

Abstract
Degradation of three-dimensional hydrogels is known to regulate many cellular behaviors. Accordingly, several elegant approaches have been used to render hydrogels degradable by cell-secreted proteases. However, existing hydrogel systems are limited in their ability to simultaneously and quantitatively tune two aspects of hydrogel degradability: cleavage rate (the rate at which individual chemical bonds are cleaved) and degraded hydrogel architecture (the network structure during degradation). Using standard peptide engineering approaches, we alter the proteolytic kinetics of the polymer cleavage rate to tune gel degradation time from less than 12 h to greater than 9 days. Independently, we vary the cross-linker functionality to achieve network architectures that initially have identical molecular weight between cross-links but upon degradation are designed to release between 5% and 100% of the polymer. Confirming the biological relevance of both parameters, formation of vascular-like structures by endothelial cells is regulated both by bond cleavage rate and by degraded hydrogel architecture. This strategy to fine-tune different aspects of hydrogel degradability has applications in cell culture, regenerative medicine, and drug delivery.
Graphical Abstract
Hydrogel-based materials that mimic the native extracellular matrix (ECM) have been employed as tissue engineering scaffolds,1,2 injectable carriers for stem cell therapies,3 and three-dimensional (3D) cell culture platforms.4 In the native cellular microenvironment, the ECM provides biochemical and biophysical cues that regulate cell phenotype. In synthetic systems, material properties such as stiffness, viscoelasticity, adhesive ligand interactions, and growth factor presentation influence cellular behaviors including proliferation, differentiation, and migration.5 By embedding cells within 3D hydrogels, researchers can recapitulate aspects of the native cellular microenvironment that are absent from traditional two-dimensional (2D) culture.6 A particular advantage provided by 3D culture is the ability to study matrix degradation and remodeling. While 2D culture permits unrestricted spreading, proliferation, and migration, matrix remodeling of 3D hydrogels is required to facilitate these same behaviors, which are crucial to processes such as blood vessel sprouting and cancer metastasis.6
The degradation of bulk hydrogel materials is a function of both the cleavage rate of individual chemical bonds in the polymers comprising the network and how the individual polymers are connected within the network (i.e., the network architecture). The simplest method to alter the degradation rate of a hydrogel is to increase the cross-linking density of the network, either by increasing the polymer content or by decreasing the molecular weight between cross-links. Both approaches result in a smaller network mesh size, which decreases transport through the hydrogel. This decreased transport is thought to be the primary cause for decreased degradation. However, this change in network mesh size will also alter the biomaterial mechanical properties, which are well-known to impact the biology of encapsulated cells.5,6 As an alternative approach, another common method to control the degradation rate of hydrogels is to incorporate peptides into the network that are targets for cell-secreted proteases. Our group and others have demonstrated that changing either the concentration of these peptides or their amino acid target sequences, which dictate their rate of proteolytic cleavage, can be used to control the rate of gel degradation.7–10
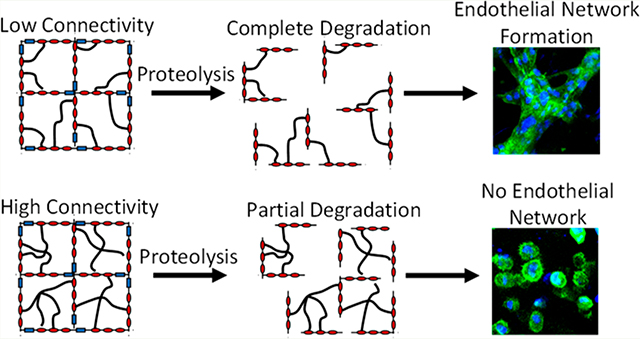
While all of these degradation strategies have been widely useful for a number of interesting biomaterial designs, the network structure of the hydrogel during degradation has been a typically overlooked parameter. Depending on the initial network connectivity, cleavage of chemical bonds within the hydrogel can result in the release of polymer fragments. This loss of polymer content can open up space within the hydrogel to enable cell spreading, migration, proliferation, and cell–cell contact. Thus, it is important to develop new strategies to control the network structure during degradation, without altering the initial polymer content, the initial hydrogel mesh size, or the rate of chemical bond cleavage. To accomplish this goal, we combine the use of protease-targeted peptides with synthetic cross-linkers of varying functionality to control the initial network connectivity. We reasoned that networks formed with bifunctional cross-linkers would have a much greater release of polymer fragments during degradation than networks formed with tetrafunctional cross-linkers, even though both networks have similar polymer concentration, similar mesh size, and similar rate of chemical bond cleavage (Figure 1).
Figure 1.

Simplified schematic for an idealized hydrogel network illustrating control of degraded network architecture by varying cross-linker functionality. After a given number of bonds are cleaved, (A) significant fragments of a network formed with bifunctional cross-linkers are released from the hydrogel, whereas (B) the same extent of bond cleavage in a network formed with tetrafunctional cross-linkers results in no fragment release.
To experimentally test this idea, we describe a modular approach using protein engineering to independently tune chemical bond cleavage rate and degraded network architecture in response to the proteolytic enzyme urokinase plasminogen activator (uPA). The hydrogels are comprised of engineered elastin-like proteins (ELPs) with alternating elastin-like structural domains and variable domains that contain uPA cleavage sites or cell-adhesive sites (Figures 2A and S1). The previously reported uPA-cleavable domains were derived from a combinatorial peptide screen and were shown to have different proteolytic kinetics in response to uPA treatment,9,11 thereby enabling control of the chemical bond cleavage rate independent of the hydrogel network connectivity. The elastin-like domains contain lysine residues that are functionalized with azides via aqueous diazo transfer to permit cross-linking of the hydrogels by the bioorthogonal strain-promoted azide–alkyne cycloaddition (SPAAC) reaction (Figures S2 and S3).12 The ELPs were designed such that three potential cross-linking sites were located between each uPA-cleavable domain. To control the initial network architecture, we cross-linked the ELPs using multiarm poly(ethylene glycol) (PEG) cross-linkers with different degrees of functionality (Figures 2B,C and S4). The molecular weights of the PEG cross-linkers were chosen such that the average PEG molecular weight between two ELP cross-link points was ~10 kDa (Figure 2B). Thus, all hydrogel formulations have approximately identical cross-link densities (~7.2 mM), identical concentrations of cleavable peptides (~2.6 mM), and identical molecular weights between cross-links (~13.5 kDa). We hypothesized that controlling the network connectivity in this way would allow control of the hydrogel architecture during degradation. Thus, hydrogel networks formed with lower functionality cross-linkers are expected to release more polymer fragments and eventually lose their percolating architecture. In contrast, hydrogel networks formed with higher functionality cross-linkers will release fewer polymer fragments and can maintain a percolating network architecture, even after cleavage of all proteolytically degradable sites (Figure 2C).
Figure 2.

(A) Engineered elastin-like proteins (ELPs) enable simultaneous tuning of peptide bond cleavage rate, cell adhesivity, and hydrogel network architecture through modular design. Primary amines on the ELPs are functionalized with azides to permit cross-linking via SPAAC. (B) Multiarm PEGs functionalized with bicyclononynes serve as cross-linkers to control network connectivity while keeping molecular weight between cross-links (and hence mesh size) constant. (C) The maximal degradable fraction of the network is controlled by altering cross-linker functionality. Increasing the cross-linker functionality increases the redundant connections within the network, decreasing the fraction of the network able to be released and retaining the percolating achitecture.
The probability of a cross-linked network remaining after chemical bond cleavage can be estimated from percolation theory. The critical gelation point (pg), i.e., the percent of productive cross-links needed to form a percolating network, can be estimated from the Flory–Stockmayer equation (Table S1)13,14
| (1) |
where r is the stoichiometric ratio of the reactive groups; fA is the number of reactive groups on hydrogel component A (the azides on the ELPs); and f B is the number of reactive groups on hydrogel component B (the bicyclononynes (BCN) on the PEG cross-linkers). Estimating the degree of azide functionalization by NMR reveals that ~2.75 potential cross-linking sites exist between each uPA cleavage site (Figure S2). In our system, this means that for a bifunctional PEG cross-linker (PEG-bis-BCN) pg is ~0.89 after all the proteolytic sites have been cleaved (Table S1). Given that this theoretical treatment assumes ideal network connectivity, the estimate that 89% cross-linking efficiency is sufficient to maintain a percolating network is likely too low. This led us to hypothesize that a bifunctional cross-linker may permit complete loss of network percolation. In contrast, an 8-arm cross-linker would still yield a predicted pg of ~0.34 after full proteolysis, suggesting that an 8-arm cross-linker would largely retain the percolating architecture even after complete proteolysis.
Hydrogel stiffness is known to influence cellular behavior,5,6 so the stiffness of the hydrogels post-cross-linking should ideally be similar for all networks studied to prevent introduction of a confounding variable. As predicted by ideal elastic theory,15 controlling PEG molecular weight to ensure equivalent cross-link density in all formulations resulted in hydrogels with similar shear moduli on the order of 103 Pa, independent of network architecture or proteolytic cleavage kinetics (Figure S5A). This choice of PEG molecular weights also ensured that the total polymer content of the hydrogels was constant across all conditions. As expected, all hydrogel formulations exhibited predominantly elastic mechanical properties (Figure S5B,C).
Another potential confounding variable is local heterogeneity within the network architecture, resulting in variations in cross-link density at the microscopic scale that can impact hydrogel mesh size and thus alter the diffusion of proteases and affect hydrogel degradation. Step-growth cross-linked polymer networks are reported to form nearly homogeneous networks,16 in contrast to chain-growth networks that are highly heterogeneous.17 In addition to being highly cytocompatible, SPAAC cross-linking has been shown to yield approximately ideal step-growth networks.18 Thus, we do not expect significant heteroegeneities to be present in the gels used in this study.
To quantify the effects of proteolytic cleavage kinetics and initial network architecture on the release of polymer fragments over time, the uPA-cleavable ELPs were labeled with a Cy5 fluorescent dye. Labeled ELP hydrogels were then incubated with human uPA in phosphate-buffered saline (PBS) at 37 °C. At designated time points, a sample of the buffer was collected for fluorescence measurements to assess release of material from the hydrogel network. After the final time point, any remaining hydrogel material was treated with 2.5% (w/v) trypsin to fully degrade the gels, allowing for normalization to total fluorescence signal to calculate the fraction of the gel that had been released in response to uPA.
Hydrogels were prepared using the u1 ELP sequence (Figure 2A), which was previously shown to facilitate rapid proteolysis.9 Consistent with our predictions from percolation theory, u1 ELP gels cross-linked with bifunctional PEGs degraded completely, with no percolating gel network remaining after 12 h (Figure 3A). In u1 ELP hydrogels cross-linked with PEGs of higher functionality, the fraction of the network that was released reached a plateau value, instead of completely degrading as with the bifunctional cross-linker (Figure 3A). As predicted, increasing the degree of cross-linker functionality resulted in a decrease in the total degradable fraction of the network, from ~30% in 3-arm PEG cross-linked gels to ~16% in 4-arm cross-linked gels to ~5% in 8-arm cross-linked gels. Thus, even after complete proteolysis of all uPA-cleavable sites, hydrogels cross-linked by PEGs with a functionality greater than 3 retained a percolating gel architecture. Importantly, this control over degraded hydrogel architecture is afforded while retaining similar starting material properties, including hydrogel mesh size, protein and PEG weight fraction, mechanical stiffness, concentration of uPA-degradable peptides, and protein amino acid sequence.
Figure 3.

Fraction of the original protein hydrogel material that is released over time during uPA-mediated bond cleavage of (A) u1 and (B) u2 ELP hydrogels cross-linked by PEGs with varying functionality. Right panels show rescaled y-axes to visualize differences among hydrogels with a PEG functionality of 3 or greater. Data are mean ± SD, n = 4.
Controlling hydrogel network degradation and fragment release by varying cross-linker functionality was generalizable to hydrogels with different proteolytic cleavage kinetics. To demonstrate this, hydrogels were prepared using the u2 ELP sequence (Figure 2A), which is less susceptible to uPA-mediated cleavage and has a slower chemical bond cleavage rate.9 As expected, u2 gels released hydrogel fragments at a slower rate than the u1 gels for all cross-linker functionalities tested (Figure 3B). Slowly degrading u2 ELP gels exhibited a similar dependence of fraction released on cross-linker functionality to the u1 gels. At each time point, the gel fraction released was observed to decrease with increasing cross-linker functionality, ranging from substantial material released from bifunctional PEG cross-linked gels to minimal material released from 8-arm PEG cross-linked gels (Figure 3B). Due to the slower degradation kinetics of the u2 gels, the fraction of the network released did not reach a plateau value over the course of the experiment. If provided additional time to degrade, we expect that the maximal fraction of material able to be released from the u2 networks would be dictated by the cross-linker functionality, similar to the u1 gels.
Having established a system that permitted independent tuning of the chemical bond cleavage kinetics and the connectivity of the hydrogel architecture, we next sought to determine how simultaneous tuning of these two parameters influences cellular behavior. Vasculogenesis requires matrix degradation and remodeling to permit endothelial cell migration and organization into new vessels.19 In vitro, endothelial migration is known to require matrix degradation,10,20,21 so we employed a 3D endothelial network formation assay to interrogate the influence of chemical bond cleavage kinetics and degraded hydrogel architecture. As a model endothelial cell line, we chose the murine brain microvascular cell line bEnd.3, which is known to secrete high levels of uPA.22 We have previously demonstrated high viability and phenotypic maintenance of endothelial cells encapsulated within SPAAC-cross-linked ELP hydrogels.12 Dispersed cell suspensions of bEnd.3 cells were encapsulated within u1 and u2 ELP hydrogels with varying cross-linker functionality. Cell-adhesive ELPs containing an extended RGD integrin-binding domain from human fibronectin23 were included in these gels to permit cell spreading and migration. The RGD ELPs were modified with a low degree of azide substitution (less than 2 per protein) to allow incorporation via reaction with excess BCN groups without appreciably altering network connectivity. The total RGD concentration was kept constant at ~1 mM.
After 5 days, the embedded cells were fixed and stained to assess cell spreading and endothelial network formation by confocal microscopy. Validating that both the chemical bond cleavage kinetics and the degraded hydrogel architecture are important parameters, endothelial network formation only occurred in hydrogels that had rapid bond cleavage and full disruption of the percolating gel architecture (Figure 4). Within these gels, the cells became elongated and formed multiple cell–cell contacts with a typical branching-type morphology. In contrast, in gels that had slower bond cleavage rates that still resulted in full disruption of the percolating gel architecture, only some cell spreading and clustering was observed. The chemical bonds in these gels likely cleave too slowly to permit endothelial network assembly within the culture duration assessed. Further, in gels with rapid bond cleavage rates but an architecture that results in release of only ~30% of the gel material, the cells also only exhibited moderate spreading. In this case, the remaining percolating hydrogel architecture likely inhibited cellular migration to block endothelial network assembly. In all other hydrogel formulations, negligible cell spreading was observed, and cells retained a rounded morphology with limited cell–cell contact.
Figure 4.

(A) Confocal microscopy of brain microvascular cells embedded in ELP hydrogels reveals that endothelial network formation depends on both the chemical bond cleavage kinetics and the degraded hydrogel architecture. Insets are higher magnification images to assess cell spreading. Representative images were selected from 3 biological replicates. Blue: nuclei (DAPI), Green: F-actin (phalloidin). (B) “Phase diagram” summarizing how endothelial cell phenotype is affected by chemical bond cleavage rate and degraded hydrogel architecture.
While only the fully degradable, bifunctionally cross-linked hydrogels enabled endothelial network formation, it is not clear that total disruption of the percolating architecture is necessary for formation of vascular-like structures. In the trifunctionally cross-linked hydrogels that exhibited a maximal fraction degraded of only ~30%, there is significant cell spreading and local regions of high cell–cell contact. Further increasing the fraction degraded above 30% may enable larger-scale organization of the endothelial cells into tube-like structures without requiring complete degradation of the local hydrogel architecture.
By harnessing the modular design afforded by protein engineering, we have developed hydrogels with tunable bulk degradation properties through simultaneous, independent, and quantitative control of chemical bond cleavage kinetics and degraded hydrogel architecture. Both of these parameters play a role in enabling endothelial network formation in vitro, as endothelial network formation was observed only in gels with rapid proteolytic cleavage kinetics that could fully disrupt the percolating hydrogel architecture. We anticipate that other cell types of therapeutic interest, such as neural progenitor cells24 and mesenchymal stem cells,25 may also exhibit changes in cellular behavior in response to both chemical bond cleavage kinetics and degraded hydrogel architecture, making materials with control over these properties of interest to the tissue engineering and regenerative medicine communities. These systems may additionally find applications in drug delivery to simultaneously control the rate and amount of drug release. Furthermore, the design principles employed in this study can be used to modulate changes in hydrogel swelling upon degradation. Varying the identity of the protease cleavage sequence does not alter the initial swelling properties of the gels (Figure S6), and protease-degradable PEG hydrogels are known to swell in proportion to the number of cross-links cleaved.8 The system described herein may thus be useful for controlling the rate and extent of hydrogel swelling, with possible applications in device fabrication, such as materials for stimulus-responsive valves in microfluidic devices.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Kyle Lampe and Amy Proctor for assistance with protein expression and purification. This work was supported by funding from the NIH (F31 EB020502 to C.M.M.; U19 AI116484, R21 HL138042 to S.C.H.), the NSF (DMR 1508006 to S.C.H.), CIRM (RT3-07948 to S. C.H.), and the Siebel Scholars Foundation (C.M.M.). Part of this work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-1542152.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmacrolett.8b00664.
Experimental details and supporting figures (PDF)
REFERENCES
- (1).Lee KY; Mooney DJ Hydrogels for Tissue Engineering. Chem. Rev 2001, 101, 1869–1879. [DOI] [PubMed] [Google Scholar]
- (2).Ratner BD; Bryant SJ Biomaterials: Where We Have Been and Where We Are Going. Annu. Rev. Biomed. Eng 2004, 6, 41–75. [DOI] [PubMed] [Google Scholar]
- (3).Marquardt LM; Heilshorn SC Design of Injectable Materials to Improve Stem Cell Transplantation. Curr. Stem Cell Rep 2016, 2, 207–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Tibbitt MW; Anseth KS Hydrogels as Extracellular Matrix Mimics for 3D Cell Culture. Biotechnol Bioeng.2009, 103, 655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Madl CM; Heilshorn SC Engineering Hydrogel Microenvironments to Recapitulate the Stem Cell Niche. Annu. Rev. Biomed. Eng 2018, 20, 21–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Baker BM; Chen CS Deconstructing the Third Dimension – How 3D Culture Microenvironments Alter Cellular Cues. J. Cell Sci 2012, 125, 3015–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lutolf MP; Raeber GP; Zisch AH; Tirelli N; Hubbell JA Cell-Responsive Synthetic Hydrogels. Adv. Mater 2003, 15, 888–892. [Google Scholar]
- (8).Lutolf MP; Lauer-Fields JL; Schmoekel HG; Metters AT; Weber FE; Fields GB; Hubbell JA Synthetic Matrix Metalloproteinase-Sensitive Hydrogels for the Conduction of Tissue Regeneration: Engineering Cell-Invasion Characteristics. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 5413–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Straley KS; Heilshorn SC Dynamic, 3D-Pattern Formation within Enzyme-Responsive Hydrogels. Adv. Mater 2009, 21, 4148–4152. [Google Scholar]
- (10).Trappmann B; Baker BM; Polacheck WJ; Choi CK; Burdick JA; Chen CS Matrix Degradability Controls Multicellularity of 3D Cell Migration. Nat. Commun 2017, 8, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Harris JL; Backes BJ; Leonetti F; Mahrus S; Ellman JA; Craik CS Rapid and General Profiling of Protease Specificity by Using Combinatorial Fluorogenic Substrate Libraries. Proc. Natl. Acad. Sci. U. S. A 2000, 97, 7754–7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Madl CM; Katz LM; Heilshorn SC Bio-Orthogonally Crosslinked, Engineered Protein Hydrogels with Tunable Mechanics and Biochemistry for Cell Encapsulation. Adv. Funct. Mater 2016, 26, 3612–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Flory PJ Molecular Size Distribution in Three Dimensional Polymers. I. Gelation. J. Am. Chem. Soc 1941, 63, 3083–3090. [Google Scholar]
- (14).Stockmayer WH Theory of Molecular Size Distribution and Gel Formation in Branched-Chain Polymers. J. Chem. Phys 1943, 11, 45–55. [Google Scholar]
- (15).Flory PJ Principles of Polymer Chemistry; Cornell University Press: Itacha, NY, 1953. [Google Scholar]
- (16).Matsunaga T; Sakai T; Akagi Y; Chung U.-i.; Shibayama M SANS and SLS Studies on Tetra-Arm PEG Gels in As-Prepared and Swollen States. Macromolecules 2009, 42, 6245–6252. [Google Scholar]
- (17).de Molina PM; Lad S; Helgeson ME Heterogeneity and Its Influence on the Properties of Difunctional Poly(Ethylene Glycol) Hydrogels: Structure and Mechanics. Macromolecules 2015, 48, 5402–5411. [Google Scholar]
- (18).DeForest CA; Polizzotti BD; Anseth KS Sequential Click Reactions for Synthesizing and Patterning Three-Dimensional Cell Microenvironments. Nat. Mater 2009, 8, 659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).van Hinsbergh VWM; Engelse MA; Quax PHA Pericellular Proteases in Angiogenesis and Vasculogenesis. Arterioscler., Thromb., Vasc. Biol 2006, 26, 716–728. [DOI] [PubMed] [Google Scholar]
- (20).Zisch AH; Lutolf MP; Ehrbar M; Raeber GP; Rizzi SC; Davies N; Schmökel H; Bezuidenhout D; Djonov V; Zilla P; Hubbell JA Cell-Demanded Release of VEGF from Synthetic, Biointeractive Cell Ingrowth Matrices for Vascularized Tissue Growth. FASEB J.2003, 17, 2260–2262. [DOI] [PubMed] [Google Scholar]
- (21).Turturro MV; Christenson MC; Larson JC; Young DA; Brey EM; Papavasiliou G MMP-Sensitive PEG Diacrylate Hydrogels with Spatial Variations in Matrix Properties Stimulate Directional Vascular Sprout Formation. PLoS One 2013, 8, No. e58897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Montesano R; Pepper MS; Möhle-Steinlein U; Risau W; Wagner EF; Orci L Increased Proteolytic Activity Is Responsible for the Aberrant Morphogenetic Behavior of Endothelial Cells Expressing the Middle T Oncogene. Cell 1990, 62, 435–445. [DOI] [PubMed] [Google Scholar]
- (23).Straley KS; Heilshorn SC Independent Tuning of Multiple Biomaterial Properties Using Protein Engineering. Soft Matter 2009, 5, 114–124. [Google Scholar]
- (24).Madl CM; LeSavage BL; Dewi RE; Dinh CB; Stowers RS; Khariton M; Lampe KJ; Nguyen D; Chaudhuri O; Enejder A; Heilshorn SC Maintenance of Neural Progenitor Cell Stemness in 3D Hydrogels Requires Matrix Remodelling. Nat. Mater 2017, 16, 1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Khetan S; Guvendiren M; Legant WR; Cohen DM; Chen CS; Burdick JA Degradation-Mediated Cellular Traction Directs Stem Cell Fate in Covalently Crosslinked Three-Dimensional Hydrogels. Nat. Mater 2013, 12, 458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.