

Abstract
The thiol (−SH) functional group is found in a number of drug compounds and confers a unique combination of useful properties. Thiol-containing drugs can reduce radicals and other toxic electrophiles, restore cellular thiol pools, and form stable complexes with heavy metals such as lead, arsenic, and copper. Thus, thiols can treat a variety of conditions by serving as radical scavengers, GSH prodrugs, or metal chelators. Many of the compounds discussed here have been in use for decades, yet continued exploration of their properties has yielded new understanding in recent years, which can be used to optimize their clinical application and provide insights into the development of new treatments. The purpose of this narrative review is to highlight the biochemistry of currently used thiol drugs within the context of developments reported in the last five years. More specifically, this review focuses on thiol drugs that represent the standard of care for their associated conditions, including N-acetylcysteine, 2,3-meso-dimercaptosuccinic acid, British anti-Lewisite, D-penicillamine, amifostine, and others. Reports of novel dosing regimens, delivery strategies, and clinical applications for these compounds were examined with an eye toward emerging approaches to address a wide range of medical conditions in the future.
Keywords: Thiol, heavy metals, toxicity, radioprotectant, glutathione, antioxidant, chelation, ROS
Graphical Abstract
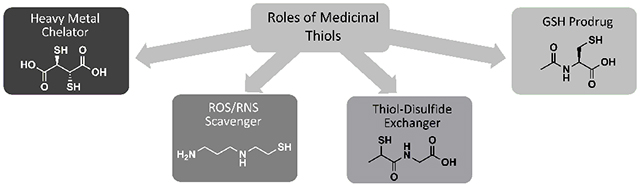
1. INTRODUCTION
Oxidative stress arises from an overabundance of reactive oxygen and/or nitrogen species (ROS/RNS) in the body and is associated with a wide range of pathological states, including inflammation, aging, and cancer [1]. In general, the term “reactive species” encompasses both free radicals (molecules containing unpaired electrons) and non-radical electrophiles [2]. Small amounts of certain reactive species are generated during respiration and are required for normal cell signaling, differentiation, and proliferation [3], but if left unchecked, reactive species are capable of oxidizing vital cellular components, such as proteins, lipids, and nucleic acids, resulting in dysfunction and dysregulation. Antioxidants help maintain appropriate levels of reactive species, often by directly reducing them to less reactive products or by catalyzing reactions that keep ROS/RNS levels under control. The latter are known as antioxidant enzymes and include superoxide dismutases (SOD), catalases (CAT), peroxidases, and many others [4–6].
Thiols are compounds that possess a sulfhydryl (−SH) functional group. Several key chemical properties enable thiols to play unique and essential roles in redox biology. Thiols are ‘soft’ electron donors, and readily form complexes with ions of metals such as copper, lead, and mercury. As a result, thiol-containing drugs are used as chelators of heavy metal ions, removing them from the body and preventing them from blocking enzyme activity and promoting oxidation [7]. Thiols and their conjugate bases, thiolates, are also good nucleophiles and are therefore reactive toward electrophilic species, including ROS/RNS [8]. Thus, thiol drugs are used as ROS/RNS scavengers to prevent them from oxidizing proteins, lipids, and DNA. One of the most important antioxidants in the body is glutathione (GSH), shown in (Fig. 1). GSH is the most abundant non-protein thiol, and it is crucial for maintaining favorable redox status in cells [2]. Thiol-containing drugs can perform some of the ROS/RNS scavenging roles of GSH, and drugs that are metabolized to cysteine can promote GSH synthesis. These are considered GSH prodrugs. Thiols can be crosslinked by the formation of a disulfide bond (-S-S-) upon oxidation, and they can then be reduced back to thiol form via a thiol-disulfide exchange reaction, which entails nucleophilic attack by the thiol on the disulfide [8]. This principle underlies the use of thiols as mucolytics and antiurolithics.
Fig. (1).
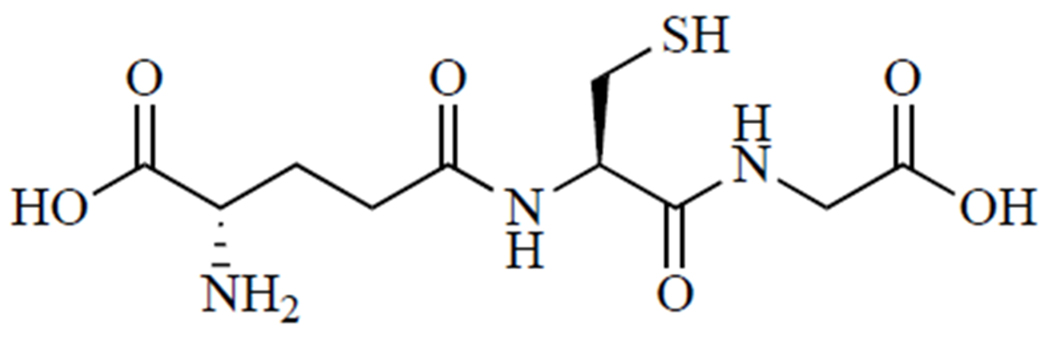
Glutathione (GSH, γ-glutamylcysteinylglycine) is the body’s most abundant and important nonprotein thiol.
The many-faceted biochemistry of thiol compounds underpins their complex and varied roles in human health and disease. In recent years, the impetus to repurpose existing approved medications [9] has renewed interest in thiol-containing drugs, many of which have been applied in a limited scope for decades. However, the full realization of their potential as useful treatments depends upon the continued investigation of these roles. As a result, this narrative review seeks to highlight discoveries and improvements in the last five years that build upon foundational research in the application of medicinal thiols. Promising experimental/novel thiols will be included where appropriate, but areas in which thiol drugs represent the current standard of care will be emphasized, including acetaminophen overdose, heavy metal toxicity, and radiation poisoning.
2. ACETAMINOPHEN OVERDOSE
Acetaminophen, also known as Tylenol, paracetamol, or N-acetyl-p-aminophenol (APAP), is used by approximately 43 million adults in the USA every week [10]. However, it poses a serious risk of hepatotoxicity at supratherapeutic doses and as a result, accounts for the majority of drug-induced liver injuries [11–13] and about 39% of all liver failures [14]. The only FDA-approved treatment for this all-too-common condition is N-acetylcysteine, a thiol drug that supports liver GSH levels during the acute oxidative challenge of APAP overdose.
2.1. Toxicity of Acetaminophen
At recommended doses, more than 90% of APAP is metabolized via glucuronidation by UDP-glucuronosyl transferase or via sulfonation by sulfotransferase to generate non-toxic products that can be excreted [15, 16], and approximately 2% of ingested APAP is eliminated in urine without undergoing any metabolic processing [16]. The remaining APAP is oxidized to the electrophilic metabolite N-acetyl-p-benzoquinone imine (NAPQI), in two successive single-electron transfer steps by microsomal cytochrome P450 enzymes [17]. With sufficient GSH available, the quinone imine alkylates GSH to form glutathione-S-conjugates [18], from which nontoxic mercapturates are produced and then excreted in the urine [16, 19, 20].
However, supratherapeutic doses of APAP can generate enough NAPQI to exhaust hepatic GSH stores in several ways. The oxidation of APAP to NAPQI results in the production of ROS, including H2O2, which can then be converted to hydroxyl radical via Fenton reaction [17]. GSH can scavenge these radical species directly and can be used in other antioxidative processes, such as glutathione peroxidase-dependent reduction of H2O2. If NAPQI production exceeds the glutathionylation capacity of remaining GSH stores, excess NAPQI can undergo Michael addition to cysteine residues on nearby proteins [17–18], including enzymes found in the mitochondria or cytoplasm. If left untreated, downstream effects, such as calpain activation, decreased ATP production, disruption of mitochondrial membrane potential, and a broad loss of enzymatic activity culminate in hepatocytic apoptosis or necrosis, leading to liver dysfunction or failure [21, 22].
2.2. Standard of Care
Treatment for APAP overdose is determined based on the level of APAP in the blood and time since ingestion. If less than 4 hours has passed since APAP ingestion, efforts will be made to block APAP absorption into the bloodstream by inducing vomiting, gastric lavage, or activated charcoal gavage [23]. Activated charcoal can be given up to 4 hours post-APAP ingestion [16]. However, most patients present to the emergency department long after ingestion, at which time APAP is already in the bloodstream and undergoing metabolism in the liver. In this case, N-acetylcysteine, shown in (Fig. 2), is the only FDA-approved treatment. It is highly effective and may be administered orally or intravenously. Patients who refuse oral N-acetylcysteine and those who are unconscious or unable to absorb N-acetylcysteine intestinally are given IV N-acetylcysteine. If the patient does not respond to N-acetylcysteine and liver failure is imminent, the only recourse is liver transplant [16].
Fig. (2).
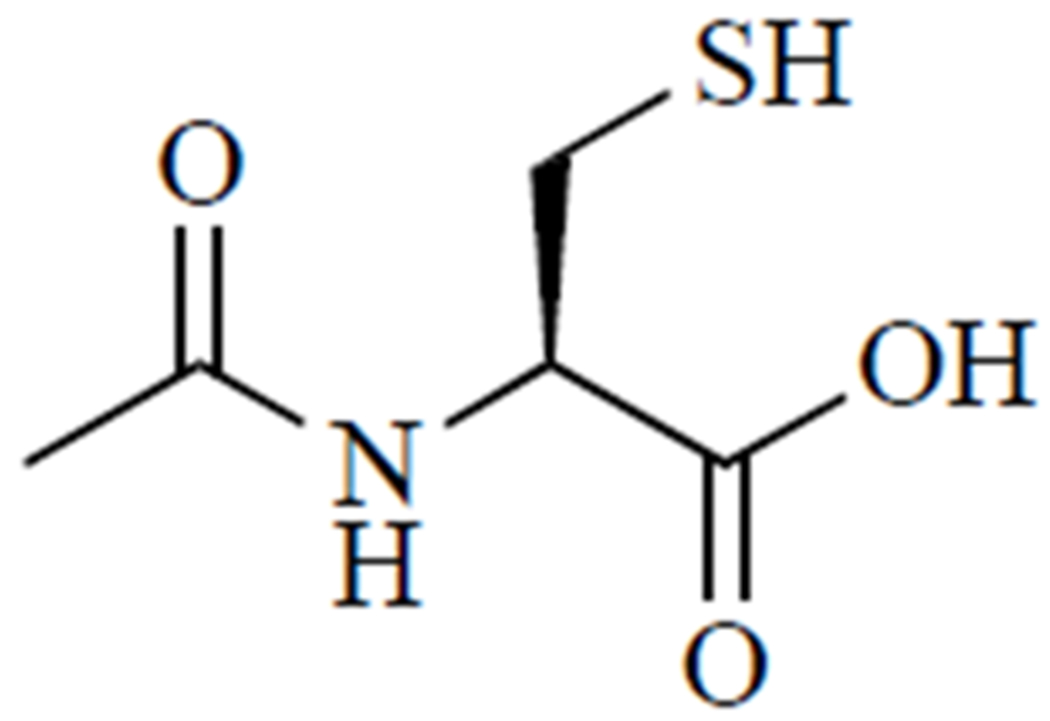
N-acetylcysteine.
N-acetylcysteine treats APAP-induced hepatoxicity by acting as a GSH prodrug. N-acetylcysteine is metabolized to cysteine, the rate-limiting amino acid in GSH synthesis [2, 20, 24]. Supporting GSH synthesis in the liver increases its capacity to detoxify reactive species and prevent further reaction of protein thiols with NAPQI. Further, treatment with N-acetylcysteine can restore Krebs cycle intermediates depleted by reactive APAP metabolites, reviving ATP production [20, 25].
2.3. Addressing N-acetylcysteine’s Shortcomings
Preventing acute liver failure requires initiating treatment early enough after the ingestion of APAP. A Rumack-Matthew nomogram is a predictive tool that allows physicians to determine the likelihood of hepatotoxicity based on blood APAP levels and the time since ingestion. However, it can be inaccurate if the patient has repeatedly experienced APAP poisoning, has a pre-existing liver condition, has abused alcohol, or the time of ingestion is unknown [16]. Prompt initiation of treatment is critical since N-acetylcysteine is most effective when it is administered before NAPQI forms adducts with cysteine residues in vital proteins [15, 16].
Outcomes are generally favorable if patients present to the ER within 8 hours post-ingestion. However, increases in hepatotoxicity were observed with treatment delays within 8-16 hours after ingestion [26]. In a study conducted by Smilkstein et al. [27], 6.1% of APAP overdose patients suffered from hepatotoxicity when N-acetylcysteine was administered less than 10 hours after APAP ingestion, compared to 26.4% when treatment was delayed more than 10 hours post-ingestion. The effects of treatment delay are exacerbated in cases of staggered overdose (repeated ingestion of high doses in a short period of time). N-acetylcysteine may be less effective in these patients, which makes up a significant proportion of APAP overdose cases (24.3% in one cohort study [28]). For example, N-acetylcysteine failed to improve hepatotoxicity, as indicated by serum alanine aminotransferase (ALT) and histological evaluation of liver slices, in both young and old C57BL/6 mice subjected to a staggered overdose. While N-acetylcysteine increased GSH in these animals, that alone was not protective against the hepatological stress of the repeated subacute dosage, which highlights the need for treatments to address this type of overdose [29].
For APAP overdose, N-acetylcysteine is available in two forms: oral and IV. Both forms exhibit similar effectiveness in a clinical setting [30]. Oral N-acetylcysteine requires a 140 mg/kg loading dose, followed by 70 mg/kg every 4 hours for 72 hours. The length of this treatment contributes to longer hospital stays and higher overall healthcare expenses for oral N-acetylcysteine [31]. IV N-acetylcysteine was approved by the FDA in 2004 and represented an attractive alternative to oral N-acetylcysteine. This 21-hour treatment requires shorter hospital stays (median of 5 days) and lower overall healthcare costs ($7,608 median cost) [31]. However, the dosing regimen for IV N-acetylcysteine is a relatively complicated three-bag process, requiring different mg/kg dosing, additive volumes, and infusion rates for each step [32]. As a result, the IV N-acetylcysteine regimen is error-prone. One study reported a medication error rate of 33% in the administration of IV N-acetylcysteine. More disturbingly, a similar error rate was observed in pediatric patients [33]. In addition to being error-prone, IV N-acetylcysteine triggers an anaphylactoid reaction in a significant number of patients (up to 28.5% in some populations) [34, 35]. Clearly, while safe and effective for the majority of patients, N-acetylcysteine for APAP overdose leaves much to be desired in terms of effectiveness and tolerability.
2.3.1. Improvements to Existing Regimen
APAP is commonly used for the treatment of fever and minor aches and pains, as well as long-term management of chronic pain. N-acetylcysteine is frequently prescribed concurrently as a mucolytic. Consequently, APAP and N-acetylcysteine are often, albeit inadvertently, prescribed together without adverse effects. However, N-acetylcysteine had not been studied as an adjuvant to APAP formulations to protect against liver damage until a prospective, randomized, double-blind controlled trial concluded that N-acetylcysteine was able to preserve GSH without interfering with the analgesic efficacy of APAP. In the future, the inclusion of N-acetylcysteine in APAP formulations could significantly reduce the incidence of APAP-related toxicity [36].
There have also been efforts to investigate alternative dosing protocols for N-acetylcysteine to increase tolerability and effectiveness. For example, UK medical guidelines have lowered the serum APAP threshold for initiation of treatment in order to reduce failure rates. This potential benefit must be weighed against the increased risk of anaphylaxis and healthcare costs for patients that might have recovered without N-acetylcysteine [34]. It has also been suggested that N-acetylcysteine can be administered in cases where liver toxicity is probable, but the time of ingestion or serum APAP concentration cannot be resolved [16]. Here, the potential benefit of N-acetylcysteine and risk of liver damage likely outweigh risks of short- or long-term side effects [16, 37].
Abbreviated N-acetylcysteine dosing procedures may be better tolerated and achieve similar outcomes while reducing healthcare costs. One study reported that for patients with high APAP serum levels, a shortened IV procedure with a higher loading dose was better tolerated than the standard three-bag, 20-hour procedure, except for patients with adverse reactions, in whom slower infusion rates reduce likelihood and severity [23].
For repeated supratherapeutic ingestion, Australian medical guidelines allow the discontinuation of N-acetylcysteine after 8 hours if ALT is normal or stable and APAP serum levels are low at this time [38]. Since pharmacokinetic models suggest the presence of N-acetylcysteine for several hours post-administration [39], it may confer protective effects even after patients are released [23]. In order to investigate the validity of these guidelines, a retrospective study was conducted with 91 cases, 39 of which received the abbreviated regimen. The study concluded that the adjusted guidelines reduce hospital stay and free up beds, with very little risk to patients, but it was limited by the lack of follow-up and the small number of patients. It also highlights the need for more standardization and evidence-based approaches to setting N-acetylcysteine treatment guidelines, especially with respect to reporting of side effects, which is highly dependent on study methodology [40].
2.3.2. Alternatives to N-acetylcysteine
Developing new treatments also requires better preclinical models [41]. The widely available C57BL/6 mouse is ubiquitous in preclinical research, but the two major strains respond differently to APAP overdose. Both strains (N and J) exhibit GSH depletion and cytochrome P450 activation following supratherapeutic APAP doses, but detrimental effects observed in the N strain are more extensive than those reported in the J strain. Because strain differences can influence study outcome, this limits the validity of comparisons amongst APAP toxicity studies conducted with this model [42]. However, researchers may be able to take advantage of the differential susceptibility to APAP in order to study the efficacy of treatment in more vulnerable or robust systems, provided that the experimental design and reporting are rigorous and transparent.
A commonality amongst proposed alternatives to N-acetylcysteine is their ability to support hepatic GSH levels, be it through sparing or regenerating GSH by thiol-disulfide exchange or providing substrates for de novo GSH synthesis. As such, these compounds either possess thiol groups or can supply thiol groups indirectly via enzymatic conversion to thiol-containing species [15]. Some compounds of interest include tiopronin, British anti-Lewisite (BAL) [43], methionine [44], dihydrolipoic acid [45], and cysteamine [43, 46]. The higher reactivity of cysteine compared to N-acetylcysteine precludes its use as a treatment [24]. A systematic review conducted by Chiew et al. [23] concluded that N-acetylcysteine was more effective than cysteamine and BAL, and methionine was demonstrated to be comparable to N-acetylcysteine in efficacy and safety while being much cheaper [23, 47].
Other novel compounds, such as N-acetylcysteine amide [48], cysteamine and ethyl cysteine amides of Trolox [49], ψ-GSH [50], and N,N’-bis(2-mercaptoethyl)isophthalamide [51], have been developed to overcome limitations of N-acetylcysteine. For example, our research has evaluated the efficacy of an N-acetylcysteine analogue, N-acetylcysteine amide, in cell and animal models of oxidative stress-related conditions, including APAP toxicity. As shown in (Fig. 3), instead of a carboxylate group, N-acetylcysteine amide possesses an amide group that, unlike carboxylate, is uncharged at a physiological pH [52]. Thus, N-acetylcysteine amide exhibits increased lipophilicity and may be more bioavailable than N-acetylcysteine, while still providing cysteine for GSH synthesis. In the human hepatoma HepRG cell line [53] and in C57BL/6 mice [48], N-acetylcysteine amide exhibited a greater capacity to restore hepatic GSH and increase survival compared to N-acetylcysteine. Another proposed alternative to N-acetylcysteine is ψ-GSH, which is orally bioavailable and metabolism-resistant. Orally administered ψ-GSH was shown to be as effective as intraperitoneal N-acetylcysteine in a mouse model of APAP overdose [50]. Proponents of these alternatives cite better safety profiles and bioavailability, characteristics that could allow for cheaper, simpler, and better-tolerated treatments for APAP overdose [33, 34].
Fig. (3).
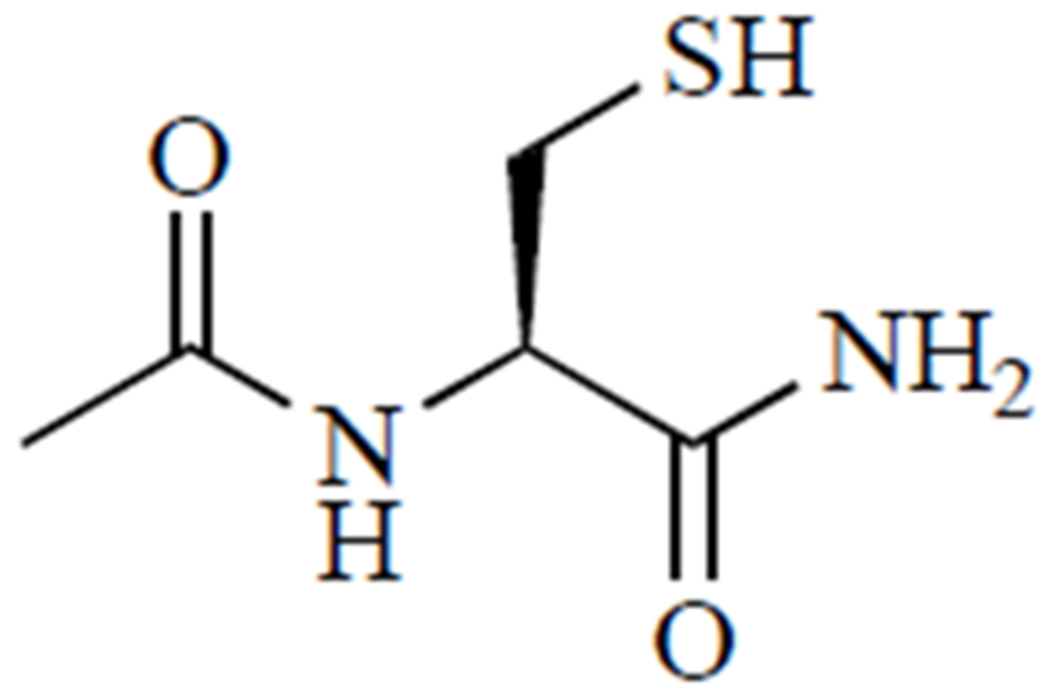
N-acetylcysteine amide.
3. HEAVY METAL TOXICITY
According to Hard-Soft Acid Base theory, metal ions such as Hg2+, Cu+, and Pb2+ are considered ‘soft’ Lewis acids; Cu2+ and As3+ are considered borderline between ‘hard’ and ‘soft’ [7]. Soft and borderline metal ions exhibit higher affinities for ‘soft’ ligands, such as thiols and thiolates, than ‘hard’ metal ions like Mg2+ or Fe3+ do [7, 54]. Further, complexation with a multidentate ligand is entropically favored over complexation with multiple ligands that each possess one of the donor groups present on the multidentate ligand [54–55]. For these reasons, thiol-containing drugs, including dithiols like BAL and meso-2,3-dimercaptosuccinic acid and aminothiols like D-penicillamine, are essential in the treatment of heavy metal toxicity.
3.1. Lead
While the incidence of lead poisoning has decreased over the past several decades [56], concerns have resurfaced since the Flint, Michigan water crisis surged to the forefront of national attention in the United States. Of particular concern are the effects of lead on children, in whom intestinal absorption of lead ranges from 40-50%, compared to 3-10% in adults [57]. Children in economically struggling areas, such as Flint, are already at a higher risk for developmental delays, and unacceptably high levels of lead in drinking water place this vulnerable demographic at an even greater disadvantage [58].
3.1.1. Toxicity of Lead
Lead usually enters the body through inhalation or ingestion, resulting in long-term accumulation. Up to 99% of absorbed lead persists in the blood for over a month, after which it moves to soft tissues and bones [59]. Lead(II) is in the most stable oxidation state in the physiological environment, and while Pb2+ exhibits a higher affinity for S-donor-containing ligands, it still forms complexes with hydroxide ions, which can impact the effectiveness of chelation strategies [60].
Lead toxicity occurs through several pathways. First, the ability of Pb2+ ions to replace crucial Zn2+ and Ca2+ ions in the biological milieu plays an important role in lead toxicity. Lead disrupts calcium homeostasis, which upregulates calmodulin and phosphodiesterase enzymes [61]. Lead can also replace zinc ions in enzyme active sites [62]. Additionally, lead affects membrane integrity by promoting lipid oxidation [63, 64]. Lead may activate phospholipase A2, which increases the arachidonic acid content of membranes [65] and thereby increases the average chain length and degree of unsaturation in membrane fatty acid content. Polyunsaturated fatty acids present multiple sites of increased electron density for attack by electrophilic reactive species [2]. By increasing average membrane lipid chain-length and degree of unsaturation, lead makes membranes more vulnerable to oxidation. Indeed, lead-induced fatty acid oxidation increases in severity for longer fatty acyl groups with higher degrees of unsaturation [63]. Another way in which lead may damage membranes is through binding with phosphatidylcholine, a common membrane lipid component [64]. In addition, lead inhibits hemoglobin synthesis. Lead binds to the thiol moiety in the active site of δ-aminolevulinic acid (δ-ALA) dehydratase, which catalyzes the condensation of two δ -ALA molecules in hemoglobin synthesis [63]. This increases levels of circulating δ-ALA, which facilitates ROS generation since the enol form of δ-ALA autoxidizes to form superoxide radicals. Lead may also interact with oxyhemoglobin to generate ROS [67]. These ROS attack the already vulnerable RBC membranes [68], leading to ‘oxidative hemolysis’ that results in the anemia often observed in children with lead poisoning [63]. Lead also disrupts antioxidant enzymes, further decreasing the capacity of cells to detoxify reactive species. Glutathione reductase is another enzyme in which lead complexation with thiol groups in its active site inhibits its catalytic activity, specifically, the regeneration of glutathione disulfide (GSSG) to its active reduced form GSH. Lead may also form a complex with selenium in the active site of glutathione peroxidase and interfere with the reduction of peroxides [69]. The heme group of catalase may be affected by lead as well [63, 70]. Because of its effect on membrane lipids and its ability to inhibit hemoglobin synthesis, lead is particularly detrimental to red blood cells (RBCs) [64].
3.1.2. Standard of Care
The current standard of care for heavy metal poisoning is chelation therapy. In many cases, the preferred treatment for lead toxicity is meso-2,3-dimercaptosuccinic acid due to its favorable safety profile and ability to be administered orally [71]. While meso-2,3-dimercaptosuccinic acid (Fig. 4, left) is water-soluble, BAL (Fig. 4, right) is a highly lipophilic and cell-permeable chelator that is indicated when neurological symptoms of lead poisoning are present. Treatment with BAL is often accompanied by the administration of Ca-Na2EDTA [72]. Because of its lipophilic nature, BAL is formulated in peanut oil and must be given via intramuscular injection. EDTA (ethylenediaminetetraacetic acid) contains only nitrogen and oxygen ligand donors, and thus exhibits less specificity for Pb2+ ions [64].
Fig. (4).
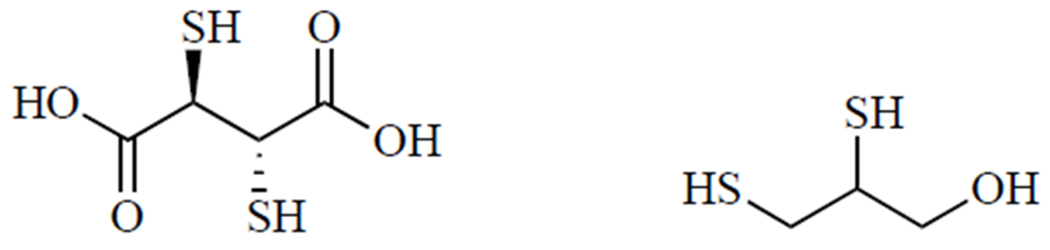
meso-2,3-Dimercaptosuccinic acid (left) and BAL (right).
3.1.3. Limitations of Current Therapies
EDTA is a highly hydrophilic molecule, and as such, removes lead most efficiently from the extracellular environment [72]. Since it cannot cross the blood-brain barrier, it is not effective at removing lead already accumulated in the brain, and it may transfer lead from other tissues to the brain [73]. Finally, its lack of specificity results in the chelation of essential metal ions and trace metal ions, including those that are essential to enzyme activity [64]. BAL, in contrast, is highly lipophilic and can remove intracellular lead. However, it requires painful intramuscular administration and has a very unfavorable side effect profile that includes allergic reactions, nephrotoxicity, and hypertension [57]. meso-2,3-Dimercaptosuccinic acid is a selective chelator of toxic heavy metals like lead, arsenic, and mercury. It effectively removes lead from soft tissues and has not been shown to redistribute it to the brain [71]. Even so, meso-2,3-dimercaptosuccinic acid suffers from serious bioavailability limitations. When administered orally, it has a bioavailability of about 20%, with most of the drug bound to protein in plasma, and its hydrophilicity limits its access to heavy metal ions in intracellular compartments [74]. Further, it has been shown that chelation therapy fails to improve cognitive functioning in lead-poisoned children [75].
3.1.4. Understanding Current Treatments
Clearly, none of these chelators are appropriate for all situations, as each has serious limitations. However, the lack of available patients for rigorous clinical trials has impeded a thorough evaluation of current and potential chelation strategies [76]. While meso-2,3-dimercaptosuccinic acid is the standard treatment for lead poisoning in the United States, EDTA is used more often in some other countries. In the first highly rigorous, open-label therapeutic trial of lead poisoning treatment, 37 patients with blood lead levels greater than 40 μg/dL were randomized to receive two five-day courses of either meso-2,3-dimercaptosuccinic acid or EDTA, with a 10-day intervening rest period. meso-2,3-Dimercaptosuccinic acid was shown to be more effective during the first course, but not in the second course [77]. This study sheds light on the importance of considering patient adherence and side effects such as the depletion of essential metal ions by EDTA [64]; however, it was limited by the small sample size, which can be difficult to increase due to the nature of this condition.
In the absence of large-scale clinical studies, in silico models can advance understanding of how chelators interact with heavy metals in the body. One study by Kaviani et al. [78] employed density functional theory to model lead, mercury, and cadmium chelation by meso-2,3-dimercaptosuccinic acid and compared the interactions between the different metals and the chelator. Bond length calculations show that lead interacts more strongly than the other metals, but frontier molecular orbital energies of the complexes indicate that meso-2,3-dimercaptosuccinic acid is a good candidate for chelation of all three metals. Models can also be used to predict the effects of chelation on lead concentrations in the body. Toxicokinetic models for lead levels in different tissues after chelation are lacking; however, van Eijkeren et al. [79] developed a mathematical model that incorporated the effects of chelation therapy on lead levels in different compartmentalized areas of the body. The model was able to predict blood lead levels accurately, but it struggled with urine levels. It worked for both inhalation and ingestion exposure under “real-life” constraints in which patient lead exposure information was not available to the clinicians responsible for the treatment. This model could be further developed for predicting how many rounds of chelation therapy are needed or which agent would be most effective for each patient. An important caveat for modeling lead complexation is the consideration of competition with hydroxyl ions, which can affect lead complexation in aqueous environments at pH ≥ 5 [60].
3.1.5. Promising Alternatives and Adjuvants
New chelators and adjuvants are being explored to overcome the limitations of currently used therapies. For example, a monoisoamyl ester of meso-2,3-dimercaptosuccinic acid has been developed to increase its lipophilicity and the ability to target intracellular lead accumulation. Monoisoamyl meso-2,3-dimercaptosuccinic acid encapsulated in copolymeric nanoparticles has already demonstrated greater ability to remove arsenic from the brain, blood, and other tissues, with improved histological outcomes and indicators of oxidative stress [80], and this strategy may hold great promise for addressing lead toxicity as well [72, 73]. In addition to chemical modification of the chelation agent, nanoplatforms may also be able to enhance the effectiveness of chelators by increasing circulation time and cell penetration, preventing metabolic degradation and targeting release sites [80]. A study by Zhai et al. [74] investigated the use of mesoporous silica nanoparticles (MSNs) to deliver meso-2,3-dimercaptosuccinic acid covalently bonded to their surface via redox-sensitive disulfide bond to form MSN-meso-2,3-dimercaptosuccinic acid nanoparticles. It was hypothesized that upon entering a highly reducing environment, such as the cytosol, meso-2,3-dimercaptosuccinic acid would be released through a thiol-disulfide exchange between GSH and MSN-meso-2,3-dimercaptosuccinic acid, producing either free reduced meso-2,3-dimercaptosuccinic acid or MSN-SH, both of which can act as effective chelating agents for Pb2+ and Hg2+. In vitro results confirmed the ability of MSNs to release meso-2,3-dimercaptosuccinic acid intracellularly and maintain cell viability even at very high concentrations. One problematic aspect of this approach, however, may be that the reduction of disulfide bond by GSH could exacerbate the GSH shortage precipitated by elevated Pb2+ levels [63].
It has been proposed that chelation alone may not be sufficient to address the downstream oxidative effects of lead poisoning [64]. Adjuvants, such as antioxidant compounds, may be able to redress the detrimental effects of lead toxicity while the chelating agent removes it from the system and prevents further harm. For example, the antioxidant flavonolignan silymarin was investigated in a rat model of lead toxicity, where it was shown that the combination of silymarin and meso-2,3-dimercaptosuccinic acid resulted in the greatest reduction in blood lead levels and protection of renal tubule cells [81]. Drugs such as N-acetylcysteine, captopril, and α-lipoic acid have been proposed as standalone or adjuvant therapies for lead poisoning. While their capacity to act as effective chelators is debatable [54, 82], many in vitro, in vivo, and human studies suggest they would be extremely valuable as antioxidants, when used in conjunction with conventional chelators. N-acetylcysteine, already FDA-approved for the treatment of acetaminophen overdose, has garnered some interest as a potential therapy for lead poisoning. In PC-12 cells [83], CD-1 mice [84], Wistar rat pups [85], and in chronically exposed human workers [86], N-acetylcysteine exhibited the ability to improve various indices of oxidative stress following lead exposure. Lipoic acid is considered a ‘universal antioxidant’ since it is both fat- and water-soluble. It may have utility as an antioxidant in mitigating the effects of lead poisoning [62], as has been demonstrated in Chinese hamster ovary (CHO) cells and Fischer 344 rats [87]. Captopril appears to have antioxidative effects as well in lead-exposed Fischer 344 rats [88] and Swiss albino mice [89]. In lenses of lead-exposed rats, treatment with captopril resulted in lower indices of oxidative stress than treatment with lipoic acid [90]. However, in general, these antioxidants failed to demonstrate superior ability to reduce lead concentrations when compared to approved chelators but provided beneficial effects in remediating oxidative stress brought on by lead exposure [68, 91]. In studies where antioxidants were applied as adjuvants to standard chelating agents, combined treatments resulted in more favorable outcomes than either agent alone [57, 81, 92, 93].
3.2. Arsenic and BAL
BAL was originally synthesized during World War II to treat exposure to arsenic-containing chemical warfare agent Lewisite [7, 54]. Also known as dimercaprol, this dithiol compound, in a racemic mixture, is currently FDA-approved for the treatment of arsenic, gold, mercury, and lead poisoning [94]. However, it has multiple adverse side effects and may redistribute arsenic to the brain and testes [80, 95]. When absorbed, arsenic(V) is quickly reduced to the more toxic arsenic(III) by arsenate reductases or nonenzymatically by GSH [96]. The toxic effects of arsenic are primarily related to its affinity for sulfur-containing compounds, such as the dihydrolipoic acid cofactor of pyruvate dehydrogenase and Cys-rich motifs on proteins, such as metallochaperones and zinc fingers [95, 96].
BAL and other dithiols can compete with proteins for arsenic binding, leading to water-soluble complexes that can be excreted in urine [95]. Multi-thiol ligands are preferred over monothiols, and the arrangement of thiol groups also plays a role, with vicinal dithiols exhibiting the highest binding strength due to the formation of a five-member ring with arsenic(III) that minimizes ring strain [96]. Density functional theory calculations augmented by solvent-assisted proton exchange revealed that thermodynamic stability of As(III) complexes is the highest for complexes with S-As-S bond angles of less than 100 degrees, which is best accomplished by vicinal dithiols such as BAL [95]. Further, NMR analysis of H3AsO3:BAL mixtures with varying ratios showed that three major complexes existed: [As(OH)BAL—H]−, [AsBAL2]−, and [As2BAL3—H]−, with the bidentate [As(OH)BAL—H]− species dominating as the proportion of As increased. Stability constants for these complexes were calculated and compared to those reported for other dithiol compounds in the literature, showing that BAL forms the most stable complexes with arsenic(III) in solution at pH 7-8 [96]. These studies provide crucial insights into BAL’s mechanism of action and may aid in the design of more effective chelating agents.
3.3. Copper and d-Penicillamine
Copper is an essential trace element that is primarily stored in the liver but is also found in muscles, kidneys, and brain. It is found in the active sites of several enzymes, including cytochrome c oxidase, dopamine-β-hydroxylase, and Cu/Zn SOD [97–98]. Wilson’s disease (WD) disrupts copper metabolism: specifically, it is the result of an autosomal recessive mutation in the gene for the P-type ATPase, ATP7B [99]. When this transporter is dysfunctional, copper builds up in the liver, and free copper is released into the bloodstream and then excreted in urine [100]. If not excreted efficiently, it builds up in the brain and cornea [101]. This deficiency in copper metabolism may result in neurological deterioration and liver failure if left untreated [102], although some patients may remain asymptomatic early in life [99].
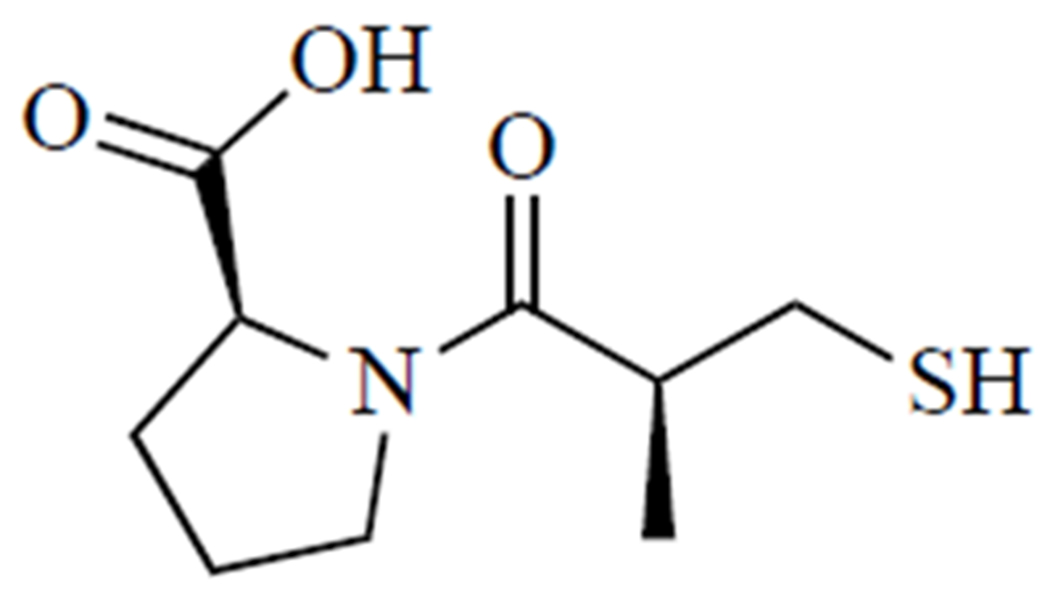
Treatment for Wilson’s disease usually proceeds in two phases. First, treatment is aimed at increasing copper excretion via active chelation, using either d-penicillamine (Fig. 5), a thiol degradation product of penicillin [103], or trientine. Once symptoms are under control and copper levels in urine stabilize, the maintenance phase is commenced, which entails switching to a lower dose of chelator or zinc supplementation, if possible [101]. Zinc salts are used for blocking copper absorption [104]. It is believed that d-penicillamine forms copper complexes in a molar ratio of 1 copper ion to 2 d-penicillamine molecules, which means that it takes about 1 g of d-penicillamine to eliminate 200 mg of copper [104]. Treatment with d-penicillamine is associated with a host of adverse side effects: hypersensitivity, suppression of bone marrow, skin toxicity, nephrotoxicity, and autoimmune diseases [97], as well as urinary abnormalities that may result from nephrotoxicity of the drug or prolonged exposure to copper before treatment was initiated [105].
Fig. (5).
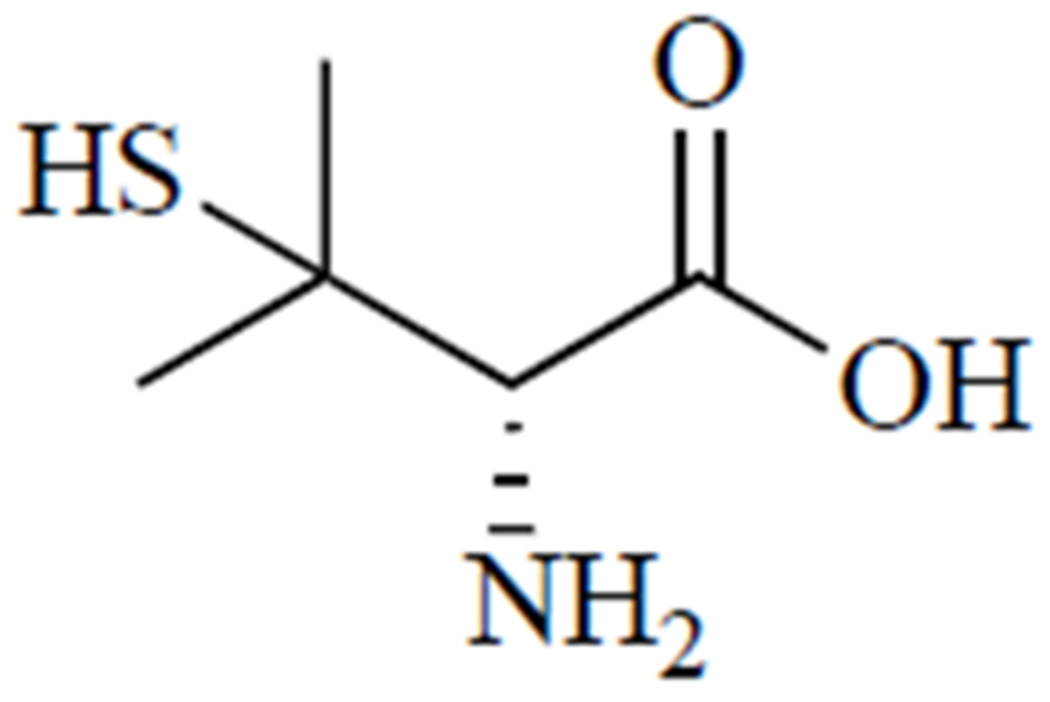
D-penicillamine.
d-penicillamine was FDA-approved in 1956 [106], but recent work has focused on understanding the interaction of d-penicillamine and copper ions in the body to optimize its usage and to uncover characteristics necessary for the development of improved chelation agents. A critical step in this process is determining the oxidation state of copper in various physiological environments, which is vital to understanding its coordination chemistry under these conditions [107]. While copper(II) is the more stable oxidation state in aqueous solutions, copper(I) is also known to play a role in many biological contexts [108–109]. However, the instability of Cu+ in aqueous solution outside the physiological environment has impeded efforts to elucidate the nature of its complexes with thiol-containing ligands [108]. Fortunately, a study performed by Konigsberger et al. [108] shed light on this important topic, confirming that Cu(I) is in the predominant oxidation state in blood plasma, aqueous humor, and lens. Further, this study showed that at physiological pH and molar ratios of Cu+: ligand ranging from 1:1 to 1:3, Cu+ is almost equally distributed between negatively charged binuclear and mononuclear complexes. As a ‘soft’ Lewis acid, Cu+ readily forms stable complexes with ‘soft’ ligand donors, such as thiol groups found on cysteine residues throughout the body or thiol-containing drugs like d-penicillamine [107, 108]. In this case, d-penicillamine is a bidentate ligand, with nitrogen and sulfur atoms acting as ligand donors [97, 109]. Interestingly, d-penicillamine in the presence of Cu2+ may enhance the anticancer effects of cisplatin and radiation therapy. A study by Sciegienka et al. [110] demonstrated that d-penicillamine in the presence of Cu2+ increases breast and lung carcinoma cell death via H2O2-derived ROS. d-penicillamine was also investigated as an adjuvant in a Phase II trial in glioblastoma [111]. The generation of ROS in the presence of Cu2+ and d-penicillamine was also demonstrated by Katerji et al. [97] in a neuronal cell line, which may help explain its ineffectiveness in treating Wilson’s-associated neurological symptoms. While well-tolerated, d-penicillamine did not significantly enhance the antitumor effects of chemoradiotherapy. As a result, further investigations with physiologically relevant copper speciation may be warranted to clarify the redox biology of d-penicillamine and exploit its full therapeutic potential in the future.
To fully understand the action of d-penicillamine, it is also necessary to consider the competing effects of other biological ligands that could retard the ability of medicinal chelators to sequester and eliminate excess copper from the body, such as metalloproteins. Because the vast majority of copper ions in the body are protein-bound, Smirnova et al. [106] compared the ability of d-penicillamine and other chelators to compete for Cu+ complexed to two well-known copper-binding proteins, metallothionein and Cox17. ESI-MS monitoring of Cox17 and metallothionein demetallation revealed that d-penicillamine was a far less effective chelator of protein-bound copper than meso-2,3-dimercaptosuccinic acid, BAL, or dihydrolipoic acid. This study also examined the relationship between copper-binding and chemical structure and reported a highly linear correlation between the number of sulfur atoms in a molecule and especially the number of atoms separating the thiol ligand donors.
4. RADIATION POISONING
4.1. Demand for Radioprotective Agents
Ionizing radiation produces free radicals that damage DNA, resulting in tumorigenesis or cell death if repair is unsuccessful [6, 112–115]. Exposure to ionizing radiation can occur under a variety of circumstances and may be deliberate (radiotherapy), incidental (medical imaging and flying aircraft), or unforeseen (nuclear attack, “dirty” bomb, or powerplant meltdown). In the first two scenarios, exposure is tightly controlled, and risks are mitigated where practical [6, 112, 116, 117]. In cases where exposure to radiation is anticipated, a radioprotectant can minimize the damage to healthy tissues. Since there is a direct relationship between the amount of radiation exposure and cell death, radiation therapy can cause massive cell death, whereas infrequent medical imaging shows little impact after 24 hours [117]. As a result, different exposure risks present different challenges for the development of chemical radioprotectants.
4.2. Radiation as a Cancer Treatment
Radiation therapy (RT) is a relatively noninvasive and cost-effective method for battling cancer. It accounts for 40% of remission cases and has been shown to cure certain skin, prostate, lung, cervix, head, and neck cancers without the aid of other forms of treatment such as chemotherapy or immunotherapy [112]. The goal of RT in cancer treatment is to destroy the genetic information of cancer cells and, thus, their ability to proliferate while minimizing damage to healthy cells [112, 118, 119]. Tumor cells are particularly vulnerable to radiation-induced damage, as their limited antioxidant defenses, reduced capacity for genomic repair, and higher reproductive rate accelerate cell death through apoptosis, necrosis, or mitotic catastrophe [112, 113].
4.3. Radiation-induced Genomic Instability
Radiation can damage nucleic acids either directly or indirectly through the generation of electrophilic radical species that attack sites of increased electron density, such as lone pairs or double bonds on purine or pyrimidine bases. This results in altered bases, single-stranded breaks, or double-stranded breaks (DSBs). DNA damage activates signal transduction pathways that cause cell cycle arrest, allowing the cell time to repair the damaged DNA before it can proliferate. If the damage to DNA is too severe or lethal mutations are created, the cell enters apoptosis [112, 113]. Delayed cell death after radiation therapy is often attributed to mitotic catastrophe [113]. Senescence, or the loss of proliferative ability accompanied by irreversible cell cycle arrest, and autophagy can also occur as a result of radiation exposure [112, 113]. These different mechanisms of cell death may contribute to RT resistance of some tumors and could allow for better targeting of the tumor microenvironment to increase tumor cells’ susceptibility [113].
4.4. Standard of Care
Amifostine, also known as WR-2721 (Fig. 6), is one of only three FDA-approved radioprotectant drugs. It is a phosphorothioate that must be dephosphorylated to generate the active form, 2-((aminopropyl)amino)ethanethiol (WR-1065), an aminothiol that is purported to scavenge radicals generated by radiation [120]. While radical scavenging undoubtedly plays a role, several other mechanisms may contribute to the radioprotective effects of this drug.
Fig. (6).
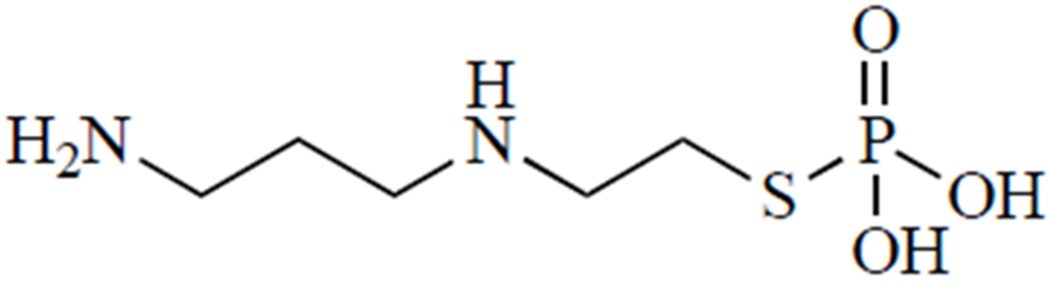
Amifostine.
4.4.1. Possible Mechanisms of Action
Alkaline phosphatase (ALP) enzymes quickly metabolize amifostine to WR-1065. However, amifostine affords a greater degree of radioprotection to healthy cells, an effect that may be explained by differential ALP expression between healthy and tumor cells. An in vitro study comparing the effects of amifostine in normal human dermal fibroblasts and MCF7 human breast adenocarcinoma cells found that ALP activity was indeed lower, but ALP mRNAs were elevated in the MCF7 cells. This result points to dysfunctional translation machinery that prevents the cancerous cells from metabolizing amifostine and thus deprives them of the radioprotection provided by the active metabolite [121]. How might amifostine confer radioprotection in the first place? One of the primary sources of radiation resistance in tumors is their propensity for hypoxia. In the absence of oxygen, radiation does not produce as many reactive species [122]. Using EPR oximetry, Ueno et al. [123] investigated the effect of amifostine on oxygenation of healthy tissues, demonstrating that amifostine significantly decreases tissue pO2 for up to 4 days post-administration. Thus, in healthy cells, where amifostine is more effectively converted into WR-1065, the resulting lower pO2 may inhibit ROS/RNS formation in response to radiation exposure. Although the mechanism of hypoxia was not addressed, it may be a result of hypotension, a common side effect of amifostine, a hypothesis that has not yet been explored.
4.4.2. Effectiveness of Amifostine
A number of recent theoretical and animal studies and clinical trials have investigated the use of amifostine as a radioprotectant. For example, buildup factors (parameters used in the prediction of radiation shielding effectiveness) were calculated for a variety of purported radioprotectants, and results indicated that cysteine and amifostine exhibit the greatest potential for moderate-level γ-radiation protection [124]. In addition, recent animal studies reported disparities in the degree of radioprotection afforded by amifostine in different tissues. In a rat model of radiation-induced bone weakening, amifostine showed significant protection of bone mineral, but not of collagen [125], and in a study of RT-induced retinopathy in female rats, it was concluded that amifostine was more effective at preserving the inner and outer plexiform layers than the retina at 12 hours post-irradiation [126]. In humans, a meta-analysis suggested that amifostine was the only intervention that prevented moderate to severe xerostomia, salivary flow, and overall quality of life in RT patients [127]. Another study showed that amifostine significantly reduced the risk of grade 2 or higher esophageal toxicity and pulmonary toxicity. Furthermore, there was no evidence of tumor protection by amifostine, the prospect of which remains an obstacle to wider usage, despite much evidence to the contrary [128].
4.4.3. Improvements to Existing Regimen
Intravenous injection is the most common route of administration for amifostine. It is inconvenient and costly, and rapid dephosphorylation results in high peak plasma concentrations of WR-1065 when administered via this route, which has been hypothesized to increase the likelihood of adverse side effects that include vomiting, nausea, diarrhea, and hypotension [119, 129]. Thus, alternative routes of administration are of interest, since the radioprotective benefits of amifostine would outweigh the costs and risks if better-tolerated formulations were available. Subcutaneous (SC) injection has been proposed, but it leaves a rash at the injection site and neither slows the release of WR-1065 nor exhibits greater effectiveness in human patients [130, 131]. Therefore, several alternative routes of administration were investigated in a murine model by Ranganathan et al. [130]. When compared to SC, oral amifostine resulted in approximately 8-fold lower peak plasma concentrations, and peak levels were reached much later after administration. These results suggest that an oral formulation may be able to achieve therapeutically relevant plasma levels that are still low enough to avoid adverse side effects. In addition, an oral formulation would be cheaper and more convenient, which may enable more patients to take full advantage of its radioprotective capabilities.
While amifostine is approved as a radioprotectant, its use as a post-exposure treatment for acute radiation syndrome has not been well established. One strategy for improving its effectiveness in these conditions is to slow its metabolism to WR-1065. Currently, the pharmacokinetic profile of amifostine precludes its use as a post-treatment for radiation exposure, which may last for hours under circumstances such as dirty bomb blast exposure or nuclear plant meltdown [132]. Nanomaterials offer promising means of overcoming the pharmacokinetic limitations of the neat drug. For example, poly (lactic glycolic acid) nanoparticles containing amifostine or WR-1065 have been shown to improve 30-day survival, hematopoietic progenitor survival, and jejunal crypt cell survival following irradiation in animal models. Other nanomaterials, such as chitosan and solid-lipid nanoparticles, may also be promising drug delivery platforms for radioprotectants [133]. CCM-amifostine, a polyethylene glycol micelle containing amifostine, was examined in C57BL/6 mice exposed to radiation for 4 hours, a time-frame designed to mimic exposure during a “dirty bomb” attack or powerplant meltdown. CCM-amifostine significantly improved RBC and lymphocyte counts and survival compared to animals treated with the neat drug. In comparing the pharmacokinetic profiles of the neat drug and CCM micelle-delivered drug, the CCM micelles resulted in increased plasma concentrations of WR-1065 up to 6 hours post-administration [132]. These very promising results highlight the potential for drug delivery strategies to optimize the release profiles of radioprotectants for the challenges of different exposure scenarios. Combining amifostine with other pharmacological modifiers, such as selenium, prostaglandin E2, misoprostol, or β-glucan, may mitigate side effects and enhance radioprotection [134].
4.4.4. Alternatives to Amifostine
Due to its side-effect profile, there is considerable interest in developing better-tolerated alternatives to amifostine. Captopril, a thiol-containing angiotensin-converting enzyme (ACE) inhibitor, may be a good candidate because the renin-angiotensin axis plays an important role in the proliferation of hematopoietic cells following radiation exposure [135]. Captopril may be a better-tolerated radioprotectant than amifostine in some cases, and it has been shown to reduce cardiac and pulmonary toxicity following thoracic RT [136, 137]. Further, captopril is FDA-approved and routinely and safely prescribed. A study by McCart et al. [138] demonstrated that inhibitors of hematopoietic progenitor proliferation such as captopril may, somewhat counterintuitively, protect against radiation-induced DNA damage. By slowing proliferation, these agents effectively allow more time for DNA repair in important progenitor cells and may, therefore, be beneficial in accelerating the recovery of blood cell populations. In a mouse model of radiation exposure complicated by severe skin burns, however, captopril actually reduced survival [139]. This scenario can be particularly challenging, as depletion of immune cell populations greatly increases the risk of infection and slows wound healing in these patients [140].
Both N-acetylcysteine and N-acetylcysteine amide have also been explored as alternatives to amifostine. While N-acetylcysteine is already FDA-approved, generally considered safe, and available in both oral and inhalable dosing forms, N-acetylcysteine amide may more easily pass through cell membranes [52], and thus more effectively protect intracellular structures including nucleic acids and mitochondria. In X-ray-exposed CHO cells, both the drugs were able to increase GSH and cysteine levels significantly compared to untreated cells, but N-acetylcysteine amide was effective at a wider range of concentrations than N-acetylcysteine [141]. Building on a previous work by our group [142], Neal et al. [143] demonstrated that both d- and l-isomers of N-acetylcysteine exerted similar antioxidant effects following radiation exposure, which implies that its radioprotective capabilities are the result of radical scavenging, since only the l-isomer contributes to GSH synthesis in vivo [144, 145]. N-acetylcysteine has also been shown to protect against DSBs in human RBCs following radiation exposure ex vivo [146]. Additionally, in a very small prospective controlled trial [147], a multi-antioxidant supplement containing N-acetylcysteine protected against DNA DSBs measured after 99mTc bone scans for cancer staging. This limited “proof-of-concept” trial may provide the impetus for further investigation in trials with optimized antioxidant dosages. Further, because the supplement would be relatively safe and cost-effective as a prophylactic, it could be used by medical personnel, emergency workers, pilots, and even astronauts who are routinely exposed to radiation [129, 147]. Indeed, different types of exposure or radiation may require specific radioprotectors. For example, α particles, with their extremely short mean free path, pose a greater threat to cytoplasmic components like mitochondria than to the nucleus [148]. In an in vitro model of inhaled α particle exposure, Wu et al. [149] showed that autophagy serves a cytoprotective role by destroying dysfunctional mitochondria that produce ROS and dysregulate the energy balance of the cell. However, N-acetylcysteine, which was shown to inhibit autophagy, may result in delayed DNA repair and decreased survival.
5. OTHER CURRENTLY APPROVED THIOL DRUGS
d-penicillamine has been prescribed as a treatment for Rheumatoid Arthritis (RA) since the 1970s when it was regarded as a prototypical disease-modifying anti-rheumatic drug, or a drug that can halt or reverse joint degeneration, as opposed to treatments that modify symptoms only [150]. However, treatment with d-penicillamine or gold salts was superseded by methotrexate in the 1980s, and more recently, by biologics such as TNF inhibitors beginning in the 1990s [151]. As a result, only a small minority of patients use d-penicillamine to manage RA [152], but a few recent studies have shed light on its purported side effects and mechanism of action. While a new prospective observational study concluded that there was no evidence of membranous lesions in adult patients with RA [153], a retrospective cohort study demonstrated that RA patients using methotrexate and d-penicillamine were at increased risk of developing non-melanoma skin cancer [154]. Recently, a study by Brancaleone et al. [155] demonstrated that d-penicillamine is a potent inhibitor of cystathionine-γ-lyase, an enzyme that contributes to H2S production, which has been shown to play a pro-inflammatory role in RA [156].
N-(2-mercaptopropionyl)glycine, or tiopronin (Fig. 7), is best known as a treatment for cystinuria, where it prevents the formation of cystine kidney stones by forming soluble cysteine-tiopronin disulfides [157]. There are limited clinical trials for this rare disease [158], but an epidemiological study in the UK reported that tiopronin had fewer side effects than d-penicillamine [157]. Tiopronin is also an effective antioxidant and Cu2+ chelator [159] and has therefore been explored for a number of oxidative stress-related conditions, most notably cataracts [160, 161].
Fig. (7).
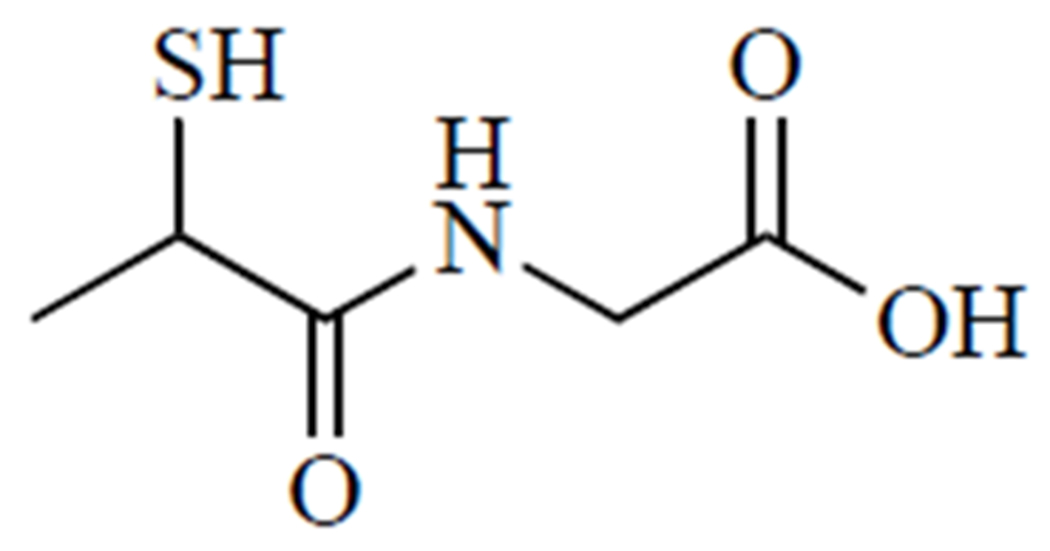
Tiopronin.
Captopril (Fig. 8) was the first angiotensin-converting enzyme (ACE) inhibitor; it is fast-acting but has a short half-life [162]. ACE inhibitors significantly reduce the risk of stroke, coronary heart disease, and heart failure [163]. An important component of captopril’s inhibitory activity is conferred by the interaction of the thiol group with the zinc ion located in the active site of the ACE enzyme [164]. In comparison to other ACE inhibitors, a recent nation-wide retrospective cohort analysis in Taiwan [165] reported that captopril had the highest overall increased risk of mortality, but patients recovering from cardiovascular events were most likely to receive captopril due to its short time to peak plasma concentration [162]. The addition of etamicastat, a dopamine-β-hydroxylase inhibitor, improved its anti-hypertensive effects in a rodent model [166].
Fig. (8).
Captopril.
6. FUTURE DIRECTIONS FOR THIOL DRUGS
Thiol drugs are indispensable and fill varied but important niches in healthcare (Table 1). However, there is considerable interest in repurposing certain thiol drugs, such as N-acetylcysteine, d-penicillamine, and meso-2,3-dimercaptosuccinic acid, as their antioxidant and metal chelating properties, may prove beneficial in conditions for which there is no effective treatment. For example, thiol affinity for metals may be useful outside of chelation therapy for heavy metal poisoning: Metallo-β-lactamases, which have evolved as a bacterial defense against β-lactam antibiotics, incorporate a metal ion such as zinc in their active site [167]. Thiol compounds like captopril, tiopronin, and thiorphan are able to bind this metal ion and block the active site [168]. Indeed, replacing the thiol group with carboxylate obliterated the inhibitory effects of captopril [169], an indicator that thiol-containing drugs may play a crucial role in combating antibiotic resistance. Another emerging area of thiol drug investigation is in relation to psychiatric and neurodegenerative conditions, including administration of N-acetylcysteine or its analogue N-acetylcysteine amide for schizophrenia, bipolar disorder, addiction, depression, epilepsy, traumatic brain injury, HIV dementia, and others [170–172]. Captopril has also shown neuroprotective effects against lipopolysaccharide-induced memory impairment and Alzheimer’s-related neuroinflammation in rodent models [173, 174]. While more rigorous trials and further studies are needed before they become widely accepted [171], the prospect of relatively safe and affordable treatments for these conditions is appealing and highlights the need for continued exploration of the rich biochemistry and therapeutic potential of medicinal thiols.
Table 1.
Summary of medicinal thiols and their current and potential uses.
| Common Name | Structure | Primary Use | Primary Mechanism | Other Uses Covered in this Review |
|---|---|---|---|---|
| N-Acetyl-L-cysteine | 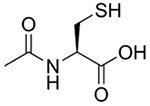 |
APAP overdose [16, 18, 21, 23, 25, 29, 32, 34, 36, 38–42, 45–51, 53] | Acts as GSH prodrug by supplying cysteine | Lead toxicity [82, 85–86] Radioprotection [129, 146–147] Psychiatric/Neurologic Disorders [170, 171] |
| Amifostine, WR-2721 | Radioprotection [113, 119, 121–134] | Scavenges free radicals formed during RT | - | |
| Captopril | 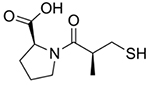 |
Hypertension [163, 165–166] | Blocks activation of angiotensin, a vasoconstrictor | Lead toxicity [89] Radioprotection [136–139] Metallo-β-lactamase inhibition [167–169] Alzheimer’s/dementia [173, 174] |
| Dimercaprol, BAL in oil |  |
Heavy metal toxicity As, Au, Hg, and Pb [71–72, 94–96] | Chelates heavy metals, facilitating their removal | - |
| 2,3-meso- Dimercaptosuccinic acid |
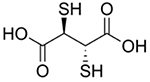 |
Lead poisoning [71–72, 74, 76–81] | Chelates Pb to form soluble, excretable complexes | - |
| N-(2-Mercaptopropionyl)-glycine, Tiopronin, Thiola | 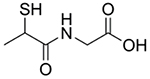 |
Cystinuria [157–158] | Forms soluble cysteine-tiopronin disulfides with excess cysteine in urine | Metallo-β-lactamase inhibition [167] Copper chelation [159] |
| D-Penicillamine | 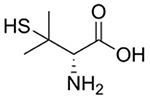 |
Wilson’s disease [97, 99, 101–102, 104, 106, 108, 110] | Chelates Cu to form soluble, excretable complexes | Rheumatoid arthritis [151–155] Chemotherapy adjuvant [98] |
CONCLUSION
Thiol-containing drugs represent a collection of widely used and indispensable medicines: their numerous indications include acetaminophen overdose, heavy metal toxicity, and radiation exposure. These diverse applications are made possible by the chemistry of the −SH moiety, which enables these compounds to chelate heavy metals, scavenge ROS, and support endogenous antioxidant systems. While many medicinal thiols have been in use for decades, a wealth of recent research demonstrates continued interest in elucidating their mechanisms of action and further exploration of their therapeutic potential in an ever-broadening range of conditions, including antibiotic resistance and neurodegenerative diseases. Indeed, repurposing, employing delivery strategies, and optimizing treatment regimens of existing thiol drugs hold much promise for advancing our understanding of these conditions and improving patient outcomes.
ACKNOWLEDGEMENTS
Declared none.
FUNDING
This work was supported by the NEI of the National Institutes of Health under award number R15EY029813 (Crossref Funder ID http://dx.doi.org/10.13039/100000053) and the Richard K. Vitek/FCR Endowment at Missouri University of Science and Technology. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
LIST OF ABBREVIATIONS
- ACE
Angiotensin-Converting Enzyme
- ALP
Alkaline Phosphatase
- ALT
Alanine Aminotransferase
- APAP
N-acetyl-p-aminophenol
- ATP7B
P-type ATPase
- BAL
British anti-Lewisite
- CAT
Catalases
- CHO
Chinese Hamster Ovary
- CT
Computed Tomography
- DSB
Double Stranded Break
- EDTA
Ethylenediaminetetraacetic Acid
- EPR
Electron Paramagnetic Resonance
- ESI-MS
Electrospray Ionization Mass Spectrometry
- FDA
United States Food and Drug Administration
- GSH
Glutathione
- GSSG
Glutathione Disulfide
- IV
Intravenous
- MSN
Mesoporous Silica Nanoparticle
- NAPQI
N-acetyl-p-benzoquinone Imine
- NMR
Nuclear Magnetic Resonance
- pO2
Partial Pressure of Oxygen
- RA
Rheumatoid Arthritis
- RBC
Red Blood Cell
- ROS/RNS
Reactive Oxygen and/or Nitrogen Species
- RT
Radiation Therapy
- SC
Subcutaneous
- SOD
Superoxide Dismutases
- TNF
Tumor Necrosis Factor
- 99mTc
Technetium-99m
- UDP
Uridine Diphosphate
- UK
United Kingdom
- WD
Wilson’s Disease
- WR-1065
2-((aminopropyl)amino)ethanethiol
- WR-2721
Amifostine
- δ-ALA
δ-Aminolevulinic Acid
Footnotes
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- [1].Laher I Systems Biology of Free Radicals and Antioxidants; Springer: Berlin, Heidelberg, 2014. [ 10.1007/978-3-642-30018-9] [DOI] [Google Scholar]
- [2].Gutteridge JMC; Halliwell B Free Radicals in Biology and Medicine, 5th ed; Oxford University Press: New York, 2015. [Google Scholar]
- [3].Ratliff BB; Abdulmahdi W; Pawar R; Wolin MS Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal, 2016, 25(3), 119–146. [ 10.1089/ars.2016.6665] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Quiñonez-Flores CM; González-Chávez SA; Del Río Nájera D; Pacheco-Tena C Oxidative Stress Relevance in the Pathogenesis of the Rheumatoid Arthritis: A Systematic Review. BioMed Res. Int, 2016, 2016, 6097417 [ 10.1155/2016/6097417] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schieber M; Chandel NS ROS function in redox signaling and oxidative stress. Curr. Biol, 2014, 24(10), R453–R462. [ 10.1016/j.cub.2014.03.034] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Smith TA; Kirkpatrick DR; Smith S; Smith TK; Pearson T; Kailasam A; Herrmann KZ; Schubert J; Agrawal DK Radioprotective agents to prevent cellular damage due to ionizing radiation. J. Transl. Med, 2017, 15(1), 232 [ 10.1186/s12967-017-1338-x] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Crichton E; Ward RJ; Hider RC Metal Chelation in Medicine; The Royal Society of Chemistry: Cambridge, UK, 2016, Vol. 8, p. 322 [ 10.1039/9781782623892] [DOI] [Google Scholar]
- [8].Huxtable RJ Biochemistry of Sulfur; , 1986. [ 10.1007/978-1-4757-9438-0] [DOI] [Google Scholar]
- [9].Nosengo N Can you teach old drugs new tricks? Nature, 2016, 554(7607), 314–316. [ 10.1038/534314a] [DOI] [PubMed] [Google Scholar]
- [10].Blieden M; Paramore LC; Shah D; Ben-Joseph R A perspective on the epidemiology of acetaminophen exposure and toxicity in the United States. Expert Rev. Clin. Pharmacol, 2014, 7(3), 341–348. [ 10.1586/17512433.2014.904744] [DOI] [PubMed] [Google Scholar]
- [11].Lee WM Acetaminophen and the U.S. Acute Liver Failure Study Group. Acetaminophen and the U.S. Acute Liver Failure Study Group: lowering the risks of hepatic failure. Hepatology, 2004, 40(1), 6–9. [ 10.1002/hep.20293] [DOI] [PubMed] [Google Scholar]
- [12].Larson AM; Polson J; Fontana RJ; Davern TJ; Lalani E; Hynan LS; Reisch JS; Schiadt FV; Ostapowicz G; Shakil AO; Lee WM Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multi-center, prospective study. Hepatology, 2005, 42(6), 1364–1372. [ 10.1002/hep.20948] [DOI] [PubMed] [Google Scholar]
- [13].Schiødt FV; Atillasoy E; Shakil AO; Schiff ER; Caldwell C; Kowdley KV; Stribling R; Crippin JS; Flamm S; Somberg KA; Rosen H; McCashland TM; Hay JE; Lee WM Etiology and outcome for 295 patients with acute liver failure in the United States. Liver Transpl. Surg, 1999, 5(1), 29–34. [ 10.1002/lt.500050102] [DOI] [PubMed] [Google Scholar]
- [14].Nourjah P; Ahmad SR; Karwoski C; Willy M Estimates of acetaminophen (Paracetomal)-associated overdoses in the United States. Pharmacoepidemiol. Drug Saf., 2006, 15(6), 398–405. [ 10.1002/pds.1191] [DOI] [PubMed] [Google Scholar]
- [15].Heard KJ Acetylcysteine for acetaminophen poisoning. N. Engl. J. Med, 2008, 359(3), 285–292. [ 10.1056/NEJMct0708278] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yoon E; Babar A; Choudhary M; Kutner M; Pyrsopoulos N Acetaminophen-Induced Hepatotoxicity: a Comprehensive Update. J. Clin. Transl. Hepatol, 2016, 4(2), 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Macherey A-C; Dansette PM Biotransformations Leading to Toxic Metabolites: Chemical AspectThe Practice of Medicinal Chemistry, 3rd ed; Wermuth CG, Ed.; Academic Press, 2011. [Google Scholar]
- [18].Klopčič I; Poberžnik M; Mavri J; Dolenc MS A quantum chemical study of the reactivity of acetaminophen (paracetamol) toxic metabolite N-acetyl-p-benzoquinone imine with deoxyguanosine and glutathione. Chem. Biol. Interact, 2015, 242, 407–414. [ 10.1016/j.cbi.2015.11.002] [DOI] [PubMed] [Google Scholar]
- [19].James LP; Mayeux PR; Hinson JA Acetaminophen-induced hepatotoxicity. Drug Metab. Dispos, 2003, 31(12), 1499–1506. [ 10.1124/dmd.31.12.1499] [DOI] [PubMed] [Google Scholar]
- [20].Saito C; Zwingmann C; Jaeschke H Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology, 2010, 51(1), 246–254. [ 10.1002/hep.23267] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ghanem CI; Perez MJ; Manautou JE; Mottino AD Acetaminophen from liver to brain: New insights into drug pharmacological action and toxicity. Pharmacol. Res, 2016, 109, 119–131. [ 10.1016/j.phrs.2016.02.020] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kim JW; Ryu SH; Kim S; Lee HW; Lim MS; Seong SJ; Kim S; Yoon YR; Kim KB Pattern recognition analysis for hepatotoxicity induced by acetaminophen using plasma and urinary 1H NMR-based metabolomics in humans. Anal. Chem, 2013, 85(23), 11326–11334. [ 10.1021/ac402390q] [DOI] [PubMed] [Google Scholar]
- [23].Chiew AL; Gluud C; Brok J; Buckley NA Interventions for paracetamol (acetaminophen) overdose. Cochrane Database Syst. Rev, 2018, 2CD003328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Atkuri KR; Mantovani JJ; Herzenberg LA; Herzenberg LA N-Acetylcysteine--a safe antidote for cysteine/glutathione deficiency. Curr. Opin. Pharmacol, 2007, 7(4), 355–359. [ 10.1016/j.coph.2007.04.005] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chan JCY; Soh ACK; Kioh DYQ; Li J; Verma C; Koh SK; Beuerman RW; Zhou L; Chan ECY Reactive Metabolite-induced Protein Glutathionylation: A Potentially Novel Mechanism Underlying Acetaminophen Hepatotoxicity. Mol. Cell. Proteomics, 2018, 17(10), 2034–2050. [ 10.1074/mcp.RA118.000875] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Marzullo L An update of N-acetylcysteine treatment for acute acetaminophen toxicity in children. Curr. Opin. Pediatr, 2005, 17(2), 239–245. [ 10.1097/01.mop.0000152622.05168.9e] [DOI] [PubMed] [Google Scholar]
- [27].Smilkstein MJ; Knapp GL; Kulig KW; Rumack BH Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N. Engl. J. Med, 1988, 319(24), 1557–1562. [ 10.1056/NEJM198812153192401] [DOI] [PubMed] [Google Scholar]
- [28].Craig DG; Bates CM; Davidson JS; Martin KG; Hayes PC; Simpson KJ Staggered overdose pattern and delay to hospital presentation are associated with adverse outcomes following paracetamol-induced hepatotoxicity. Br. J. Clin. Pharmacol, 2012, 73(2), 285–294. [ 10.1111/j.1365-2125.2011.04067.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kane AE; Huizer-Pajkos A; Mach J; McKenzie C; Mitchell SJ; de Cabo R; Jones B; Cogger V; Le Couteur DG; Hilmer SN N-Acetyl cysteine does not prevent liver toxicity from chronic low-dose plus subacute high-dose paracetamol exposure in young or old mice. Fundam. Clin. Pharmacol, 2016, 30(3), 263–275. [ 10.1111/fcp.12184] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Green JL; Heard KJ; Reynolds KM; Albert D Oral and Intravenous Acetylcysteine for Treatment of Acetaminophen Toxicity: A Systematic Review and Meta-analysis. West. J. Emerg. Med, 2013, 14(3), 218–226. [ 10.5811/westjem.2012.4.6885] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Blackford MG; Felter T; Gothard MD; Reed MD Assessment of the clinical use of intravenous and oral N-acetylcysteine in the treatment of acute acetaminophen poisoning in children: a retrospective review. Clin. Ther, 2011, 33(9), 1322–1330. [ 10.1016/j.clinthera.2011.08.005] [DOI] [PubMed] [Google Scholar]
- [32].Pauley KA; Sandritter TL; Lowry JA; Algren DA Evaluation of an Alternative Intravenous N-Acetylcysteine Regimen in Pediatric Patients. J. Pediatr. Pharmacol. Ther, 2015, 20(3), 178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hayes BD; Klein-Schwartz W; Doyon S Frequency of medication errors with intravenous acetylcysteine for acetaminophen overdose. Ann. Pharmacother, 2008, 42(6), 766–770. [ 10.1345/aph.1K685] [DOI] [PubMed] [Google Scholar]
- [34].Stine JG; Lewis JH Current and future directions in the treatment and prevention of drug-induced liver injury: a systematic review. Expert Rev. Gastroenterol. Hepatol, 2016, 10(4), 517–536. [ 10.1586/17474124.2016.1127756] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Schmidt LE Identification of patients at risk of anaphylactoid reactions to N-acetylcysteine in the treatment of paracetamol over-dose. Clin. Toxicol. (Phila.), 2013, 51(6), 467–472. [ 10.3109/15563650.2013.799677] [DOI] [PubMed] [Google Scholar]
- [36].Pickering G; Macian N; Papet I; Duale C; Coudert C; Pereira B N-acetylcysteine prevents glutathione decrease and does not interfere with paracetamol antinociceptive effect at therapeutic dosage: a randomized double-blind controlled trial in healthy subjects. Fundam. Clin. Pharmacol, 2019, 33(3), 303–311. [ 10.1111/fcp.12437] [DOI] [PubMed] [Google Scholar]
- [37].Kelly GS Clinical applications of N-acetylcysteine. Altern. Med. Rev, 1998, 3(2), 114–127. [PubMed] [Google Scholar]
- [38].Chiew AL; Fountain JS; Graudins A; Isbister GK; Reith D; Buckley NA Summary statement: new guidelines for the management of paracetamol poisoning in Australia and New Zealand. Med. J. Aust, 2015, 203(5), 215–218. [ 10.5694/mja15.00614] [DOI] [PubMed] [Google Scholar]
- [39].Wong A; Landersdorfer C; Graudins A Pharmacokinetic modelling of modified acetylcysteine infusion regimens used in the treatment of paracetamol poisoning. Eur. J. Clin. Pharmacol, 2017, 73(9), 1103–1110. [ 10.1007/s00228-017-2277-4] [DOI] [PubMed] [Google Scholar]
- [40].Wong A; Gunja N; McNulty R; Graudins A Analysis of an 8-hour acetylcysteine infusion protocol for repeated supratherapeutic ingestion (RSTI) of paracetamol. Clin. Toxicol. (Phila.), 2018, 56(3), 199–203. [ 10.1080/15563650.2017.1359620] [DOI] [PubMed] [Google Scholar]
- [41].Du K; Ramachandran A; Jaeschke H Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol, 2016, 10, 148–156. [ 10.1016/j.redox.2016.10.001] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Duan L; Davis JS; Woolbright BL; Du K; Cahkraborty M; Weemhoff J; Jaeschke H; Bourdi M Differential susceptibility to acetaminophen-induced liver injury in sub-strains of C57BL/6 mice: 6N versus 6J Food Chem. Toxicol, 2016, 95(Pt B), 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hughes RD; Gazzard BG; Hanid MA; Trewby PN; Murray-Lyon IM; Davis M; Williams R; Bennet JR Controlled trial of cysteamine and dimercaprol after paracetamol overdose. BMJ, 1977, 2(6099), 1395 [ 10.1136/bmj.2.6099.1395] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hamlyn AN; Lesna M; Record CO; Smith PA; Watson AJ; Meredith T; Volans GN; Crome P Methionine and cysteamine in paracetamol (acetaminophen) overdose, prospective controlled trial of early therapy. J. Int. Med. Res, 1981, 9(3), 226–231. [ 10.1177/030006058100900314] [DOI] [PubMed] [Google Scholar]
- [45].Castaneda-Arriaga R; Perez-Gonzalez A; Galano A Chemical Protectors against the Toxic Effects of Paracetamol (Acetaminophen) and Its Meta Analogue: Preventing Protein Arylation. ACS Omega, 2018, 3(12), 18582–18591. [ 10.1021/acsomega.8b02943] [DOI] [Google Scholar]
- [46].Koyama R; Mizuta R Acrolein scavengers, cysteamine and N-benzylhydroxylamine, reduces the mouse liver damage after acetaminophen overdose. J. Vet. Med. Sci, 2017, 75(12), 1903–1905. [ 10.1292/jvms.16-0325] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Buckley NA; Dawson AH; Isbister GK Treatments for paracetamol poisoning. BMJ, 2016, 353, i2579 [ 10.1136/bmj.i2579] [DOI] [PubMed] [Google Scholar]
- [48].Khayyat A; Tobwala S; Hart M; Ercal N N-acetylcysteine amide, a promising antidote for acetaminophen toxicity. Toxicol. Lett, 2016, 241, 133–142. [ 10.1016/j.toxlet.2015.11.008] [DOI] [PubMed] [Google Scholar]
- [49].Theodosis-Nobelos P; Athanasekou C; Rekka EA Dual antioxidant structures with potent anti-inflammatory, hypolipidemic and cytoprotective properties. Bioorg. Med. Chem. Lett, 2017, 27(21), 4800–4804. [ 10.1016/j.bmcl.2017.09.054] [DOI] [PubMed] [Google Scholar]
- [50].More SS; Nugent J; Vartak AP; Nye SM; Vince R Hepatoprotective Effect of ψ-Glutathione in a Murine Model of Acetaminophen-Induced Liver Toxicity. Chem. Res. Toxicol, 2017, 30(3), 777–784. [ 10.1021/acs.chemrestox.6b00291] [DOI] [PubMed] [Google Scholar]
- [51].Nilsson JLA; Blomgren A; Nilsson UJ; Högestätt ED; Grundemar L N,N’-Bis(2-mercaptoethyl)isophthalamide Binds Electrophilic Paracetamol Metabolites and Prevents Paracetamol-Induced Liver Toxicity. Basic Clin. Pharmacol. Toxicol, 2018, 123(5), 589–593. [ 10.1111/bcpt.13058] [DOI] [PubMed] [Google Scholar]
- [52].Ates B; Abraham L; Ercal N Antioxidant and free radical scavenging properties of N-acetylcysteine amide (NACA) and comparison with N-acetylcysteine (NAC). Free Radic. Res, 2008, 42(4), 372–377. [ 10.1080/10715760801998638] [DOI] [PubMed] [Google Scholar]
- [53].Tobwala S; Khayyat A; Fan W; Ercal N Comparative evaluation of N-acetylcysteine and N-acetylcysteineamide in acetaminophen-induced hepatotoxicity in human hepatoma HepaRG cells. Exp. Biol. Med. (Maywood), 2015, 240(2), 261–272. [ 10.1177/1535370214549520] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Baran EJ Chelation therapies: a chemical and biochemical perspective. Curr. Med. Chem, 2010, 17(31), 3658–3672. [ 10.2174/092986710793213760] [DOI] [PubMed] [Google Scholar]
- [55].Chemistry Coordination. A Century of Progress; American Chemical Society, 1994, Vol. 565, . [Google Scholar]
- [56].Advisory Committee for Childhood Lead Poisoning Prevention. Low level lead exposure harms children: a renewed call for primary prevention (A report of the Advisory Committee on Childhood Lead Poisoning Prevention); Centers for Disease Control and Prevention; Atlanta: GA, 2012. [Google Scholar]
- [57].Jomova K; Valko M Advances in metal-induced oxidative stress and human disease. Toxicology, 2011, 283(2-3), 65–87. [ 10.1016/j.tox.2011.03.001] [DOI] [PubMed] [Google Scholar]
- [58].Hanna-Attisha M; LaChance J; Sadler RC; Champney Schnepp A Elevated Blood Lead Levels in Children Associated With the Flint Drinking Water Crisis: A Spatial Analysis of Risk and Public Health Response. Am. J. Public Health, 2016, 106(2), 283–290. [ 10.2105/AJPH.2015.303003] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gillis BS; Arbieva Z; Gavin IM Analysis of lead toxicity in human cells. BMC Genomics, 2012, 13, 344 [ 10.1186/1471-2164-13-344] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Farkas E; Buglyó P Lead(II) Complexes of Amino Acids, Peptides, and Other Related Ligands of Biological Interest. Met. Ions Life Sci, 2017, 17, 17 [ 10.1515/9783110434330-008] [DOI] [PubMed] [Google Scholar]
- [61].Kern M; Wisniewski M; Cabell L; Audesirk G Inorganic lead and calcium interact positively in activation of calmodulin. Neurotoxicology, 2000, 21(3), 353–363. [PubMed] [Google Scholar]
- [62].Flora G; Gupta D; Tiwari A Toxicity of lead: A review with recent updates. Interdiscip. Toxicol, 2012, 5(2), 47–58. [ 10.2478/v10102-012-0009-2] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ercal N; Gurer-Orhan H; Aykin-Burns N Toxic metals and oxidative stress part I: mechanisms involved in metal-induced oxidative damage. Curr. Top. Med. Chem, 2001, 1(6), 529–539. [ 10.2174/1568026013394831] [DOI] [PubMed] [Google Scholar]
- [64].Gurer H; Ercal N Can antioxidants be beneficial in the treatment of lead poisoning? Free Radic. Biol. Med, 2000, 29(10), 927–945. [ 10.1016/S0891-5849(00)00413-5] [DOI] [PubMed] [Google Scholar]
- [65].Kasperczyk A; Prokopowicz A; Dobrakowski M; Pawlas N; Kasperczyk S The effect of occupational lead exposure on blood levels of zinc, iron, copper, selenium and related proteins. Biol. Trace Elem. Res, 2012, 150(1-3), 49–55. [ 10.1007/s12011-012-9490-x] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Monteiro HP; Abdalla DS; Augusto O; Bechara EJ Free radical generation during delta-aminolevulinic acid autoxidation: induction by hemoglobin and connections with porphyrinpathies. Arch. Biochem. Biophys, 1989, 271(1), 206–216. [ 10.1016/0003-9861(89)90271-3] [DOI] [PubMed] [Google Scholar]
- [67].Monteiro HP; Abdalla DS; Faljoni-Alàrio A; Bechara EJ Generation of active oxygen species during coupled autoxidation of oxyhemoglobin and delta-aminolevulinic acid. Biochim. Biophys. Acta, 1986, 881(1), 100–106. [ 10.1016/0304-4165(86)90102-9] [DOI] [PubMed] [Google Scholar]
- [68].Gürer H; Ozgünes H; Neal R; Spitz DR; Erçal N Antioxidant effects of N-acetylcysteine and succimer in red blood cells from lead-exposed rats. Toxicology, 1998, 128(3), 181–189. [ 10.1016/S0300-483X(98)00074-2] [DOI] [PubMed] [Google Scholar]
- [69].Othman AI; El Missiry MA Role of selenium against lead toxicity in male rats. J. Biochem. Mol. Toxicol, 1998, 12(6), 345–349. [] [DOI] [PubMed] [Google Scholar]
- [70].Tobwala S; Wang H-J; Carey J; Banks W; Ercal N Effects of Lead and Cadmium on Brain Endothelial Cell Survival, Monolayer Permeability, and Crucial Oxidative Stress Markers in an in Vitro Model of the Blood-Brain Barrier. Toxics, 2014, 2(2), 258–275. [ 10.3390/toxics2020258] [DOI] [Google Scholar]
- [71].Aaseth J; Skaug MA; Cao Y; Andersen O Chelation in metal intoxication--Principles and paradigms. J. Trace Elem. Med. Biol, 2015 31, 260–266. [ 10.1016/j.jtemb.2014.10.001] [DOI] [PubMed] [Google Scholar]
- [72].Björklund G; Mutter J; Aaseth J Metal chelators and neurotoxicity: lead, mercury, and arsenic. Arch. Toxicol, 2017, 91(12), 3787–3797. [ 10.1007/s00204-017-2100-0] [DOI] [PubMed] [Google Scholar]
- [73].Flora SJ; Pachauri V Chelation in metal intoxication. Int. J. Environ. Res. Public Health, 2010, 7(7), 2745–2788. [ 10.3390/ijerph7072745] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Zhai H; Wang Y; Wang M; Liu S; Yu F; Gao C; Li G; Wu Q Construction of a Glutathione-Responsive and Silica-Based Nanocomposite for Controlled Release of Chelator Dimercaptosuccinic Acid. Int. J. Mol. Sci, 2018, 19(12)E3790 [ 10.3390/ijms19123790] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kosnett MJ Chelation for heavy metals (arsenic, lead, and mercury): protective or perilous? Clin. Pharmacol. Ther, 2010, 88(3), 412–415. [ 10.1038/clpt.2010.132] [DOI] [PubMed] [Google Scholar]
- [76].Andersen O; Aaseth J A review of pitfalls and progress in chelation treatment of metal poisonings. J. Trace Elem. Med. Biol, 2016. 38, 74–80. [ 10.1016/j.jtemb.2016.03.013] [DOI] [PubMed] [Google Scholar]
- [77].Sakthithasan K; Lévy P; Poupon J; Garnier R A comparative study of edetate calcium disodium and dimercaptosuccinic acid in the treatment of lead poisoning in adults. Clin. Toxicol. (Phila.), 2018, 56(11), 1143–1149. [ 10.1080/15563650.2018.1478424] [DOI] [PubMed] [Google Scholar]
- [78].Kaviani S; Shahab S; Sheikhi M; Ahmadianarog M DFT study on the selective complexation of meso-2,3-dimercaptosuccinic acid with toxic metal ions (Cd2+, Hg2+ and Pb2+) for pharmaceutical and biological applications. J. Mol. Struct, 2019, 1176, 901–907. [ 10.1016/j.molstruc.2018.09.027] [DOI] [Google Scholar]
- [79].van Eijkeren JC; Olie JD; Bradberry SM; Vale JA; de Vries I; Clewell HJ III; Meulenbelt J; Hunault CC Modeling the effect of succimer (DMSA; dimercaptosuccinic acid) chelation therapy in patients poisoned by lead. Clin. Toxicol. (Phila.), 2017, 55(2), 133–141. [ 10.1080/15563650.2016.1263855] [DOI] [PubMed] [Google Scholar]
- [80].Yadav A; Flora SJ Nano drug delivery systems: a new paradigm for treating metal toxicity. Expert Opin. Drug Deliv, 2016, 13(6), 831–841. [ 10.1517/17425247.2016.1160890] [DOI] [PubMed] [Google Scholar]
- [81].Alcaraz-Contreras Y; Mendoza-Lozano RP; Martínez-Alcaraz ER; Martínez-Alfaro M; Gallegos-Corona MA; Ramírez-Morales MA; Vázquez-Guevara MA Silymarin and dimercaptosuccinic acid ameliorate lead-induced nephrotoxicity and genotoxicity in rats. Hum. Exp. Toxicol, 2016, 35(4), 398–403. [ 10.1177/0960327115591373] [DOI] [PubMed] [Google Scholar]
- [82].Sisombath NS; Jalilehvand F Similarities between N-Acetylcysteine and Glutathione in Binding to Lead(II) Ions. Chem. Res. Toxicol, 2015, 28(12), 2313–2324. [ 10.1021/acs.chemrestox.5b00323] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Aykin-Burns N; Franklin EA; Ercal N Effects of N-acetylcysteine on lead-exposed PC-12 cells. Arch. Environ. Contam. Toxicol, 2005, 49(1), 119–123. [ 10.1007/s00244-004-0025-0] [DOI] [PubMed] [Google Scholar]
- [84].Penugonda S; Ercal N Comparative evaluation of N-acetylcysteine (NAC) and N-acetylcysteine amide (NACA) on glutamate and lead-induced toxicity in CD-1 mice. Toxicol. Lett, 2011, 201(1), 1–7. [ 10.1016/j.toxlet.2010.11.013] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Pedroso TF; Oliveira CS; Fonseca MM; Oliveira VA; Pereira ME Effects of Zinc and N-Acetylcysteine in Damage Caused by Lead Exposure in Young Rats. Biol. Trace Elem. Res, 2017, 180(2), 275–284. [ 10.1007/s12011-017-1009-z] [DOI] [PubMed] [Google Scholar]
- [86].Kasperczyk S; Dobrakowski M; Kasperczyk A; Romuk E; Rykaczewska-Czerwińska M; Pawlas N; Birkner E Effect of N-acetylcysteine administration on homocysteine level, oxidative damage to proteins, and levels of iron (Fe) and Fe-related proteins in lead-exposed workers. Toxicol. Ind. Health, 2016, 32(9), 1607–1618. [ 10.1177/0748233715571152] [DOI] [PubMed] [Google Scholar]
- [87].Gurer H; Ozgunes H; Oztezcan S; Ercal N Antioxidant role of alpha-lipoic acid in lead toxicity. Free Radic. Biol. Med, 1999, 27(1-2), 75–81. [ 10.1016/S0891-5849(99)00036-2] [DOI] [PubMed] [Google Scholar]
- [88].Gurer H; Neal R; Yang P; Oztezcan S; Ercal N Captopril as an antioxidant in lead-exposed Fischer 344 rats. Hum. Exp. Toxicol, 1999, 18(1), 27–32. [ 10.1177/096032719901800104] [DOI] [PubMed] [Google Scholar]
- [89].Aldahmash BA; El-Nagar DM Antioxidant effects of captopril against lead acetate-induced hepatic and splenic tissue toxicity in Swiss albino mice. Saudi J. Biol. Sci, 2016, 23(6), 667–673. [ 10.1016/j.sjbs.2016.05.005] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Neal R; Cooper K; Kellogg G; Gurer H; Ercal N Effects of some sulfur-containing antioxidants on lead-exposed lenses. Free Radic. Biol. Med, 1999, 26(1-2), 239–243. [ 10.1016/S0891-5849(98)00214-7] [DOI] [PubMed] [Google Scholar]
- [91].Neal R; Cooper K; Gurer H; Ercal N Effects of N-acetylcysteine and 2,3-dimercaptosuccinic acid on lead induced oxidative stress in rat lenses. Toxicology, 1998, 130(2-3), 167–174. [ 10.1016/S0300-483X(98)00104-8] [DOI] [PubMed] [Google Scholar]
- [92].Flora SJ; Pande M; Kannan GM; Mehta A Lead induced oxidative stress and its recovery following co-administration of melatonin or N-acetylcysteine during chelation with succimer in male rats Cell. Mol. Biol. (Noisy-le-grand), 2004, 50, OL543–51. [PubMed] [Google Scholar]
- [93].Ercal N; Treeratphan P; Hammond TC; Matthews RH; Grannemann NH; Spitz DR In vivo indices of oxidative stress in lead-exposed C57BL/6 mice are reduced by treatment with meso-2,3-dimercaptosuccinic acid or N-acetylcysteine. Free Radic. Biol. Med, 1996, 21(2), 157–161. [ 10.1016/0891-5849(96)00020-2] [DOI] [PubMed] [Google Scholar]
- [94].Ioannou PV; Vachliotis DG; Chrissanthopoulos A Chelation therapy: The interaction of British Anti-Lewisite (BAL) with some heavy metal cations of p and d blocks. Main Group Chem, 2017, 16(2), 125–139. [ 10.3233/MGC-170231] [DOI] [Google Scholar]
- [95].Harper LK; Bayse CA Modeling the chelation of As(III) in lewisite by dithiols using density functional theory and solvent-assisted proton exchange. J. Inorg. Biochem, 2015, 153, 60–67. [ 10.1016/j.jinorgbio.2015.10.004] [DOI] [PubMed] [Google Scholar]
- [96].Szekeres LI; Gyurcsik B; Kiss T; Kele Z; Jancsó A Interaction of Arsenous Acid with the Dithiol-Type Chelator British Anti-Lewisite (BAL): Structure and Stability of Species Formed in an Unexpectedly Complex System. Inorg. Chem, 2018, 57(12), 7191–7200. [ 10.1021/acs.inorgchem.8b00894] [DOI] [PubMed] [Google Scholar]
- [97].Katerji M; Barada K; Jomaa M; Kobeissy F; Makkawi AK; Abou-Kheir W; Usta J Chemosensitivity of U251 Cells to the Co-treatment of D-Penicillamine and Copper: Possible Implications on Wilson Disease Patients. Front. Mol. Neurosci, 2017, 10, 10 [ 10.3389/fnmol.2017.00010] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Gaur K; Vázquez-Salgado A; Duran-Camacho G; Dominguez-Martinez I; Benjamín-Rivera J; Fernández-Vega L; Carmona Sarabia L; Cruz García A; Pérez-Deliz F; Méndez Román J; Vega-Cartagena M; Loza-Rosas S; Rodriguez Acevedo X; Tinoco A Iron and Copper Intracellular Chelation as an Anticancer Drug Strategy. Inorganics, 2018, 6(4), 126 [ 10.3390/inorganics6040126] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Pfeiffenberger J; Beinhardt S; Gotthardt DN; Haag N; Freissmuth C; Reuner U; Gauss A; Stremmel W; Schilsky ML; Ferenci P; Weiss KH Pregnancy in Wilson’s disease: Management and outcome. Hepatology, 2018, 67(4), 1261–1269. [ 10.1002/hep.29490] [DOI] [PubMed] [Google Scholar]
- [100].Roberts EA; Schilsky ML American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology, 2008, 47(6), 2089–2111. [ 10.1002/hep.22261] [DOI] [PubMed] [Google Scholar]
- [101].Poujois A; Woimant F Wilson’s disease: A 2017 update. Clin. Res. Hepatol. Gastroenterol, 2018, 42(6), 512–520. [ 10.1016/j.clinre.2018.03.007] [DOI] [PubMed] [Google Scholar]
- [102].Hachmöller O; Zibert A; Zischka H; Sperling M; Groba SR; Grünewald I; Wardelmann E; Schmidt HH; Karst U Spatial investigation of the elemental distribution in Wilson’s disease liver after d-penicillamine treatment by LA-ICP-MS. J. Trace Elem. Med. Biol, 2017, 44, 26–31. [ 10.1016/j.jtemb.2017.05.008] [DOI] [PubMed] [Google Scholar]
- [103].Rasheed S; Sánchez SS; Yousuf S; Honoré SM; Choudhary MI Drug repurposing: In-vitro anti-glycation properties of 18 common drugs. PLoS One, 2018, 13(1)e0190509 [ 10.1371/journal.pone.0190509] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Członkowska A; Litwin T Wilson disease - currently used anti-copper therapy. Handb. Clin. Neurol, 2017, 142, 181–191. [ 10.1016/B978-0-444-63625-6.00015-X] [DOI] [PubMed] [Google Scholar]
- [105].Helmy H; Fahmy M; Abdel Aziz H; Ghobrial C; Abdel Hameed N; El-Karaksy H Urinary abnormalities in children and adolescents with Wilson disease before and during treatment with d-penicillamine. J. Gastroenterol. Hepatol, 2019. [ 10.1111/jgh.14653] [DOI] [PubMed] [Google Scholar]
- [106].Smirnova J; Kabin E; Järving I; Bragina O; Tõugu V; Plitz T; Palumaa P Copper(I)-binding properties of de-coppering drugs for the treatment of Wilson disease. α-Lipoic acid as a potential anti-copper agent. Sci. Rep, 2018, 8(1), 1463 [ 10.1038/s41598-018-19873-2] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Conry RR Copper: Inorganic & Coordination Chemistry Based in part on the article Copper: Inorganic & Coordination Chemistry by Rebecca R Conry & Kenneth D Karlin which appeared in the Encyclopedia of Inorganic Chemistry. Encyclopedia of Inorganic Chemistry, 1st ed; King RB; Crabtree RH; Lukehart CM; Atwood DA; Scott RA, Eds.; John Wiley & Sons, Ltd, 2006. [ 10.1002/0470862106.ia052] [DOI] [Google Scholar]
- [108].Königsberger LC; Königsberger E; Hefter G; May PM Formation constants of copper(I) complexes with cysteine, penicillamine and glutathione: implications for copper speciation in the human eye. Dalton Trans, 2015, 44(47), 20413–20425. [ 10.1039/C5DT02129D] [DOI] [PubMed] [Google Scholar]
- [109].Rubino JT; Franz KJ Coordination chemistry of copper proteins: how nature handles a toxic cargo for essential function. J. Inorg. Biochem, 2012, 107(1), 129–143. [ 10.1016/j.jinorgbio.2011.11.024] [DOI] [PubMed] [Google Scholar]
- [110].Sciegienka SJ; Solst SR; Falls KC; Schoenfeld JD; Klinger AR; Ross NL; Rodman SN; Spitz DR; Fath MA D-penicillamine combined with inhibitors of hydroperoxide metabolism enhances lung and breast cancer cell responses to radiation and carboplatin via H2O2-mediated oxidative stress. Free Radic. Biol. Med, 2017, 108, 354–361. [ 10.1016/j.freeradbiomed.2017.04.001] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Brem S; Grossman SA; Carson KA; New P; Phuphanich S; Alavi JB; Mikkelsen T; Fisher JD New Approaches to Brain Tumor Therapy CNS Consortium. Phase 2 trial of copper depletion and penicillamine as antiangiogenesis therapy of glioblastoma. Neuro-oncol, 2005, 7(3), 246–253. [ 10.1215/S1152851704000869] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Baskar R; Lee KA; Yeo R; Yeoh KW Cancer and radiation therapy: current advances and future directions. Int. J. Med. Sci, 2012, 9(3), 193–199. [ 10.7150/ijms.3635] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Kim BM; Hong Y; Lee S; Liu P; Lim JH; Lee YH; Lee TH; Chang KT; Hong Y Therapeutic Implications for Overcoming Radiation Resistance in Cancer Therapy. Int. J. Mol. Sci, 2015, 16(11), 26880–26913. [ 10.3390/ijms161125991] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Abbotts R; Wilson DM III Coordination of DNA single strand break repair. Free Radic. Biol. Med, 2017, 107, 228–244. [ 10.1016/j.freeradbiomed.2016.11.039] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Ceccaldi R; Rondinelli B; D’Andrea AD Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol, 2016, 26(1), 52–64. [ 10.1016/j.tcb.2015.07.009] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Cadet J; Sage E; Douki T Ultraviolet radiation-mediated damage to cellular DNA. Mutat. Res, 2005, 571(1-2), 3–17. [ 10.1016/j.mrfmmm.2004.09.012] [DOI] [PubMed] [Google Scholar]
- [117].Kuefner M; Brand M; Engert C; Schwab S; Uder M Radiation Induced DNA Double-Strand Breaks in Radiology. RöFo - Fortschritte auf dem Gebiet der Röntgenstrahlen und der bildge-benden Verfahren, 2015, 187(10), 872–878. [ 10.1055/s-0035-1553209] [DOI] [PubMed] [Google Scholar]
- [118].Gerber DE; Chan TA Recent advances in radiation therapy. Am. Fam. Physician, 2008, 78(11), 1254–1262. [PubMed] [Google Scholar]
- [119].Kamran MZ; Ranjan A; Kaur N; Sur S; Tandon V Radioprotective Agents: Strategies and Translational Advances. Med. Res. Rev, 2016, 36(3), 461–493. [ 10.1002/med.21386] [DOI] [PubMed] [Google Scholar]
- [120].Brenner B; Wasserman L; Beery E; Nordenberg J; Schechter J; Gutman H; Fenig E Variable cytotoxicity of amifostine in malignant and non-malignant cell lines. Oncol. Rep, 2003, 10(5), 1609–1613. [ 10.3892/or.10.5.1609] [DOI] [PubMed] [Google Scholar]
- [121].Hofer M; Falk M; Komůrková D; Falková I; Bačiková A; Klejdus B; Pagáčová E; Štefančíková L; Weiterová L; Angelis KJ; Kozubek S; Dušek L; Galbavý Š Two New Faces of Amifostine: Protector from DNA Damage in Normal Cells and Inhibitor of DNA Repair in Cancer Cells. J. Med. Chem, 2016, 59(7), 3003–3017. [ 10.1021/acs.jmedchem.5b01628] [DOI] [PubMed] [Google Scholar]
- [122].Graham K; Unger E Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomedicine, 2018, 13, 6049–6058. [ 10.2147/IJN.S140462] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Ueno M; Matsumoto S; Matsumoto A; Manda S; Nakanishi I; Matsumoto KI; Mitchell JB; Krishna MC; Anzai K Effect of amifostine, a radiation-protecting drug, on oxygen concentration in tissue measured by EPR oximetry and imaging. J. Clin. Biochem. Nutr, 2017, 60(3), 151–155. [ 10.3164/jcbn.15-130] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Kavaz E; Perişanoğlu U; Ekinci N; Özdemir Y Determination of energy absorption and exposure buildup factors by using G-P fitting approximation for radioprotective agents. Int. J. Radiat. Biol, 2016, 92(7), 380–387. [ 10.1080/09553002.2016.1175681] [DOI] [PubMed] [Google Scholar]
- [125].Felice PA; Gong B; Ahsan S; Deshpande SS; Nelson NS; Donneys A; Tchanque-Fossuo C; Morris MD; Buchman SR Raman spectroscopy delineates radiation-induced injury and partial rescue by amifostine in bone: a murine mandibular model. J. Bone Miner. Metab, 2015, 33(3), 279–284. [ 10.1007/s00774-014-0599-1] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Akkus Yildirim B; Çetin E; Topkan E; Ozyigit G; Cengiz M; Surucu S; Usubutun A; Akyol F Prevention of Radiation-Induced Retinopathy with Amifostine in Wistar Albino Rats. Retina, 2015, 35(7), 1458–1464. [ 10.1097/IAE.0000000000000493] [DOI] [PubMed] [Google Scholar]
- [127].Ferraiolo DM; Veitz-Keenan A Insufficient evidence for interventions to prevent dry mouth and salivary gland dysfunction post head and neck radiotherapy. Evid. Based Dent, 2018, 19(1), 30–31. [ 10.1038/sj.ebd.6401295] [DOI] [PubMed] [Google Scholar]
- [128].Devine A; Marignol L Potential of Amifostine for Chemoradiotherapy and Radiotherapy-associated Toxicity Reduction in Advanced NSCLC: A Meta-Analysis. Anticancer Res, 2016, 36(1), 5–12. [PubMed] [Google Scholar]
- [129].McLaughlin MF; Donoviel DB; Jones JA Novel Indications for Commonly Used Medications as Radiation Protectants in Spaceflight. Aerosp. Med. Hum. Perform, 2017, 88(7), 665–676. [ 10.3357/AMHP.4735.2017] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Ranganathan K; Simon E; Lynn J; Snider A; Zhang Y; Nelson N; Donneys A; Rodriguez J; Buchman L; Reyna D; Lipka E; Buchman SR Novel Formulation Strategy to Improve the Feasibility of Amifostine Administration. Pharm. Res, 2018, 35(5), 99 [ 10.1007/s11095-018-2386-5] [DOI] [PubMed] [Google Scholar]
- [131].Lawrie TA; Green JT; Beresford M; Wedlake L; Burden S; Davidson SE; Lal S; Henson CC; Andreyev HJN Interventions to reduce acute and late adverse gastrointestinal effects of pelvic radiotherapy for primary pelvic cancers. Cochrane Database Syst. Rev, 2018, 1 CD012529 [ 10.1002/14651858.CD012529.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Chen CH; Kuo ML; Wang JL; Liao WC; Chang LC; Chan LP; Lin J CCM-AMI, a Polyethylene Glycol Micelle with Amifostine, as an Acute Radiation Syndrome Protectant in C57BL/6 Mice. Health Phys, 2015, 109(3), 242–248. [ 10.1097/HP.0000000000000326] [DOI] [PubMed] [Google Scholar]
- [133].Xie J; Wang C; Zhao F; Gu Z; Zhao Y Application of Multifunctional Nanomaterials in Radioprotection of Healthy Tissues. Adv. Healthc. Mater, 2018, 7(20)e1800421 [ 10.1002/adhm.201800421] [DOI] [PubMed] [Google Scholar]
- [134].Hofer M; Hoferová Z; Depeš D; Falk M Combining Pharmacological Countermeasures to Attenuate the Acute Radiation Syndrome-A Concise Review. Molecules, 2017, 22(5)E834 [ 10.3390/molecules22050834] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Rodgers KE; Dizerega GS Contribution of the Local RAS to Hematopoietic Function: A Novel Therapeutic Target. Front. Endocrinol. (Lausanne), 2013, 4, 157 [ 10.3389/fendo.2013.00157] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].van der Veen SJ; Ghobadi G; de Boer RA; Faber H; Cannon MV; Nagle PW; Brandenburg S; Langendijk JA; van Luijk P; Coppes RP ACE inhibition attenuates radiation-induced cardiopulmonary damage. Radiother. Oncol, 2015, 114(1), 96–103. [ 10.1016/j.radonc.2014.11.017] [DOI] [PubMed] [Google Scholar]
- [137].Small W Jr; James JL; Moore TD; Fintel DJ; Lutz ST; Movsas; Suntharalingam M; Garces YI; Ivker R; Moulder J; Pugh S; Berk LB Utility of the ACE Inhibitor Captopril in Mitigating Radiation-associated Pulmonary Toxicity in Lung Cancer: Results From NRG Oncology RTOG 0123. Am. J. Clin. Oncol, 2018, 41(4), 396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].McCart EA; Lee YH; Jha J; Mungunsukh O; Rittase WB; Summers TA Jr; Muir J; Day RM Delayed Captopril Administration Mitigates Hematopoietic Injury in a Murine Model of Total Body Irradiation. Sci. Rep, 2019, 9(1), 2198 [ 10.1038/s41598-019-38651-2] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Islam A; Bolduc DL; Zhai M; Kiang JG; Swift JM Captopril Increases Survival after Whole-Body Ionizing Irradiation but Decreases Survival when Combined with Skin-Burn Trauma in Mice. Radiat. Res, 2015, 184(3), 273–279. [ 10.1667/RR14113.1] [DOI] [PubMed] [Google Scholar]
- [140].Kiang JG; Jiao W; Cary LH; Mog SR; Elliott TB; Pellmar TC; Ledney GD Wound trauma increases radiation-induced mortality by activation of iNOS pathway and elevation of cytokine concentrations and bacterial infection. Radiat. Res, 2010, 173(3), 319–332. [ 10.1667/RR1892.1] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Wu W; Abraham L; Ogony J; Matthews R; Goldstein G; Ercal N Effects of N-acetylcysteine amide (NACA), a thiol antioxidant on radiation-induced cytotoxicity in Chinese hamster ovary cells. Life Sci, 2008, 82(21-22), 1122–1130. [ 10.1016/j.lfs.2008.03.016] [DOI] [PubMed] [Google Scholar]
- [142].Ercal N; Luo X; Matthews RH; Armstrong DW In vitro study of the metabolic effects of D-amino acids. Chirality, 1996, 8(1), 24–29. [] [DOI] [PubMed] [Google Scholar]
- [143].Neal R; Matthews RH; Lutz P; Ercal N Antioxidant role of N-acetyl cysteine isomers following high dose irradiation. Free Radic. Biol. Med, 2003, 34(6), 689–695. [ 10.1016/S0891-5849(02)01372-2] [DOI] [PubMed] [Google Scholar]
- [144].Corcoran GB; Wong BK Role of glutathione in prevention of acetaminophen-induced hepatotoxicity by N-acetyl-L-cysteine in vivo: studies with N-acetyl-D-cysteine in mice. J. Pharmacol. Exp. Ther, 1986, 238(1), 54–61. [PubMed] [Google Scholar]
- [145].Yin J; Ren W; Yang G; Duan J; Huang X; Fang R; Li C; Li T; Yin Y; Hou Y; Kim SW; Wu G L-Cysteine metabolism and its nutritional implications. Mol. Nutr. Food Res, 2016, 60(1), 134–146. [ 10.1002/mnfr.201500031] [DOI] [PubMed] [Google Scholar]
- [146].Brand M; Sommer M; Ellmann S; Wuest W; May MS; Eller A; Vogt S; Lell MM; Kuefner MA; Uder M Influence of Different Antioxidants on X-Ray Induced DNA Double-Strand Breaks (DSBs) Using γ-H2AX Immunofluorescence Microscopy in a Preliminary Study. PLoS One, 2015, 10(5)e0127142 [ 10.1371/journal.pone.0127142] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Velauthapillai N; Barfett J; Jaffer H; Mikulis D; Murphy K Antioxidants Taken Orally prior to Diagnostic Radiation Exposure Can Prevent DNA Injury. J. Vasc. Interv. Radiol, 2017, 28(3), 406–411. [ 10.1016/j.jvir.2016.10.022] [DOI] [PubMed] [Google Scholar]
- [148].Council NR Health Effects of Exposure to Radon: BEIR VI; The National Academies Press: Washington, DC, 1999, p. 516. [PubMed] [Google Scholar]
- [149].Wu J; Zhang B; Wuu YR; Davidson MM; Hei TK Targeted cytoplasmic irradiation and autophagy. Mutat. Res, 2017, 806, 88–97. [ 10.1016/j.mrfmmm.2017.02.004] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Buer JK To stop the erosion of hope: the DMARD category and the place of semantics in modern rheumatology. Inflammopharmacology, 2017, 25(2), 185–190. [ 10.1007/s10787-017-0320-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Mahlich J; Sruamsiri R Treatment patterns of rheumatoid arthritis in Japanese hospitals and predictors of the initiation of biologic agents. Curr. Med. Res. Opin, 2017, 33(1), 101–107. [ 10.1080/03007995.2016.1239191] [DOI] [PubMed] [Google Scholar]
- [152].Machado-Alba JE; Ruiz AF; Machado-Duque ME Effectiveness of treatment with biologic- and disease-modifying antirheumatic drugs in rheumatoid arthritis patients in Colombia. Int. J. Clin. Pract, 2016, 70(6), 506–511. [ 10.1111/ijcp.12809] [DOI] [PubMed] [Google Scholar]
- [153].Muthukumar P; Dhanapriya J; Gopalakrishnan N; Dineshkumar T; Sakthirajan R; Balasubramaniyan T Evaluation of renal lesions and clinicopathologic correlation in rheumatoid arthritis. Saudi J. Kidney Dis. Transpl, 2017, 28(1), 44–50. [ 10.4103/1319-2442.198118] [DOI] [PubMed] [Google Scholar]
- [154].Lange E; Blizzard L; Venn A; Francis H; Jones G Disease-modifying anti-rheumatic drugs and non-melanoma skin cancer in inflammatory arthritis patients: a retrospective cohort study. Rheumatology (Oxford), 2016, 55(9), 1594–1600. [ 10.1093/rheumatology/kew214] [DOI] [PubMed] [Google Scholar]
- [155].Brancaleone V; Esposito I; Gargiulo A; Vellecco V; Asimakopoulou A; Citi V; Calderone V; Gobbetti T; Perretti M; Papapetropoulos A; Bucci M; Cirino G D-Penicillamine modulates hydrogen sulfide (H2S) pathway through selective inhibition of cystathionine-γ-lyase. Br. J. Pharmacol, 2016, 173(9), 1556–1565. [ 10.1111/bph.13459] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Muniraj N; Stamp LK; Badiei A; Hegde A; Cameron V; Bhatia M Hydrogen sulfide acts as a pro-inflammatory mediator in rheumatic disease. Int. J. Rheum. Dis, 2017, 20(2), 182–189. [ 10.1111/1756-185X.12472] [DOI] [PubMed] [Google Scholar]
- [157].Rhodes HL; Yarram-Smith L; Rice SJ; Tabaksert A; Edwards N; Hartley A; Woodward MN; Smithson SL; Tomson C; Welsh GI; Williams M; Thwaites DT; Sayer JA; Coward RJ Clinical and genetic analysis of patients with cystinuria in the United Kingdom. Clin. J. Am. Soc. Nephrol, 2015, 10(7), 1235–1245. [ 10.2215/CJN.10981114] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Malieckal DA; Modersitzki F; Mara K; Enders FT; Asplin JR; Goldfarb DS Effect of increasing doses of cystine-binding thiol drugs on cystine capacity in patients with cystinuria. Urolithiasis, 2019, 47(6), 549–555. [ 10.1007/s00240-019-01128-y] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Castañeda-Arriaga R; Vivier-Bunge A; Raul Alvarez-Idaboy J Primary antioxidant and metal-binding effects of tiopronin: A theoretical investigation of its action mechanism. Comput. Theor. Chem, 2016, 1077, 48–57. [ 10.1016/j.comptc.2015.10.012] [DOI] [Google Scholar]
- [160].Jiang T-Y; Sun C-S; Shen X; Wang T-Y; Wang S-L Development of a poloxamer analogs/bioadhesive polymers-based in situ gelling ophthalmic delivery system for tiopronin. J. Appl. Polym. Sci, 2009, 114(2), 775–783. [ 10.1002/app.30520] [DOI] [Google Scholar]
- [161].Ichikawa H; Imaizumi K; Tazawa Y; Obara Y; Ishikawa Y; Tobari I; Tanabe Y Effect of tiopronin on senile cataracts. A double-blind clinical study. Ophthalmologica, 1980, 180(5), 293–298. [ 10.1159/000308990] [DOI] [PubMed] [Google Scholar]
- [162].Kubo SH; Cody RJ Clinical pharmacokinetics of the angiotensin converting enzyme inhibitors. A review. Clin. Pharmacokinet, 1985, 10(5), 377–391. [ 10.2165/00003088-198510050-00001] [DOI] [PubMed] [Google Scholar]
- [163].Thomopoulos C; Parati G; Zanchetti A Effects of blood pressure lowering on outcome incidence in hypertension: 4. Effects of various classes of antihypertensive drugs--overview and metaanalyses. J. Hypertens, 2015, 33(2), 195–211. [ 10.1097/HJH.0000000000000447] [DOI] [PubMed] [Google Scholar]
- [164].Natesh R; Schwager SL; Evans HR; Sturrock ED; Acharya KR Structural details on the binding of antihypertensive drugs captopril and enalaprilat to human testicular angiotensin I-converting enzyme. Biochemistry, 2004, 43(27), 8718–8724. [ 10.1021/bi049480n] [DOI] [PubMed] [Google Scholar]
- [165].Chang CH; Lin JW; Caffrey JL; Wu LC; Lai MS Different Angiotensin-converting enzyme inhibitors and the associations with overall and cause-specific mortalities in patients with hypertension. Am. J. Hypertens, 2015, 28(6), 823–830. [ 10.1093/ajh/hpu237] [DOI] [PubMed] [Google Scholar]
- [166].Igreja B; Pires NM; Bonifácio MJ; Loureiro AI; Fernandes-Lopes C; Wright LC; Soares-da-Silva P Blood pressure-decreasing effect of etamicastat alone and in combination with antihypertensive drugs in the spontaneously hypertensive rat. Hypertens. Res, 2015, 38(1), 30–38. [ 10.1038/hr.2014.143] [DOI] [PubMed] [Google Scholar]
- [167].Büttner D; Kramer JS; Klingler FM; Wittmann SK; Hartmann MR; Kurz CG; Kohnhäuser D; Weizel L; Brüggerhoff A; Frank D; Steinhilber D; Wichelhaus TA; Pogoryelov D; Proschak E Challenges in the Development of a Thiol-Based Broad-Spectrum Inhibitor for Metallo-β-Lactamases. ACS Infect. Dis, 2018, 4(3), 360–372. [ 10.1021/acsinfecdis.7b00129] [DOI] [PubMed] [Google Scholar]
- [168].Brem J; van Berkel SS; Zollman D; Lee SY; Gileadi O; McHugh PJ; Walsh TR; McDonough MA; Schofield CJ Structural Basis of Metallo-β-Lactamase Inhibition by Captopril Stereoisomers. Antimicrob. Agents Chemother, 2015, 60(1), 142–150. [ 10.1128/AAC.01335-15] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Yusof Y; Tan DTC; Arjomandi OK; Schenk G; McGeary RP Captopril analogues as metallo-β-lactamase inhibitors. Bioorg. Med. Chem. Lett, 2016, 26(6), 1589–1593. [ 10.1016/j.bmcl.2016.02.007] [DOI] [PubMed] [Google Scholar]
- [170].Minarini A; Ferrari S; Galletti M; Giambalvo N; Perrone D; Rioli G; Galeazzi GM N-acetylcysteine in the treatment of psychiatric disorders: current status and future prospects. Expert Opin. Drug Metab. Toxicol, 2017, 13(3), 279–292. [ 10.1080/17425255.2017.1251580] [DOI] [PubMed] [Google Scholar]
- [171].Deepmala; Slattery J; Kumar N; Delhey L; Berk M; Dean O; Spielholz C; Frye R Clinical trials of N-acetylcysteine in psychiatry and neurology: A systematic review. Neurosci. Biobehav. Rev, 2015, 55, 294–321. [ 10.1016/j.neubiorev.2015.04.015] [DOI] [PubMed] [Google Scholar]
- [172].Price TO; Uras F; Banks WA; Ercal N A novel antioxidant N-acetylcysteine amide prevents gp120- and Tat-induced oxidative stress in brain endothelial cells. Exp. Neurol, 2006, 201(1), 193–202. [ 10.1016/j.expneurol.2006.03.030] [DOI] [PubMed] [Google Scholar]
- [173].Abareshi A; Hosseini M; Beheshti F; Norouzi F; Khazaei M; Sadeghnia HR; Boskabady MH; Shafei MN; Anaeigoudari A The effects of captopril on lipopolysaccharide induced learning and memory impairments and the brain cytokine levels and oxidative damage in rats. Life Sci, 2016, 167, 46–56. [ 10.1016/j.lfs.2016.10.026] [DOI] [PubMed] [Google Scholar]
- [174].Torika N; Asraf K; Roasso E; Danon A; Fleisher-Berkovich S Angiotensin Converting Enzyme Inhibitors Ameliorate Brain Inflammation Associated with Microglial Activation: Possible Implications for Alzheimer’s Disease. J. Neuroimmune Pharmacol, 2016, 11(4), 774–785. [ 10.1007/s11481-016-9703-8] [DOI] [PubMed] [Google Scholar]