G, Wirsching HG, Knobbe-Thomsen CB & Weller M Advances in the molecular genetics of gliomas - implications for classification and therapy. Nat Rev Clin Oncol 14, 434–452, doi: 10.1038/nrclinonc.2016.204 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Ceccarelli M et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 164, 550–563, doi: 10.1016/j.cell.2015.12.028 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leeper HE et al. IDH mutation, 1p19q codeletion and ATRX loss in WHO grade II gliomas. Oncotarget 6, 30295–30305, doi: 10.18632/oncotarget.4497 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wiestler B et al. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol 126, 443–451, doi: 10.1007/s00401-013-1156-z (2013). [DOI] [PubMed] [Google Scholar]
- 49.Capper D et al. DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474, doi: 10.1038/nature26000 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Christensen BC et al. DNA methylation, isocitrate dehydrogenase mutation, and survival in glioma. J Natl Cancer Inst 103, 143–153, doi: 10.1093/jnci/djq497 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Turcan S et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483, doi: 10.1038/nature10866 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hovestadt V et al. Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol 125, 913–916, doi: 10.1007/s00401-013-1126-5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerson SL MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer 4, 296–307, doi: 10.1038/nrc1319 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Wick W et al. MGMT testing—the challenges for biomarker-based glioma treatment. Nature Reviews Neurology 10, 372, doi: 10.1038/nrneurol.2014.100 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Hegi ME et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352, 997–1003, doi: 10.1056/NEJMoa043331 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Weller M et al. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J Clin Oncol 27, 5743–5750, doi: 10.1200/JCO.2009.23.0805 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Gilbert MR et al. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol 31, 4085–4091, doi: 10.1200/JCO.2013.49.6968 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malmstrom A et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol 13, 916–926, doi: 10.1016/S1470-2045(12)70265-6 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Stupp R et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10, 459–466, doi: 10.1016/S1470-2045(09)70025-7 (2009). [DOI] [PubMed] [Google Scholar]
- 60.Baumert BG et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033–26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol 17, 1521–1532, doi: 10.1016/S1470-2045(16)30313-8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bell EH et al. Association of MGMT Promoter Methylation Status With Survival Outcomes in Patients With High-Risk Glioma Treated With Radiotherapy and Temozolomide: An Analysis From the NRG Oncology/RTOG 0424 Trial. JAMA Oncol, doi: 10.1001/jamaoncol.2018.1977 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leu S et al. IDH/MGMT-driven molecular classification of low-grade glioma is a strong predictor for long-term survival. Neuro-Oncology 15, 469–479, doi: 10.1093/neuonc/nos317 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wick W et al. Prognostic or predictive value of MGMT promoter methylation in gliomas depends on IDH1 mutation. Neurology 81, 1515–1522, doi: 10.1212/WNL.0b013e3182a95680 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Yang P et al. IDH mutation and MGMT promoter methylation in glioblastoma: results of a prospective registry. Oncotarget 6, 40896–40906, doi: 10.18632/oncotarget.5683 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nguyen HN et al. Human TERT promoter mutation enables survival advantage from MGMT promoter methylation in IDH1 wild-type primary glioblastoma treated by standard chemoradiotherapy. Neuro Oncol 19, 394–404, doi: 10.1093/neuonc/now189 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Perry JR et al. Short-Course Radiation plus Temozolomide in Elderly Patients with Glioblastoma. N Engl J Med 376, 1027–1037, doi: 10.1056/NEJMoa1611977 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Wick W et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol 13, 707–715, doi: 10.1016/S1470-2045(12)70164-X (2012). [DOI] [PubMed] [Google Scholar]
- 68.Hegi ME et al. MGMT Promoter Methylation Cutoff with Safety Margin for Selecting Glioblastoma Patients into Trials Omitting Temozolomide: A Pooled Analysis of Four Clinical Trials. Clin Cancer Res, doi: 10.1158/1078-0432.CCR-18-3181 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reis GF et al. CDKN2A loss is associated with shortened overall survival in lower-grade (World Health Organization Grades II-III) astrocytomas. J Neuropathol Exp Neurol 74, 442–452, doi: 10.1097/NEN.0000000000000188 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shirahata M et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol 136, 153–166, doi: 10.1007/s00401-018-1849-4 (2018). [DOI] [PubMed] [Google Scholar]
- 71.Korshunov A et al. Integrated molecular characterization of IDH-mutant glioblastomas. Neuropathol Appl Neurobiol 45, 108–118, doi: 10.1111/nan.12523 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Ostrom QT et al. The epidemiology of glioma in adults: a “state of the science” review. Neuro Oncol 16, 896–913, doi: 10.1093/neuonc/nou087 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ostrom QT et al. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol 15 Suppl 2, ii1–56, doi: 10.1093/neuonc/not151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Arora RS et al. Age-incidence patterns of primary CNS tumors in children, adolescents, and adults in England. Neuro Oncol 11, 403–413, doi: 10.1215/15228517-2008-097 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee CH, Jung KW, Yoo H, Park S & Lee SH Epidemiology of primary brain and central nervous system tumors in Korea. J Korean Neurosurg Soc 48, 145–152, doi: 10.3340/jkns.2010.48.2.145 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dobes M et al. Increasing incidence of glioblastoma multiforme and meningioma, and decreasing incidence of Schwannoma (2000–2008): Findings of a multicenter Australian study. Surg Neurol Int 2, 176, doi: 10.4103/2152-7806.90696 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gousias K et al. Descriptive epidemiology of cerebral gliomas in northwest Greece and study of potential predisposing factors, 2005–2007. Neuroepidemiology 33, 89–95, doi: 10.1159/000222090 (2009). [DOI] [PubMed] [Google Scholar]
- 78.Forman D et al. Cancer incidence in five continents, Volume X. Lyon, Geneva: International Agency for Research on Cancer; Distributed by WHO Press. (2014).
- 79.Leece R et al. Global incidence of malignant brain and other central nervous system tumors by histology, 2003–2007. Neuro Oncol 19, 1553–1564, doi: 10.1093/neuonc/nox091 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jacobs D et al. Leveraging Ethnic Group Incidence Variation to Investigate Genetic Susceptibility to Glioma: A Novel Candidate SNP Approach. 3, doi: 10.3389/fgene.2012.00203 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ostrom QT, Cote DJ, Ascha M, Kruchko C & Barnholtz-Sloan JS Adult Glioma Incidence and Survival by Race or Ethnicity in the United States From 2000 to 2014. JAMA Oncol, e181789, doi: 10.1001/jamaoncol.2018.1789 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Noone AM et al. SEER Cancer Statistics Review, 1975–2015, <https://seer.cancer.gov/csr/1975_2015/> (2018).
- 83.Little MP et al. Mobile phone use and glioma risk: comparison of epidemiological study results with incidence trends in the United States. BMJ 344, e1147, doi: 10.1136/bmj.e1147 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Deltour I et al. Mobile phone use and incidence of glioma in the Nordic countries 1979–2008: consistency check. Epidemiology 23, 301–307, doi: 10.1097/EDE.0b013e3182448295 (2012). [DOI] [PubMed] [Google Scholar]
- 85.Surawicz TS et al. Descriptive epidemiology of primary brain and CNS tumors: Results from the Central Brain Tumor Registry of the United States, 1990–1994. Neuro-Oncology 1, 14–25, doi: 10.1093/neuonc/1.1.14 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ostrom QT et al. Sex-specific glioma genome-wide association study identifies new risk locus at 3p21.31 in females, and finds sex-differences in risk at 8q24.21. Sci Rep 8, 7352, doi: 10.1038/s41598-018-24580-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Crocetti E et al. Epidemiology of glial and non-glial brain tumours in Europe. European Journal of Cancer 48, 1532–1542, doi: 10.1016/j.ejca.2011.12.013 (2012). [DOI] [PubMed] [Google Scholar]
- 88.Ho VK et al. Changing incidence and improved survival of gliomas. Eur J Cancer 50, 2309–2318, doi: 10.1016/j.ejca.2014.05.019 (2014). [DOI] [PubMed] [Google Scholar]
- 89.Benson VS, Kirichek O, Beral V & Green J Menopausal hormone therapy and central nervous system tumor risk: large UK prospective study and meta-analysis. Int J Cancer 136, 2369–2377, doi: 10.1002/ijc.29274 (2015). [DOI] [PubMed] [Google Scholar]
- 90.Kabat GC, Park Y, Hollenbeck AR, Schatzkin A & Rohan TE Reproductive Factors and Exogenous Hormone Use and Risk of Adult Glioma in Women in the NIH-AARP Diet and Health Study. International journal of cancer. Journal international du cancer 128, 944–950, doi: 10.1002/ijc.25413 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zong H et al. Reproductive factors in relation to risk of brain tumors in women: an updated meta-analysis of 27 independent studies. Tumour Biol 35, 11579–11586, doi: 10.1007/s13277-014-2448-1 (2014). [DOI] [PubMed] [Google Scholar]
- 92.Wigertz A et al. Risk of brain tumors associated with exposure to exogenous female sex hormones. Am J Epidemiol 164, 629–636, doi: 10.1093/aje/kwj254 (2006). [DOI] [PubMed] [Google Scholar]
- 93.Sanai N, Mirzadeh Z & Berger MS Functional outcome after language mapping for glioma resection. N Engl J Med 358, 18–27, doi: 10.1056/NEJMoa067819 (2008). [DOI] [PubMed] [Google Scholar]
- 94.Sanai N, Polley MY, McDermott MW, Parsa AT & Berger MS An extent of resection threshold for newly diagnosed glioblastomas. J Neurosurg 115, 3–8, doi: 10.3171/2011.2.JNS10998 and 10.3171/2011.7.JNS10238 (2011). [DOI] [PubMed] [Google Scholar]
- 95.Marko NF et al. Extent of resection of glioblastoma revisited: personalized survival modeling facilitates more accurate survival prediction and supports a maximum-safe-resection approach to surgery. J Clin Oncol 32, 774–782, doi: 10.1200/JCO.2013.51.8886 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jakola AS et al. Comparison of a strategy favoring early surgical resection vs a strategy favoring watchful waiting in low-grade gliomas. JAMA 308, 1881–1888, doi: 10.1001/jama.2012.12807 (2012). [DOI] [PubMed] [Google Scholar]
- 97.Claus EB et al. Survival and low-grade glioma: the emergence of genetic information. Neurosurg Focus 38, E6, doi: 10.3171/2014.10.FOCUS12367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stupp R, van den Bent MJ & Hegi ME Optimal role of temozolomide in the treatment of malignant gliomas. Curr Neurol Neurosci Rep 5, 198–206 (2005). [DOI] [PubMed] [Google Scholar]
- 99.Dubrow R et al. Time trends in glioblastoma multiforme survival: the role of temozolomide. Neuro-Oncology 15, 1750–1761, doi: 10.1093/neuonc/not122 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Johnson DR, Ma DJ, Buckner JC & Hammack JE Conditional probability of long-term survival in glioblastoma: a population-based analysis. Cancer 118, 5608–5613, doi: 10.1002/cncr.27590 (2012). [DOI] [PubMed] [Google Scholar]
- 101.Darefsky AS, King JT Jr. & Dubrow R Adult glioblastoma multiforme survival in the temozolomide era: a population-based analysis of Surveillance, Epidemiology, and End Results registries. Cancer 118, 2163–2172, doi: 10.1002/cncr.26494 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Koshy M et al. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J Neurooncol 107, 207–212, doi: 10.1007/s11060-011-0738-7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chinot OL et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med 370, 709–722, doi: 10.1056/NEJMoa1308345 (2014). [DOI] [PubMed] [Google Scholar]
- 104.Gilbert MR et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 370, 699–708, doi: 10.1056/NEJMoa1308573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Porter KR, McCarthy BJ, Berbaum ML & Davis FG Conditional survival of all primary brain tumor patients by age, behavior, and histology. Neuroepidemiology 36, 230–239, doi: 10.1159/000327752 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Farah P et al. Conditional survival after diagnosis with malignant brain and central nervous system tumor in the United States, 1995–2012. Journal of Neuro-Oncology 128, 419–429, doi: 10.1007/s11060-016-2127-8 (2016). [DOI] [PubMed] [Google Scholar]
- 107.Lindor NM et al. Concise handbook of familial cancer susceptibility syndromes - second edition. J Natl Cancer Inst Monogr, 1–93, doi: 10.1093/jncimonographs/lgn001 (2008). [DOI] [PubMed] [Google Scholar]
- 108.Kyritsis AP, Bondy ML, Rao JS & Sioka C Inherited predisposition to glioma. Neuro Oncol 12, 104–113, doi: 10.1093/neuonc/nop011 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.D’Angelo F et al. The molecular landscape of glioma in patients with Neurofibromatosis 1. Nat Med 25, 176–187, doi: 10.1038/s41591-018-0263-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hartmann C et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 118, 469–474, doi: 10.1007/s00401-009-0561-9 (2009). [DOI] [PubMed] [Google Scholar]
- 111.Hayes J et al. Genomic analysis of the origins and evolution of multicentric diffuse lower-grade gliomas. Neuro Oncol 20, 632–641, doi: 10.1093/neuonc/nox205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ohgaki H & Kleihues P The definition of primary and secondary glioblastoma. Clin Cancer Res 19, 764–772, doi: 10.1158/1078-0432.CCR-12-3002 (2013). [DOI] [PubMed] [Google Scholar]
- 113.Robertson LB et al. Survey of familial glioma and role of germline p16INK4A/p14ARF and p53 mutation. Fam Cancer 9, 413–421, doi: 10.1007/s10689-010-9346-5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Malmer B, Gronberg H, Bergenheim AT, Lenner P & Henriksson R Familial aggregation of astrocytoma in northern Sweden: an epidemiological cohort study. Int J Cancer 81, 366–370 (1999). [DOI] [PubMed] [Google Scholar]
- 115.Wrensch M et al. Familial and personal medical history of cancer and nervous system conditions among adults with glioma and controls. Am J Epidemiol 145, 581–593 (1997). [DOI] [PubMed] [Google Scholar]
- 116.Hemminki K, Tretli S, Sundquist J, Johannesen TB & Granstrom C Familial risks in nervous-system tumours: a histology-specific analysis from Sweden and Norway. Lancet Oncol 10, 481–488, doi: 10.1016/S1470-2045(09)70076-2 (2009). [DOI] [PubMed] [Google Scholar]
- 117.Malmer B et al. Genetic epidemiology of glioma. Br J Cancer 84, 429–434, doi: 10.1054/bjoc.2000.1612 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.de Andrade M et al. Segregation analysis of cancer in families of glioma patients. Genet Epidemiol 20, 258–270, doi: 10.1002/1098-2272(200102)20:2<258::AID-GEPI8>3.0.CO;2-N (2001). [DOI] [PubMed] [Google Scholar]
- 119.Bainbridge MN et al. Germline mutations in shelterin complex genes are associated with familial glioma. J Natl Cancer Inst 107, 384, doi: 10.1093/jnci/dju384 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wrensch M et al. Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat Genet 41, 905–908, doi: 10.1038/ng.408 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kinnersley B et al. Genome-wide association study identifies multiple susceptibility loci for glioma. Nat Commun 6, 8559, doi: 10.1038/ncomms9559 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sanson M et al. Chromosome 7p11.2 (EGFR) variation influences glioma risk. Hum Mol Genet 20, 2897–2904, doi: 10.1093/hmg/ddr192 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jenkins RB et al. A low-frequency variant at 8q24.21 is strongly associated with risk of oligodendroglial tumors and astrocytomas with IDH1 or IDH2 mutation. Nat Genet 44, 1122–1125, doi: 10.1038/ng.2388 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Enciso-Mora V et al. Deciphering the 8q24.21 association for glioma. Hum Mol Genet 22, 2293–2302, doi: 10.1093/hmg/ddt063 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Eckel-Passow JE et al. Using germline variants to estimate glioma and subtype risks. Neuro Oncol 21, 451–461, doi: 10.1093/neuonc/noz009 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Labreche K et al. Diffuse gliomas classified by 1p/19q co-deletion, TERT promoter and IDH mutation status are associated with specific genetic risk loci. Acta Neuropathol 135, 743–755, doi: 10.1007/s00401-018-1825-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Killedar A et al. A Common Cancer Risk-Associated Allele in the hTERT Locus Encodes a Dominant Negative Inhibitor of Telomerase. PLoS Genet 11, e1005286, doi: 10.1371/journal.pgen.1005286 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Telomeres Mendelian Randomization C et al. Association Between Telomere Length and Risk of Cancer and Non-Neoplastic Diseases: A Mendelian Randomization Study. JAMA Oncol 3, 636–651, doi: 10.1001/jamaoncol.2016.5945 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ostrom QT, Gittleman H, Stetson L, Virk S & Barnholtz-Sloan J in Intracranial Gliomas. Part I – Surgery Vol. 30 (ed Muragaki Y Chernov MF, Kesari S, McCutcheon IE) 1–11 (Karger, 2018). [DOI] [PubMed] [Google Scholar]
- 130.Wang LE et al. Polymorphisms of DNA repair genes and risk of glioma. Cancer Res 64, 5560–5563, doi: 10.1158/0008-5472.CAN-03-2181 (2004). [DOI] [PubMed] [Google Scholar]
- 131.Preston DL et al. Tumors of the nervous system and pituitary gland associated with atomic bomb radiation exposure. J Natl Cancer Inst 94, 1555–1563 (2002). [DOI] [PubMed] [Google Scholar]
- 132.Sadetzki S et al. Long-term follow-up for brain tumor development after childhood exposure to ionizing radiation for tinea capitis. Radiat Res 163, 424–432 (2005). [DOI] [PubMed] [Google Scholar]
- 133.Neglia JP et al. New primary neoplasms of the central nervous system in survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst 98, 1528–1537, doi: 10.1093/jnci/djj411 (2006). [DOI] [PubMed] [Google Scholar]
- 134.Pearce MS et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: a retrospective cohort study. Lancet 380, 499–505, doi: 10.1016/S0140-6736(12)60815-0 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Humans, I. W. G. o. t. E. o. C. R. t. Non-ionizing radiation, Part 2: Radiofrequency electromagnetic fields. IARC Monogr Eval Carcinog Risks Hum 102, 1–460 (2013). [PMC free article] [PubMed] [Google Scholar]
- 136.Grayson JK Radiation exposure, socioeconomic status, and brain tumor risk in the US Air Force: a nested case-control study. Am J Epidemiol 143, 480–486 (1996). [DOI] [PubMed] [Google Scholar]
- 137.Cardis E et al. The INTERPHONE study: design, epidemiological methods, and description of the study population. Eur J Epidemiol 22, 647–664, doi: 10.1007/s10654-007-9152-z (2007). [DOI] [PubMed] [Google Scholar]
- 138.Vila J et al. Occupational exposure to high-frequency electromagnetic fields and brain tumor risk in the INTEROCC study: An individualized assessment approach. Environ Int 119, 353–365, doi: 10.1016/j.envint.2018.06.038 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Amirian ES et al. Approaching a Scientific Consensus on the Association between Allergies and Glioma Risk: A Report from the Glioma International Case-Control Study. Cancer Epidemiol Biomarkers Prev 25, 282–290, doi: 10.1158/1055-9965.EPI-15-0847 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Linos E, Raine T, Alonso A & Michaud D Atopy and risk of brain tumors: a meta-analysis. J Natl Cancer Inst 99, 1544–1550, doi: 10.1093/jnci/djm170 (2007). [DOI] [PubMed] [Google Scholar]
- 141.Disney-Hogg L et al. Impact of atopy on risk of glioma: a Mendelian randomisation study. BMC Med 16, 42, doi: 10.1186/s12916-018-1027-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wiemels JL et al. Reduced immunoglobulin E and allergy among adults with glioma compared with controls. Cancer Res 64, 8468–8473, doi: 10.1158/0008-5472.CAN-04-1706 (2004). [DOI] [PubMed] [Google Scholar]
- 143.Wiemels JL et al. History of allergies among adults with glioma and controls. Int J Cancer 98, 609–615 (2002). [DOI] [PubMed] [Google Scholar]
- 144.Schwartzbaum J et al. Association between prediagnostic IgE levels and risk of glioma. J Natl Cancer Inst 104, 1251–1259, doi: 10.1093/jnci/djs315 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wiemels JL et al. IgE, allergy, and risk of glioma: update from the San Francisco Bay Area Adult Glioma Study in the temozolomide era. Int J Cancer 125, 680–687, doi: 10.1002/ijc.24369 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Brooks WH, Roszman TL, Mahaley MS & Woosley RE Immunobiology of primary intracranial tumours. II. Analysis of lymphocyte subpopulations in patients with primary brain tumours. Clin Exp Immunol 29, 61–66 (1977). [PMC free article] [PubMed] [Google Scholar]
- 147.Dix AR, Brooks WH, Roszman TL & Morford LA Immune defects observed in patients with primary malignant brain tumors. J Neuroimmunol 100, 216–232 (1999). [DOI] [PubMed] [Google Scholar]
- 148.Grossman SA et al. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin Cancer Res 17, 5473–5480, doi: 10.1158/1078-0432.CCR-11-0774 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hughes MA, Parisi M, Grossman S & Kleinberg L Primary brain tumors treated with steroids and radiotherapy: low CD4 counts and risk of infection. Int J Radiat Oncol Biol Phys 62, 1423–1426, doi: 10.1016/j.ijrobp.2004.12.085 (2005). [DOI] [PubMed] [Google Scholar]
- 150.Bambury RM et al. The association of pre-treatment neutrophil to lymphocyte ratio with overall survival in patients with glioblastoma multiforme. J Neurooncol 114, 149–154, doi: 10.1007/s11060-013-1164-9 (2013). [DOI] [PubMed] [Google Scholar]
- 151.Dubinski D et al. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro Oncol 18, 807–818, doi: 10.1093/neuonc/nov280 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gabrusiewicz K et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 1, doi: 10.1172/jci.insight.85841 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gielen PR et al. Increase in both CD14-positive and CD15-positive myeloid-derived suppressor cell subpopulations in the blood of patients with glioma but predominance of CD15-positive myeloid-derived suppressor cells in glioma tissue. J Neuropathol Exp Neurol 74, 390–400, doi: 10.1097/NEN.0000000000000183 (2015). [DOI] [PubMed] [Google Scholar]
- 154.Mason M et al. Neutrophil-lymphocyte ratio dynamics during concurrent chemoradiotherapy for glioblastoma is an independent predictor for overall survival. J Neurooncol 132, 463–471, doi: 10.1007/s11060-017-2395-y (2017). [DOI] [PubMed] [Google Scholar]
- 155.Chongsathidkiet P et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med 24, 1459–1468, doi: 10.1038/s41591-018-0135-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wiencke JK et al. Epigenetic biomarkers of T-cells in human glioma. Epigenetics 7, 1391–1402, doi: 10.4161/epi.22675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wiencke JK et al. Immunomethylomic approach to explore the blood neutrophil lymphocyte ratio (NLR) in glioma survival. Clin Epigenetics 9, 10, doi: 10.1186/s13148-017-0316-8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ruder AM et al. The Upper Midwest Health Study: industry and occupation of glioma cases and controls. Am J Ind Med 55, 747–755, doi: 10.1002/ajim.22085 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yiin JH et al. The Upper Midwest Health Study: a case-control study of pesticide applicators and risk of glioma. Environ Health 11, 39, doi: 10.1186/1476-069X-11-39 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Li HX et al. A Meta-Analysis of Association Between Pesticides Exposure and Glioma Risk in Adults. J Craniofac Surg 26, e672–673, doi: 10.1097/SCS.0000000000001707 (2015). [DOI] [PubMed] [Google Scholar]
- 161.Wiedmann MKH et al. Overweight, obesity and height as risk factors for meningioma, glioma, pituitary adenoma and nerve sheath tumor: a large population-based prospective cohort study. Acta Oncol 56, 1302–1309, doi: 10.1080/0284186X.2017.1330554 (2017). [DOI] [PubMed] [Google Scholar]
- 162.Kitahara CM, Gamborg M, Rajaraman P, Sorensen TI & Baker JL A prospective study of height and body mass index in childhood, birth weight, and risk of adult glioma over 40 years of follow-up. Am J Epidemiol 180, 821–829, doi: 10.1093/aje/kwu203 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Niedermaier T et al. Body mass index, physical activity, and risk of adult meningioma and glioma: A meta-analysis. Neurology 85, 1342–1350, doi: 10.1212/WNL.0000000000002020 (2015). [DOI] [PubMed] [Google Scholar]
- 164.Braganza MZ et al. Cigarette smoking, alcohol intake, and risk of glioma in the NIH-AARP Diet and Health Study. Br J Cancer 110, 242–248, doi: 10.1038/bjc.2013.611 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Li HX et al. Cigarette smoking and risk of adult glioma: a meta-analysis of 24 observational studies involving more than 2.3 million individuals. Onco Targets Ther 9, 3511–3523, doi: 10.2147/OTT.S99713 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Inskip PD, Mellemkjaer L, Gridley G & Olsen JH Incidence of intracranial tumors following hospitalization for head injuries (Denmark). Cancer Causes Control 9, 109–116 (1998). [DOI] [PubMed] [Google Scholar]
- 167.Nygren C et al. Primary brain tumors following traumatic brain injury--a population-based cohort study in Sweden. Cancer Causes Control 12, 733–737 (2001). [DOI] [PubMed] [Google Scholar]
- 168.Chen YH, Keller JJ, Kang JH & Lin HC Association between traumatic brain injury and the subsequent risk of brain cancer. J Neurotrauma 29, 1328–1333, doi: 10.1089/neu.2011.2235 (2012). [DOI] [PubMed] [Google Scholar]
- 169.Munch TN, Gortz S, Wohlfahrt J & Melbye M The long-term risk of malignant astrocytic tumors after structural brain injury--a nationwide cohort study. Neuro Oncol 17, 718–724, doi: 10.1093/neuonc/nou312 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]