

Abstract
Objective
To determine serologic characteristics, frequency, phenotype, paraneoplastic associations, and electrodiagnostic and histopathologic features accompanying contactin-1 autoimmunity.
Methods
Archived sera known to produce synaptic tissue-based immunofluorescence patterns were reevaluated, and contactin-1 specificity was confirmed by recombinant protein assays. Screening of 233 chronic/relapsing demyelinating neuropathies for additional cases was performed.
Results
We identified 10 contactin-1 IgG seropositive cases. Frequency of contactin-1 immunoglobulin (Ig) G among tested Mayo Clinic chronic/relapsing demyelinating neuropathies was 2%. Sensory predominant presentations (n = 9, 90%), neuropathic pain (n = 6, 60%), and subacute progression (n = 5, 50%) were commonly encountered among contactin-1 neuropathies. Two patients had chronic immune sensory polyradiculopathy-like phenotype at presentation. Electrodiagnostic studies were consistent with demyelination (slowed conduction velocities and/or prolonged distal latencies) without conduction block. Markedly elevated CSF protein (median 222 mg/dL, range 69–960 mg/dL), thickening/gadolinium enhancement of nerve roots (4/5), and subperineural edema on nerve biopsy (4/4) were other characteristic features. Three cases were diagnosed with paraneoplastic demyelinating neuropathies (thymoma, n = 1; breast cancer, n = 1; plasmacytoma, n = 1). Four of the 9 patients treated with IV immunoglobulin demonstrated initial clinical improvement, but the favorable response was sustained in only 1 case (median follow-up, 60 months). Sustained clinical stabilization or improvement was observed among 3 of the 6 cases in whom second-line therapies (rituximab, cyclophosphamide, and azathioprine) were used.
Conclusion
Contactin-1 IgG has a distinct sensory predominant presentation commonly associated with neuropathic pain, with demyelinating changes on electrophysiologic studies. A paraneoplastic cause should be considered. Testing of contactin-1 IgG among cases with similar presentations may guide immunotherapy selection, especially second-line immunotherapy consideration.
A minority of chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) cases have been demonstrated to have antibodies targeting paranodal antigens such as neurofascin-155 and contactin-1.1–5 Herein, we provide a retrospective clinical review of 10 contactin-1 neuropathy cases identified: (1) in the course of evaluation of consecutively acquired specimens in the Neuroimmunology Laboratory, Mayo Clinic, and (2) by screening serums from a CIDP cohort.
Methods
Standard protocol approvals, registrations, and patient consents
The Mayo Clinic Institutional Review Board (#08–006647) approved human specimen acquisition and chart retrospective review.
Study population and laboratory methods
As previously described,6 between January 1, 1993, and June 1, 2019, the Mayo Clinic Neuroimmunology Laboratory tested 616,025 serum and CSF specimens submitted for service testing for autoimmune neurologic disorders by tissue-based indirect immunofluorescence assay (IFA). Of those, 368 samples (serum, n = 334; CSF, n = 34) produced diffuse neural-restricted synaptic staining by IFA. From that specimen cohort, sera from 4 patients with available medical records produced an identical unique staining pattern (supplementary figure 1A, links.lww.com/NXI/A261). Contactin-1 was determined to be the autoantigen (previously described3) by immunoprecipitation and mass spectrometry (supplementary methods, links.lww.com/NXI/A262). Antigen specificity was confirmed by Western blot, cell-based assays (transfected HEK293 cells; Euroimmun [supplementary figure 1B, links.lww.com/NXI/A261]), and confocal microscopy.
An additional 5 patients were identified among 233 stored specimens from patients diagnosed with chronic/relapsing demyelinating neuropathy were tested (CIDP, n = 225 [sera, n = 210; CSF, n = 15]; chronic immune sensory polyradiculopathy [CISP], n = 8). Another contactin-1 immunoglobulin (Ig) G-seropositive case (Western blot, Washington University Laboratory) with insufficient sample for cell-based assay testing was included.
We also evaluated stored sera from 39 patients with monophasic acute inflammatory demyelinating polyradiculoneuropathy (AIDP, n = 25) or polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes (POEMS, n = 14). Clinical outcome was assessed by the Inflammatory Neuropathy Cause and Treatment disability score.7
Data availability
All methods are available above, and data are published in this article.
Results
Demographic and clinical findings
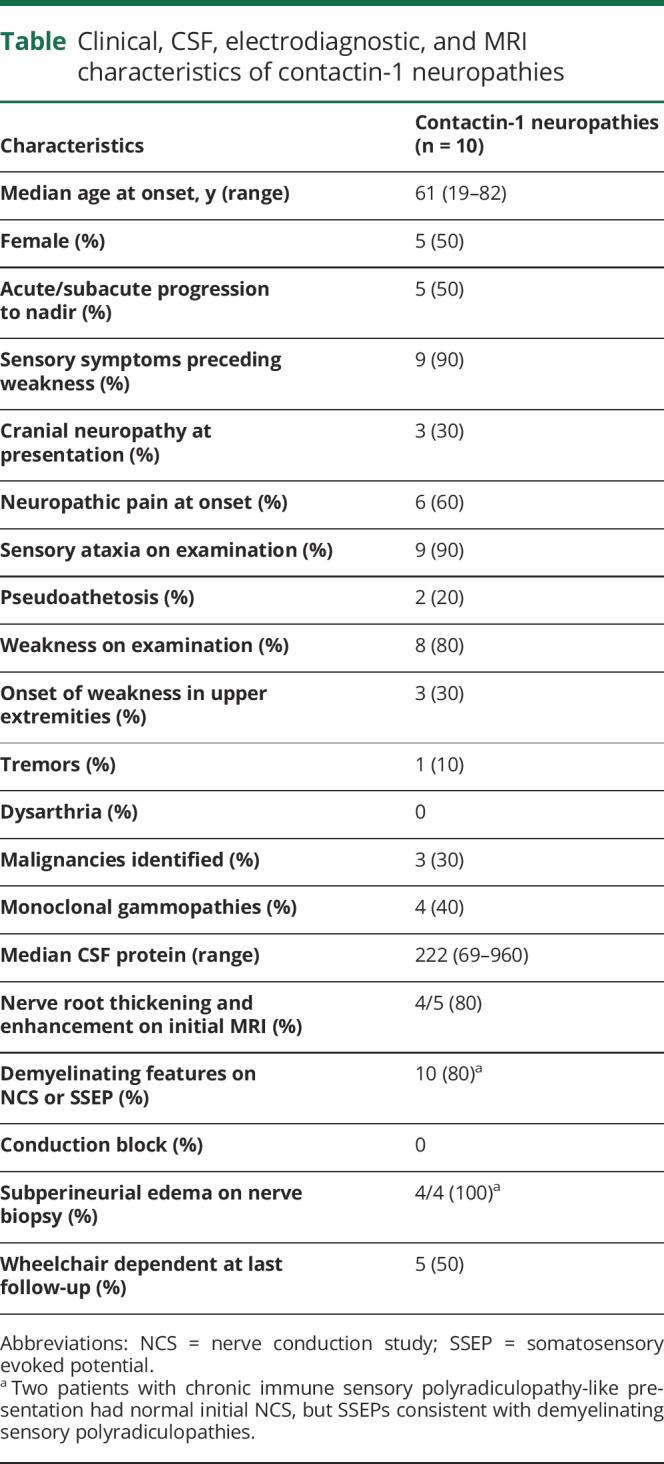
Five of the 10 contactin-1 IgG-seropositive cases were men, and the median symptom onset age was 61 years (range, 19–82 years). The frequency of contactin-1 seropositivity among Mayo Clinic acquired demyelinating neuropathy cohort was 2% (5/233).
All 10 had inflammatory demyelinating neuropathy diagnosis. Symptom onset to nadir was <8 weeks in 5 of the 10 patients, leading to their initial diagnosis of AIDP (n = 4) or subacute inflammatory demyelinating polyradiculoneuropathy (n = 1).8 Two of the 10 cases were diagnosed of CISP-like phenotype at initial presentation (table).
Table.
Clinical, CSF, electrodiagnostic, and MRI characteristics of contactin-1 neuropathies
Paraproteinemic and oncologic associations
Four of the 10 cases had monoclonal gammopathies (IgM, n = 2; IgG, n = 2) detected by serum protein electrophoresis. Bone marrow biopsy in 1 patient demonstrated clonal proliferation consistent with plasmacytoma. A mediastinal lymph node in another patient led to a diagnosis of Rosai-Dorfman disease. In addition, solid tumors were detected in 2 of the 9 patients within 6 months onset of neuropathy symptoms (breast cancer, n = 1; thymoma, n = 1).
Sensory predominant presentations
Nine patients reported initial sensory symptoms (numbness and paresthesia) preceding the onset of weakness. Weakness was noticed a few days later by 7 of those 9 patients (median interval 14 days; range 1–4 weeks). However, 2 patients lacked objective evidence of weakness on neurologic examination; a diagnosis of CISP (figure 1, A and B) was suspected.9 Weakness in 3 cases started in the arms (asymmetric) and spread to a lower extremity. Three patients additionally reported cranial nerve palsies at initial presentation (cranial nerve [CN] VII, n = 1; CN III and XII, n = 1; CN V, n = 1). Nine patients had sensory ataxia with proprioception loss noted on examination. Pseudoathetosis was observed in 2 cases. Six patients reported burning neuropathic pain involving the extremities or face at disease onset.
Figure 1. Somatosensory evoked potentials (A and B) and MRIs (C–F) of 2 contactin-1 IgG patients demonstrating favorable response to immunotherapy.

Right median somatosensory evoked potential demonstrating prolongation of N9 (arrow) to N13 (arrowhead) interval consistent with cervical sensory root involvement (A), which improves after treatment with IV immunoglobulin (B). Enhancement of the cauda equina on (C) sagittal and (D) axial postgadolinium T1 sequences, which resolves postimmunotherapy (E and F). IVIG = IV immunoglobulin; IVMP = intravenous methylprednisolone; MMF = mycophenolate mofetil.
CSF and radiology evaluations
CSF analysis at initial presentation (n = 7) demonstrated markedly elevated protein (median 222 mg/dL, range 69–960 mg/dL). One patient had lymphocytic pleocytosis (median 1 cell/mm3, range 0–68 cell/mm3). Diffuse enhancement and thickening of nerve roots were detected in 4 of 5 patients who had MRI studies performed at initial presentation (figure 1, C–F).
Neurophysiologic and pathologic studies
Electrodiagnostic studies were consistent with demyelinating polyradiculoneuropathy (slowed conduction velocities and/or prolonged distal latencies by nerve conduction study [NCS]). Two patients with CISP-like presentation had normal initial NCS. Both of them had prolonged somatosensory evoked potential (SSEP; figure 1, A and B). None of the 10 available NCS reports demonstrated conduction block.
Closely approximated osmium-fixed teased nerve fibers in 1 case's fascicular root biopsy demonstrated widened nodes of Ranvier with demyelination (figure 2A); sural nerve biopsy from 2 patients (figure 2, C–E) demonstrated axonal degeneration, including empty nerve strands (decreased density of myelinated fibers). Semithin preparations of fascicular root and both sural nerve biopsies showed subperineurial edema (figure 2, D–F) without onion bulb formation10 or significant epineurial or endoneurial inflammatory infiltrates.11 One patient with enlarged proximal nerves on MRI underwent femoral nerve segment resection, but biopsy only revealed significant subperineurial and endoneurial edema without evidence of nerve sheath neoplasm.
Figure 2. Nerve pathology of contactin-1 IgG neuropathy is distinct from classic-CIDP.

Three example patients with contactin-1 autoimmunity are shown (A–F). Closely approximated osmium-fixed teased nerve fibers from a fascicular biopsy demonstrate widened nodes of Ranvier with demyelination (A) compared with 2 sural nerve biopsies (C and F) where axonal injury is prominent including with early (asterisk) and late (arrowhead) degenerative stages including with empty nerve fibers (arrow) (E). Semithin sections stained with toluidine blue in all patients had distinct prominent subperineural edema (asterisk) without onion bulb formation as is typical in CIDP. Axonal degeneration was most prominently seen in (D, F-arrowheads). CIDP = chronic inflammatory demyelinating polyradiculoneuropathy.
Immunotherapy and clinical outcomes
All except 1 patient (9/10) was treated with intravenous immunoglobulin (IVIG). Other first-line therapies included plasmapheresis (4/9) and IV corticosteroids (2/9). Four of the 9 patients who received IVIG as first-line immunotherapy improved, but 3 of those 4 relapsed within 4 weeks and became refractory to retreatment with IVIG. Among 3 patients with undetectable subtype anti-contactin IgG4 in our series, 2 received IVIG and 1 received plasmapheresis as first-line immunotherapy. None of these 3 cases had a sustained response to first-line therapy and had a relapsing or progressive course.
Six of the 10 contactin-1 neuropathy cases received second-line immunosuppressive agents (rituximab, n = 2; mycophenolate mofetil, n = 2; cyclophosphamide, n = 1; azathioprine, n = 1). Two of those patients stabilized (cyclophosphamide and azathioprine) and 1 (infused with rituximab) improved. Five patients were using a wheelchair for ambulation at last follow-up. The median follow-up period was 60 months (range, 4–180 months).
Discussion
This study demonstrates a 2% frequency of contactin-1 IgG seropositivity in a US-based tertiary care cohort of patients with diverse immune-mediated demyelinating neuropathies, similar to the frequency reported in a Japanese population (2%).12 In addition, our systematic screening of specimens referred for diverse clinical indications confirms that contactin-1 IgG is highly specific for demyelinating neuropathy.
Most of the seropositive patients we identified had a distinctive sensory predominant presentation, some with neuropathic pain. Prolonged SSEPs (figure 1, A and B) consistent with the patient's sensory ataxia localized to sensory roots. However, involvement of dorsal root ganglia as previously reported cannot be excluded.12 One-third of our cases had cranial nerve involvement, and half of the cases had aggressive disease progression. Oncologic associations in 3 cases included thymoma, breast cancer, and plasmacytoma. One was diagnosed with Rosai-Dorfman disease. Previous descriptions of patients with contactin-1 neuropathy have demonstrated electrophysiologic characteristics mimicking seronegative CIDP.12 However, our electrophysiologic studies demonstrated only slowing of conduction velocity and no conduction blocks. Electrophysiologic features we encountered have been typically associated with POEMS syndrome rather than CIDP.13
The nerve biopsy finding of subperineural edema is supportive of altered blood-nerve barrier and suggests polyradicular localization.9,14 Consistent with a previously reported sural nerve biopsy from a single case of autoimmune contactin-1 neuropathy,15 the teased preparation of a fascicular root biopsy of one of our patients revealed segmental demyelination (figure 2A) and absence of onion bulbs.
The majority of cases either failed to improve following IVIG therapy or soon relapsed after initial transient improvement, but 1 patient had sustained clinical improvement (figure 1, A and B). The long-term wheelchair dependence of 5 of our 10 cases underscores the higher morbidity associated with autoimmune contactin-1 neuropathy compared with idiopathic CIDP.16 Retrospective design and small number of seropositive cases limit our assessment of optimal chronic immunotherapy selection. However, the described phenotypic, electrodiagnostic, and histopathologic features we describe may guide clinicians in considering serologic evaluation for contactin-1 IgG in patients with demyelinating neuropathy.
Glossary
- AIDP
acute inflammatory demyelinating polyradiculoneuropathy
- CIDP
chronic inflammatory demyelinating polyradiculoneuropathy
- CISP
chronic immune sensory polyradiculopathy
- CN
cranial nerve
- IFA
immunofluorescence assay
- Ig
immunoglobulin
- IVIG
intravenous immunoglobulin
- NCS
nerve conduction study
- SSEP
somatosensory evoked potential
Appendix. Authors
Study funding
This work was funded by the Center for Individualized Medicine and Euroimmun AG. The Center for Individualized Medicine and Euroimmun AG provided Neuroimmunology fellowship grant for Josephe A. Honorat.
Disclosure
D. Dubey has a patent pending for Kelch-like protein 11 as a marker of neurological autoimmunity and has received research support from Grifols, Center of Multiple Sclerosis and Autoimmune Neurology, and Center for Clinical and Translational Science. D. Dubey has consulted for UCB and Astellas. All compensation for consulting activities is paid directly to Mayo Clinic. J.A. Honorat has a patent pending for septin-5-IgG as biomarker of autoimmune neurological disease. S. Shelly reports no disclosures. C.J. Klein reports honorarium from Akcea for teaching on TTR amyloid and Fabry disease and is a consultant for Pfizer Pharmaceuticals on TTR amyloidosis. L. Komorowski is employed by Euroimmun AG. J.R. Mills reports no disclosures. S. Brakopp and C. Probst are employed by Euroimmun AG. V.A. Lennon has a patent application pending for septin-5 IgG as a biomarker of autoimmune neurologic disease. She has a financial interest in the following intellectual property: “Marker for Neuromyelitis Optica.” A patent has been issued for this technology, and it has been licensed to commercial entities. They have received cumulative royalties of greater than the federal threshold for significant financial interest from the licensing of these technologies, but receive no royalties from the sale of these tests by Mayo Medical Laboratories. S.A. Pittock receives personal fees and nonfinancial support from Alexion Pharmaceuticals and MedImmune, Inc. He receives grant/research support from Alexion, Grifols, MedImmune, and Autoimmune Encephalitis Alliance. He received personal compensation for attending the UCB Advisory Board Meeting in Stockholm, Sweden, on September 10, 2019. He is a named inventor on filed patents that relate to functional AQP4/NMO-IgG assays and NMO-IgG as a cancer marker and has a patent pending for MAP1B, Septin 5, Kelch-like protein 11, and GFAP as markers of neurological autoimmunity and paraneoplastic disorders. All compensation for consulting activities is paid directly to Mayo Clinic. A. McKeon has patent pendings for MAP1B, Kelch-like protein 11, PDE10A, GFAP, and Septins 5 and 7 as markers of neurological autoimmunity and paraneoplastic disorders; has consulted for Grifols, MedImmune, Inc, and Euroimmun; and has received research support from MedImmune, Inc and Euroimmun but has not received personal compensation. Go to Neurology.org/NN for full disclosures.
References
- 1.Querol L, Devaux J, Rojas-Garcia R, Illa I. Autoantibodies in chronic inflammatory neuropathies: diagnostic and therapeutic implications. Nat Rev Neurol 2017;13:533–547. [DOI] [PubMed] [Google Scholar]
- 2.Doppler K, Appeltshauser L, Wilhelmi K, et al. . Destruction of paranodal architecture in inflammatory neuropathy with anti-contactin-1 autoantibodies. J Neurol Neurosurg Psychiatry 2015;86:720–728. [DOI] [PubMed] [Google Scholar]
- 3.Querol L, Nogales-Gadea G, Rojas-Garcia R, et al. . Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann Neurol 2013;73:370–380. [DOI] [PubMed] [Google Scholar]
- 4.Hu W, Xin Y, He Z, Zhao Y. Association of neurofascin IgG4 and atypical chronic inflammatory demyelinating polyneuropathy: a systematic review and meta-analysis. Brain Behav 2018;8:e01115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cortese A, Lombardi R, Briani C, et al. . Antibodies to neurofascin, contactin-1, and contactin-associated protein 1 in CIDP: clinical relevance of IgG isotype. Neurol Neuroimmunol Neuroinflamm 2020;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Honorat JA, Lopez-Chiriboga AS, Kryzer TJ, et al. . Autoimmune gait disturbance accompanying adaptor protein-3B2-IgG. Neurology 2019;93:e954–e963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breiner A, Barnett C, Bril V. INCAT disability score: a critical analysis of its measurement properties. Muscle Nerve 2014;50:164–169. [DOI] [PubMed] [Google Scholar]
- 8.Joint Task Force of the E, the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society--First Revision. J Peripher Nerv Syst 2010;15:1–9. [DOI] [PubMed] [Google Scholar]
- 9.Sinnreich M, Klein CJ, Daube JR, Engelstad J, Spinner RJ, Dyck PJ. Chronic immune sensory polyradiculopathy: a possibly treatable sensory ataxia. Neurology 2004;63:1662–1669. [DOI] [PubMed] [Google Scholar]
- 10.Tracy JA, Dyck PJ, Klein CJ, Engelstad JK, Meyer JE, Dyck PJB. Onion-bulb patterns predict acquired or inherited demyelinating polyneuropathy. Muscle Nerve 2019;59:665–670. [DOI] [PubMed] [Google Scholar]
- 11.Rizzuto N, Morbin M, Cavallaro T, Ferrari S, Fallahi M, Galiazzo Rizzuto S. Focal lesions area feature of chronic inflammatory demyelinating polyneuropathy (CIDP). Acta Neuropathol 1998;96:603–609. [DOI] [PubMed] [Google Scholar]
- 12.Miura Y, Devaux JJ, Fukami Y, et al. . Contactin 1 IgG4 associates to chronic inflammatory demyelinating polyneuropathy with sensory ataxia. Brain 2015;138:1484–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sung JY, Kuwabara S, Ogawara K, Kanai K, Hattori T. Patterns of nerve conduction abnormalities in POEMS syndrome. Muscle Nerve 2002;26:189–193. [DOI] [PubMed] [Google Scholar]
- 14.Uceyler N, Necula G, Wagemann E, Toyka KV, Sommer C. Endoneurial edema in sural nerve may indicate recent onset inflammatory neuropathy. Muscle Nerve 2016;53:705–710. [DOI] [PubMed] [Google Scholar]
- 15.Koike H, Kadoya M, Kaida KI, et al. . Paranodal dissection in chronic inflammatory demyelinating polyneuropathy with anti-neurofascin-155 and anti-contactin-1 antibodies. J Neurol Neurosurg Psychiatry 2017;88:465–473. [DOI] [PubMed] [Google Scholar]
- 16.Kuwabara S, Misawa S, Mori M, Tamura N, Kubota M, Hattori T. Long term prognosis of chronic inflammatory demyelinating polyneuropathy: a five year follow up of 38 cases. J Neurol Neurosurg Psychiatry 2006;77:66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All methods are available above, and data are published in this article.