

Translesion synthesis is a cancer adaptation that suppresses replication stress–induced gaps that limit fitness.
Abstract
The replication stress response, which serves as an anticancer barrier, is activated not only by DNA damage and replication obstacles but also oncogenes, thus obscuring how cancer evolves. Here, we identify that oncogene expression, similar to other replication stress–inducing agents, induces single-stranded DNA (ssDNA) gaps that reduce cell fitness. DNA fiber analysis and electron microscopy reveal that activation of translesion synthesis (TLS) polymerases restricts replication fork slowing, reversal, and fork degradation without inducing replication gaps despite the continuation of replication during stress. Consistent with gap suppression (GS) being fundamental to cancer, we demonstrate that a small-molecule inhibitor targeting the TLS factor REV1 not only disrupts DNA replication and cancer cell fitness but also synergizes with gap-inducing therapies such as inhibitors of ATR or Wee1. Our work illuminates that GS during replication is critical for cancer cell fitness and therefore a targetable vulnerability.
INTRODUCTION
The replication stress response is activated in response to DNA lesions or intrinsic replication fork barriers and is critical to ensure the accurate transmission of genetic material to daughter cells. In response to sustained replication stress, replication forks slow and remodel into reversed fork structures. This local fork response is thought to confer a signal to arrest DNA replication throughout the cell (1, 2). Cells either undergo replication stress–associated senescence or engage in DNA repair or other transactions that help restart stalled DNA replication forks. The replication stress response is also induced by oncogenes, making it a critical barrier to cancer (3–8). However, the oncogene-inducing lesion that limits fitness and is eventually overcome in cancer remains unknown. Confounding this understanding, oncogenes have been shown to both accelerate and slow replication (9–11). These disparate findings could reflect distinct experimental systems or kinetics of analysis (3, 5, 7). Thus, it will be critical to further address how cellular replication responds in the immediate aftermath of replication stress induced by drugs or oncogene expression.
A pathway known for tolerating DNA damage that interferes with replicative polymerases is translesion synthesis (TLS) (12). TLS polymerases are recruited to bypass replication blocking lesions when replicative polymerases are not functional or physically blocked (13). TLS polymerases have been implicated in bypassing DNA damage induced by chemotherapies such as cisplatin, providing a reason for TLS inhibition in cancer therapy (14–16). While most appreciated in bypass of DNA adducts, emerging evidence reveals that TLS polymerases are activated in the absence of DNA damage and are required for replication of DNA structures enhanced by DNA replication stress such as G-quadruplexes (G4s), which limit replication progression and promote single-stranded DNA (ssDNA) gap formation (12, 17–19). Conceivably, ssDNA gaps underlie the mechanism of action of genotoxic therapies, and gap suppression (GS) is the key factor that confers resistance (20, 21).
Here, we propose that the primary function of TLS polymerases is GS, to confer chemoresistance and overcome oncogene-induced replication stress (8). Specifically, we show that TLS polymerases maintain continuous replication to limit ssDNA gaps induced by replication stress, oncogenes, or chemotherapy. Moreover, we identify several cancer cell lines that are dependent on TLS polymerases for replication and fitness, suggesting that a TLS polymerase rewiring is essential for cancer initiation and/or evolution. A small-molecule inhibitor targeting the C-terminal domain of the TLS factor REV1 (REV1-CT), which inhibits its interaction with other TLS polymerases, not only disrupts DNA replication and cancer cell fitness but also synergizes with gap-inducing therapies. This work implicates that GS is the fundamental mechanism of overcoming the anticancer barrier and that TLS inhibition is critical for therapy response.
RESULTS
TLS polymerases limit replication fork slowing during stress
To test the hypothesis that TLS polymerases avoid the replication stress response (Fig. 1A), we sought to study replication fork dynamics using DNA fiber spreading analysis in cells with enhanced TLS polymerase activity (hereafter called pro-TLS cells). TLS polymerases are favored when the DNA helicase FANCJ is disrupted for either its DNA damage–induced acetylation or BRCA1 binding (22, 23). Here, we used the FANCJ-BRCA1 interaction defective mutant, FANCJS990A, that promotes TLS polymerase focal accumulation and dependence on TLS factors for chemotherapy resistance (23). We complemented FANCJ knockout (K/O) U2OS osteosarcoma cancer cells and FANCJ-null FA-J patient immortalized fibroblast cells with FANCJS990A (pro-TLS), FANCJWT (control), or vector (V). As expected, we found that both FANCJWT and FANCJS990A elevated mitomycin C (MMC) resistance as compared with vector (Fig. 1B and fig. S1, A and B) (23, 24). To track the actively replicating fork, cells were labeled with sequential pulses of 5-iodo-2′-deoxyuridine (IdU) and 5-chloro-2′-deoxyuridine (CldU), and the DNA tract lengths were measured. DNA fiber spreading analysis revealed that under unchallenged conditions, vector-, control-, or pro-TLS U2OS– or FA-J–complemented cell lines had similar tract lengths, indicating that TLS polymerase induction did not affect normal replication progression (Fig. 1C and fig. S1C). However, when CldU labeling was coincident with 0.5 mM hydroxyurea (HU), a dose that does not completely deplete nucleotide pools but activates replication stress (25), control cells had an expected reduction in tract lengths as compared with untreated control, suggesting replication fork slowing during stress (Fig. 1C and fig. S1D). Similarly, following treatment with ultraviolet (UV) radiation, control cells had a significant shorter tract length correlative of replication fork slowing (Fig. 1C and fig. S1D). Notably, the DNA tracts in the pro-TLS U2OS or FA-J cells failed to fully shorten both during HU and after UV and were significantly longer than the control (Fig. 1C and fig. S1D), indicating changes in replication fork progression were neither replication stress nor cell type specific. Further verifying that TLS polymerases contribute to the unrestrained replication during stress, tracts fully shortened when the pro-TLS cells were treated with the TLS inhibitor (TLSi) that targets the C-terminal domain of REV1 (REV1-CT) and prevents protein-protein interactions (PPIs) between this domain and the REV1 interacting regions (RIRs) present in multiple other TLS polymerases, including polη, polι, polκ, and polζ. Disruption of the REV1 scaffolding function effectively inhibits the function of these TLS polymerases and disrupts the TLS pathway in a manner specific to REV1 (Fig. 1C) (26, 27). In contrast, the TLSi alone did not alter the length of DNA tracts in unchallenged conditions (fig. S1C).
Fig. 1. TLS polymerases limit replication fork slowing, reversal, degradation, and gap induction.
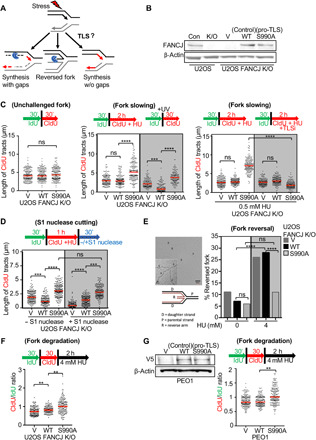
(A) Model to test whether TLS promotes unrestrained replication without gaps upon stress. (B) Western blot analysis with the indicated antibodies (Abs) of whole-cell extract (WCE) from U2OS control and FANCJ K/O cells complemented with vector (V), wild type (FANCJWT), and the FANCJ-BRCA1 binding–deficient mutant (FANCJS990A). (C and D) Schematic and quantification of CldU tract length under unchallenged condition or following coincubation with 0.5 mM HU or 2 J/m2 UV or 20 μM TLSi or after S1 nuclease treatment. (E) Schematic representation of reversed fork structure and quantification following a 2-hour HU treatment. Number of reversed forks analyzed; untreated, ~70 (V), 94 (WT), and 84 (S990A); HU treated, ~167 (V), 130 (WT), and 176 (S990A). (F) Schematic and quantification of CldU/IdU ratio following 4 mM HU. (G) Western blot analysis with the indicated Abs of WCE from PEO1 cells expressing V5 tagged—(V), (FANCJWT), and (FANCJS990A). Schematic and quantification of CldU/IdU ratio following HU treatment. Each dot represents 1 fiber and at least 200 fibers quantified from two independent experiments. Bars represent the means ± SD. Statistical analysis was performed according to two-tailed Mann-Whitney test. All P values are described in the “Statistical methods.”
The longer tracts and the failure to slow replication in the pro-TLS cells could stem from a more rapid restart of stalled forks, repriming and/or the firing of new origins upon stress. To address this question, we labeled cells with IdU, arrested replication with high-dose HU (4 mM), and following release from HU, labeled with CldU. Dual-labeled tracts were greatly diminished in the vector FA-J cells, and new origins were aberrantly activated (fig. S1E), corroborating the role of FANCJ in replication restart and regulating new origin firing (28, 29). In contrast, FA-J cells complemented with either wild type (WT) or S990A had comparable levels of dual-labeled replication tracts consistent with control and pro-TLS cells, promoting replication restart following fork arrest (fig. S1E). Together, these findings suggest that TLS polymerases limit fork slowing, but upon a full replication arrest, TLS polymerases do not alter replication restart or dormant origin firing.
TLS polymerases promote replication fork progression during stress without ssDNA gap induction
Failure to slow replication during stress is associated with fork degradation, genomic instability, and low fitness (1, 24, 30). We reasoned that TLS polymerase–dependent replication during stress could avoid this outcome by suppressing ssDNA gaps, a replication stress–associated lesion (24, 31). To test this hypothesis, DNA fiber assays were performed in the presence or absence of S1 nuclease treatment. S1 nuclease degrades DNA fibers with ssDNA gaps that occur within the labeled replication tracts, not easily observable in the standard DNA fiber assay (32). We observed that pro-TLS cells generated significantly longer tracts that were maintained even after S1 nuclease treatment (Fig. 1D and fig. S1F). These findings further indicate that the failure to slow replication during stress is not associated with repriming or new origin firing. Moreover, these findings indicate that in response to stress, TLS polymerases not only disrupt fork slowing, but replication continues without generating ssDNA gaps. Further validating TLS polymerases as the mechanism of fork elongation during stress, replication tract lengthening does not occur during stress in cells expressing the TLS inactivating mutant FANCJS990A+K52R (pro-TLS + helicase dead) as found for the FANCJK52R (helicase dead) mutant (fig. S1F) (23, 33). Notably, in response to the UV or HU (1 hour) fork slowing assay, replication tracts in FANCJ-deficient cells are longer than WT, and the longer HU tracts shorten with S1 consistent with ssDNA gaps due to either repriming and/or new origin firing (Fig. 1D and fig. S1F). However, in the HU (2 hours) fork slowing assay, replication tracts in FANCJ-deficient cells and WT are similar (Fig. 1C), suggesting that longer-gapped replication tracts in the FANCJ-deficient cells eventually undergo fork degradation.
TLS polymerases avoid fork reversal and degradation
We predicted that continued replication during stress would limit replication fork reversal. To directly visualize and investigate the frequency of reversed fork intermediates in cells with or without TLS induction, we analyzed the fine replication fork architecture by using psoralen cross-linking coupled to electron microscopy (EM). Following treatment with 4 mM HU to ensure nucleotide depletion and replication fork stalling, we found a significant accumulation of the reversed fork structures in the control cells (~28% reversed forks), whereas pro-TLS cells exhibited significantly lower frequency (~11%) of fork reversal events (Fig. 1E). Collectively, these results suggest that TLS restricts fork reversal.
We reasoned that a reduction in fork reversal by TLS would in turn prevent fork degradation and enhance fork protection. To access fork degradation, we analyzed the ratio of CldU to IdU tract lengths following sequential pulses with IdU and CldU followed by HU treatment (Fig. 1F) (34). Compared with control, the pro-TLS cells had a modestly enhanced CldU-to-IdU tract length ratio, consistent with less fork degradation upon stress (Fig. 1F and fig. S1G). Moreover, pro-TLS cells also maintained fork integrity following a prolonged period of replication stress. In addition, in fork degradation–prone BRCA2-deficient PEO1 ovarian cancer cells (34–38), ectopic expression of the pro-TLS FANCJ mutant not only promoted unrestrained replication and enhanced fork protection but also conferred cisplatin resistance as compared with control (Fig. 1G and fig. S1H). Together, these findings indicate that TLS provides fork protection through suppression of fork remodeling similar to the loss of fork remodelers (36, 38, 39).
TLS polymerases disrupt the global replication stress response without ssDNA induction
During stress, fork slowing and remodeling promote the global arrest of DNA replication (Fig. 1A) (2). Thus, TLS polymerases counteracting fork slowing in response to stress could also limit the global arrest of DNA replication. To test this idea, an asynchronous population of the control or the pro-TLS U2OS cell lines was either left untreated or treated with varying doses of HU while also being labeled with 5-ethynyl-2′-deoxyuridine (EdU) to track active replication. Replication was quantified by scoring the number of EdU-positive cells. Under unperturbed conditions, the number of EdU-positive cells was similar between the control and pro-TLS U2OS cells, further suggesting that TLS does not impact the global replication in unchallenged conditions (Fig. 2A). However, upon HU treatment, we observed that the number of EdU-positive cells was significantly reduced in the control U2OS cells, mimicking the replication fork slowing as studied in the DNA fiber assay (Fig. 2A). In contrast, the pro-TLS U2OS cells continued to incorporate EdU not only in low-dose HU but also following UV treatment (fig. S2C), further validating that TLS polymerases promote replication during stress.
Fig. 2. TLS polymerases promote global replication during stress and suppress ssDNA gaps.
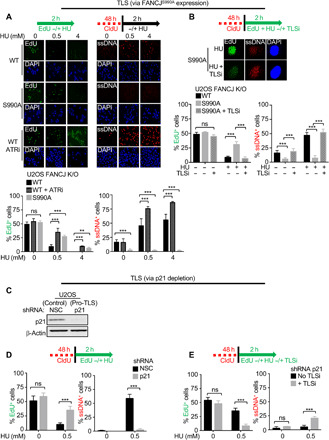
(A) Schematic, representative images, and quantification of EdU- and ssDNA-positive cells. For EdU assay, cells were labeled with EdU alone either for 30 min or for 2 hours with varying doses of HU −/+ 5 μM ATRi. For ssDNA detection, cells were first labeled with CldU for 48 hours followed by a 2-hour HU treatment. Staining for EdU was performed by ClickiT chemistry, and for ssDNA using CldU-specific Ab under nondenaturing conditions. Percent EdU- and ssDNA-positive cells were quantified from over 300 cells counted from multiple fields. (B) Schematic, representative images (63×), and quantification of EdU- and ssDNA-positive cells following initial labeling with CldU for 48 hours followed by EdU treatment with or without 0.5 mM HU −/+ 20 μM TLSi. (C) Western blot analysis with the indicated Abs of WCE from U2OS cells expressing shRNA against NSC or p21. (D and E) Schematic and quantification of EdU- and ssDNA-positive cells following initial labeling with CldU for 48 hours followed by EdU treatment with or without 0.5 mM HU −/+ 20 μM TLSi. Bars represent the means ± SD. Statistical analysis was performed according to two-tailed Mann-Whitney test. All P values are described in the “Statistical methods.”
Similar to our findings with TLS polymerase activity, inhibition of the checkpoint kinase ATR enables replication during stress (Fig. 2A and fig. S2G) (2). Given that ATR inhibition is toxic to cells (40), we considered that a key difference between TLS activation and ATR inhibition was ssDNA gap induction. To test whether replication during stress differed by ssDNA gap induction, we performed nondenaturing immunofluorescence following incorporation of CldU to visualize ssDNA regions that are positive for anti-CldU staining. In the presence of HU, we observed that the ATR inhibitor (ATRi) leads to widespread global ssDNA gaps, whereas by comparison to control, the pro-TLS cells appeared resistant to gap formation (Fig. 2A and fig. S2G). Collectively, these findings indicate that TLS, unlike ATR inhibition, promotes replication during stress without genome wide ssDNA gap induction.
To verify that unrestrained replication without ssDNA gaps is a distinct feature of TLS polymerases and not limited to a pro-TLS phenotype driven by FANCJ, we depleted the negative regulator of TLS, p21 (41), in the parental U2OS cell line. To confirm TLS polymerase induction, which is associated with TLS polymerase foci formation (13, 42), cells were transfected with either enhanced green fluorescent protein (eGFP)–polη or eGFP-REV1, and foci formation was evaluated in untreated cells or following either UV or MMC treatment. In response to either stress, we confirmed that polη and Rev1 foci formation were enriched not only in the pro-TLS U2OS FANCJS990A cells (23) but also in the p21-depleted cells (fig. S2, A and B). Furthermore, depletion of p21 led to continuous “ungapped” replication during HU treatment as compared with the control (Fig. 2, C and D, and fig. S2D). In either pro-TLS system, FANCJS990A, or p21 depletion, treatment with TLSi disrupted EdU incorporation and induced ssDNA gaps (Fig. 2, B and E, and fig. S2E). Notably, in the pro-TLS cells under unchallenged conditions or in control cells, the TLSi alone did not interfere with EdU incorporation nor induce ssDNA gaps (Fig. 2B and fig. S2F), suggesting that the addition of stress was a prerequisite to induce TLS-dependent replication. Collectively, these findings indicate that TLS polymerases are a robust mechanism for continuation of replication during stress without ssDNA gap formation.
Oncogene expression induces ssDNA gaps and reduces cell fitness
Oncogene activation is associated with replication stress that serves as a barrier to cancer (3–9, 43). Given our findings, we sought to test the hypothesis that oncogene-induced stress can be offset by TLS. To test this hypothesis, we generated cells stably infected with either empty vector or CCNE1 vector that encodes the oncogene cyclin E1 in a doxycycline inducible manner (DOX-ON system) (Fig. 3A). As previously reported, we observed that cyclin E1 expression did not alter EdU incorporation (Fig. 3B and fig. S3A) (10, 11, 44). However, there was a significant induction of genome-wide ssDNA, as well as loss of clonogenic capacity, both of which were suppressed by TLS polymerase activation as achieved by FANCJS990A mutant (Fig. 3, B and C, and fig. S3A). Similar findings were observed in another well-established U2OS cyclin E1 inducible system (TET-OFF system) (44). Upon cyclin E1 overexpression (OE), as compared with the normal ectopic levels (NE), EdU incorporation was unhindered, but ssDNA was induced and clonogenic survival was reduced unless counteracted by TLS achieved by p21 depletion (Fig. 3, D to F, and fig. S3C). Furthermore, coincubation with TLSi restored cyclin E1–induced ssDNA gaps and reduced the clonogenic capacity of the pro-TLS cells, but had no effect on colonies without any oncogene induction (Fig. 3, C and F, and fig. S3B). Collectively, these findings indicate that TLS polymerases buffer oncogene-induced stress to facilitate continuous replication without ssDNA gap induction and promote survival.
Fig. 3. TLS polymerases overcome oncogene-induced stress response and promotes cell fitness.
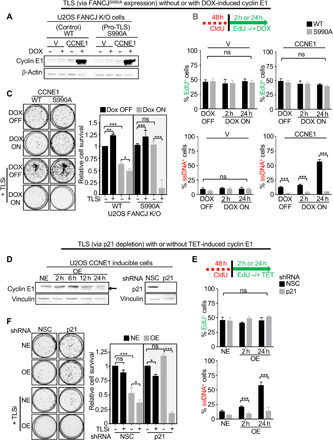
(A) Western blot analysis with the indicated Abs of WCE from U2OS FANCJWT or FANCJS990A cells with pInducer vector(V) or CCNE1(cyclin E1). Cyclin E1 expression induced with doxycycline (DOX) (1 μg/ml) for 24 hours. (B) Schematic and quantification of EdU- and ssDNA-positive cells. EdU and ssDNA staining were performed as described in Fig. 2. (C) Representative images and quantification of colony formation following indicated treatments. (D) Western blot analysis with the indicated Abs of WCE from U2OS cyclin E1 inducible cell line expressing shRNA against NSC or p21. NE, normal level of cyclin E1 (Tet ON); OE, cyclin E1 overexpression (Tet OFF). Arrow indicates cyclin E1 overexpression. (E) Schematic and quantification of EdU- and ssDNA-positive cells. (F) Representative images and quantification of the colony formation in NSC- versus p21-depleted U2OS cyclin E1 NE or OE cells following indicated treatments. Experiments were performed in biological triplicate. Bars represent the means ± SD. Statistical analysis was performed according to two-tailed Mann-Whitney test. All P values are described in the “Statistical methods.”
Cancer cells show TLS polymerase dependence
If TLS polymerases overcome the loss of fitness due to oncogene expression, then cancer evolution could favor TLS polymerase activation. To identify a possible pro-TLS rewiring in cancer, we tested the ability of distinct cancer cell types to replicate during stress. We found that replication robustly continued in the breast cancer cell line MCF7, the endometrial cancer cell line HeLa, the colon cancer cell line HCT15, the lung cancer cell lines A549 and NCI-H522, and the leukemia cell line MOLT-4 following HU treatment (Fig. 4A and fig. S4, A and B). Moreover, the TLSi curtailed replication during stress and induced ssDNA gaps in these cell lines (Fig. 4A and fig. S4B). Notably, MCF7 cells also showed a flattened morphology suggestive of senescence (Fig. 4A). HeLa cells halted replication and induced ssDNA even in the absence of HU (Fig. 4A), consistent with a pro-TLS phenotype even in unchallenged conditions. In contrast, similar to U2OS cells, the immortalized retinal pigment epithelial (RPE) cell line ceased to replicate in low-dose HU (fig. S4A).
Fig. 4. TLS polymerases subvert the replication stress response to promote cancer fitness.
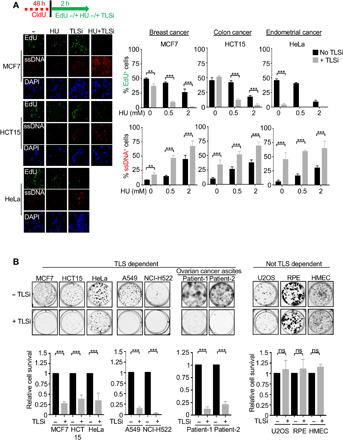
(A) Schematic, representative images, and quantification of EdU- and ssDNA-positive cells following initial labeling with CldU for 48 hours followed by treatment with either EdU alone for 30 min or for 2 hours with or without 0.5 mM HU −/+ 20 μM TLSi. EdU and ssDNA staining was performed as described in Fig. 2. (B) Representative images and quantification of the colony formation with and without the continuous presence of 20 μM TLSi across the different cell lines. Experiments were performed in biological triplicate. Bars represent the means ± SD. Statistical analysis was performed according to two-tailed Mann-Whitney test. All P values are described in the “Statistical methods.”
Consistent with a TLS polymerase rewiring, cancer cell lines with TLS polymerase–dependent replication lost clonogenic capacity upon treatment with the TLSi (Fig. 4B), whereas the TLSi did not affect the colony-forming capacity of cells not dependent on TLS polymerases, such as RPE, U2OS, and the human mammary epithelial cell line HMEC (Fig. 4B). Moreover, early passage ovarian cancer ascites cells from two different patients were also highly sensitive to the TLSi treatment (Fig. 4B). In addition, TLS polymerase–dependent Hela cancer cells showed dependence on the TLS factor FANCJ for replication and cellular fitness. Namely, FANCJ K/O in HeLa cells exhibited significantly reduced DNA replication and impaired clonogenic capacity (fig. S5, A and B). As compared with the control HeLa cells, p21 levels were also observed to be elevated in the FANCJ K/O HeLa cells (fig. S5A), consistent with FANCJ promoting TLS in part through p21 suppression. p21 depletion in the FANCJ K/O HeLa cells improved replication, fitness, and suppressed ssDNA gaps, unless REV1 was inhibited (fig. S5, A, C, and D). Together, these findings reveal that distinct cancer cell lines rely on TLS polymerases for continuous replication and fitness, indicating replication gaps as a cancer vulnerability.
Gap-inducing therapies are also evaded by TLS polymerases
Currently, there is a major clinical effort to treat cancer by induction of replication stress through inhibition of ATR or the mitotic checkpoint kinase Wee1 (45). Given that these drugs induce replication gaps (Fig. 2A) (8, 40, 46), we considered that if gaps were the sensitizing lesion, then activated TLS polymerases could also interfere with their effectiveness. Compared with the non-TLS cells, pro-TLS cells conferred greater clonogenic survival following treatment with either the ATRi or the Wee1 inhibitor (Wee1i) (Fig. 5, A and B, and fig. S6, A to D). Similarly, the pro-TLS cancer cell line HCT15 showed resistance to both ATRi and Wee1i (Fig. 5, A and B, and fig. S6, A to D) (45). However, when coincubated with the TLSi, the pro-TLS U2OS cells, with either FANCJS990A or p21 depletion, or the HCT15 cancer cell lines were resensitized, suggesting a more potent therapeutic response when TLSi is used in combination with ATRi or Wee1i (Fig. 5, A and B, and fig. S6, A to D). Collectively, these findings demonstrate that TLS polymerases overcome replication stress from oncogene expression that explains the prevalence of cancer cells rewired to depend on TLS polymerases for replication and fitness. This TLS polymerase rewiring mitigates the effectiveness of drugs such as ATRi and Wee1i that induces gaps, suggesting the greater clinical potential of targeting TLS factors as a cancer therapy (Fig. 5C).
Fig. 5. TLS polymerases as a gap suppression mechanism in cancer that can be resensitized by using the TLSi.
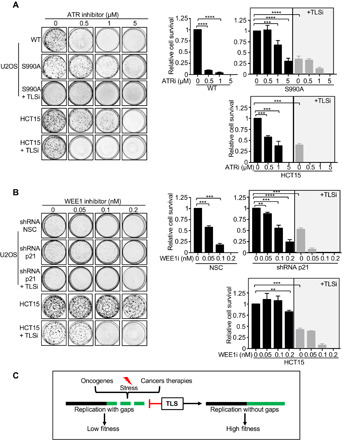
(A and B) Representative images and quantification of colony formation after dose-dependent treatment with ATRi (μM) and WEE1i (nM) alone or in combination with 20 μM TLSi across the different cell lines. (C) Model summarizing that TLS polymerases are a replication stress avoidance mechanism in cancer. Experiments were performed in biological triplicate. Bars represent the means ± SD. Statistical analysis was performed according to two-tailed Mann-Whitney test. All P values are described in the “Statistical methods.”
DISCUSSION
It has been a long-standing mystery how cancer cells ultimately overcome the replication stress response initiated by oncogenes. It has also been a challenge to understand the variation in the response of cancer cells to chemotherapy or different drugs being clinically tested that inhibit checkpoint kinases, such as ATR or Wee1 (46). On the basis of our work, we propose that rewired replication that favors TLS polymerases is an essential adaptation to blunt oncogene-induced replication stress that otherwise rapidly induces ssDNA gaps and limits cell fitness. The pro-TLS rewiring also counteracts therapies that we and others demonstrate induce gaps such ATR or Wee1 inhibitors (Fig. 5C) (8, 40, 46–49). Critically, a small-molecule inhibitor targeting the TLS factor, REV1 (27, 50), not only effectively disrupts TLS polymerase–dependent DNA replication and cancer cell fitness but also synergizes with gap-inducing therapies. Collectively, we propose a new model for TLS polymerases in cancer and therapy response, distinct from lesion bypass to a mechanism of GS.
Mechanistically, we uncover that during replication stress, TLS polymerases curtail the slowing and remodeling of replication forks and the global replication arrest response while also suppressing ssDNA gaps. GS may be critical for cell fitness as gaps that persist are toxic and drive apoptosis (46, 51). Correspondingly, TLS factors not only tolerate oncogenic stress but are also elevated in cancer to alleviate replication stress (8, 12, 52, 53). Notably, despite being a tumor suppressor, FANCJ is overexpressed in many cancers (54–56). Excess FANCJ could propel TLS because there is insufficient BRCA1 binding and/or acetylation to limit its TLS activity (22, 23). The pro-TLS FANCJ could disrupt secondary DNA fork structures, fork remodeling factors, or nucleases to limit replication fork “jumping” that leads to gap formation. FANCJ could also promote TLS polymerases by stabilizing G-quadruplex secondary structures that are platforms for REV1 mobilization (57). TLS polymerases are also likely further licensed because FANCJ suppresses p21, a negative regulator of TLS (41). Similar to FANCJ, other Fanconi anemia (FA) genes modulate TLS, which could be fundamental to preventing FA.
Our study challenges the model that replication fork slowing and reversal is a unifying response to genotoxic stress (31), but rather indicates that the response varies depending on the type of genotoxic agent, kinetics of analysis, and cellular context. For example, U2OS, RPE, and HMEC cells rapidly arrest replication in response to low-dose HU, whereas MCF7 and HeLa cells continue to replicate. However, U2OS cells similar to other cancer cells continue to replicate in the immediate aftermath of oncogene expression or treatment with inhibitors ATR or Wee1 as also found for PARPi (58). While we envision that the relative toxicity of this continued replication relates to the abundance of replication gaps that are counteracted by TLS polymerases, we cannot exclude that there are other toxic DNA structures that interfere with the completion of DNA replication and, thus, reduce cell fitness. A key decision point to slow or continue replication during stress is regulated by proliferating cell nuclear antigen (PCNA). PCNA monoubiquitylation promotes TLS, whereas PCNA polyubiquitylation provides a nexus for fork remodelers that are essential for slowing and the reversal of replication forks during stress (39). As such, loss of fork remodelers, similar to loss of PCNA ubiquitination, reduces cell fitness and ssDNA gaps accumulate (24, 59). ATRi also disrupts fork reversal, and gaps develop (2). These findings suggest that a failure to slow and remodel replication forks into reversed fork structures is not a productive means for tumorigenesis, at least when replication ssDNA gaps form. Correspondingly, a failure to slow and reverse replication in response to stress is linked to genomic instability and enhanced cell death (1). While gaps are avoided by pausing replication in response to stress, reversed forks are susceptible to nucleolytic processing. Fork degradation causes genomic instability in BRCA-deficient cancer (34). Thus, we propose that cancer cells avoid the vulnerability of reversed forks and replication gaps by favoring TLS polymerases.
In summary, our data reveal that TLS polymerases subvert the replication stress response by restricting ssDNA gaps and that this activity is targetable in cancer to reduce fitness. Our data also highlight the clinical importance of identifying TLS polymerase–dependent cancers, especially those that lack current treatment options such as ovarian that are sensitive to the TLSi in vitro. Leveraging this vulnerability by TLS inhibition alone or in conjunction with gap-inducing therapies that include inhibitors of ATR, Wee1 and PARP as well as cisplatin ideally will improve efficacy (20, 21). Conceivably, targeting TLS factors in cancer will also reduce the ability of the cancer to mutate and, therefore, retain its vulnerability to other therapies. As the TLSi restricts replication, gaps likely form due to a greater replication fork uncoupling (60). An important future goal will be to identify biomarkers that signify TLS polymerase–dependent cancers. A reasonable place to begin is with cancers expressing oncogenes such as cyclin E, CDC25A, KRAS, MOS, and MYC. Identifying TLS polymerase–dependent cancers will also be facilitated by uncovering the core factors and modulators driving the TLS polymerase rewiring. In addition to OE of TLS factors, TLS polymerases may also provide mutational signatures or strong gene dependencies [i.e., cancer cell line encyclopedia (CCLE)]. Collectively, our findings highlight the importance of replication gaps as a cancer vulnerability in a wide range of cancers.
MATERIALS AND METHODS
Cell culture and cell lines
U2OS, PEO1, HeLa, MCF7, HCT15, and A549 cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin and streptomycin (100 U/ml each). U2OS cells with inducible OE of cyclin E (U2OS-CE) were maintained in DMEM supplemented with 10% FBS (Invitrogen, catalog no. 10500), penicillin-streptomycin-glutamine (Invitrogen, catalog no. 10378-016), G418 (400 μg/ml) (Invitrogen, catalog no. 10131-027), puromycin (1 μg/ml) (Sigma-Aldrich, catalog no. P8833), and tetracycline (2 μg/ml) (Sigma-Aldrich, catalog no. T7660). Right before the experiment, the cells were split into two aliquots. One aliquot was cultured in media without tetracycline to induce cyclin E OE (OE cells) and the other in media with doxycycline (1 μg/ml) to maintain low levels of ectopic cyclin E expression (NE cells). MOLT-4 and NCI-H522 cell lines were grown in RPMI supplemented with 10% FBS and penicillin and streptomycin (100 U/ml each). HMEC cell line was grown in DMEM supplemented with 15% FBS and penicillin and streptomycin (100 U/ml each). FA-J cells (EUFA30-F) were immortalized with human telomerase reverse transcriptase (hTERT) and cultured as previously described (61). Stable FA-J pOZ–complemented cell lines were generated and selected as previously described. U2OS and HeLa FANCJ K/O CRISPR cell lines were generated as previously described (24). Stable U2OS FANCJ K/O– and PEO1 pLenti–complemented cell lines were generated by blasticidin selection (5 μg/ml). Stable HeLa and U2OS shRNA knockdown cell lines were generated by puromycin selection (0.25 and 0.5 μg/ml, respectively).
Human subjects
Malignant ovarian cancer cells were recovered from ascitic fluids from patients with ovarian cancer by the University of Massachusetts Medical School (UMMS) Biorepository and Tissue Bank. Patient consent was obtained prior to specimen collection under a UMMS Institutional Review Board (IRB)–approved protocol (H4721). Malignant ovarian cancer cells were recovered from ascitic fluids by centrifugation at 200g and cryopreserved in RPMI media supplemented with 10% FBS and 10% dimethyl sulfoxide (DMSO). Cells were slowly frozen at −80°C in an isopropanol bath overnight and stored long term in the vapor phase of a liquid nitrogen freezer. The consent process included conditions for sharing deidentified samples and information with other investigators. No identifiable information will be shared at any time per Health Insurance Portability and Accountability Act guidelines.
Plasmid and shRNA constructs
The WT and S990A FANCJ pLentiviral vectors were a gift from J. Chen. The WT and S990A pOZ vectors were generated as previously described. HeLa FANCJ K/O and U2OS cells were infected with pLK0.1 vector containing shRNAs against nonsilencing control (NSC) or one of three shRNAs against p21/CDKN1A (A) (target region: 3′UTR—CGCTCTACATCTTCTGCCTTA), (B) (CDS—GAGCGATGGAACTTCGACTTT), and (C) (CDS—GTCACTGTCTTGTACCCTTGT). pInducer20 empty vector and pInducer20 cyclin E1 plasmids were obtained from Addgene. The FANCJ K/O U2OS cells complemented with FANCJWT or FANCJS990A were further infected with the respected virus to express the empty vector or cyclin E1 in a doxycycline inducible manner. shRNAs were obtained from the UMMS shRNA core facility. The eGFP–pol-η and eGFP-Rev1 constructs were used as described earlier (62, 63).
Drugs and reagents
The following drugs were used in the course of this study: Cisplatin (Sigma-Aldrich) was prepared as a 1-mM solution in saline per the manufacturer’s instructions. MMC (Sigma-Aldrich) was prepared by dissolving 0.5 mg/ml in water. HU (Sigma-Aldrich) was prepared fresh in complete media prior to experiments per the manufacturer’s instructions. The ATRi, VE-821 (Selleckchem) and Wee1i, MK-1775 (Selleckchem) were prepared as a 15 and 5 mM solutions in DMSO, respectively. CldU and IdU were obtained from Sigma-Aldrich. Click-iT EdU Alexa Fluor 488 Imaging Kit was obtained from Invitrogen. Concentration and duration of treatment are indicated in the corresponding figures and sections.
Immunoblotting and antibodies
Cells were harvested, lysed, and processed for Western blot analysis as described previously using 150 mM NETN lysis buffer [20 mM tris (pH 8.0), 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 1 mM phenyl-methyl-sulfonyl fluoride, leupeptin (10 mg/ml), and aprotinin (10 mg/ml)]. For cell fractionation, we isolated cytoplasmic and soluble nuclear fractions with the NE-PER Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol; to isolate the chromatin fraction, the insoluble pellet was resuspended in radioimmunoprecipitation assay (RIPA) buffer and sonicated in a Bioruptor according to the manufacturer’s protocol (medium power, 20 min, 30 s on, 30 s off at 4°C). Proteins were separated using SDS–polyacrylamide gel electrophoresis and electrotransferred to nitrocellulose membranes. Membranes were blocked in 5% nonfat dry milk phosphate-buffered saline (PBS)/Tween and incubated with primary antibody (Ab) for overnight at 4°C. Abs for Western blot analysis included anti–β-actin (Sigma-Aldrich), anti-FANCJ (E67), anti-vinculin (Sigma-Aldrich), anti–cyclin E1 (Abcam), and anti-p21 (BD Pharmingen). Membranes were washed and incubated with horseradish peroxidase–linked secondary Abs (Amersham) for 1 hour at room temperature (RT) and detected by chemiluminescence (Amersham).
Immunofluorescence
Immunofluorescence was performed as described previously (33). Cells were grown on coverslips in 10 μM 5-bromo-2′-deoxyuridine (BrdU) for 48 hours before the treatment with drugs. Cells were then treated with the aforementioned drugs for 2 hours. After treatment, cells were washed with PBS and preextracted with 0.5% Triton X-100 made in PBS on ice. Cells were then fixed using 4% formalin for 15 min at RT. Fixed cells were then incubated with primary Abs against BrdU (Abcam) at 37°C for 1 hour. Cells were washed and incubated with secondary Abs for 1 hour at RT. After washing, coverslips were mounted onto glass slides using Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories). For EdU labeling, staining was carried out with Click-iT EdU imaging kit (Invitrogen) according to the manufacturer’s instructions. For visualization of pol-η and Rev1 foci, cells were transfected with either eGFP–pol-η or eGFP-Rev1, incubated for overnight, seeded on coverslips, and again incubated for overnight and examined 4 hours after UV or 48 hours after MMC. Cells were first permeabilized with 0.5% Triton X-100 made in PBS on ice and then fixed with 4% formalin and foci counted.
DNA fiber assays
To directly visualize replication fork dynamics, we established single molecular DNA fiber analysis. In this assay, progressing replication forks in cells were labeled by sequential incorporation of two different nucleotide analogs, IdU (50 μM) and CldU (50 μM), into nascent DNA strands for the indicated time and conditions. After nucleotide analogs were incorporated in vivo, the cells were collected, washed, spotted (2.5 μl of 105 cells/ml PBS cell suspension), and lysed on positively charged microscope slides (Globe Scientific, #1358 W) by 7.5 μl of spreading buffer [0.5% SDS, 200 mM tris-HCl (pH 7.4), and 50 mM EDTA] for 8 min at RT. Individual DNA fibers were released and spread by tilting the slides at a 45°. After air drying, the fibers were fixed by 3:1 methanol:acetic acid at RT for 3 min. After air drying again, fibers were rehydrated in PBS, denatured with 2.5 M HCl for 30 min, washed with PBS, and blocked with blocking buffer (3% BSA and 0.1%Trition in PBS) for 1 hour. Next, slides were incubated for 2.5 hours with primary Abs (IdU: 1:100, mouse monoclonal anti-BrdU, Becton Dickinson 347580; CldU: 1:100, rat monoclonal anti-BrdU, Abcam 6326) diluted in blocking buffer, washed several times in PBS, and then incubated with secondary Abs (IdU: 1:200, goat anti-mouse, Alexa 488; CldU: 1:200, goat anti-rat, Alexa 594) in blocking buffer for 1 hour. After washing and air drying, the slides were mounted with Prolong (Invitrogen, P36930). Last, the visualization of green and/or red signals by fluorescence microscopy (Axioplan 2 imaging, Zeiss) will provide information about the active replication directionality at the single molecular level.
S1 nuclease fiber assay
As described previously, cells were exposed to 50 μM IdU to label replication forks, followed by 50 μM CldU with 0.5 mM HU for 1 hour. Subsequently, cells were permeabilized with CSK (cytoskeletal) buffer [100 mM NaCl, 10 mM MOPS (3-(N-morpholino) propanesulfonic acid), 3 mM MgCl2 (pH 7.2), 300 mM sucrose, 0.5% Triton X-100] at RT for 8 min, followed by S1 nuclease (20 U/ml) in S1 buffer [30 mM sodium acetate (pH 4.6), 10 mM zinc acetate, 5% glycerol, and 50 mM NaCl] for 30 min at 37°C. Last, cells were collected by scraping, pelleted, and resuspended in 100 to 500 μl of PBS; 2 μl of cell suspension was spotted on a positively charged slide and lysed and processed as described in the “DNA fiber assays” section.
Electron microscopy
For the EM analysis of replication intermediates, 5 × 106 to 10 × 106 U2OS FANCJ K/O CRISPR cells, transfected with either FANCJWT or FANCJS990A, were harvested immediately after treatment with HU at a 4 mM concentration for 2 hours. Genomic DNA was cross-linked by three rounds of incubation in 4,5′,8-trimethylpsoralen (10 μg/ml; Sigma-Aldrich) and 3 min of irradiation with 366-nm UV light on a precooled metal block (64, 65). Cells were lysed and genomic DNA was isolated from the nuclei by proteinase K (Roche) digestion and phenol-chloroform extraction. DNA was purified by isopropanol precipitation, digested with PvuII HF in the proper buffer for 3 to 5 hours at 37°C, and replication intermediates were enriched on a benzoylated naphthoylated DEAE-cellulose (Sigma-Aldrich) column. EM samples were prepared by spreading the DNA on carbon-coated grids in the presence of benzyl-dimethyl-alkylammonium chloride and visualized by platinum rotary shadowing. Images were acquired on a transmission electron microscope (JEOL 1400 EX) with side-mounted camera (AMTXR41 supported by AMT software v601) and analyzed with ImageJ (National Institutes of Health). EM analysis allows distinguishing duplex DNA—which is expected to appear as a 10-nm-thick fiber after the platinum/carbon coating step necessary for EM visualization—from ssDNA, which has a reduced thickness of 5 to 7 nm. Internal ssDNA gaps behind forks are scored by measuring ssDNA regions located in the daughter arms of three-way junction fork structures and excluding ssDNA discontinuities present at fork junctions. The criteria used for the unequivocal assignment of reversed forks include the presence of a rhomboid structure at the junction itself to provide a clear indication that the junction is opened up and that the four-way junction structure is not simply the result of the occasional crossing of two DNA molecules (66). In addition, the length of the two arms corresponding to the newly replicated duplex should be equal (b = c), whereas the length of the parental arm and the regressed arm can vary (a ≠ b = c ≠ d). Conversely, canonical Holliday junction structures will be characterized by arms of equal length (a = b, c = d). EM analysis was performed by calculating the percentage of either reversed replication forks or internal ssDNA gaps in each sample.
Viability assays
Cells were seeded onto 96-well plates (500 cells per well, performed in triplicates for each experiment) and incubated overnight. Next day, the cells were treated with increasing dose of MMC for 1 hour in serum-free media or cisplatin or TLSi and maintained in complete media for 5 days. Percent survival was measured using CellTitre-Glo viability assay (Promega) photometrically in a microplate reader (Beckman Coulter DTX 880 Multimode Detector).
Colony formation assay
For colony formation assays, either 500 or 1000 cells per well were seeded into six-well plates and were treated continuously with or without different drugs as mentioned in the respective figures. Once the colonies had developed, the cells were fixed with 90% methanol and then stained with 0.05% (w/v) crystal violet solution. Plates were then imaged using ChemiDoc Touch Imaging system (Bio-Rad), and the number of colonies was counted using the Cell Profiler software version 3.1.5 from Broad Institute.
Statistical methods
Statistical differences in DNA fiber assay, S1 nuclease assay, immunofluorescence, and colony forming assays were determined using a two-tailed Mann-Whitney test. Statistical analysis was performed using Excel and GraphPad Prism (version 7.0). In all cases, ns indicates not significant (P > 0.01), **P < 0.01, *P < 0.001, and ****P < 0.0001.
Supplementary Material
Acknowledgments
We thank all the members of the Cantor laboratory for the helpful discussions. We thank M. Green for providing the HCT15, A549, MOLT-4, and NCI-H522 cell lines, and T. D. Halazonetis for providing the U2OS cyclin E1 inducible cell line. We thank O. D. Scharer, K. Son, and R. Woodgate for the helpful suggestions. We also thank K. Simin and the University of Massachusetts Medical School (UMMS) Biorepository and Tissue Bank for providing the human patient ovarian cancer ascites sample. Funding: The work in the S.B.C. laboratory was supported by R01 CA176166-01A1, R01 CA225018-01A1, and the Basser Center for BRCA research, as well as charitable contributions from the Lipp Family Foundation. The work in the A.V. laboratory is supported by NIH grant R01GM108648 and by DOD BRCP Breakthrough Award BC151728. The work in the M.K.H. laboratory is supported by the University of Connecticut Research Foundation and NIH/NCI grant R01 CA 233959-01A1. The University of Massachusetts Medical School (UMMS) Biorepository and Tissue Bank is supported by the National Center for Advancing Translational Sciences of the NIH under award number UL1-TR001453. Author contributions: S.B.C., S.N., M.K.H., and A.V. designed the experiments. S.N., J.A.C., K.C., M.P., E.B., and J.J. performed the experiments. S.N. and J.A.C. analyzed the data. A.M.Z. generated the TLSi reagent. S.B.C. and S.N. wrote the manuscript. S.B.C., M.K.H., and A.V. supervised the research. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/24/eaaz7808/DC1
REFERENCES AND NOTES
- 1.Neelsen K. J., Lopes M., Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 16, 207–220 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Mutreja K., Krietsch J., Hess J., Ursich S., Berti M., Roessler F. K., Zellweger R., Patra M., Gasser G., Lopes M., ATR-mediated global fork slowing and reversal assist fork traverse and prevent chromosomal breakage at dna interstrand cross-links. Cell Rep. 24, 2629–2642.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartkova J., Rezaei N., Liontos M., Karakaidos P., Kletsas D., Issaeva N., Vassiliou L.-V. F., Kolettas E., Niforou K., Zoumpourlis V. C., Takaoka M., Nakagawa H., Tort F., Fugger K., Johansson F., Sehested M., Andersen C. L., Dyrskjot L., Ørntoft T., Lukas J., Kittas C., Helleday T., Halazonetis T. D., Bartek J., Gorgoulis V. G., Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Mailand N., Falck J., Lukas C., Syljuåsen R. G., Welcker M., Bartek J., Lukas J., Rapid destruction of human Cdc25A in response to DNA damage. Science 288, 1425–1429 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Bartkova J., Hořejší Z., Koed K., Krämer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J. M., Lukas C., Ørntoft T., Lukas J., Bartek J., DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Gorgoulis V. G., Vassiliou L.-V. F., Karakaidos P., Zacharatos P., Kotsinas A., Liloglou T., Venere M., DiTullio R. A. Jr., Kastrinakis N. G., Levy B., Kletsas D., Yoneta A., Herlyn M., Kittas C., Halazonetis T. D., Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907–913 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Di Micco R., Fumagalli M., Cicalese A., Piccinin S., Gasparini P., Luise C., Schurra C., Garre’ M., Nuciforo P. G., Bensimon A., Maestro R., Pelicci P. G., d’Adda di Fagagna F., Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444, 638–642 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Yang Y., Gao Y., Mutter-Rottmayer L., Zlatanou A., Durando M., Ding W., Wyatt D., Ramsden D., Tanoue Y., Tateishi S., Vaziri C., DNA repair factor RAD18 and DNA polymerase Polκ confer tolerance of oncogenic DNA replication stress. J. Cell Biol. 216, 3097–3115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neelsen K. J., Zanini I. M., Herrador R., Lopes M., Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J. Cell Biol. 200, 699–708 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santoni-Rugiu E., Falck J., Mailand N., Bartek J., Lukas J., Involvement of Myc activity in a G1/S-promoting mechanism parallel to the pRb/E2F pathway. Mol. Cell. Biol. 20, 3497–3509 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alevizopoulos K., Vlach J., Hennecke S., Amati B., Cyclin E and c-Myc promote cell proliferation in the presence of p16INK4a and of hypophosphorylated retinoblastoma family proteins. EMBO J. 16, 5322–5333 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tonzi P., Huang T. T., Role of Y-family translesion DNA polymerases in replication stress: Implications for new cancer therapeutic targets. DNA Repair 78, 20–26 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kannouche P. L., Lehmann A. R., Ubiquitination of PCNA and the polymerase switch in human cells. Cell Cycle 3, 1011–1013 (2004). [PubMed] [Google Scholar]
- 14.Doles J., Oliver T. G., Cameron E. R., Hsu G., Jacks T., Walker G. C., Hemann M. T., Suppression of Rev3, the catalytic subunit of Polζ, sensitizes drug-resistant lung tumors to chemotherapy. Proc. Natl. Acad. Sci. U.S.A. 107, 20786–20791 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie K., Doles J., Hemann M. T., Walker G. C., Error-prone translesion synthesis mediates acquired chemoresistance. Proc. Natl. Acad. Sci. U.S.A. 107, 20792–20797 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wojtaszek J. L., Chatterjee N., Najeeb J., Ramos A., Lee M., Bian K., Xue J. Y., Fenton B. A., Park H., Li D., Hemann M. T., Hong J., Walker G. C., Zhou P., A small molecule targeting mutagenic translesion synthesis improves chemotherapy. Cell 178, 152–159.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papadopoulou C., Guilbaud G., Schiavone D., Sale J. E., Nucleotide pool depletion induces G-quadruplex-dependent perturbation of gene expression. Cell Rep. 13, 2491–2503 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang S., Naiman K., Thrall E. S., Kath J. E., Jergic S., Dixon N. E., Fuchs R. P., Loparo J. J., A gatekeeping function of the replicative polymerase controls pathway choice in the resolution of lesion-stalled replisomes. Proc. Natl. Acad. Sci. U.S.A. 116, 25591–25601 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallo D., Brown G. W., Post-replication repair: Rad5/HLTF regulation, activity on undamaged templates, and relationship to cancer. Crit. Rev. Biochem. Mol. Biol. 54, 301–332 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Cong K., Kousholt A. N., Peng M., Panzarino N. J., Lee W. T. C., Nayak S., Krais J., Calvo J., Bere M., Rothenberg E., Johnson N., Jonkers J., Cantor S. B., PARPi synthetic lethality derives from replication-associated single-stranded DNA gaps. bioRxiv , 781989 (2019). [Google Scholar]
- 21.Panzarino N. J., Krais J., Peng M., Mosqueda M., Nayak S., Bond S., Calvo J., Cong K., Doshi M., Bere M., Ou J., Deng B., Zhu L. J., Johnson N., Cantor S. B., Replication gaps underlie BRCA-deficiency and therapy response. bioRxiv , 781955 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie J., Peng M., Guillemette S., Quan S., Maniatis S., Wu Y., Venkatesh A., Shaffer S. A., Brosh R. M. Jr., Cantor S. B., FANCJ/BACH1 acetylation at lysine 1249 regulates the DNA damage response. PLOS Genet. 8, e1002786 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie J., Litman R., Wang S., Peng M., Guillemette S., Rooney T., Cantor S. B., Targeting the FANCJ–BRCA1 interaction promotes a switch from recombination to polη-dependent bypass. Oncogene 29, 2499–2508 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng M., Cong K., Panzarino N. J., Nayak S., Calvo J., Deng B., Zhu L. J., Morocz M., Hegedus L., Haracska L., Cantor S. B., Opposing roles of FANCJ and HLTF protect forks and restrain replication during stress. Cell Rep. 24, 3251–3261 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koç A., Wheeler L. J., Mathews C. K., Merrill G. F., Hydroxyurea arrests DNA replication by a mechanism that preserves basal dNTP pools. J. Biol. Chem. 279, 223–230 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Korzhnev D. M., Hadden M. K., Targeting the translesion synthesis pathway for the development of anti-cancer chemotherapeutics. J. Med. Chem. 59, 9321–9336 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Sail V., Rizzo A. A., Chatterjee N., Dash R. C., Ozen Z., Walker G. C., Korzhnev D. M., Hadden M. K., Identification of small molecule translesion synthesis inhibitors that target the Rev1-CT/RIR protein–protein interaction. ACS Chem. Biol. 12, 1903–1912 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raghunandan M., Chaudhury I., Kelich S. L., Hanenberg H., Sobeck A., FANCD2, FANCJ and BRCA2 cooperate to promote replication fork recovery independently of the Fanconi Anemia core complex. Cell Cycle 14, 342–353 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwab R. A., Nieminuszczy J., Shin-ya K., Niedzwiedz W., FANCJ couples replication past natural fork barriers with maintenance of chromatin structure. J. Cell Biol. 201, 33–48 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lossaint G., Larroque M., Ribeyre C., Bec N., Larroque C., Décaillet C., Gari K., Constantinou A., FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol. Cell 51, 678–690 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Zellweger R., Dalcher D., Mutreja K., Berti M., Schmid J. A., Herrador R., Vindigni A., Lopes M., Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 208, 563–579 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quinet A., Carvajal-Maldonado D., Lemacon D., Vindigni A., DNA fiber analysis: Mind the gap! Methods Enzymol. 591, 55–82 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Cantor S. B., Bell D. W., Ganesan S., Kass E. M., Drapkin R., Grossman S., Wahrer D. C. R., Sgroi D. C., Lane W. S., Haber D. A., Livingston D. M., BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105, 149–160 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M., Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakai W., Swisher E. M., Jacquemont C., Chandramohan K. V., Couch F. J., Langdon S. P., Wurz K., Higgins J., Villegas E., Taniguchi T., Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 69, 6381–6386 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kolinjivadi A. M., Sannino V., De Antoni A., Zadorozhny K., Kilkenny M., Técher H., Baldi G., Shen R., Ciccia A., Pellegrini L., Krejci L., Costanzo V., Smarcal1-mediated fork reversal triggers Mre11-dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol. Cell. 67, 867–881.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mijic S., Zellweger R., Chappidi N., Berti M., Jacobs K., Mutreja K., Ursich S., R. Chaudhuri A., Nussenzweig A., Janscak P., Lopes M., Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 8, 859 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taglialatela A., Alvarez S., Leuzzi G., Sannino V., Ranjha L., Huang J.-W., Madubata C., Anand R., Levy B., Rabadan R., Cejka P., Costanzo V., Ciccia A., Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol. Cell. 68, 414–430.e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vujanovic M., Krietsch J., Raso M. C., Terraneo N., Zellweger R., Schmid J. A., Taglialatela A., Huang J.-W., Holland C. L., Zwicky K., Herrador R., Jacobs H., Cortez D., Ciccia A., Penengo L., Lopes M., Replication fork slowing and reversal upon DNA damage require PCNA polyubiquitination and ZRANB3 DNA translocase activity. Mol. Cell. 67, 882–890.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Couch F. B., Bansbach C. E., Driscoll R., Luzwick J. W., Glick G. G., Betous R., Carroll C. M., Jung S. Y., Qin J., Cimprich K. A., Cortez D., ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 27, 1610–1623 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Avkin S., Sevilya Z., Toube L., Geacintov N., Chaney S. G., Oren M., Livneh Z., p53 and p21 regulate error-prone DNA repair to yield a lower mutation load. Mol. Cell 22, 407–413 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Tissier A., Kannouche P., Reck M.-P., Lehmann A. R., Fuchs R. P. P., Cordonnier A., Co-localization in replication foci and interaction of human Y-family members, DNA polymerase polη and REVl protein. DNA Repair 3, 1503–1514 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Dominguez-Sola D., Ying C. Y., Grandori C., Ruggiero L., Chen B., Li M., Galloway D. A., Gu W., Gautier J., Dalla-Favera R., Non-transcriptional control of DNA replication by c-Myc. Nature 448, 445–451 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Lukas J., Herzinger T., Hansen K., Moroni M. C., Resnitzky D., Helin K., Reed S. I., Bartek J., Cyclin E-induced S phase without activation of the pRb/E2F pathway. Genes Dev. 11, 1479–1492 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Bukhari A. B., Lewis C. W., Pearce J. J., Luong D., Chan G. K., Gamper A. M., Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J. Clin. Invest. 129, 1329–1344 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Forment J. V., O’Connor M. J., Targeting the replication stress response in cancer. Pharmacol. Ther. 188, 155–167 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Beck H., Nähse V., Larsen M. S. Y., Groth P., Clancy T., Lees M., Jørgensen M., Helleday T., Syljuåsen R. G., Sørensen C. S., Regulators of cyclin-dependent kinases are crucial for maintaining genome integrity in S phase. J. Cell Biol. 188, 629–638 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toledo L. I., Altmeyer M., Rask M.-B., Lukas C., Larsen D. H., Povlsen L. K., Bekker-Jensen S., Mailand N., Bartek J., Lukas J., ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Zheng H., Shao F., Martin S., Xu X., Deng C.-X., WEE1 inhibition targets cell cycle checkpoints for triple negative breast cancers to overcome cisplatin resistance. Sci. Rep. 7, 43517 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dash R. C., Ozen Z., Rizzo A. A., Lim S., Korzhnev D. M., Hadden M. K., Structural approach to identify a lead scaffold that targets the translesion synthesis polymerase Rev1. J. Chem. Inf. Model. 58, 2266–2277 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Nur-E-Kamal A., Li T.-K., Zhang A., Qi H., Hars E. S., Liu L. F., Single-stranded DNA induces ataxia telangiectasia mutant (ATM)/p53-dependent DNA damage and apoptotic signals. J. Biol. Chem. 278, 12475–12481 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Gao Y., Mutter-Rottmayer E., Greenwalt A. M., Goldfarb D., Yan F., Yang Y., Martinez-Chacin R. C., Pearce K. H., Tateishi S., Major M. B., Vaziri C., A neomorphic cancer cell-specific role of MAGE-A4 in trans-lesion synthesis. Nat. Commun. 7, 12105 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schleicher E. M., Galvan A. M., Imamura-Kawasawa Y., Moldovan G.-L., Nicolae C. M., PARP10 promotes cellular proliferation and tumorigenesis by alleviating replication stress. Nucleic Acids Res. 46, 8908–8916 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eelen G., Vanden Bempt I., Verlinden L., Drijkoningen M., Smeets A., Neven P., Christiaens M. R., Marchal K., Bouillon R., Verstuyf A., Expression of the BRCA1-interacting protein Brip1/BACH1/FANCJ is driven by E2F and correlates with human breast cancer malignancy. Oncogene 27, 4233–4241 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Gupta I., Ouhtit A., Al-Ajmi A., Rizvi S. G. A., al-Riyami H., Al-Riyami M., Tamimi Y., BRIP1 overexpression is correlated with clinical features and survival outcome of luminal breast cancer subtypes. Endocr. Connect. 7, 65–77 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hampton O. A., Den Hollander P., Miller C. A., Delgado D. A., Li J., Coarfa C., Harris R. A., Richards S., Scherer S. E., Muzny D. M., Gibbs R. A., Lee A. V., Milosavljevic A., A sequence-level map of chromosomal breakpoints in the MCF-7 breast cancer cell line yields insights into the evolution of a cancer genome. Genome Res. 19, 167–177 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu C. G., Spies M., G-quadruplex recognition and remodeling by the FANCJ helicase. Nucleic Acids Res. 44, 8742–8753 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maya-Mendoza A., Moudry P., Merchut-Maya J. M., Lee M., Strauss R., Bartek J., High speed of fork progression induces DNA replication stress and genomic instability. Nature 559, 279–284 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Thakar T., Leung W., Nicolae C. M., Clements K. E., Shen B., Bielinsky A.-K., Moldovan G.-L., PCNA ubiquitination protects stalled replication forks from DNA2-mediated degradation by regulating Okazaki fragment maturation and chromatin assembly. bioRxiv, 759985 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ghosal G., Chen J., DNA damage tolerance: A double-edged sword guarding the genome. Transl. Cancer Res. 2, 107–129 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Litman R., Peng M., Jin Z., Zhang F., Zhang J., Powell S., Andreassen P. R., Cantor S. B., BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 8, 255–265 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Kannouche P., Broughton B. C., Volker M., Hanaoka F., Mullenders L. H., Lehmann A. R., Domain structure, localization, and function of DNA polymerase η, defective in xeroderma pigmentosum variant cells. Genes Dev. 15, 158–172 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hicks J. K., Chute C. L., Paulsen M. T., Ragland R. L., Howlett N. G., Guéranger Q., Glover T. W., Canman C. E., Differential roles for DNA polymerases eta, zeta, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links. Mol. Cell. Biol. 30, 1217–1230 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lemaçon D., Jackson J., Quinet A., Brickner J. R., Li S., Yazinski S., You Z., Ira G., Zou L., Mosammaparast N., Vindigni A., MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 8, 860 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thangavel S., Berti M., Levikova M., Pinto C., Gomathinayagam S., Vujanovic M., Zellweger R., Moore H., Lee E. H., Hendrickson E. A., Cejka P., Stewart S., Lopes M., Vindigni A., DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 208, 545–562 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Neelsen K. J., Chaudhuri A. R., Follonier C., Herrador R., Lopes M., Visualization and interpretation of eukaryotic DNA replication intermediates in vivo by electron microscopy. Methods Mol. Biol. 1094, 177–208 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/24/eaaz7808/DC1