

Abstract
Objective
To assess the safety of ocrelizumab (OCR) shorter duration infusion in patients with MS.
Methods
ENSEMBLE PLUS is a randomized, double-blind substudy to the single-arm ENSEMBLE study (NCT03085810). In ENSEMBLE, patients with early stage relapsing-remitting MS received OCR 600 mg initially as two 300 mg IV infusions 2 weeks apart and subsequently as a single 3.5-hour 600 mg infusion every 24 weeks for 192 weeks. In ENSEMBLE PLUS, OCR 600 mg administered over the approved 3.5-hour infusion time (conventional duration) is compared with a 2-hour infusion (shorter duration). The primary end point was the proportion of patients with infusion-related reactions (IRRs) after the first randomized dose (assessed during and up to 24 hours postinfusion).
Results
From November 1, 2018, to September 27, 2019, 580 patients were randomized 1:1 to the conventional or shorter infusion group. After the first randomized dose, 67 of 291 patients (23.1%) in the conventional and 71 of 289 patients (24.6%) in the shorter infusion group experienced IRRs. Most IRRs were mild or moderate in both groups; one patient in each group experienced a severe IRR, and in both groups, 98.6% (136 of 138) of all IRRs resolved without sequelae. No IRRs were serious, life-threatening, or fatal. No IRR-related discontinuation occurred. During the first randomized dose, 14 of 291 (4.8%) and 25 of 289 (8.7%) patients in the conventional and shorter infusion groups, respectively, had IRRs leading to infusion slowing/interruption.
Conclusion
The frequency and severity of IRRs were similar between conventional and shorter OCR infusions. Shortening the infusion time to 2 hours reduces the total infusion site stay time and lessens the overall patient and site staff burden.
Classification of evidence
This interventional study provides Class I evidence that the frequency and severity of IRRs were similar at the first randomized dose using OCR (600 mg) infusions of conventional and shorter duration in patients with relapsing-remitting MS.
Clinical trial identifier number
Ocrelizumab (OCR) is a humanized anti-CD20 monoclonal antibody approved uniquely for both relapsing and primary progressive MS.1,2 The current OCR infusion schedule, including mandatory premedication 1-hour preinfusion and 1-hour postinfusion observations, requires an on-site stay of 5.5–6 hours. Shortened infusion times can minimize the treatment burden for patients, reduce the time required at the infusion site, and lead to decreased workloads for site staff, without compromising patient safety.3–5 Here, we describe the primary findings from the ENSEMBLE PLUS study evaluating the safety, including infusion-related reactions (IRRs), of a shorter vs conventional infusion of OCR in patients with early relapsing-remitting MS (RRMS).
Methods
Trial design and patients
The ENSEMBLE PLUS substudy is a prospective, multicenter, randomized, double-blind phase IIIb study designed to evaluate the safety of a shorter duration infusion of OCR in patients with early stage RRMS enrolled in the main ENSEMBLE study. In ENSEMBLE, treatment-naïve patients (age 18–55 years) with a confirmed diagnosis of RRMS,6 disease duration ≤3 years, one or more relapses/signs of MRI activity in the previous 12 months, and an Expanded Disability Status Scale score of 0–3.5 (inclusive) received OCR 600 mg infusions every 24 weeks for 192 weeks (up to 8 doses), with mandatory premedication. Patients with a previous serious OCR-related IRR were excluded from the substudy. The target enrollment was 700 patients in the ENSEMBLE PLUS substudy, which included 150 patients already enrolled in the main ENSEMBLE study plus 550 newly enrolled patients.
In all patients, the first dose of OCR was administered, per label, as an initial dose of two 300 mg infusions, separated by 14 days (figure 1A). Randomization was performed with the use of an interactive web response system in permuted blocks (block size = 4). Randomization to either the conventional or shorter infusion group occurred at week 24 for newly enrolled patients. For patients already enrolled in the main ENSEMBLE study, randomization occurred at their next scheduled infusion (week 48, 72, 96, or 120). Patients eligible to take part in this substudy were randomized (1:1) into conventional 3.5-hour and shorter 2-hour infusion groups stratified by region (United States, Canada, and Australia vs the rest of the world) and dose at which the patient is randomized. Patients received 600 mg OCR in 500 mL 0.9% sodium chloride infused over approximately 3.5 hours in the conventional infusion group (with a mimic switch infusion at approximately 2 hours) or 2 hours, followed by a 100 mL 0.9% sodium chloride infusion over the remaining 1.5 hours in the shorter infusion group, every 24 weeks for the remainder of the study duration (figure 1B). Blood samples were only collected at the first OCR infusion postrandomization and 30 minutes after the completion of the shorter and conventional infusion, representing the peak concentration (Cmax) of OCR.
Figure 1. ENSEMBLE PLUS (A) study design and (B) infusion schedule.

The ENSEMBLE PLUS primary end point is the proportion of patients with IRRs after the first randomized dose (frequency and severity assessed during and 24 hours postinfusion). aRandomization of new patients at week 24. AH = antihistamine; IRR = infusion-related reaction; IVMP = methylprednisolone; PK = pharmacokinetic assessment.
Patients, site personnel, and sponsor employees remained blinded during the study. Infusions were preloaded and placed into standardized infusion cover bags on an infusion rack; the infusion administration pump was covered and operated only by an unblinded infusion nurse.
Standard protocol approvals, registrations, and patient consents
The relevant institutional review boards/ethics committees approved the trial protocols (NCT03085810). All patients provided written informed consent. The Steering Committee and study investigators gathered the data, and the sponsor performed the data analyses. The author and Steering Committee agreed to submit the manuscript for publication.
Study objectives
The primary research question provides Class I evidence to determine if the frequency and severity of IRRs after the first randomized dose differed using OCR (600 mg) infusions of conventional and shorter duration in patients with RRMS. The primary end point was the proportion of patients with IRRs during or within 24 hours after the first randomized dose using shorter vs conventional duration OCR infusion groups (IRRs assessed during and 24 hours postinfusion). Secondary end points include the severity and symptoms of IRRs, IRRs leading to treatment discontinuation, the proportion of patients with IRRs overall, and the overall safety.
Safety reporting
IRRs were classified as occurring during infusion or within 24 hours after the end of the infusion (collected by follow-up telephone contact). IRR events occurring in a patient at both time points (during and postinfusion) were reported as 2 separate IRRs per infusion. Safety was assessed through the monitoring and recording of adverse events (AEs) and serious AEs. AEs were defined as all AEs including IRRs and serious MS relapses, but excluding nonserious MS relapses. AEs were reported from the first randomized dose onward up to the interim clinical cutoff date (iCCOD).
Statistical methods
The proportion of patients with IRRs that occurred during or within 24 hours after the first randomized dose of OCR were summarized as point estimates of the between-treatment difference and associated symmetric 2-sided 95% CIs. All summaries of IRRs are based on the intent-to-treat (ITT) population; overall AEs are based on the safety population. Analyses are based on patients who had completed the 24-hour evaluation period after the first randomized dose by the prespecified iCCOD of September 27, 2019. A descriptive analysis (mean and median in patients with/without IRR) was used to describe any association between the Cmax of OCR vs IRR maximum intensity.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and from the corresponding author on reasonable request.
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://vivli.org/). Further details on the Roche criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche Global Policy on the Sharing of Clinical Study Information and how to request access to related clinical study documents, see roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm.
Results
Patient disposition and analysis population
A total of 586 patients were enrolled in the ongoing ENSEMBLE PLUS study by the prespecified iCCOD (183 from the main ENSEMBLE study and 403 newly enrolled patients) across 21 countries. Of the 586 patients enrolled, 580 patients were randomized (1:1), stratified by region and dose at which the patient is randomized, to the conventional infusion group (N = 291) or shorter infusion group (N = 289, figure 2). Two patients (0.7%) were withdrawn from the shorter infusion group. All patients received the full 600 mg dose in each group. Baseline demographics and disease characteristics were well-balanced across conventional and shorter infusion groups; most patients were female (181 of 291 [62.2%]/186 of 289 [64.4%]), with a mean age (SD) of 34.0 (8.5)/34.2 (8.8) years and a mean (SD) duration since relapsing MS diagnosis of 1.1 (0.6)/(0.7) years. There was a slight imbalance in the proportion of patients with prefirst randomized dose IRRs (n = 71/291 [24.4%] in the conventional and n = 78/289 [27.0%] in the shorter infusion groups).
Figure 2. Patient disposition and analysis of population.

One patient disclosed that they were pregnant after randomization but before receiving any study treatment. Per protocol, treatment is withheld from patients who become pregnant during the study. There was also one withdrawal from the conventional infusion group because of an adverse event (depressive symptom) that was considered unrelated to the study treatment but because of concurrent illness of depression. A discontinuation visit had not been scheduled or undertaken for the patient at the time of CCOD; hence, this patient could not be included in any of the tables which display discontinuation. Other: accidental unblinding. CCOD = clinical cutoff date.
Shorter infusion time summary
All patients received at least one randomized OCR infusion, except for one patient in the conventional infusion group. In the conventional infusion group, 236 of 291 (81.1%), 54 of 291 (18.6%), and zero patients received one, 2, and 3 randomized doses, respectively; this was 233 of 289 (80.6%), 55 of 289 (19.0%), and 1 of 289 (0.3%) patients in the shorter infusion group. Overall, the median (range) infusion time was 215 (195–350) and 120 (109–255) minutes in the conventional and shorter infusion groups, respectively.
Infusion-related reactions
The incidence of IRRs at the first randomized dose (primary end point) in patients was comparable between the conventional (n = 67/291 [23.1%]) and shorter (n = 71/289 [24.6%]) infusion groups (stratified difference in proportions [95% CI]: 2.0% [−4.7% to 8.7%], table). Of patients experiencing IRRs, the onset of IRR symptoms occurred during infusion for n = 27 of 67 (40.3%) and n = 40 of 71 (56.3%) patients and within 24 hours postinfusion for n = 48/67 (71.6%) and n = 40/71 (56.3%) patients, in conventional and shorter infusion groups, respectively. Most IRRs were mild (grade 1; n = 46/67 [68.7%] and n = 47/71 [66.2%]) or moderate (grade 2; n = 21/67 [31.3%] and n = 23/71 [32.4%]) in the conventional and shorter infusion groups, respectively. One severe IRR (grade 3) occurred in the shorter (fatigue at first randomized dose) and in the conventional (laryngeal inflammation at second randomized dose) infusion groups. No IRRs were life-threatening, serious, or fatal, and >98% (136/138) of all IRRs resolved without sequelae in both groups. The most frequent symptoms associated with IRRs in both groups were throat irritation, fatigue, and headache, with the most common treatments being paracetamol, diphenhydramine hydrochloride, and chlorpheniramine (table). Overall, n = 14 of 291 (4.8%) and n = 25 of 289 (8.7%) patients in the conventional and shorter infusion groups had IRRs leading to temporary infusion interruption/slowing, respectively (table). There was no correlation between peak serum OCR concentration and observed IRRs.
Table.
Summary of (1) primary end point (proportion of patients with IRRs after the first randomized dose) and severity, (2) IRRs at the first randomized dose leading to intervention in OCR infusion, (3) symptoms and management of IRRs, and (4) AEs

Adverse events
Overall, the AE profile between conventional and shorter infusion groups was balanced; the most common AEs in either infusion group were IRRs (table). One patient (0.3%) from the conventional infusion group withdrew because of an AE (depressive symptom). Serious AEs occurred in n = 3 (1.0%) patients in both groups (conventional infusion: events, n = 1 typhoid fever and intraductal papilloma of breast, n = 2 depressive symptoms; shorter infusion: events, n = 1 urinary tract infection, peripheral edema, and hypotension).
Discussion
This primary analysis of the ENSEMBLE PLUS study shows that the frequency, severity, and symptoms of IRRs were similar between conventional and shorter OCR infusion periods. During the first randomized dose, there was a moderately higher incidence of IRRs leading to infusion slowing/interruption in the shorter (25 of 289 patients; 8.7%) vs conventional (14 of 291 patients; 4.8%) infusion group. Overall, AEs were consistent with the known safety profile of OCR,7–9 and no new safety signals were observed with a shorter infusion time. The safety profile of OCR remains unchanged. Shortening the OCR infusion time to 2 hours may reduce the total site stay time and lessen the burden on patients and site staff, which is of particular importance in light of the current coronavirus disease 2019 pandemic.
Acknowledgment
The author thanks all patients, their families, and the investigators who participated in this trial (including the ENSEMBLE PLUS study Steering Committee, which provided study oversight). The author also thanks the independent data monitoring committee for performing data analysis and safety monitoring, and is grateful to Regine Buffels and Jad Abdul Samad (of F. Hoffmann-La Roche Ltd) for additional critical review of this manuscript and technical advice.
Glossary
- AE
adverse event
- iCCOD
interim clinical cutoff
- IRR
infusion-related reaction
- ITT
intent-to-treat
- OCR
ocrelizumab
- RRMS
relapsing-remitting MS
Appendix 1. Author

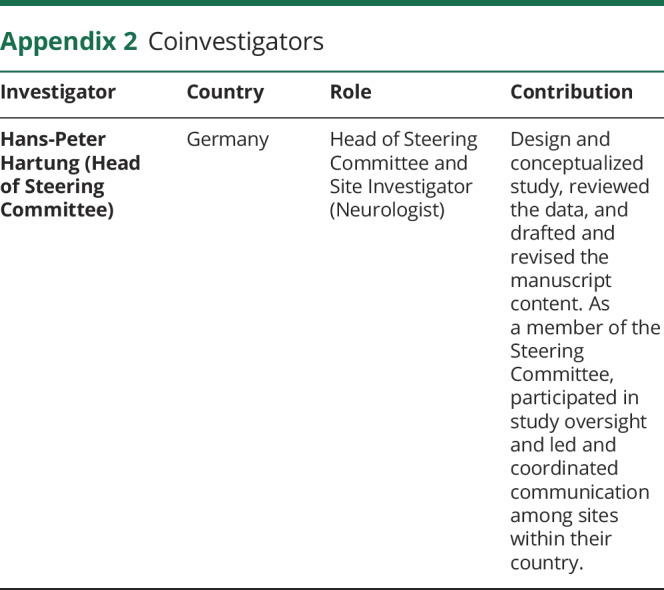
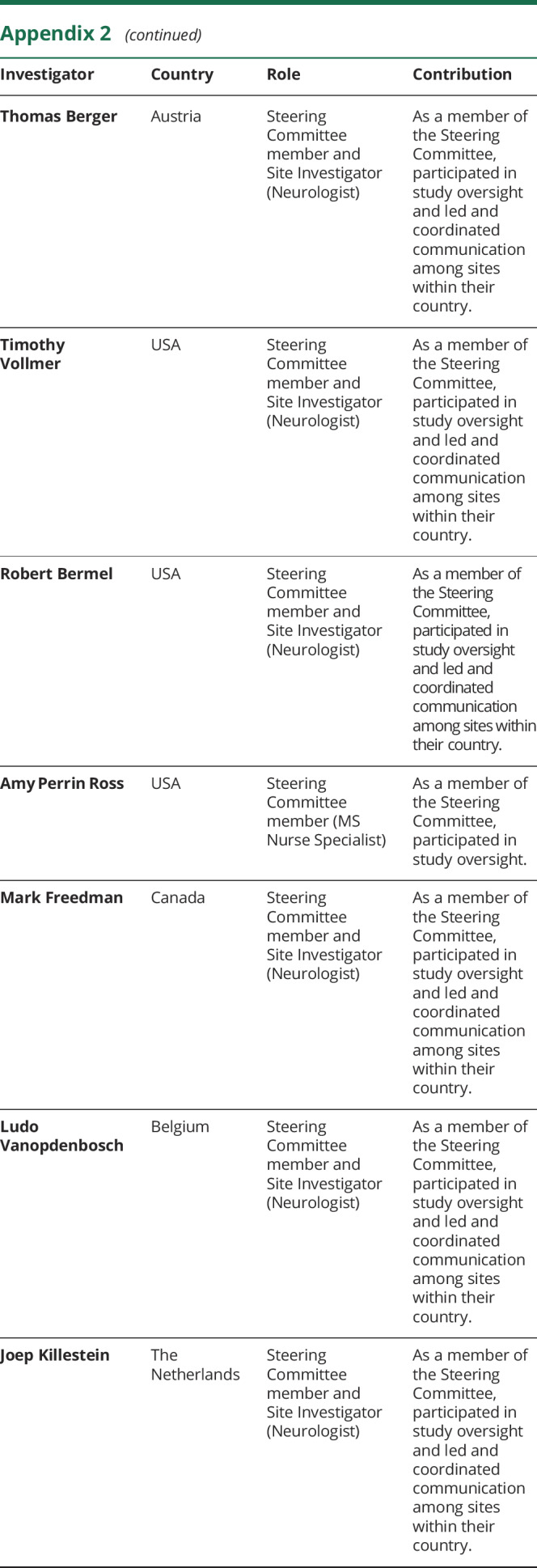
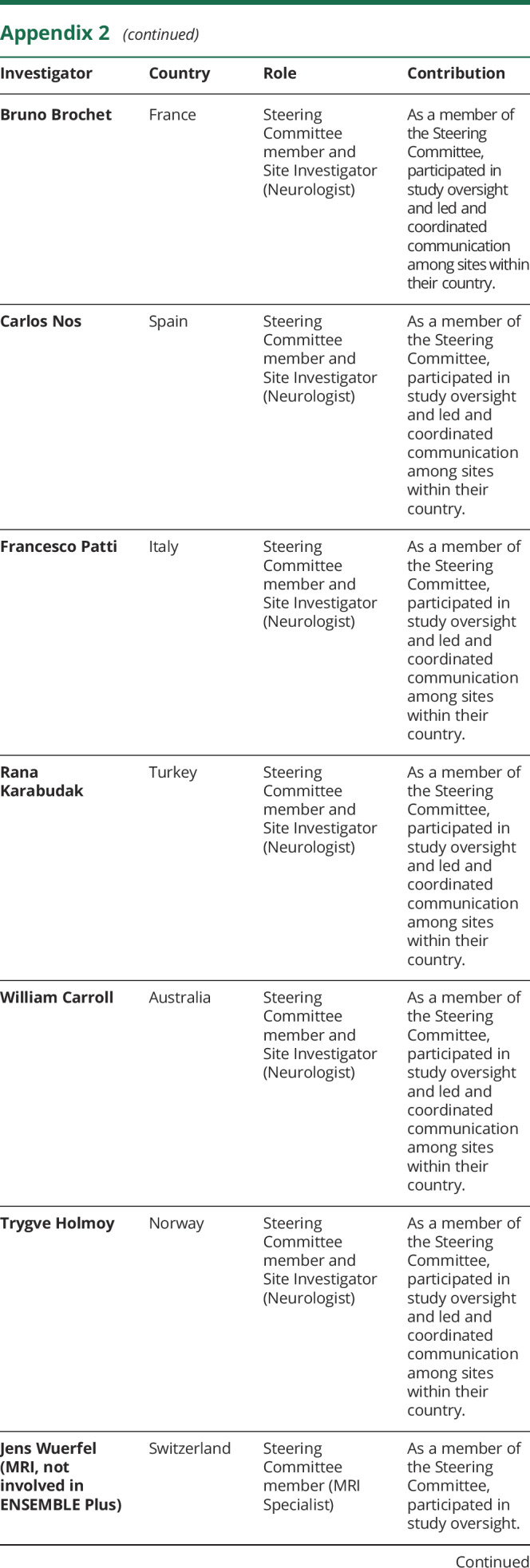
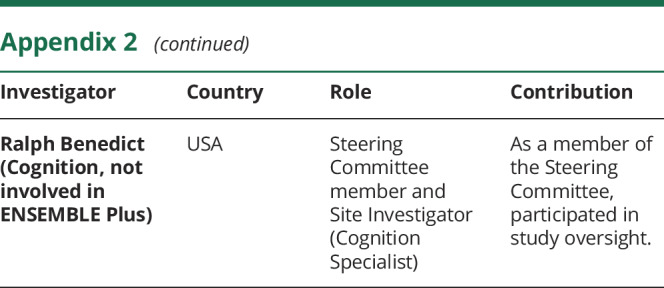
Appendix 2. Coinvestigators
Study funding
This research was funded by F. Hoffmann-La Roche Ltd, Basel, Switzerland. This manuscript was submitted by the author on behalf of the ENSEMBLE PLUS principle investigators. The author had full editorial control of the manuscript and provided their final approval of all content. The first author wrote the first draft of the manuscript with assistance from an independent medical writer funded by F. Hoffmann-La Roche Ltd. Medical editorial assistance was provided by Terence Smith of Articulate Science and was funded by F. Hoffmann-La Roche Ltd.
Disclosure
H-P Hartung has received honoraria for serving on steering and data monitoring committees from Bayer Healthcare, Biogen, Celgene BMS, GeNeuro, MedImmune, Merck, Novartis, Roche, Sanofi Genzyme, Teva, TG Therapeutics, and Viela Bio with approval by the Rector of Heinrich-Heine University Düsseldorf. Go to Neurology.org/NN for full disclosures.
References
- 1.Genentech. Ocrevus (package insert). South San Francisco: Genentech, Inc.; 2019. [Google Scholar]
- 2.Roche. Ocrevus (summary of product characteristics). Welwyn Garden City: Roche Products Limited; 2019. [Google Scholar]
- 3.Pritchard CH, Greenwald MW, Kremer JM, et al. . Safety of infusing rituximab at a more rapid rate in patients with rheumatoid arthritis: results from the RATE-RA study. BMC Musculoskelet Disord 2014;15:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sehn LH, Donaldson J, Filewich A, et al. . Rapid infusion rituximab in combination with corticosteroid-containing chemotherapy or as maintenance therapy is well tolerated and can safely be delivered in the community setting. Blood 2007;109:4171–4173. [DOI] [PubMed] [Google Scholar]
- 5.Tuthill M, Crook T, Corbet T, King J, Webb A. Rapid infusion of rituximab over 60 min. Eur J Haematol 2009;82:322–325. [DOI] [PubMed] [Google Scholar]
- 6.Polman CH, Reingold SC, Banwell B, et al. . Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011;69:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayer L, Kappos L, Racke MK, et al. . Ocrelizumab infusion experience in patients with relapsing and primary progressive multiple sclerosis: results from the phase 3 randomized OPERA I, OPERA II, and ORATORIO studies. Mult Scler Relat Disord 2019;30:236–243. [DOI] [PubMed] [Google Scholar]
- 8.Hauser SL, Bar-Or A, Comi G, et al. . Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med 2017;376:221–234. [DOI] [PubMed] [Google Scholar]
- 9.Montalban X, Hauser SL, Kappos L, et al. . Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 2017;376:209–220. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and from the corresponding author on reasonable request.
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://vivli.org/). Further details on the Roche criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche Global Policy on the Sharing of Clinical Study Information and how to request access to related clinical study documents, see roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm.