

Incomplete annotation has the greatest impact on brain-specific genes and consequently, brain-related disorders.
Abstract
Growing evidence suggests that human gene annotation remains incomplete; however, it is unclear how this affects different tissues and our understanding of different disorders. Here, we detect previously unannotated transcription from Genotype-Tissue Expression RNA sequencing data across 41 human tissues. We connect this unannotated transcription to known genes, confirming that human gene annotation remains incomplete, even among well-studied genes including 63% of the Online Mendelian Inheritance in Man–morbid catalog and 317 neurodegeneration-associated genes. We find the greatest abundance of unannotated transcription in brain and genes highly expressed in brain are more likely to be reannotated. We explore examples of reannotated disease genes, such as SNCA, for which we experimentally validate a previously unidentified, brain-specific, potentially protein-coding exon. We release all tissue-specific transcriptomes through vizER: http://rytenlab.com/browser/app/vizER. We anticipate that this resource will facilitate more accurate genetic analysis, with the greatest impact on our understanding of Mendelian and complex neurogenetic disorders.
INTRODUCTION
Genetic and transcriptomic studies are fundamentally reliant on accurate and complete human gene annotation, being defined as the genetic coordinates of all transcripts of a given gene. Among other analyses, this is required for the quantification of expression or splicing from RNA sequencing (RNA-seq) experiments, interpretation of significant genome-wide association study (GWAS) signals, and variant interpretation from genetic tests. As our understanding of transcriptomic complexity improves, it is apparent that existing gene annotation principally originating from four sources (RefSeq, GENCODE, Ensembl, AceView) remains incomplete (1–4). Comparison of these different existing gene annotation databases reveals that more than 17,000 Ensembl genes fall into intronic or intergenic regions according to the AceView database, and the choice of reference annotation greatly influences the performance of variant interpretation software, such as VEP and ANNOVAR (5, 6). Thus, this evidence suggests that incomplete annotation may cause pathogenic variants to be overlooked within exonic regions that are yet to be annotated as well as limiting our understanding of risk loci.
Despite accumulating evidence that the map of the human transcriptome remains incomplete, it is not yet fully understood which tissues and consequently diseases are most affected. The extent to which this poses an issue is unlikely to be equal across all types of tissues or cells. In particular, the fact that the human brain harbors longer transcripts, higher transcript diversity, and higher cellular heterogeneity than other tissues might be expected to make identifying all transcripts from this tissue more challenging (7, 8). Moreover, the difficulties of accessing brain tissue and dependence on postmortem tissue may also limit the quantity of high-quality, brain-specific data inputted into gene annotation pipelines to date. Several analyses of bulk RNA-seq data derived from human brain tissues have discovered transcription originating from intronic or intergenic regions (henceforth termed unannotated) (9–11). For example, Jaffe and colleagues found that as much as 41% of transcription in the human frontal cortex was unannotated (11) . In combination, these factors lead to specific challenges in fully capturing the transcriptome of the human brain and suggest that improvements to gene annotation may have a disproportionate impact on the understanding of neurological diseases.
In this study, we address this issue by leveraging transcriptomic data available through the Genotype-Tissue Expression (GTEx) Consortium to identify previously unannotated exons of known genes. Distinct from existing de novo assembly approaches, such as that implemented by Pertea and colleagues leading to the development of the CHESS database, our analytic approach was focused on the detection of unannotated exons among known genes rather than the assembly of previously unidentified transcripts (12). This conservative approach was adopted because of the well-recognized challenges in accurately calling transcripts from short-read sequencing data and because the major aim of this study was to improve the annotation of genes already known to contribute to neurological disease (13, 14). With this in mind, we defined transcription in an annotation-agnostic manner using RNA-seq data from 13 regions of the human central nervous system (CNS) and a further 28 nonbrain tissues. Specifically, we defined unannotated transcription in a tissue-specific manner to allow comparison between tissues. We found that this unannotated transcription although widespread is most prevalent in human brain. We provide evidence to suggest that the exons that we discover are likely to be functionally important on the basis of their tissue and cell-type specific expression, the significant depletion of genetic variation within humans, and their protein coding potential. Last, by combining unannotated transcription with junction read data, defined as reads that have a gapped alignment to the genome, we link these regions to known genes, focusing on those associated with Mendelian and complex neurological disorders. Overall, we improve the annotation of 13,429 genes, encompassing 1831 (63%) Online Mendelian Inheritance in Man (OMIM) genes and a further 317 genes associated with complex neurodegenerative and neuropsychiatric disease. We release our findings in an online platform vizER (www.rytenlab.com/browser/app/vizER), which allows individual genes to be queried and visualized for reannotation as well as the download of all exons we discover. We anticipate that this resource will facilitate basic and translational research targeted at Mendelian and complex neurogenetic disorders.
RESULTS
Optimizing the tissue-specific, annotation-agnostic detection of transcription
Pervasive transcription of the human genome, the presence of pre-mRNA even within polyA-selected RNA-seq libraries and variability in read depth complicates the identification of exons and transcripts using RNA-seq data (15, 16). With this in mind, we used a set of exons with the most reliable boundaries [namely, all exons from Ensembl v92 that did not overlap with any other exon (4)] to calibrate the detection of transcription from 41 GTEx tissues (17). Of available annotation databases, Ensembl was selected as it is one of the most commonly used and comprehensive annotation providers. We used the tool derfinder to perform this analysis (18). However, we noted that while derfinder enables the detection of continuous blocks of transcribed bases termed expressed regions (ERs) in an annotation-agnostic manner, the mean coverage cutoff (MCC) applied to determine transcribed bases is difficult to define and variability in read depth even across an individual exon can result in false segmentation of blocks of expressed sequence. Therefore, to improve our analysis and more accurately define ERs, we applied derfinder, but with the inclusion of an additional parameter we term the max region gap (MRG), which merges adjacent ERs (see detailed Materials and Methods). Next, we sought to identify the optimal values for MCC and MRG using our learning set of known, nonoverlapping exons.
This process involved generating 506 transcriptome definitions for each tissue using unique pairs of MCCs and MRGs, resulting in a total of 20,746 transcriptome definitions across all 41 tissues. For each of the 20,746 transcriptome definitions, all ERs that intersected nonoverlapping exons were extracted, and the absolute difference between the ER definition and the corresponding exon boundaries, termed the exon delta, was calculated (Fig. 1A). We summarized the exon delta for each transcriptome using two metrics, the median exon delta and the number of ERs with exon delta equal to 0. The median exon delta represents the overall accuracy of all ER definitions, whereas the number of ERs with exon delta equal to 0 indicates the extent to which ER definitions precisely match overlapping exon boundaries. The MCC and MRG pair that generated the transcriptome with the lowest median exon delta and highest number of ERs with exon delta equal to 0 was chosen as the most accurate transcriptome definition for each tissue. Across all tissues, 50 to 54% of the ERs tested had an exon delta = 0, suggesting that we had accurately defined most of ERs. Taking the cerebellum as an example and comparing ER definitions to those which would have been generated applying the default derfinder parameters used in the existing literature (MCC: 0.5, MRG: None equivalent to 0), we noted a 96–base pair (bp) refinement in ER size, equating to 67% of median exon size (Fig. 1, B and C). In summary, by using known exons to calibrate the detection of transcription, we generated more accurate annotation-agnostic transcriptome definitions for 13 regions of the CNS and a further 28 human tissues.
Fig. 1. Optimization of the detection of transcription.
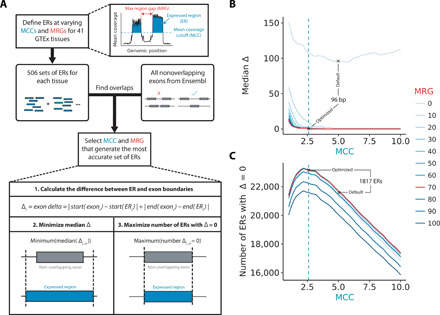
(A) Transcription in the form ERs was detected in an annotation-agnostic manner across 41 human tissues. The MCC is the number of reads supporting each base above which that base would be considered transcribed, and the MRG is the maximum number of bases between ERs below which adjacent ERs would be merged. MCC and MRG parameters were optimized for each tissue using the nonoverlapping exons from Ensembl v92 reference annotation. (B) Line plot illustrating the selection of the MCC and MRG that minimized the difference between ER and exon definitions (median exon delta). (C) Line plot illustrating the selection of the MCC and MRG that maximized the number of ERs that precisely matched exon definitions (exon delta = 0). The cerebellum tissue is plotted for (B) and (C), which is representative of the other GTEx tissues. Green and red lines indicate the optimal MCC (2.6) and MRG (70), respectively.
Unannotated transcription is most commonly observed in the CNS
To assess how much of the detected transcription was unannotated, ERs were categorized with respect to the genomic features with which they overlapped as defined by the Ensembl v92 reference annotation (exonic, intronic, and intergenic regions; fig. S1A). Those that solely overlapped intronic, or intergenic regions were classified as unannotated. We discovered 8.4 to 22 Mb of unannotated transcription across all tissues, consistent with previous reports that annotation remains incomplete (11, 12). Unannotated ERs predominantly fell into intragenic regions, suggesting that we were preferentially improving the annotation of known genes, rather than identifying entirely undiscovered genes (Fig. 2A). Although unannotated transcription was found to be ubiquitous across tissues, the abundance varied greatly between tissues (Fig. 2, B, D, and E). To investigate this further, we calculated the coefficient of variation for exonic, intronic, and intergenic ERs. We found that the levels of unannotated transcription varied 3.4 to 7.7 times more between tissues than the expression of exonic ERs (coefficient of variation of exonic ERs, 0.066 Mb; intronic ERs, 0.222 Mb; intergenic ERs, 0.481 Mb). Furthermore, focusing on a subset of unannotated ERs for which we could infer the precise boundaries of the putative exon (using intersecting junction reads), we found that more than half of these ERs were detected in only one tissue and that 86.3% were found in less than five tissues (fig. S2A). Even when restricting to ERs derived from only the 13 CNS tissues, 34.3% were specific to one CNS region (fig. S2B). This suggests that unannotated ERs are largely derived from tissue-specific transcription, potentially explaining why they had not already been discovered.
Fig. 2. Transcription detected across 41 GTEx tissues categorized by annotation feature.
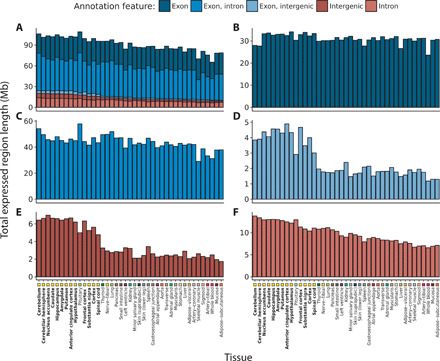
Within each tissue, the length of the ERs Mb overlapping (A) all annotation features, (B) purely exons, (C) exons and introns, (D) exons and intergenic regions, (E) purely intergenic regions, and (F) purely introns according to Ensembl v92 was computed. Tissues are plotted in descending order based on the respective total size of intronic and intergenic regions. Tissues are color-coded as indicated in the x axis, with GTEx brain regions highlighted with bold font. At least 8.4 Mb of previously unannotated transcription was discovered in each tissue, with the greatest quantity found within brain tissues (mean across brain tissues, 18.6 Mb; nonbrain, 11.2 Mb; two-sided Wilcoxon rank sum test, P = 2.35 × 10−10.
This finding lead us to hypothesize that genes highly expressed in brain would be among the most likely to be reannotated because of the difficulty of sampling human brain tissue, the cellular heterogeneity of this tissue, and the particularly high prevalence of alternative splicing (9). As we predicted, the quantity of unannotated transcription found within brain was significantly higher than nonbrain tissues (P = 2.35 × 10−10) (Fig. 2, E and F). Ranking the tissues by descending Mb of unannotated transcription demonstrated that tissues of the CNS constituted 13 of the top 14 tissues. The importance of improving annotation in the human brain tissue was most apparent when considering purely intergenic ERs and ERs that overlapped exons and extended into intergenic regions (Fig. 2, D and E).
This observation raised the question of which factors were most important in determining whether a gene was reannotated (connected to an unannotated ER). We used logistic regression to find genic properties, such as measures of structural complexity and specificity of expression to brain, that significantly changed a gene’s likelihood of reannotation. We also accounted for factors that might be expected to contribute to errors in ER identification, including whether the gene overlapped with another known gene making attribution of reads more complex. We found that the annotation of longer, brain-specific genes with higher transcript complexity were more likely to have evidence for incomplete annotation (table S1). Overlapping genes were not significantly more likely to be reannotated (taking into account gene length), suggesting that unannotated transcription is not merely a product of noise from intersecting genes. Together, these findings demonstrate that widespread unannotated transcription is found across all human tissues, the quantity of which varies extensively between tissues. CNS tissues displayed the greatest quantity of unannotated transcription, and accordingly, genes highly expressed in the human brain are most likely to be reannotated.
Validation of unannotated transcription
We recognize that a proportion of unannotated transcription may originate from technical variability or pre-mRNA contamination. Therefore, we assessed the reliability of detecting unannotated ERs across different versions of Ensembl and within an independent dataset. First, we measured how many Kb of the transcription that we detected would have been classified as unannotated with respect to Ensembl v87 but was now annotated in Ensembl v92 and found that across all tissues, an average of 68 Kb (43 to 127 Kb) had changed status. This value was 5.3 times (3.2 to 10.1 times) greater in every tissue compared to the Kb of ERs overlapping exons in Ensembl v87 that had become purely intronic or intergenic in Ensembl v92 (Fig. 3A). To further assess whether this was greater than what would be expected by chance, we compared the total Kb of unannotated ERs entering v92 annotation for each tissue to 10,000 sets of random length-matched intronic and intergenic regions. For all tissues, the total Kb of both intronic and intergenic ERs that were now annotated in Ensembl v92 was significantly higher than the total Kb distribution of the randomized negative control regions, implying a high validation rate of unannotated ERs (fig. S3). Notably, brain regions had significantly higher Kb of ERs entering Ensembl v92 annotation from Ensembl v87 than nonbrain tissues, even when subtracting the Kb of ERs leaving Ensembl v87 (P = 7.6 × 10−9), suggesting that the greater abundance of brain-specific unannotated transcription was not purely attributed to increased transcriptional noise.
Fig. 3. Validation of unannotated transcription.
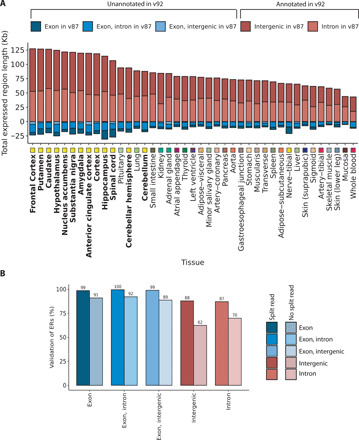
(A) The classification of ERs based on v87 and v92 of Ensembl was compared. Across all tissues, the number of intron or intergenic ERs with respect to v87 that were known to be exonic in Ensembl v92 was greater than the number of ERs overlapping exons according to v87 that were now unannotated in v92. Tissues are plotted in descending order based on the total Mb of unannotated ERs with respect to Ensembl v87 that were validated (classified as exonic in the Ensembl v92). Tissues are color-coded as indicated in the x axis, with GTEx brain regions highlighted with bold font. (B) Bar plot represents the percentage of ERs seeding from the GTEx frontal cortex that validated in an independent frontal cortex RNA-seq dataset. ERs defined in the seed tissue were requantified using coverage from the validation dataset, after which the optimized MCC was applied to determine validated ERs. Colors represent the different annotation features that the ERs overlapped, and the shade indicates whether the ER was supported by junction read(s).
While our analysis of intronic and intergenic ERs across different Ensembl versions provided a high level of confidence in the quality of ER calling, it was limited to ERs, which had already been incorporated into annotation and did not provide an overall indication of the rate of validation across all ERs. Therefore, we investigated whether our GTEx frontal cortex–derived ERs could also be discovered in an independent frontal cortex dataset reported by Labadorf and colleagues (19). As expected, ERs that overlapped with annotated exons had near-complete validation (≥89%), but importantly, 62% of intergenic and 70% of intronic ERs, respectively, were also detected in the second independent frontal cortex dataset (Fig. 3B). While this high validation rate implied that most of all ERs were reliably detected, we investigated whether a subset of ERs supported with evidence of RNA splicing as well as transcription would have even better rates of validation. Evidence of transcription is provided by the coverage data derived using derfinder, while junction reads, which are reads with a gapped alignment to the genome, provide evidence of the splicing out of an intron (fig. S1B). With this in mind, we focused our attention on the putative spliced ERs as indicated by the presence of an overlapping junction read. Consistent with expectation, we found that ERs with junction read support had higher validation rates than ERs lacking this additional feature. This increase in validation rate for ERs with junction read support was greatest for intergenic and intronic ERs with the validation rate rising to 87% for intergenic ERs and 88% for intronic ERs (as compared to 99% for ERs overlapping exons, Fig. 3B). Even when considering this set of highly validated ERs with junction read support, 1.7 to 3.8 Mb of intronic and 0.5 to 2.2 Mb of intergenic transcription was detected across all 41 tissues. Thus, in summary, most of the unannotated ERs were reliably detected and validated in an independent dataset.
Unannotated ERs are likely to be functionally important within humans
Given recent reports suggesting widespread transcriptional noise and acknowledging that transcription, even when tissue specific, does not necessarily translate to function, we investigated whether unannotated ERs were likely to be of functional significance using measures of both conservation and genetic constraint (12, 20). The degree to which a base is evolutionarily conserved across species is dependent on its functional importance, and accordingly, conservation scores have been used to aid exon identification (2). However, this measure is unable to capture genomic regions of human-specific importance. Thus, we investigated unannotated ERs not only in terms of conservation but also genetic constraint. Constraint scores, measured here as a context-dependent tolerance score (CDTS), represent the likelihood that a base is mutated within humans (21). By comparing our detected unannotated ERs to 10,000 randomized sets of length-matched intronic and intergenic regions, we found that both intronic and intergenic ERs were significantly less conserved but more constrained than expected by chance (P < 2 × 10−16; Fig. 4A). This would suggest that they have an important functional role in humans. Furthermore, considering the importance of higher-order cognitive functions in differentiating humans from other species, we separately measured the constraint of brain-specific unannotated ERs on the basis that these ERs may be the most genetically constrained of all unannotated ERs identified. We found that brain-specific unannotated ERs were even more constrained than other unannotated ERs, supporting the view that improvements in gene annotation are likely to have a disproportionate impact on our understanding of human brain diseases.
Fig. 4. Unannotated ERs collectively serve an important function for humans, and a proportion can form potentially protein-coding transcripts.
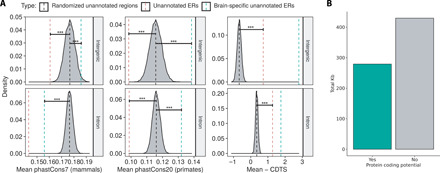
(A) Comparison of conservation (phastCons7/phastCons20) and constraint (CDTS) of intronic and intergenic ERs to 10,000 sets of random, length-matched intronic and intergenic regions. Unannotated ERs marked by the red dashed line are less conserved than expected by chance but are more constrained. Brain-specific ERs marked by the green dashed lines are among the most constrained. Data for the cerebellum shown and is representative of other GTEx tissues. ***P = <2 × 10–16. (B) The DNA sequence for ERs overlapping two junction reads was obtained and converted to amino acid sequence for all three possible frames. ERs (2168; 57%) lacked a stop codon in at least one frame and were considered potentially protein coding.
Another metric of functional importance is whether a region of the genome is translated into protein and notably the vast majority of all known Mendelian disease mutations fall within protein-coding regions. For this reason, we investigated whether unannotated ERs could potentially encode for proteins. Here, we focused on the subset of unannotated ERs that had evidence of splicing since the overlapping junction reads can be used to assign the precise boundaries of ERs, allowing us to confidently retrieve the DNA sequence and corresponding amino acid sequence for each unannotated ER. A total of 2961 ERs covering 274 Kb was found to be potentially protein coding, which represented 57% of the ERs analyzed (Fig. 4B). Among this set of ERs with protein coding potential, 758 ERs also fell within the top 20% of most constrained regions of the genome. These ERs connect to 694 genes, 30% of which are expressed specifically in the CNS (table S2). Overall, we discovered that unannotated ERs are likely to be of functional importance in humans. We also identified a subset of unannotated ERs that have protein coding potential and are highly depleted for genetic variation in humans. Together, this suggested that at least a proportion of unannotated ERs are functionally important.
Incomplete annotation limits our understanding of specific cell types and complex diseases
Given that we discovered the greatest abundance of previously unannotated transcription among brain tissues, we investigated whether this may be affecting our understanding of certain cell types within the brain more than others. We tested this by calculating whether our set of 2962 reannotated brain-specific genes were significantly enriched for cell-type specific genes when compared to the background list of 2422 brain-specific genes without reannotations. Of the 13 brain-specific cell types considered, genes specifically expressed by oligodendrocytes had the largest difference in enrichment (reannotated = <2 × 10−16; not reannotated = 0.169), suggesting that incomplete annotation was disproportionately limiting our understanding of this cell type (Fig. 5A). For example, we found that MBP, which encodes for myelin basic protein, was among those genes reannotated and with an oligodendrocyte-specific expression profile (fig. S4). We detected a 48-bp ER specific to cortex and striatal tissues (anterior cingulate cortex, cortex, frontal cortex, nucleus accumbens, and putamen), which was connected to two flanking protein-coding exons of MBP. The ER itself had protein coding potential and evidence of functional importance in humans, as demonstrated by low mammalian sequence conservation but depletion of genetic variation within humans (phasCons7: 0.06, top 20% CDTS) (Fig. 5B). MBP and oligodendroglial dysfunction have been implicated in a number of neurodegenerative disorders, including multiple system atrophy, which is characterized by myelin loss and degeneration of striatum and cortical region, as well as schizophrenia and Parkinson’s disease (22–24).
Fig. 5. Incomplete annotation of genes disproportionately affects oligodendrocytes.
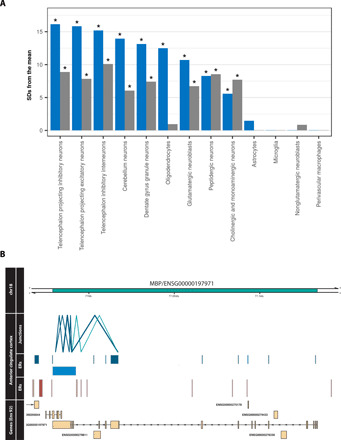
(A) Bar plot displaying the enrichment of reannotated and not reannotated genes within brain cell type–specific gene sets. Blue bars represent the reannotated genes, and gray are those without reannotations. Of all analyzed cell types, the greatest difference between enrichment of reannotated and not reannotated was observed in oligodendrocytes. “*“ represents FDR-corrected P = <0.05. (B) Previously unannotated potentially protein-coding ER discovered in MBP, with an oligodendrocyte-specific expression pattern. The two junction reads in green intersect both the unannotated ER and also the known exons of MBP.
These observations led us to postulate whether incomplete annotation could also be hindering our understanding of complex disorders. We assessed whether our list of reannotated genes was enriched for genes associated with complex forms of neurodegenerative, neuropsychiatric, or other neurological conditions. This analysis was performed by using the Systematic Target OPportunity assessment by Genetic Association Predictions (STOPGAP) database, which provides an extensive catalog of human genetic associations mapped to effector gene candidates (see detailed Materials and Methods) (25). We found that genes associated with neurodegenerative disorders were significantly overrepresented within our reannotated set (P = 0.004; table S3). In particular, important neurodegenerative disease genes such as SNCA, APOE, and CLU were among those reannotated, suggesting that despite being extensively studied, the annotation of these genes remains incomplete (complete list found in table S4). Thus, we demonstrate that incomplete annotation of brain-specific genes may be hindering our understanding of specific cell types and complex neurodegenerative disorders.
Incomplete annotation of OMIM genes may limit genetic diagnosis
Since reannotation of genes already known to cause Mendelian disease would have a direct impact on clinical diagnostic pipelines, we specifically assessed this gene set. Unannotated ERs were first connected to known genes using junction reads (fig. S1B). Next, we filtered for OMIM-morbid genes and found that 63% of this set of OMIM-morbid genes were reannotated and 14% were connected to a potentially protein-coding ER, suggesting that despite many of these genes having been extensively studied, the annotation of many OMIM-morbid genes remains incomplete (Fig. 6A). Given that OMIM-morbid genes often produce abnormalities specific to a given set of organs or systems, we investigated the relevance of unannotated transcription to disease by matching the human phenotype ontology (HPO) terms obtained from the disease corresponding to the OMIM-morbid gene, to the GTEx tissue from which ERs connected to that gene were derived. We discovered that 72% of reannotated OMIM-morbid genes had an associated unannotated ER originating from a phenotypically relevant tissue (Fig. 6B). This phenomenon was exemplified by the OMIM-morbid gene ERLIN1, which, when disrupted, is known to cause spastic paraplegia 62 (SPG62), an autosomal recessive form of spastic paraplegia, which has been reported in some families to cause not only lower limb spasticity but also cerebellar abnormalities (26). We detected a previously unannotated, cerebellar-specific ER that was intronic with respect to ERLIN1. This ER had the potential to code for a nontruncated protein and connected through intersecting junction reads to two flanking, protein-coding exons of ERLIN1, supporting the possibility of this ER being a protein-coding exon. Furthermore, this putative exon was highly conserved (phastcons7 score: 1) and was among the top 30% most constrained regions in the genome, suggesting that it is functionally important both across mammals and within humans (Fig. 6C).
Fig. 6. Reannotation of OMIM genes.
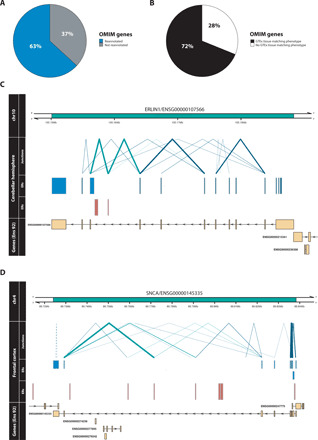
(A) A previously unannotated ER connected through a junction read was discovered for 63% of OMIM-morbid genes. (B) Comparison of the phenotype (HPO terms) associated with each reannotated OMIM-morbid gene and the GTEx tissue from which unannotated ERs were derived. Through manual inspection, HPO terms were matched to disease-relevant GTEx tissues and for 72% of reannotated OMIM genes, the associated unannotated ER was detected in the phenotype-relevant tissue. Visualized examples of reannotated OMIM-morbid genes (C) ERLIN1 and (D) SNCA. Top track represents the genomic region including the gene of interest marked in green. Second group of tracks detail the junction reads and ERs overlapping the genomic region derived from the labeled tissue. Blue ERs overlap known exonic regions, and red ERs fall within intronic or intergenic regions. Blue junction reads overlap blue ERs, while green junction reads overlap both red and blue ERs, connecting unannotated ERs to OMIM-morbid genes. Thickness of junction reads represents the proportion of samples of that tissue in which the junction read was detected. Only partially annotated junction reads (solid lines) and unannotated junction reads (dashed lines) are plotted. The last track displays the genes within the region according to Ensembl v92, with all known exons of the gene collapsed into one “meta” transcript.
Similarly, we detected a brain-specific, unannotated ER in the long intron of the gene SNCA, which encodes α-synuclein protein implicated in the pathogenesis of Mendelian and complex Parkinson’s disease. This ER connected to two flanking protein-coding exons through junction reads (Fig. 6D) and appeared to also have coding potential. While the ER sequence is not conserved within mammals (phastcons7 score: 0.09) or primates (phastcons20 score: 0.21), it is in the top 19% of most constrained regions in the genome, suggesting that it is of functional importance in humans. We validated the existence of this ER both in silico and experimentally. The expression of this ER was confirmed in silico using an independent frontal cortex dataset reported by Labadorf and colleagues (19). Using Sanger sequencing, we validated the junctions intersecting the ER and the flanking exons in RNA samples originating from pooled human frontal cortex samples (fig. S5). To gain more information about the transcript structure in which the unannotated ER was contained, we also performed Sanger sequencing from the first (ENSE00000970013) and last coding exons (ENSE00000970014) of SNCA to the unannotated ER. This implied a full transcript structure containing a minimum of 609 bp with the unannotated ER predicted to add an additional 63 amino acids (45% of existing transcript size). This example highlights the potential of incomplete annotation to both hinder genetic diagnosis and limit our understanding of a common complex neurological disease. Variants located in the unannotated ER linked to SNCA would not be captured using whole-exome sequencing and, if identified in whole-genome shotgun or through GWAS, would be misassigned as noncoding variants.
DISCUSSION
In this study, we use a pragmatic, conservative approach to identify unannotated transcription and putative unannotated exons of known genes. We find that although unannotated transcription is commonly detectable across all human tissues assayed, it is most frequently observed in the human brain. We find that the putative unannotated exons, which can be confidently assigned to a known gene using junction reads, have high replication rates (87% for intronic ERs as compared to 99% for annotated exons). Thus, our findings suggest the existence of previously unannotated exons that can be reliably detected from RNA-seq data and that might be expected to provide most insight into neurological disorders.
There are several reasons why these unannotated exons may have been previously missed from gene annotation and why they are most frequently detected in human brain. We believe that a key factor is the high cellular heterogeneity of human brain combined with the high cellular specificity of some transcripts. Lowly expressed, tissue-specific isoforms or those that are only transcribed within a cell type of proportionally low abundance may be missed from bulk RNA-seq datasets. Accordingly, we find that most of the putative unannotated exons that we detect have a restricted expression pattern across tissues and that the highest numbers are derived from human brain. Even within human brain, there appeared to be cellular biases influencing a gene’s likelihood of being reannotated. Among reannotated genes, we found a significant enrichment of genes with a cell-specific expression pattern, and this was most evident for genes specifically expressed by oligodendrocytes. We also note that the use of conservation measures in previous gene annotation pipelines may have biased exon and transcript discovery. Given that exons that are functionally important within humans might be expected to be enriched among genes of importance to human brain development, again this would predict higher rates of incomplete annotation within brain tissue. Consistent with this view, we find that, collectively, our unannotated exons are depleted for mutations within humans yet are not well conserved across other species (21). Furthermore, as we predicted, the unannotated exons identified that were connected to brain-specific genes showed the most significant depletion in mutations. Together, these findings not only explain the high yield of previously undiscovered annotation with human brain but also imply that it is likely to be disease relevant.
Given this evidence, we expect annotation to be of greatest relevance to complex and Mendelian forms of neurogenetic disease. With this in mind, it is noteworthy that 1831 OMIM genes were reannotated on the basis of our analysis of which 1111 were associated with a neurological phenotype. Some 251 of these OMIM genes had at least one associated unannotated exon with the potential to be protein coding. We highlight the example of SNCA, a gene implicated in Mendelian and complex Parkinson’s disease. We identify a previously unannotated, potentially protein coding exon of SNCA, which is validated experimentally and located in a region that is among the most depleted for mutations among humans but is poorly conserved. Furthermore, we find that genes known to cause Mendelian and complex neurodegenerative disorders are enriched among the set of genes that we reannotate. Thus, our analyses suggest that incomplete annotation is a substantial limiting factor in our understanding of both Mendelian and common complex neurological diseases.
Last, we release our results through a dedicated web resource, vizER (http://rytenlab.com/browser/app/vizER), which enables individual genes to be queried for incomplete annotation as well as the download of all the definitions of putative exons discovered in this study. We believe that vizER will be an important resource for clinical scientists in the diagnosis of Mendelian disorders, neuroscientists studying individual gene structures and functions, and, together with the emergence of larger long-read sequencing datasets, will accelerate transcript discovery particularly in human brain.
MATERIALS AND METHODS
OMIM data
Phenotype relationships and clinical synopses of all OMIM genes were downloaded using API through https://api.omim.org/ on 29 May 2018 (27). OMIM genes were filtered to exclude provisional, nondisease, and susceptibility phenotypes retaining 2898 unique genes that were confidently associated to 4034 Mendelian diseases. Phenotypic abnormality groups were linked to corresponding affected GTEx tissues through manual inspection of the HPO terms within each group by a medical specialist (17).
GTEx data
RNA-seq data in base-level coverage format for 7595 samples originating from 41 different GTEx tissues was downloaded using the R package recount version 1.4.6 (28). Cell lines, sex-specific tissues, and tissues with 10 samples or below were removed. Samples with large chromosomal deletions and duplications or large copy number variation previously associated with disease were filtered out (smafrze = “USE ME”). Coverage for all remaining samples was normalized to a target library size of 40 million 100-bp reads using the area under coverage value provided by recount2. For each tissue, base-level coverage was averaged across all samples to calculate the mean base-level coverage. GTEx junction read data, defined as reads with a noncontiguous gapped alignment to the genome, were downloaded using the recount2 resource and filtered to include only junction reads detected in at least 5% of samples for a given tissue and those that had available donor and acceptor splice sequences.
Optimizing the detection of transcription
Transcription was detected across 41 GTEx tissues using the package derfinder version 1.14.0 (18). The MCC, defined as the number of reads supporting each base above which bases were considered to be transcribed, and MRG, defined as the maximum number of bases between ERs below which adjacent ERs will be merged, were optimized. Optimization was performed using 156,674 nonoverlapping exons (defined by Ensembl v92) as the gold standard (4). Exon biotypes of all Ensembl v92 exons were compared to this set of nonoverlapping exons to ensure that we were not preferentially optimizing for one particular biotype (fig. S6). Nonoverlapping exons were selected as these definitions would be least likely to be influenced by ambiguous reads. For each tissue, we generated ERs using MCCs increasing from 1 to 10 in steps of 0.2 (46 cutoffs) and max gaps increasing from 0 to 100 in steps of 10 (11 MRGs) to produce a total of 506 unique transcriptomes. For each set of ERs, we found all ERs that intersected with nonoverlapping exons and then calculated the exon delta by summing the absolute difference between the start/stop positions of each ER and the overlapping exon (Fig. 1A). Situations in which a single ER overlapped with multiple exons were removed to avoid assigning the ER to an incorrect exon when calculating downstream optimization metrics. For each tissue, we selected the MCC and MRG, which minimized the difference between ER and “gold standard” exon definitions (median exon delta) and maximized the number of ERs that precisely matched the boundaries of exons (number of ERs with an exon delta equal to 0). All ERs that were <3 bp in width were removed as these were below the minimum size of a microexon (29).
Calculating the transcriptome size per annotation feature
ERs were classified with respect to the annotation feature (exon, intron, intergenic) with which they overlapped. A minimum of 1-bp overlap was required for an ER to be categorized as belonging to a given annotation feature. ERs overlapping multiple annotation features were labeled with a combination of each. This generated six distinct categories: “exon,” “exon, intron,” “exon, intergenic,” “exon, intergenic, intron,” “intergenic,” and “intron” (fig. S1A). ERs classified as exon, intergenic, intron were removed from all downstream analysis as these formed only 0.54% of all ERs and were presumed to be technical artifacts generated from regions of dense, overlapping gene expression. For each tissue, the length of all ERs within each annotation feature was summed generating the total Mb of ERs per annotation feature. Normalized variance of exonic, intronic, and intergenic ERs was calculated by dividing the SD of the total Mb of ERs across tissues by the mean total Mb of ERs for each annotation feature. To compare between brain and nonbrain tissues, the total Mb of intronic and intergenic ERs were first summed together to generate an overall measure of unannotated transcription abundance across brain and nonbrain tissues and then a two-sided Wilcoxon rank sum test was applied.
Annotating ERs with junction read data
Intronic and intergenic ERs were connected to known genes using reads, which we term junction reads, with a gapped alignment to the genome, presumed to be reads spanning exon-exon junctions (Fig. 2B). These exon-exon junctions are defined as noncontiguous reads that fall on the boundary between two exons of the same mRNA molecule; therefore, when aligned to the genome, these reads have a break in the middle indicating the splicing out of an intron. Junction read data were categorized into three groups: annotated junction reads, with both ends falling within known exons; partially annotated junction reads, with only one end falling within a known exon; and unannotated junction reads, with both ends within intron or intergenic regions. In this way, intron and intergenic ERs that overlapped with partially annotated junction reads were connected to known genes.
Validation of detected transcription
Transcription was validated across different versions of Ensembl and within an independent dataset. ERs that overlapped purely intronic or intergenic regions according to Ensembl v87, but fell within exons according to v92, were counted as unannotated transcription that was validated in later versions of Ensembl. Furthermore, ERs overlapping exonic regions in Ensembl v87 now classified as intronic or intergenic in v92 were measured to control for expected corrections in gene definitions. To assess whether the total Kb of validated unannotated ERs entering v92 annotation was greater than what would be expected by chance, we generated 10,000 random sets of length-matched regions for each tissue that were intronic or intergenic with respect to Ensembl. Using a one sample Wilcoxon test, we compared the total Kb of intronic and intergenic ERs entering annotation to the total Kb distribution of the randomized intronic and intergenic regions, respectively.
Validation within an independent dataset was performed using RNA-seq coverage data from 49 control frontal cortex (BA9) samples originally reported by Labadorf and colleagues and available via the recount R package version 1.4.6 (19, 28). ERs derived from the GTEx frontal cortex (BA9) data were requantified using this independent frontal cortex dataset, and those that had a mean coverage of at least 1.4 (the optimized MCC for the GTEx frontal cortex data) were counted as unannotated transcription that was validated.
Analyzing the conservation and constraint of unannotated ERs
Conservation scores in the form of phastCons7 and phastCons20 were downloaded from UCSC (30). Constraint scores generated from the genome-wide alignment of 7794 unrelated human genomes were downloaded as CDTS (21). The raw conservation and constraint scores were in bins of 1 and 10 bp, respectively; therefore, when annotating the corresponding positions of ERs, we aggregated each score as a mean across the entire genomic region of interest. To account for missing CDTS values, we calculated the coverage of each ER by dividing the number of bases annotated by the CDTS by the total length of the ER. For all downstream analysis, we filtered out ERs for which CDTS coverage was less than 80%.
To assess whether our unannotated ERs were more constrained or conserved than by expected by chance, we compared the phastCons7, phastCons20, and CDTS of unannotated ERs to 10,000 randomized length-matched sets of intronic and intergenic ERs for each tissue. For each of the 10,000 iterations, we first selected a random intronic or intergenic region that was larger than the respective ER and then selected a random segment along the randomized region that matched the length of the corresponding ER. The randomized regions were annotated with constraint scores and CDTS using the aforementioned method. The mean CDTS and phastCons scores of the unannotated ERs (split by annotation feature) were compared to the corresponding distribution of CDTS and phastCons scores of the randomized regions using a one sample, two-tailed t test. For easier interpretation when plotting, CDTS scores have been converted to their opposite sign; therefore, for both phastCons and CDTS, the higher the value, the greater the magnitude of conservation or constraint as shown in Fig. 4A.
Checking ER protein coding potential
Intronic and intergenic ERs that were intersected by two junction reads were extracted. The junction reads were used to determine the precise boundaries of the ER. The R package Biostrings version 2.46.0 was used to extract the DNA sequence corresponding to the ER genetic coordinates from the genome build hg38 (31). Since the translation frame was ambiguous without knowledge of the other exons that are part of the transcript that included the unannotated ER, we converted the DNA sequence to amino acid sequence for all three possible frames starting from the first, second, or third base. Any ER that had at least one frame that did not include a stop codon was considered to be potentially protein coding.
Gene properties influencing reannotation
All Ensembl v92 genes were marked with a 1 or a 0 depending on whether we detected a reannotation for that gene in the form of an ER connected to the gene using a junction read, with 1 representing a detected reannotation event. Details of gene length, biotype, and transcript count and whether the gene overlapped another gene were retrieved from the Ensembl v92 database. Brain specificity was assigned using the Finucane dataset and selecting the top 10% of brain-specific genes when compared to nonbrain tissues (32). Mean gene transcripts per million (TPM) was calculated by downloading tissue-specific TPM values from the GTEx portal and summarized by calculating the mean across all tissues. The list of OMIM genes (May 2018) was used to assign whether a gene was known to cause disease or not. We used a logistic regression to test whether different gene properties significantly influenced the variability of reannotation (formula = reannotated ~ brain-specific + mean TPM + overlapping gene + transcript count + gene biotype + gene length).
Sanger sequencing of unannotated junctions
Commercially purchased (Takara) frontal cortex and cerebellum RNA samples, isolated from individuals of European descent, were used for validation of unannotated junctions detected in SNCA and ERLIN1, respectively. Tissues were chosen to match the tissue in which the reannotation for each gene was detected. Reverse transcription was performed using 1 μg of RNA from each tissue and then converted to complementary DNA (cDNA) using the High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Applied Biosystems) and random primers as per manufacturer’s instructions. Primers were designed to span predicted exon-exon junctions using Primer-BLAST (National Center for Biotechnology Information) and ordered from Sigma-Aldrich (table S5). Polymerase chain reaction (PCR) was performed using FastStart PCR Master (Roche) and enzymatic cleanup of PCR products was performed using Exonuclease I (Thermo Fisher Scientific) and FastAP Thermosensitive Alkaline Phosphatase (Thermo Fisher Scientific). Sanger sequencing was performed using the BigDye Terminator Kit (Applied Biosystems), and sequences were viewed and exported using CodonCode Aligner (version 8.0.2). Sequences were blatted against the human genome (hg38) and alignment visually inspected for confirmation of validation.
Expression-weighted cell-type enrichment: Evaluating enrichment of theta-correlated genes
Expression-weighted cell-type enrichment (EWCE) was used to determine whether brain-specific genes (both reannotated and not reannotated) have higher expression within particular cell types than expected by chance (33). As our input, we used (i) neuronal and glial clusters of the CNS identified in the Linnarsson single-cell RNA-seq dataset (amounting to a subset of 114 of the original 265 clusters identified) and (ii) lists of genes split by whether or not they were reannotated, and if reannotated, by their overlap with Ensembl v92 annotation features (see table S6 for the full list of CNS neuronal clusters and genes used) (34). For each gene in the Linnarsson dataset, we estimated its cell-type specificity (the proportion of a gene’s total expression in one cell type compared to all cell types) using the “generate.celltype.data” function of the EWCE package. EWCE with the target list was run with 100,000 bootstrap replicates, which were sampled from a background list of genes that excluded all genes without a 1:1 mouse:human ortholog. We additionally controlled for transcript length and GC-content biases by selecting bootstrap lists with comparable properties to the target list. We performed the analysis with major cell-type classes (e.g., “astrocyte,” “microglia,” etc.). Data are displayed as SDs from the mean, and any values <0, which reflect a depletion of expression, are displayed as 0. P values were corrected for multiple testing using the Benjamini-Hochberg [false discovery rate (FDR)] method over all cell types and gene lists displayed.
Enrichment of reannotated genes for neurological disorder–associated genes
The STOPGAP database detailing all genes associated with 4684 GWASs was downloaded. To select which genes were associated to a GWAS, the “best gene” as determined by STOPGAP using functional evidence was used (25). The medical subject heading for each disease was used to further subgroup GWASs into four categories; neurodegenerative, neuropsychiatric, other neurological conditions, and the remaining as other (table S7). For each of the subgroups, we generated a contingency table, counting the number of genes that were reannotated or not in relation to whether they fell into that particular subgroup. For genes that were overlapping between GWASs, we classified a gene to be part of a subgroup if it was associated with at least one GWAS contained in that subgroup. A Fisher’s exact test was used to examine whether our reannotated gene list was significantly enriched for genes from any of the subgroups. FDR was used to correct for multiple testing.
Supplementary Material
Acknowledgments
Funding: S.G. was supported through the award of an Alzheimer’s Research UK PhD fellowship. R.H.R. was supported through the award of a Leonard Wolfson Doctoral Training Fellowship in Neurodegeneration. J.H. and M.R. were supported by the UK Medical Research Council (MRC), with J.H. supported by a grant (MR/N026004/) and M.R. through the award of a Tenure Track Clinician Scientist Fellowship (MR/N008324/1). J.H. was also supported by the UK Dementia Research Institute, The Wellcome Trust (202903/Z/16/Z), the Dolby Family Fund, and the NIHR. A.E.J. and L.C.-T. were supported by the R21MH109956 grant (https://projectreporter.nih.gov/project_info_description.cfm?aid=9093092). Author contributions: D.Z., S.G., and M.R. conceived and designed the study. D.Z. analyzed the data, generated figures, and together with M.R. wrote the first draft of the manuscript. R.H.R. performed analysis and generated figures for the cell type–specific section. S.G.-R. and J.A.B. developed and deployed the vizER online platform. Sanger sequence validation was performed by B.C. and W.L. T.C. helped manually associate OMIM phenotypes to GTEx tissues. S.G., L.C.-T., J.A.B., K.D., A.P., and M.R. helped guide and troubleshoot analyses. L.C.-T. and A.E.J. helped with the use of the recount2 data. D.Z., S.G., R.H.R., J.H., L.C.-T., and M.R. contributed to the critical analysis of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Code used to perform analyses in this study is publicly available via the https://github.com/dzhang32/ER_paper_2019_supp_code. The publicly available package annotatER (https://github.com/SebGuelfi/annotatER) has been used to combine junction data with ERs. The definitions of all ERs studied can be downloaded via http://rytenlab.com/browser/app/vizER. RNA sequencing data from GTEx are available via recount2: https://jhubiostatistics.shinyapps.io/recount/.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/24/eaay8299/DC1
REFERENCES AND NOTES
- 1.Thierry-Mieg D., Thierry-Mieg J., AceView: A comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. 7, S12 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harrow J., Frankish A., Gonzalez J. M., Tapanari E., Diekhans M., Kokocinski F., Aken B. L., Barrell D., Zadissa A., Searle S., Barnes I., Bignell A., Boychenko V., Hunt T., Kay M., Mukherjee G., Rajan J., Despacio-Reyes G., Saunders G., Steward C., Harte R., Lin M., Howald C., Tanzer A., Derrien T., Chrast J., Walters N., Balasubramanian S., Pei B., Tress M., Rodriguez J. M., Ezkurdia I., van Baren J., Brent M., Haussler D., Kellis M., Valencia A., Reymond A., Gerstein M., Guigo R., Hubbard T. J., GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Leary N. A., Wright M. W., Brister J. R., Ciufo S., Haddad D., Veigh R. M., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D., Astashyn A., Badretdin A., Bao Y., Blinkova O., Brover V., Chetvernin V., Choi J., Cox E., Ermolaeva O., Farrell C. M., Goldfarb T., Gupta T., Haft D., Hatcher E., Hlavina W., Joardar V. S., Kodali V. K., Li W., Maglott D., Masterson P., Mc Garvey K. M., Murphy M. R., O’Neill K., Pujar S., Rangwala S. H., Rausch D., Riddick L. D., Schoch C., Shkeda A., Storz S. S., Sun H., Thibaud-Nissen F., Tolstoy I., Tully R. E., Vatsan A. R., Wallin C., Webb D., Wu W., Landrum M. J., Kimchi A., Tatusova T., Cuccio M. D., Kitts P., Murphy T. D., Pruitt K. D., Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zerbino D. R., Achuthan P., Akanni W., Amode M. R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C. G., Gil L., Gordon L., Haggerty L., Haskell E., Hourlier T., Izuogu O. G., Janacek S. H., Juettemann T., To J. K., Laird M. R., Lavidas I., Liu Z., Loveland J. E., Maurel T., McLaren W., Moore B., Mudge J., Murphy D. N., Newman V., Nuhn M., Ogeh D., Ong C. K., Parker A., Patricio M., Riat H. S., Schuilenburg H., Sheppard D., Sparrow H., Taylor K., Thormann A., Vullo A., Walts B., Zadissa A., Frankish A., Hunt S. E., Kostadima M., Langridge N., Martin F. J., Muffato M., Perry E., Ruffier M., Staines D. M., Trevanion S. J., Aken B. L., Cunningham F., Yates A., Flicek P., Ensembl 2018. Nucleic Acids Res. 46, D754–D761 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen G., Wang C., Shi L., Qu X., Chen J., Yang J., Shi C., Chen L., Zhou P., Ning B., Tong W., Shi T., Incorporating the human gene annotations in different databases significantly improved transcriptomic and genetic analyses. RNA 19, 479–489 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCarthy D. J., Humburg P., Kanapin A., Rivas M. A., Gaulton K.; The WGS500 Consortium, Cazier J.-B., Donnelly P., Choice of transcripts and software has a large effect on variant annotation. Genome Med. 6, 26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sibley C. R., Emmett W., Blazquez L., Faro A., Haberman N., Briese M., Trabzuni D., Ryten M., Weale M. E., Hardy J., Modic M., Curk T., Wilson S. W., Plagnol V., Ule J., Recursive splicing in long vertebrate genes. Nature 521, 371–375 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sibley C. R., Blazquez L., Ule J., Lessons from non-canonical splicing. Nat. Rev. Genet. 17, 407–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yeo G., Holste D., Kreiman G., Burge C. B., Variation in alternative splicing across human tissues. Genome Biol. 5, R74 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y. E., Landback P., Vibranovski M., Long M., New genes expressed in human brains: Implications for annotating evolving genomes. Bioessays 34, 982–991 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Jaffe A. E., Shin J., Collado-Torres L., Leek J. T., Tao R., Li C., Gao Y., Jia Y., Maher B. J., Hyde T. M., Kleinman J. E., Weinberger D. R., Developmental regulation of human cortex transcription and its clinical relevance at single base resolution. Nat. Neurosci. 18, 154–161 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pertea M., Shumate A., Pertea G., Varabyou A., Chang Y.-C., Madugundu A. K., Pandey A., Salzberg S. L., CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol., 208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steijger T., Abril J. F., Engström P. G., Kokocinski F.; The RGASP Consortium, Hubbard T. J., Guigó R., Harrow J., Bertone P., Assessment of transcript reconstruction methods for RNA-seq. Nat. Methods 10, 1177–1184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jungreis I., Tress M. L., Mudge J., Sisu C., Hunt T., Johnson R., Uszczynska-Ratajczak B., Lagarde J., Wright J., Muir P., Gerstein M., Guigo R., Kellis M., Frankish A., Flicek P.; The GENCODE Consortium , Nearly all new protein-coding predictions in the CHESS database are not protein-coding. bioRxiv 2018, 360602 (2018). [Google Scholar]
- 15.ENCODE Project Consortium , Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao S., Zhang Y., Gamini R., Zhang B., von Schack D., Evaluation of two main RNA-seq approaches for gene quantification in clinical RNA sequencing: polyA+ selection versus rRNA depletion. Sci. Rep. 8, 4781 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.GTEx Consortium , The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 348, 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collado-Torres L., Nellore A., Frazee A. C., Wilks C., Love M. I., Langmead B., Irizarry R. A., Leek J. T., Jaffe A. E., Flexible expressed region analysis for RNA-seq with derfinder. Nucleic Acids Res. 45, e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Labadorf A., Hoss A. G., Lagomarsino V., Latourelle J. C., Hadzi T. C., Bregu J., MacDonald M. E., Gusella J. F., Chen J.-F., Akbarian S., Weng Z., Myers R. H., RNA sequence analysis of human huntington disease brain reveals an extensive increase in inflammatory and developmental gene expression. PLOS ONE 10, e0143563 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doolittle W. F., We simply cannot go on being so vague about ‘function’. Genome Biol. 19, 223 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Iulio J., Bartha I., Wong E. H. M., Yu H.-C., Lavrenko V., Yang D., Jung I., Hicks M. A., Shah N., Kirkness E. F., Fabani M. M., Biggs W. H., Ren B., Venter J. C., Telenti A., The human noncoding genome defined by genetic diversity. Nat. Genet. 50, 333–337 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Ettle B., Schlachetzki J. C. M., Winkler J., Oligodendroglia and myelin in neurodegenerative diseases: More than just bystanders? Mol. Neurobiol. 53, 3046–3062 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calderon D., Bhaskar A., Knowles D. A., Golan D., Raj T., Fu A. Q., Pritchard J. K., Inferring relevant cell types for complex traits by using single-cell gene expression. Am. J. Hum. Genet. 101, 686–699 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bryois J., Skene N. G., Hansen T. F., Kogelman L., Watson H. J., Liu Z.; Eating Disorders Working Group of the Psychiatric Genomics Consortium; International Headache Genetics Consortium; 23andMe Research Team, Brueggeman L., Breen G., Bulik C. M., Arenas E., Hjerling-Leffler J., Sullivan P. F., Genetic identification of cell types underlying brain complex traits yields novel insights into the etiology of Parkinson’s Disease. bioRxiv 2019, 528463 (2019). [Google Scholar]
- 25.Shen J., Song K., Slater A. J., Ferrero E., Nelson M. R., STOPGAP: A database for systematic target opportunity assessment by genetic association predictions. Bioinformatics 33, 2784–2786 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Novarino G., Fenstermaker A. G., Zaki M. S., Hofree M., Silhavy J. L., Heiberg A. D., Abdellateef M., Rosti B., Scott E., Mansour L., Masri A., Kayserili H., Al-Aama J. Y., Abdel-Salam G. M. H., Karminejad A., Kara M., Kara B., Bozorgmehri B., Ben-Omran T., Mojahedi F., Mahmoud I. G. E. D., Bouslam N., Bouhouche A., Benomar A., Hanein S., Raymond L., Forlani S., Mascaro M., Selim L., Shehata N., Al-Allawi N., Bindu P. S., Azam M., Gunel M., Caglayan A., Bilguvar K., Tolun A., Issa M. Y., Schroth J., Spencer E. G., Rosti R. O., Akizu N., Vaux K. K., Johansen A., Koh A. A., Megahed H., Durr A., Brice A., Stevanin G., Gabriel S. B., Ideker T., Gleeson J. G., Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 343, 506–511 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamosh A., Scott A. F., Amberger J., Valle D., McKusick V. A., Online Mendelian inheritance in man (OMIM). Hum. Mutat. 15, 57–61 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Collado-Torres L., Nellore A., Kammers K., Ellis S. E., Taub M. A., Hansen K. D., Jaffe A. E., Langmead B., Leek J. T., Reproducible RNA-seq analysis using recount2. Nat. Biotechnol. 35, 319–321 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irimia M., Weatheritt R. J., Ellis J. D., Parikshak N. N., Gonatopoulos-Pournatzis T., Babor M., Quesnel-Vallières M., Tapial J., Raj B., O’Hanlon D., Barrios-Rodiles M., Sternberg M. J. E., Cordes S. P., Roth F. P., Wrana J. L., Geschwind D. H., Blencowe B. J., A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 159, 1511–1523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas J. W., Touchman J. W., Blakesley R. W., Bouffard G. G., Beckstrom-Sternberg S. M., Margulies E. H., Blanchette M., Siepel A. C., Thomas P. J., McDowell J. C., Maskeri B., Hansen N. F., Schwartz M. S., Weber R. J., Kent W. J., Karolchik D., Bruen T. C., Bevan R., Cutler D. J., Schwartz S., Elnitski L., Idol J. R., Prasad A. B., Lee-Lin S. Q., Maduro V. V. B., Summers T. J., Portnoy M. E., Dietrich N. L., Akhter N., Ayele K., Benjamin B., Cariaga K., Brinkley C. P., Brooks S. Y., Granite S., Guan X., Gupta J., Haghighi P., Ho S. L., Huang M. C., Karlins E., Laric P. L., Legaspi R., Lim M. J., Maduro Q. L., Masiello C. A., Mastrian S. D., McCloskey J. C., Pearson R., Stantripop S., Tiongson E. E., Tran J. T., Tsurgeon C., Vogt J. L., Walker M. A., Wetherby K. D., Wiggins L. S., Young A. C., Zhang L. H., Osoegawa K., Zhu B., Zhao B., Shu C. L., De Jong P. J., Lawrence C. E., Smit A. F., Chakravarti A., Haussler D., Green P., Miller W., Green E. D., Comparative analyses of multi-species sequences from targeted genomic regions. Nature 424, 788–793 (2003). [DOI] [PubMed] [Google Scholar]
- 31.H. Pagès, P. Aboyoun, R. Gentleman, S. DebRoy, Biostrings: Efficient manipulation of biological strings. R Packag. version 2.46.0 (2017).
- 32.Finucane H. K., Reshef Y. A., Anttila V., Slowikowski K., Gusev A., Byrnes A., Gazal S., Loh P.-R., Lareau C., Shoresh N., Genovese G., Saunders A., Macosko E., Pollack S., Perry J. R. B., Buenrostro J. D., Bernstein B. E., Raychaudhuri S., McCarroll S., Neale B. M., Price A. L., Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 50, 621–629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skene N. G., Grant S. G. N., Identification of vulnerable cell types in major brain disorders using single cell transcriptomes and expression weighted cell type enrichment. Front. Neurosci. 10, 16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeisel A., Hochgerner H., Lönnerberg P., Johnsson A., Memic F., van der Zwan J., Häring M., Braun E., Borm L. E., La Manno G., Codeluppi S., Furlan A., Lee K., Skene N., Harris K. D., Hjerling-Leffler J., Arenas E., Ernfors P., Marklund U., Linnarsson S., Molecular architecture of the mouse nervous system. Cell 174, 999–1014.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/24/eaay8299/DC1