

Real-time detection of photo-induced bond cleavage of a single molecule was enabled by plasmon-enhanced spectroscopy.
Abstract
Determining structural transformations of single molecules (SMs) is an important fundamental scientific endeavor. Optical spectroscopies are the dominant tools used to unravel the physical and chemical features of individual molecules and have substantially contributed to surface science and biotechnology. In particular, Raman spectroscopy can identify reaction intermediates and reveal underlying reaction mechanisms; however, SM Raman experiments are subject to intrinsically weak signal intensities and considerable signal attenuation within the spectral dispersion systems of the spectrometer. Here, to monitor the structural transformation of an SM on the millisecond time scale, a plasmonic nanocavity substrate has been used to enable Raman vibrational and fluorescence spectral signals to be simultaneously collected and correlated, which thus allows a detection of photo-induced bond cleavage between the xanthene and phenyl group of a single rhodamine B isothiocyanate molecule in real time. This technique provides a novel method for investigating light-matter interactions and chemical reactions at the SM level.
INTRODUCTION
Single-molecule (SM) spectroscopy is of immense scientific interest due to its ability to reveal significant fundamental physical and chemical properties of individual molecules (1, 2), with applications such as the super-high–resolution imaging of proteins (3), the chemical recognition of the inner molecular structure of an SM (4), and strong plasmonic coupling in nanocavities (5). Real-time monitoring of changes in the chemical structure of individual molecules in heterogeneous environment can provide profound insights into the nanoscale realm (6, 7). In addition, more intriguing is identifying the intermediates/products and determining the underlying mechanisms of SM chemical reactions, such as surface-catalyzed and photochemical reactions (8–12). However, the signals from SM reactions are weak and typically hidden within ensemble-averaging experimental results. Although optical SM spectroscopies have been extensively developed to probe individual molecules, these techniques usually work by recording the fluorescence photon number, due to low signal-to-noise ratios (SNRs), at ambient temperature (13, 14). Thus, to receive the signal intensity trajectory over time for SMs and study single-turnover events, fluorescence photons are counted on an electronic detector within a fixed spectral region (13, 14). In contrast with typical spectroscopies, which measure wavelength-integrated signals, SM emission spectroscopy can further provide rewarding spectral information for individual species analysis, but it requires large signal enhancements and long integration times due to the considerable SNR attenuation within the spectral dispersion and collection systems (13).
Fluorescence is an emission spectroscopy that is sensitive to electronic structure of molecules and is capable of probing molecular conformational transformations (14, 15), while Raman spectroscopy is also capable of identifying key reaction intermediates by providing “fingerprint” chemical bond vibrational information (16–19). Thus, simultaneously detecting and correlating emission spectroscopic information with vibrational modes from the same SM can significantly improve the understanding of light-matter interactions and can even probe SM chemical reaction mechanisms—an important step for SM science. However, SM Raman spectroscopy requires higher signal enhancements due to the intrinsically lower scattering cross section of SMs (for resonant molecules, a Raman signal enhancement of up to 1.0 × 107–fold is required) (20–22).
Here, to overcome the inherent difficulty associated with SM detection, a plasmonic nanocavity substrate was used to generate a large enhancement in local electric fields. This method can be used to obtain SM emission spectra with rich vibronic features on the millisecond time scale, which enables spectral correlation analysis between electronic structure and bonding information. Thus, a photo-induced carbon─carbon bond cleavage reaction between xanthene and phenyl groups of a single rhodamine B isothiocyanate (RITC) molecule was probed in real time.
RESULTS
Ag nanocavity substrate for simultaneous plasmon-enhanced SM Raman and fluorescence spectroscopy
Figure 1A shows the schematic diagram of plasmon-enhanced SM spectroscopy enabled by a nanocavity composed of a Ag film coated with a 2-nm silica layer using atomic layer deposition (ALD) as the substrate with Ag shell-isolated nanoparticles (SHINs) deposited on top. RITC fluorescent probe molecules were covalently functionalized onto the substrate via isothiocyanate-amino chemistry (see text S1 and fig. S1 for the detailed description of the isothiocyanate-amino reaction). The average number density of RITC on the nanocavity substrate is ~3 × 10−5 to 14 × 10−5 dyes/nm2, which was achieved by using extremely low densities of the surface amino anchoring group and dye concentrations (see text S1 for calculation of number density of dyes on surface). A 532-nm continuous-wave laser was used to excite the plasmon, and a confocal Raman microscope was used to simultaneously acquire the fluorescence emission and resonant Raman scattering signals (see fig. S2 for experimental setup).
Fig. 1. Real-time detection of the structural transformation of an SM in a nanocavity substrate.
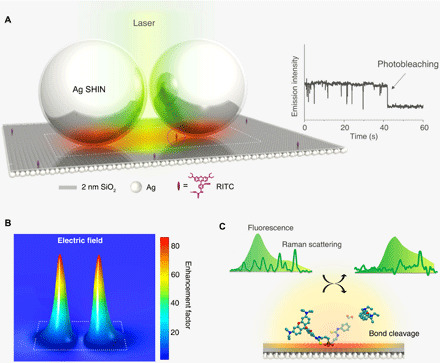
(A) Schematic diagram of plasmon-enhanced SM spectroscopy at room temperature. The SM in the nanocavity is denoted by the purple dot circle. Right inset shows the characteristic emission intensity trajectory of a single RITC molecule, showing blinking behavior and a single-step photobleaching event. (B) FEM simulation of the electric field distribution in the nanocavity substrate, where the thickness of silica shell coated on Ag nanospheres is 2 nm, and the white frame marks the location of the dimer model shown in (A). (C) Application of the plasmonic substrate in the real-time detection of the photo-induced cleavage reaction of a surface RITC molecule by correlating simultaneous fluorescence and Raman spectroscopic signals.
Typically, the far-field emission from a fluorophore is susceptible to being quenched by large nonradiative energy losses to the metal surface (14). However, with our method, the dielectric shell of the Ag SHIN prevents surface quenching of fluorescence signal intensity while still generating the plasmonic “hotspot” for enhancing quantum yield (23, 24). As shown on the right of Fig. 1A, discrete intensity fluctuations over time (“blinking” behavior) in the fluorescence emission intensity of a single RITC molecule in the nanocavity and a single-step photobleaching event (see fig. S3 for the corresponding spectra) show direct evidence of SM spectroscopy (14, 20, 25). However, as shown by finite-element method (FEM) calculations in fig. S4, the electric field and Raman enhancement in the hotspot are around 30-fold and 4 × 105–fold for a nanocavity substrate constructed with 10-nm shell Ag SHINs, which is not suitable to observe the SM vibrational spectroscopy. Therefore, to further enhance the local field intensity for vibrational Raman features, the size of nanocavity can be tuned by changing the dielectric shell thickness of SHIN from 10 to 2 nm (see fig. S5 for electron microscopy characterizations of SHINs). As a result, the electric field enhancement in a smaller nanocavity using 2-nm shell Ag SHIN is up to ~80-fold (Fig. 1B), enabling simultaneous collection of SM fluorescence and Raman signals and detection of a photo-induced bond cleavage reaction in real time (shown schematically in Fig. 1C).
Figure 2A shows the plasmon-enhanced SM emission spectrum of RITC in the nanocavity, where the plasmonic hotspot offers >1.0 × 107 Raman signal enhancement for SM resonant Raman scattering measurement. Therefore, several vibrational features can be distinguished from the fluorescence spectrum, i.e., the resonant Raman modes of the xanthene group of RITC, which are in good agreement with the literature (26) and of ensemble RITC experimental results in fig. S6. During illumination, both resonant Raman features and fluorescence band fluctuate over time before simultaneously disappearing after 37.30 s due to photo-induced degradation of the xanthene group (Fig. 2B). Moreover, there is a strong proportional correlation between the intensity fluctuations of the Raman peaks (gray area in Fig. 2A) and the fluorescence background (light red area in Fig. 2A). Correlating the intensities of the two strongest Raman peaks for the aromatic stretching modes (26) of the RITC xanthene group at 1534 and 1656 cm−1 with the corresponding fluorescence background gives R2 values of ~0.87 and ~0.83 (Fig. 2, C and D). These well-correlated Raman and fluorescence intensities can be attributed to the local field enhancement mechanism within the plasmonic hotspot, which further confirms that the Raman and fluorescence bands arise from the same SM (27–29). For comparison, two more representative SM spectra with Raman vibrational features are shown in fig. S7. In addition, Fig. 2E shows a Gaussian fit of the emission intensity distribution of 34 SM events observed using the 2-nm shell Ag SHIN substrate, which indicates the reproducible observation of RITC SMs. Compared with Ag SHINs with 10-nm shell, the thinner dielectric shell results in a stronger plasmonic coupling effect between the Ag core and the film, offering advantages in revealing vibronic features of SMs at room temperature and in real time.
Fig. 2. Plasmon-enhanced SM emission spectroscopy of RITC.
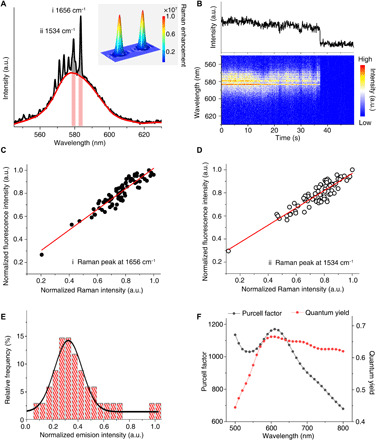
(A) Average plasmon-enhanced SM emission spectrum with visible resonant Raman vibrational bands of a single RITC molecule before photobleaching; the red curve represents the fluorescence band background. Inset: FEM simulation of the corresponding Raman signal enhancement distribution in the nanocavity using a Ag SHIN dimer with 2-nm silica shell. (B) Overview of emission spectra from the same molecule observed in (A): integrated emission spectral peak intensity trajectories (upper panel) and color-coded contour plot of time-dependent emission spectra (lower panel); spectral acquisition time = 50 ms. (C and D) Time-dependent intensity correlation of Raman peaks at 1656 and 1534 cm−1 [gray areas shown in (A)] with fluorescence background beneath them [light red areas shown in (A)], respectively. For a higher SNR, each data point corresponds to an accumulation of 10 spectra, and the red lines represent the linear fit. (E) Emission intensity distribution from 34 SM events. The black curve represents the Gaussian fit of results. (F) FEM simulations of the Purcell factor and quantum yield in nanocavities. a.u., arbitrary units.
Especially, an SM platform with a large spontaneous emission rate is a suitable candidate as a single-photon source for nanophotonics, and a fast emission rate can be achieved in a nanocavity due to the Purcell effect (5, 30). However, because of the remarkable nonradiative decay on the metal surface, it is desirable to have both a high quantum efficiency and a fast emission rate (30). To further understand the interaction between SM and optic field, FEM simulations of the Purcell factor and quantum yield in the nanocavity were carried out. As shown in Fig. 2F, the Purcell factor of a single emitter on the proposed substrate is around 1100, while the quantum yield remains higher than 0.65, which suggests an overwhelming dominance of fast radiative decay processes in the competition with nonradiative decay. Therefore, the largely enhanced emission decay rate indicates a possibility of applying the nanocavity substrate as a single-photon source at room temperature.
Application in real-time detection of SM reaction
Probing the intricate reaction pathways of an individual molecule is of profound importance for understanding intrinsic mechanisms and local environmental effects at the SM level (10, 11). In particular, photo-induced carbon─carbon bond cleavage reactions play a vital role in organic and catalysis chemistries (31). However, real-time structural characterization of SM photochemical reactions is challenging at room temperature and under ambient conditions. To apply the proposed method for SM reaction analysis, we further probe an SM photo-induced cleavage reaction by simultaneously correlating fluorescence spectrum and Raman features. Figure 3A shows the SM spectrum of an RITC molecule experiencing a photochemical reaction before the final photobleaching, where the vibrational modes are distinguished in the inset. As shown by the emission spectra contour and intensity trajectories in Fig. 3 (B and C), both the intensities of the fluorescence band and the corresponding Raman peaks fluctuate slightly from illumination onset (highlighted by the dashed pink rectangles) followed by a sudden signal drop to the background (“off” state) at 12.65 s and signal recovery at 15.05 s, until the final photobleaching event (highlighted by the pink solid rectangle). Similarly, strong intensity correlations (R2 = 0.95 and 0.94, as shown in fig. S8) between Raman peaks (~1654 and ~1519 cm−1, respectively) and the fluorescence band beneath along with the single-step photobleaching event indicate that the spectral bands are simultaneously coming from the same SM.
Fig. 3. Real-time detection of an SM reaction by plasmon-enhanced spectroscopy.
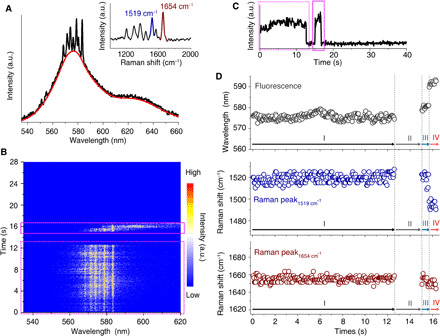
(A) Average spectrum of a single RITC molecule before photobleaching. The red curve represents the fluorescence background, and the inset shows the Raman spectrum obtained by subtracting the fluorescence band. (B and C) Corresponding contour plot of time-dependent emission spectra and the emission intensity trajectory, where the spectral acquisition time was 50 ms. In (B) and (C), dotted and solid pink rectangles mark the emissions before and after the off period. (D) Corresponding time-dependent peak-shift plots of ~1654 cm−1 (lower) and ~1519 cm−1 (middle) Raman peaks and the fluorescence band (upper).
As is known, fluorescence is sensitive to molecular electronic structures. In Fig. 3D, discrete changes in the fluorescence band at 12.65, 15.05, and 15.70 s indicate four different molecular states due to SM structural evolution, denoted by I, II, III, and IV. Therefore, simultaneous spectral shifts in the Raman peaks and the fluorescence band were analyzed to determine these chemical structures. For structure I, the fluorescence band slightly varies around 576 nm, while frequency changes of Raman peaks around 1519 and 1654 cm−1 are also negligible, characteristic of an SM (20). For structure II, we notice that the off period lasts ~2.4 s, and both the fluorescence and resonant Raman signals disappear. Unlike triplet-state blinking of SMs, which has a typical lifetime on the millisecond time scale, this long off period can be probably ascribed to the structural transformation of the cationic form of RITC into the nonfluorescent/resonant lactonic form due to the basic local environment via slight hydrolysis of SiO2 shell of the Ag SHIN (32). The emission intensity then recovered at 15.05 s with a slightly shifted fluorescence band at around 580 nm, while the Raman peaks did not change significantly (structure III).
Typically, the rhodamine dyes tend to decompose via N-dealkylation (e.g., the removal of N-ethyl groups of xanthene segment) under illumination, which results in a fluorescence shift toward shorter wavelength in ensemble experiments (33, 34). However, for structure IV in our SM experiment, a large shift in the fluorescence band from ~580 to ~591 nm and a concomitant shift in the symmetrical C─C stretching vibration modes from 1519 and 1654 cm−1 to 1500 and 1652 cm−1 occur, respectively. This suggests a possible bond cleavage reaction that influences the xanthene segment of the molecule producing the resulting shifts. For structures III and IV, observation of resonant Raman C─C stretching vibrations indicates that the xanthene segment of the molecule is not destroyed. From the literature, visible light irradiation can induce C─C bond cleavage in the pyronin analogs (35). Hence, we suggest that the phenyl carboxylic group attached to the anthracene of RITC undergoes cascading carboxylic group removal followed by a phenyl breaking reaction.
To prove the proposed reaction pathway, in Fig. 4A, representative SM spectra of the four structures, along with the ensemble spectra of RITC and the proposed product after removing the phenyl carboxylic group, i.e., pyronine B (Pyr B), are depicted. Because the molecule probed was in the plasmonic hotspot, the different states of this molecule can be distinguished by the changes in surface-enhanced spectral features. First, in the slightly basic local environment, RITC can readily form the nonfluorescent/resonant lactone, structure II, according to an inner nucleophilic addition reaction (32) (proposed pathway is shown in Fig. 4D). During laser illumination, the ensuing cascade ring-opening and decarboxylation processes result in structure III with a slightly red-shifted fluorescence band and unchanged xanthene resonant Raman frequencies, which are consistent with our density functional theory (DFT) calculations (fig. S9) and the literature (36). Last, as shown in Fig. 4 (A and D), structure III undergoes a subsequent phenyl group removal reaction, producing structure IV with a concomitant fluorescent red-shift and significant variation of Raman spectral bands that match well with the plasmon-enhanced emission spectra of Pyr B (structure IV is the cation of Pyr B, which represents the chromophore in Pyr B).
Fig. 4. Identifying the SM reaction intermediate and product by simultaneous Raman and fluorescence spectroscopy.
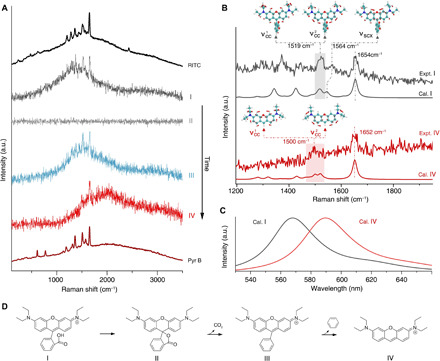
(A) Average spectra of SM spectroscopy of RITC during ~11.5 to 12.60 s (I), ~12.65 to 15 s (II), ~15.05 to 15.65 s (III), and ~15.70 to 16.3 s (IV). These spectra represent four distinct states during the structural evolution of an RITC molecule until the final photobleaching of the xanthene group. The SM event corresponds to the one shown in Fig. 3. Ensemble surface-enhanced emission spectra of RITC and Pyr B are presented at the top and bottom for reference, respectively. (B) Experimental and calculated resonant Raman features of structures I and IV. The insets illustrate the vibrational normal modes of νcc and νscx that are associated with the symmetrical stretching of C─C in side phenyls and the stretching mode of central xanthene accompanying stretching of the C─C bond between central xanthene and carboxyphenyl, respectively. (C) DFT-calculated fluorescence spectra of structures I and IV. (D) Proposed reaction pathway of a single RITC molecule (I) experiencing structural transformation to a lactonic form (II) and the subsequent bond cleavage reactions (III and IV), where the thiourea anchoring group attached to the substrate is omitted.
Comparing the aromatic stretching modes in structures I and IV in Fig. 4B, the peak at 1654 cm−1 slightly shifts to 1652 cm−1, while a noteworthy shift is also observed for the peak at 1519 cm−1. DFT calculations show two near-degenerate symmetrical stretching modes of the C─C bonds in side phenyls ( and with the calculated frequencies of 1513 and 1521 cm−1; see inset illustrations in Fig. 4B), which contribute to the observed peak at around 1519 cm−1 in the RITC spectrum. Because of the C─C bond cleavage reaction removing the phenyl group, that has considerable normal mode distribution in the carboxyphenyl shifts to 1500 cm−1, which is responsible for the appearance of a broad Raman band at ~1500 cm−1 in structure IV. Meanwhile, remains around 1520 cm−1 because it is not directly affected by the carboxyphenyl group. What is more important is the disappearance of the stretching mode of the central xanthene (νscx) in structure IV. Because νscx is strongly associated with the stretching of C─C bond between central xanthene and carboxyphenyl (νscx is at 1564 cm−1 in structure I; see inset in Fig. 4B), providing direct evidence of removal reaction of the carboxyphenyl group. Meanwhile, the conformational transformation and removal of the carboxyphenyl group cause the fluorescence emission band to shift to ~590 nm (Fig. 4C). As revealed by the calculated molecular orbitals in fig. S10, the effect of C─C bond cleavage on the HOMO (highest occupied molecular orbital) is negligible, while the LUMO (lowest unoccupied molecular orbital) becomes more stable, which leads to a reduced energy gap in structure IV (2.75 eV) as compared with structures I (2.82 eV) and III (2.79 eV). This bond cleavage reaction is reproducible as shown by another data set in fig. S11. Hence, direct observation of an SM photochemical reaction is demonstrated, and the proposed detection method has potential as a routine laboratory SM reaction characterization platform. In addition, it has been reported that a strong electric field would affect the reaction, such as an electrostatic field at 108 V/m level (37). In this work, the strength of the optical electric field is estimated to only ~3.9 × 104 V/m. To shed the light on how to mediate the SM photochemical reactions by electric field, the laser power– and temperature-dependent experiments will be carried out in the near feature.
DISCUSSION
To conclude, here, a Ag nanoantenna-film coupling nanocavity substrate was used to determine the physical and chemical properties of an SM of RITC at room temperature. The plasmonic hotspot in the nanocavity provided a large emission intensity enhancement and a Purcell factor of around 1100. Furthermore, this substrate enabled identification of SM reaction intermediates in real time on the millisecond time scale. Spectral correlations between Raman vibrational and fluorescence emission peaks, together with the DFT calculations, were used to observe an RITC SM, which structurally transformed to a lactonic species and then underwent decarboxylation followed by a phenyl-group removal reaction, to form Pyr B. This work improves our understanding of SM chemical recognition, showing the potential of the nanocavity substrate for use as a single-photon resource. By combining with nanofluidics in the near future, it will offer a new avenue for SM biochemical analysis on smart “lab-on-a-chip” devices, providing a novel method for investigating light-matter interactions at the SM level.
MATERIALS AND METHODS
Preparation of Ag SHINs
The Ag SHINs were synthesized and then dropped onto the substrate according to the method reported previously (24). At first, ~96-nm-diameter Ag nanospheres were synthesized by a seed-mediated growth method, whereby 38 mM sodium citrate (1.5 ml) was added to 0.24 mM boiling HAuCl4 (50 ml) under vigorous stirring to get the 16-nm Au seeds. The Au seed sol was lowered by 45 times in water and then mixed with l-ascorbic acid (AA), AgClO4, and sodium citrate. The concentration of AA, AgClO4, and sodium citrate was 1.84, 1.25, and 1.25 mM, respectively.
Next, the as-prepared 96-nm Ag sol was diluted with ultrapure water two times, and then the diluted Ag sol was mixed with (3-aminopropyl) trimethoxysilane (APTMS), NaBH4, and sodium silicate solution. The amount of sodium silicate, APTMS, and NaBH4 was 0.045%, 0.22 mM, and 5.5 mM, respectively. During the synthesis, the pH value of mixture was modified by H2SO4 to be ~9.7. After that, the mixture was immediately transferred to a 90°C water bath and vigorously stirred for 60 min to get 2-nm shell. For 10-nm shell, the bath temperature was then tuned down to 60°C for 150 min. In this work, the as-prepared Ag SHINs were washed twice and concentrated in a 2-ml centrifugal tube and then diluted into 2-ml with ultrapure water.
Preparation of dye-functionalized substrate
The Ag film substrate was prepared through e-beam evaporation of a ~200-nm-thick Ag film onto an atomically flat Si(111) wafer (2.5 cm by 2.5 cm) surface. To facilitate the RITC surface modification, a deposition of ~2-nm silica layer on Ag film was performed via ALD. Then, the RITC probe was modified covalently on Ag-based substrate via amino-related chemistry, where the extremely diluted concentrations of 3-aminopropyltriethoxysilane (APTS) and RITC were used. First, the Ag-based wafer substrate was immersed in a solution of silane coupling agent (ethanol/water/acetic acid/silane = 98/3/1.2/2, v/v/v/v). A 1‰ amino density on SiO2 can be obtained under a ratio of APTS/triethoxypropylsilane = 0.001/1 (v/v). After amino-group functionalization, the substrate was washed thoroughly with ethanol and stored under vacuum conditions before RITC modification. Then, the substrate was immersed in a 0.1 to 0.02 nM RITC ethanol solution (~3 ml) for 24 hours in darkness. To promote the isothiocyanate-amino reaction, the pH was adjusted by N,N-diisopropylethylamine. To avoid physical adsorption of RITC dye molecules onto the substrate surface, thorough rinsing with ethanol was required, leaving only the RITC molecules covalently bound to the surface. Then, the 2-ml Ag SHIN sol was dropped onto the RITC-functionalized substrate surface to achieve a density of ~70% on the dye-functionalized substrate surface.
Optical spectroscopy
SM spectroscopy was carried out at room temperature and performed with a confocal Raman microscope (Alpha300 R from WITec) equipped with a front illumination electron-multiplying charge-coupled device (DU970-FI from Andor). Diffraction grating (600 grooves/mm) was chosen for spectral dispersion. For SM spectroscopy, the acquisition time was 50 ms, and the excitation laser was 532 nm (the power on the sample was ~0.3 μW), which was focused through a 60×, 1.49–numerical aperture oil objective. In Figs. 2 and 3, a first-order Savitzky-Golay filter with six-point width was used to smooth the spectra via Origin2019b software. For ensemble experiments shown in Fig. 4 and fig. S6, a Ag nanosphere substrate was used.
FEM calculation
The finite-element simulation software (COMSOL Multiphysics) was used to calculate the electric field distribution and Raman enhancement of the Ag nanoantenna-film coupling nanostructure. A dimer Ag@SiO2 nanoparticle array and a 2-nm SiO2-coated Ag film were used. The height of dye in the nanocavity was set at 2 nm. The refraction index of both dielectric SiO2 shell and spacer layer was set at 1.4525, and the Ag nanoparticle core and film were according to the literature (38). Purcell factor and quantum yield calculations were based on the literature (30, 39, 40). Briefly, the RITC dye molecule was modeled as an electric dipole source with the emission wavelength varied from 500 to 800 nm. We assumed a harmonic time-dependent dipolar oscillation for the fields produced by the dipole according to the following equations (40)
| (1) |
| (2) |
where ω is the dipole frequency, μ is the relative permeability (μ0 is the value of vacuum), is the dyadic Green’s function of the system, and p is the dipole moment. Thus, could be derived numerically by evaluating the electric field at the dipole point. Then, the spontaneous emission rate of molecules coupled with plasmonic cavity, γsp, could be derived accordingly
| (3) |
where ε0 is the permittivity of vacuum. The Purcell factor can be obtained by normalizing the γsp to the intrinsic spontaneous emission rate of molecules (without coupling to plasmonic cavity), as shown in Fig. 2F.
Ab initio calculation
All geometrical and electronic structures of ground-state molecules were optimized with DFT calculations at the B3LYP/6-31G(d, p) level (41, 42) using the Gaussian 09 suite. The effects of surrounding media were taken into consideration using the polarizable continuum model (PCM) with the integral equation formalism variant (43), where the solvent was set to water. During geometrical optimization, for species I and IV, the CS symmetry was preserved, while for species III, the C1 symmetry was adopted, which generates no imaginary frequencies in the frequency analysis of all species. In addition, to get a better agreement with the experimental fluorescence and resonant Raman spectra, one of the dihedral angles between ethyl and side phenyl in species IV was further twisted by 34° in the calculations. Scaling factors of around 0.97 (specifically, 0.976, 0.977, and 0.974 for I, III, and IV, respectively) were used to account the anharmonicity of the vibrations (44). The resonant Raman spectra were calculated by a linear coupling model (45). In practical simulations, the required displacements in the linear coupling model were acquired from the force of the excited states that were calculated using time-dependent DFT (46) at the same level with nonequilibrium PCM solvation. Meanwhile, the calculated vertical excitation energies were red-shifted by 0.44 eV to eliminate the systematic error of the approximate functional. The fluorescence spectra were calculated by the well-known mirror-image rule (47) from absorbance, where the energies of 0-0 transitions were slightly blue-shifted by 62 meV to match the experimental observations. All vibrational integrals were calculated by the DynaVib software (48).
Supplementary Material
Acknowledgments
We thank M. Meng, Y. P. Huang, J. H. Gao, C. L. Wu, G. K. Liu, and B. Ren for helpful discussion. Funding: This study was supported by the National Natural Science Foundation of China (grant nos. 21775127, 21522508, 21521004, 21427813, and 21533006) and Fundamental Research Funds for the Central Universities (20720190044). S.D. is sponsored by the Shanghai Pujiang Program (19PJ1400600). Author contributions: C.-Y.L. and J.-F.L. conceived and designed the project. C.-Y.L. and C.W. carried out the experiments. S.D. and J.Y. performed the theoretical calculations. C.-Y.L., S.D., C.W., P.M.R., Z.-Q.T., and J.-F.L. analyzed the data and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/24/eaba6012/DC1
REFERENCES AND NOTES
- 1.Nie S. M., Chiu D. T., Zare R. N., Probing individual molecules with confocal fluorescence microscopy. Science 266, 1018–1021 (1994). [DOI] [PubMed] [Google Scholar]
- 2.Moerner W. E., Orrit M., Illuminating single molecules in condensed matter. Science 283, 1670–1676 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Betzig E., Patterson G. H., Sougrat R., Lindwasser O. W., Olenych S., Bonifacino J. S., Davidson M. W., Lippincott-Schwartz J., Hess H. F., Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Zhang R., Zhang Y., Dong Z. C., Jiang S., Zhang C., Chen L. G., Zhang L., Liao Y., Aizpurua J., Luo Y., Yang J. L., Hou J. G., Chemical mapping of a single molecule by plasmon-enhanced Raman scattering. Nature 498, 82–86 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Chikkaraddy R., de Nijs B., Benz F., Barrow S. J., Scherman O. A., Rosta E., Demetriadou A., Fox P., Hess O., Baumberg J. J., Single-molecule strong coupling at room temperature in plasmonic nanocavities. Nature 535, 127–130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu H. P., Xie X. S., Single-molecule spectral fluctuations at room temperature. Nature 385, 143–146 (1997). [Google Scholar]
- 7.Tenopala-Carmona F., Fronk S., Bazan G. C., Samuel I. D. W., Penedo J. C., Real-time observation of conformational switching in single conjugated polymer chains. Sci. Adv. 4, eaao5786 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sambur J. B., Chen T. Y., Choudhary E., Chen G., Nissen E. J., Thomas E. M., Zou N., Chen P., Sub-particle reaction and photocurrent mapping to optimize catalyst-modified photoanodes. Nature 530, 77–80 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Chen T., Dong B., Chen K., Zhao F., Cheng X., Ma C., Lee S., Zhang P., Kang S. H., Ha J. W., Xu W., Fang N., Optical super-resolution imaging of surface reactions. Chem. Rev. 117, 7510–7537 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Roeffaers M. B. J., Sels B. F., Uji-I H., De Schryver F. C., Jacobs P. A., De Vos D. E., Hofkens J., Spatially resolved observation of crystal-face-dependent catalysis by single turnover counting. Nature 439, 572–575 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Guan J., Jia C., Li Y., Liu Z., Wang J., Yang Z., Gu C., Su D., Houk K. N., Zhang D., Guo X., Direct single-molecule dynamic detection of chemical reactions. Sci. Adv. 4, eaar2177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong B., Pei Y., Zhao F., Goh T. W., Qi Z., Xiao C., Chen K., Huang W., Fang N., Pei Y., Zhao F., Goh T. W., Qi Z., Xiao C., Chen K., Huang W., Fang N., In situ quantitative single-molecule study of dynamic catalytic processes in nanoconfinement. Nat. Catal. 1, 135–140 (2018). [Google Scholar]
- 13.Moerner W. E., Fromm D. P., Methods of single-molecule fluorescence spectroscopy and microscopy. Rev. Sci. Instrum. 74, 3597–3619 (2003). [Google Scholar]
- 14.J. Lakowicz, Principles of Fluorescence Spectroscopy (Springer, ed. 3, 2006). [Google Scholar]
- 15.Acuna G. P., Möller F. M., Holzmeister P., Beater S., Lalkens B., Tinnefeld P., Fluorescence enhancement at docking sites of DNA-directed self-assembled nanoantennas. Science 338, 506–510 (2012). [DOI] [PubMed] [Google Scholar]
- 16.van Schrojenstein Lantman E. M., Deckert-Gaudig T., Mank A. J. G., Deckert V., Weckhuysen B. M., Catalytic processes monitored at the nanoscale with tip-enhanced Raman spectroscopy. Nat. Nanotechnol. 7, 583–586 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Itoh T., Yamamoto Y. S., Ozaki Y., Plasmon-enhanced spectroscopy of absorption and spontaneous emissions explained using cavity quantum optics. Chem. Soc. Rev. 46, 3904–3921 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Xie W., Schlücker S., Hot electron-induced reduction of small molecules on photorecycling metal surfaces. Nat. Commun. 6, 7570 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao Y. C., Jin R., Mirkin C. A., Nanoparticles with Raman spectroscopic fingerprints for DNA and RNA detection. Science 297, 1536–1540 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Nie S. M., Emory S. R., Probing single molecules and single nanoparticles by surface-enhanced Raman scattering. Science 275, 1102–1106 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Kneipp K., Wang Y., Kneipp H., Perelman L. T., Itzkan I., Dasari R. R., Feld M. S., Single molecule detection using surface-enhanced Raman scattering (SERS). Phys. Rev. Lett. 78, 1667–1670 (1997). [Google Scholar]
- 22.Xu H., Bjerneld E. J., Käll M., Börjesson L., Spectroscopy of single hemoglobin molecules by surface enhanced Raman scattering. Phys. Rev. Lett. 83, 4357–4360 (1999). [Google Scholar]
- 23.Li J., Huang Y. F., Ding Y., Yang Z. L., Li S. B., Zhou X. S., Fan F. R., Zhang W., Zhou Z. Y., Wu D. Y., Ren B., Wang Z. L., Tian Z. Q., Shell-isolated nanoparticle-enhanced Raman spectroscopy. Nature 464, 392–395 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Li C. Y., Meng M., Huang S. C., Li L., Huang S. R., Chen S., Meng L. Y., Panneerselvam R., Zhang S. J., Ren B., Yang Z. L., Li J. F., Tian Z. Q., “Smart” Ag nanostructures for plasmon-enhanced spectroscopies. J. Am. Chem. Soc. 137, 13784–13787 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Kinkhabwala A., Yu Z., Fan S., Avlasevich Y., Müllen K., Moerner W. E., Large single-molecule fluorescence enhancements produced by a bowtie nanoantenna. Nat. Photonics 3, 654–657 (2009). [Google Scholar]
- 26.Vosgröne T., Meixner A. J., Surface- and resonance-enhanced micro-Raman spectroscopy of xanthene dyes: From the ensemble to single molecules. ChemPhysChem 6, 154–163 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Galloway C. M., Etchegoin P. G., Le Ru E. C., Ultrafast nonradiative decay rates on metallic surfaces by comparing surface-enhanced Raman and fluorescence signals of single molecules. Phys. Rev. Lett. 103, 063003 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Walter M. J., Lupton J. M., Becker K., Feldmann J., Gaefke G., Höger S., Simultaneous Raman and fluorescence spectroscopy of single conjugated polymer chains. Phys. Rev. Lett. 98, 137401 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Moskovits M., Surface-enhanced spectroscopy. Rev. Mod. Phys. 57, 783–826 (1985). [Google Scholar]
- 30.Akselrod G. M., Argyropoulos C., Hoang T. B., Ciracì C., Fang C., Huang J., Smith D. R., Mikkelsen M. H., Probing the mechanisms of large Purcell enhancement in plasmonic nanoantennas. Nat. Photonics 8, 835–840 (2014). [Google Scholar]
- 31.N. J. Turro, V. Ramamurthy, J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules (University Science Books, 2010). [Google Scholar]
- 32.Arbeloa I. L., Rohatgimukherjee K. K., Solvent effect on photophysics of the molecular forms of rhodamine B: Solvation models and spectroscopic parameters. Chem. Phys. Lett. 128, 474–479 (1986). [Google Scholar]
- 33.Hu X., Mohamood T., Ma W., Chen C., Zhao J., Oxidative decomposition of rhodamine B dye in the presence of VO2+ and/or Pt(IV) under visible light irradiation: N-deethylation, chromophore cleavage, and mineralization. J. Phys. Chem. B 110, 26012–26018 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Wilhelm P., Stephan D., Photodegradation of rhodamine B in aqueous solution via SiO2@TiO2 nano-spheres. J. Photochem. Photobiol. A 185, 19–25 (2007). [Google Scholar]
- 35.Štacko P., Šebej P., Veetil A. T., Klán P., Carbon–carbon bond cleavage in fluorescent pyronin analogues induced by yellow light. Org. Lett. 14, 4918–4921 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Luo X., Qian L., Xiao Y., Tang Y., Zhao Y., Wang X., Gu L., Lei Z., Bao J., Wu J., He T., Hu F., Zheng J., Li H., Zhu W., Shao L., Dong X., Chen D., Qian X., Yang Y., A diversity-oriented rhodamine library for wide-spectrum bactericidal agents with low inducible resistance against resistant pathogens. Nat. Commun. 10, 258 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aragonès A. C., Haworth N. L., Darwish N., Ciampi S., Bloomfield N. J., Wallace G. G., Diez-Perez I., Coote M. L., Electrostatic catalysis of a Diels–Alder reaction. Nature 531, 88–91 (2016). [DOI] [PubMed] [Google Scholar]
- 38.E. D. Palik, Handbook of Optical Constants of Solids (Academic Press, 1998), vol. 3. [Google Scholar]
- 39.Rose A., Hoang T. B., McGuire F., Mock J. J., Ciracì C., Smith D. R., Mikkelsen M. H., Control of radiative processes using tunable plasmonic nanopatch antennas. Nano Lett. 14, 4797–4802 (2014). [DOI] [PubMed] [Google Scholar]
- 40.L. Novotny, B. Hecht, Principles of Nano-Optics (Cambridge Univ. Press, 2012). [Google Scholar]
- 41.Stephens P. J., Devlin F. J., Chabalowski C. F., Frisch M. J., Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98, 11623–11627 (1994). [Google Scholar]
- 42.Ditchfield R., Hehre W. J., Pople J. A., Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 54, 724–728 (1971). [Google Scholar]
- 43.Lipparini F., Scalmani G., Mennucci B., Cancès E., Caricato M., Frisch M. J., A variational formulation of the polarizable continuum model. J. Chem. Phys. 133, 014106 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Alecu I. M., Zheng J., Zhao Y., Truhlar D. G., Computational thermochemistry: Scale factor databases and scale factors for vibrational frequencies obtained from electronic model chemistries. J. Chem. Theory Comput. 6, 2872–2887 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Macak P., Luo Y., Ågren H., Simulations of vibronic profiles in two-photon absorption. Chem. Phys. Lett. 330, 447–456 (2000). [Google Scholar]
- 46.Bauernschmitt R., Ahlrichs R., Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 256, 454–464 (1996). [Google Scholar]
- 47.Ai Y.-J., Zhang F., Chen S.-F., Luo Y., Fang W.-H., Importance of the intramolecular hydrogen bond on the photochemistry of anionic hydroquinone (FADH−) in DNA photolyase. J. Phys. Chem. Lett. 1, 743–747 (2010). [Google Scholar]
- 48.Tian G., Sun D., Zhang Y., Yu X., Franck–Condon blockade and aggregation-modulated conductance in molecular devices using aggregation-induced emission-active molecules. Angew. Chem. Int. Ed. 58, 5951–5955 (2019). [DOI] [PubMed] [Google Scholar]
- 49.G. T. Hermanson, Bioconjugate Techniques (Academic Press, 2013). [Google Scholar]
- 50.Rathor N., Panda S., Aminosilane densities on nanotextured silicon. Mater. Sci. Eng. C 29, 2340–2345 (2009). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/24/eaba6012/DC1