Desiree De Simoni
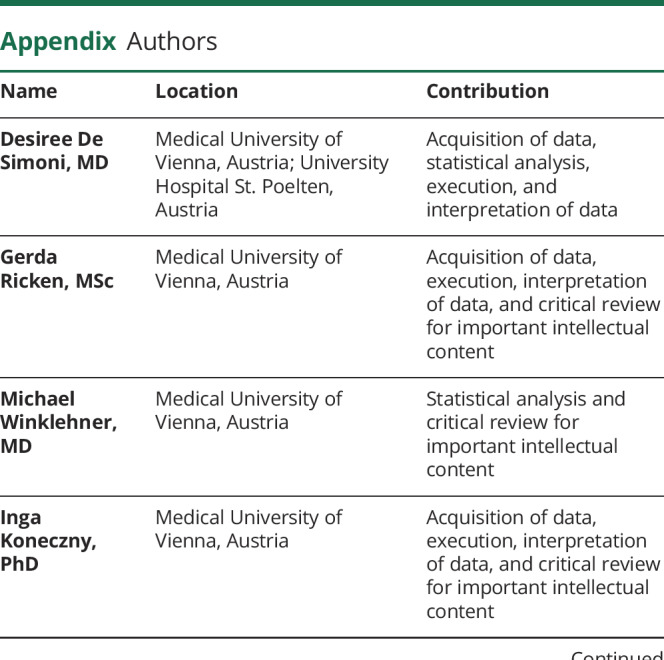
Desiree De Simoni, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Gerda Ricken
Gerda Ricken, MSc
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Michael Winklehner
Michael Winklehner, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Inga Koneczny
Inga Koneczny, PhD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Michael Karenfort
Michael Karenfort, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Ulf Hustedt
Ulf Hustedt, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Ulrich Seidel
Ulrich Seidel, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Omar Abdel-Mannan
Omar Abdel-Mannan, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Pinki Munot
Pinki Munot, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Simon Rinaldi
Simon Rinaldi, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Claudia Steen
Claudia Steen, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Michael Freilinger
Michael Freilinger, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Markus Breu
Markus Breu, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Rainer Seidl
Rainer Seidl, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Markus Reindl
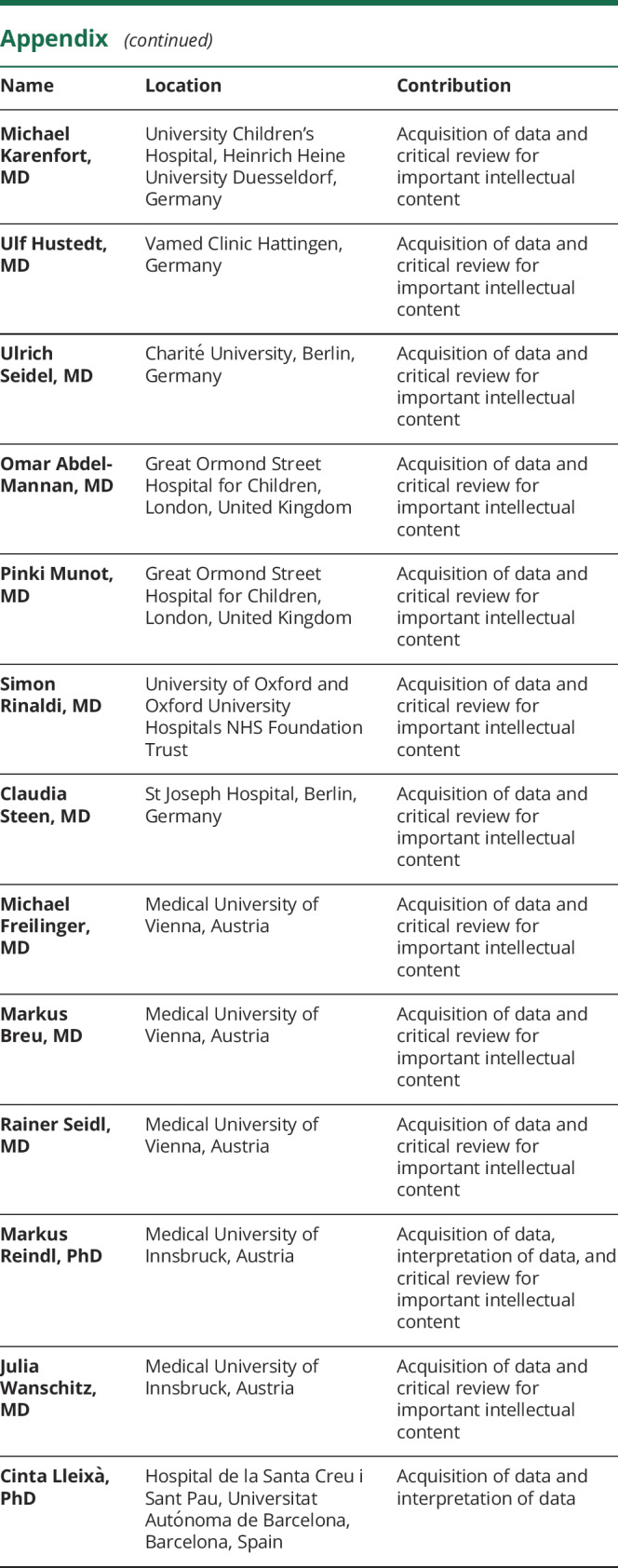
Markus Reindl, PhD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Julia Wanschitz
Julia Wanschitz, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Cinta Lleixà
Cinta Lleixà, PhD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Günther Bernert
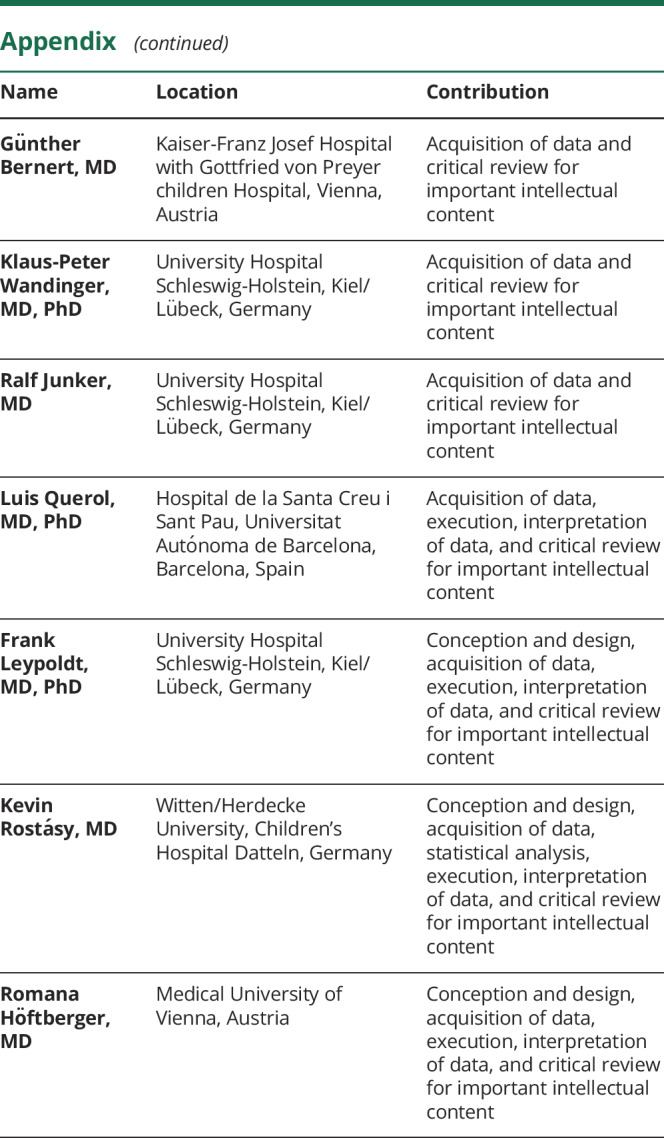
Günther Bernert, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Klaus-Peter Wandinger
Klaus-Peter Wandinger, MD, PhD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Ralf Junker
Ralf Junker, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Luis Querol
Luis Querol, MD, PhD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Frank Leypoldt
Frank Leypoldt, MD, PhD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,
Kevin Rostásy
Kevin Rostásy, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,*,✉,
Romana Höftberger
Romana Höftberger, MD
1From the Division of Neuropathology and Neurochemistry (D.D.S., G.R., M.W., I.K., R.H.), Department of Neurology, Medical University of Vienna, Austria; Department of Neurology (D.D.S.), University Hospital St. Poelten, Austria; Department of General Pediatrics, Neonatology and Pediatric Cardiology (M.K.), University Children's Hospital, Heinrich Heine University Duesseldorf, Germany; Department of Neuropediatric Rehabilitation (U.H.), Vamed Clinic Hattingen, Germany; Department of Neuropediatrics (U.S.), Charité University, Berlin, Germany; Paediatric Neurology (O.A.-M.), Great Ormond Street Hospital for Children, London, United Kingdom; Dubowitz Neuromuscular Centre (P.M.), Great Ormond Street Hospital for Children, London, United Kingdom; Nuffield Department of Clinical Neurosciences (S.R.), University of Oxford and Oxford University Hospitals NHS Foundation Trust; Department of Paediatric and Adolescent Medicine (C.S.), St Joseph Hospital, Berlin, Germany; Department of Pediatrics and Adolescent Medicine (M.F., M.B., R.S.), Medical University of Vienna, Austria; Department of Neurology (M.R., J.W.), Medical University of Innsbruck, Austria; Neuromuscular Diseases Unit (C.L., L.Q.), Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Spain; SMZ Süd (G.B.), Kaiser-Franz Josef Hospital with Gottfried von Preyer Children Hospital, Vienna, Austria; Institute of Clinical Chemistry (K.-P.W., R.J., F.L.), University Hospital Schleswig-Holstein, Kiel/Lübeck, Germany; Department of Neurology (F.L.), University Hospital Schleswig-Holstein, Kiel, Germany; and Department of Pediatric Neurology (K.R.), Witten/Herdecke University, Children's Hospital Datteln, Germany.
1,*,✉