

A system for controlled sexual conversion of malaria parasites enables the characterization of early sexual stages.
Abstract
Malaria transmission requires that some asexual parasites convert into sexual forms termed gametocytes. The initial stages of sexual development, including sexually committed schizonts and sexual rings, remain poorly characterized, mainly because they are morphologically identical to their asexual counterparts and only a small subset of parasites undergo sexual development. Here, we describe a system for controlled sexual conversion in the human malaria parasite Plasmodium falciparum, based on conditional expression of the PfAP2-G transcription factor. Using this system, ~90 percent of the parasites converted into sexual forms upon induction, enabling the characterization of committed and early sexual stages without further purification. We characterized sexually committed schizonts and sexual rings at the transcriptomic and phenotypic levels, which revealed down-regulation of genes involved in solute transport upon sexual commitment, among other findings. The new inducible lines will facilitate the study of early sexual stages at additional levels, including multiomic characterization and drug susceptibility assays.
INTRODUCTION
Malaria is a vector-borne disease caused by protozoan parasites of the genus Plasmodium. Of the five species that infect humans, Plasmodium falciparum is the most lethal. During its ~48-hour intraerythrocytic development cycle, the parasite develops through the asexual ring, trophozoite, and multinucleated schizont stages. Upon egress, up to 32 daughter merozoites are released and invade new erythrocytes. The asexual growth of the parasite in the human blood is responsible for all malaria symptoms, but asexual stages cannot infect anopheline mosquito vectors. Human to vector transmission requires that some parasites differentiate into nonreplicating sexual forms termed gametocytes, which develop through the morphologically distinct stages I to V over ~10 days. Immature gametocytes are sequestered in tissues such as the bone marrow until they are released back to the peripheral blood circulation as mature (stage V) gametocytes that are infectious to mosquitoes (1, 2). Since gametocytes are essential for transmission, they are an attractive target for intervention in the context of renewed efforts to eliminate malaria (3).
The first step for the production of gametocytes is the commitment of a subset of asexual parasites to sexual development (1, 2, 4). Commitment, defined as a cell state that irreversibly results in sexual conversion at a later point (5), is marked by expression of the master regulator PfAP2-G (6–8), a transcription factor of the ApiAp2 family (9). In asexual parasites, the pfap2-g gene is epigenetically silenced by heterochromatin containing the histone mark H3K9me3 and the heterochromatin protein 1 (HP1) (10–12). Activation of the gene, which triggers the sexual development program, requires eviction of HP1 by the gametocyte development 1 (GDV1) protein (13, 14).
Following commitment, the next step for gametocyte production is sexual conversion, marked by the expression of specific proteins absent from any replicating blood stages (5). For many years, the prevailing model was that sexually committed parasites must undergo an additional round of replication before sexual conversion (4), but recent research in P. falciparum and the murine malaria parasite Plasmodium berghei demonstrated that when AP2-G expression starts early enough in the ring stage, conversion can proceed directly without additional replication after commitment (5, 15). The two alternative sexual differentiation pathways are named next cycle conversion (NCC) and same cycle conversion (SCC) (5). The first developmental stage upon sexual conversion by either pathway is the sexual ring, also referred to as sexually or gametocyte-committed ring, or gametocyte ring (1, 13, 16–18). Sexual rings develop into stage I gametocytes and then follow sexual development until they reach maturity (stage V).
In P. falciparum, the proportion of parasites that abandon asexual growth and start differentiating into gametocytes, referred to as the sexual conversion rate, is typically low (<10%), both under culture conditions and in human infections. The level of investment into production of sexual forms varies between parasite clones and is also affected by the environment: While there is a baseline level of spontaneous pfap2-g activation and sexual conversion (6), several types of stress, including immune pressure, nutrient limitation, and drug pressure, have been proposed to stimulate conversion rates (1, 16). To date, the best established stimulus is depletion of the serum lipid lysophosphatidylcholine or choline from the culture medium (19).
There are no known differences in morphology or physical properties between sexually committed schizonts and their asexual counterparts or between sexual and asexual rings. Together with low levels of sexual conversion, this limits the ability to study these initial stages of sexual development, as it is difficult to obtain sufficient biological material and they cannot be readily separated from asexual parasites (1). To overcome this limitation, we generated parasite lines in which expression of pfap2-g can be conditionally activated to induce massive synchronous sexual conversion. Using these new parasite lines, we obtained highly pure preparations of sexually committed parasites and sexual rings that enabled the characterization of these largely unexplored parasite stages at multiple levels.
RESULTS
Conditional activation of pfap2-g leads to massive sexual conversion
To conditionally activate pfap2-g expression and trigger sexual conversion in P. falciparum, we designed a conditional activation construct that was inserted immediately upstream of the pfap2-g coding sequence [73 base pairs (bp) upstream of the start codon, i.e., between the promoter and the coding sequence] using the CRISPR-Cas9 system (20). The construct consisted of a strong constitutive calmodulin promoter (5′cam), the hdhfr selectable marker (confers resistance to the drug WR99210), and a terminator sequence. The hdhfr marker and terminator are flanked by loxP sites, such that upon recombination between the two loxP sites, the 5′cam promoter is adjacent to the pfap2-g coding sequence and is expected to control its expression. Recombination is mediated by the inducible Cre recombinase (DiCre), which is active only upon inducing dimerization with rapamycin (Fig. 1A) (21).
Fig. 1. Generation of transgenic parasite lines to conditionally activate pfap2-g expression.
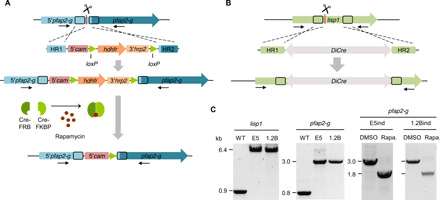
(A) Overview of the strategy to generate the inducible parasite lines. The conditional activation cassette was integrated using CRISPR-Cas9 technology. Arrows indicate the position of the primers used for diagnostic PCR. Scissors indicate the position targeted by the guide RNA, where Cas9-mediated cleavage is expected. HR refers to homology regions. The part of the pfap2-g upstream region (5′pfap2-g) presumably including the promoter of the gene remains intact after editing. (B) Schematic of the strategy to integrate the DiCre expression cassette in the lisp1 locus using CRISPR-Cas9 technology. (C) Diagnostic PCR analysis to validate the integration of the constructs and to assess recombination in samples collected 24 hours after induction with rapamycin and in DMSO-treated controls. WT, wild type.
We integrated the conditional activation construct in the E5 line, a 3D7 subclone with high levels of sexual conversion (6). In this parasite line, conversion is stimulated by choline depletion (22). In a preliminary set of experiments, the DiCre recombinase was expressed episomally. Treatment with rapamycin resulted in ~30% sexual conversion, whereas gametocytes were never observed in dimethyl sulfoxide (DMSO)–treated control cultures (fig. S1, A and B). To achieve higher conversion rates, we generated a new transgenic line in which the DiCre expression cassette was integrated at the liver specific protein 1 (lisp1) locus, which is dispensable during blood stages (Fig. 1, B and C, and fig. S1C) (23). The parasite line, termed E5 gametocyte-inducible line (E5ind), was maintained under constant WR99210 pressure to select for parasites that keep the edited pfap2-g locus in a transcriptionally active state. Treatment with rapamycin resulted in efficient excision of the floxed region, with undetectable levels of unedited locus by diagnostic polymerase chain reaction (PCR) (Fig. 1C).
To determine the optimal time for induction, we added rapamycin to tightly synchronized E5ind cultures at different stages. Induction at the late trophozoite stage resulted in maximal sexual conversion at the cycle after induction (NCC route of conversion) and maximal total amount of gametocytes (fig. S2). Conversion via the SCC route was observed when cultures were treated at 0 to 5 hours postinvasion (hpi), but it occurred at relatively low levels (<10%) (fig. S2B). The low levels of conversion by the SCC route were expected from the kinetics of DiCre activity, as excision in 50% of the parasites requires >10 hours (24). Thus, in the majority of parasites, direct conversion via the SCC route is no longer possible by the time recombination occurs and PfAP2-G is expressed (5).
On the basis of these results, we established a straightforward induction protocol in which rapamycin is added to sorbitol-synchronized cultures when the majority of parasites reach the late trophozoite/early schizont stage (~20 hours after synchronization) (Fig. 2A). Using this protocol, we consistently obtained ~90% sexual conversion (e.g., 90% of the rings at the cycle after rapamycin treatment developed as gametocytes), as determined by light microscopy analysis of Giemsa-stained smears (Fig. 2B). By starting with cultures at high parasitemia, we obtained gametocytemia levels as high as 12%. No gametocytes were observed in DMSO-treated control cultures.
Fig. 2. Sexual conversion upon conditional activation of pfap2-g.
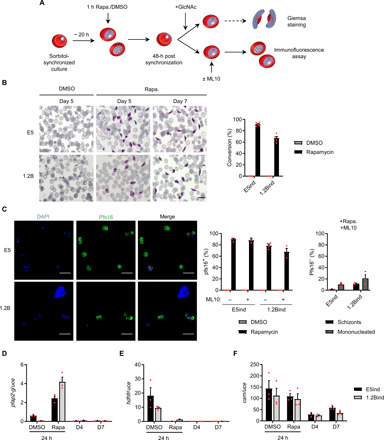
(A) Schematic of the procedure used to induce pfap2-g expression and trigger sexual conversion in the inducible lines. GlcNAc, N-acetylglucosamine. (B) Representative images of Giemsa-stained smears of the gametocytes obtained after induction and quantification of sexual conversion rates based on analysis of Giemsa-stained smears (day 5 gametocytes). Data are presented as the average and SEM of six (E5ind) or five (1.2Bind) independent experiments. Scale bar, 10 μm. (C) Representative Pfs16 IFA images and quantification of the percentage of Pfs16-positive parasites determined 3 days after rapamycin induction, in cultures treated or not with ML10. The bar chart at the right shows the distribution of schizonts and mononucleated cells (according to the number of nuclei) among Pfs16-negative parasites in ML10-treated cultures. Data are presented as the average and SEM of four (left bar chart) or three (right bar chart) independent experiments. In each experiment, >250 parasites were counted. Scale bars, 10 μm. (D to F) Transcriptional analysis of pfap2-g, hdhfr, and the endogenous cam 24 hours after rapamycin or DMSO (control) treatment and in day 4 or 7 gametocytes. Transcript levels are normalized against ubiquitin-conjugating enzyme (uce). Data are presented as the average and SEM of three independent experiments.
Similar conversion rates were estimated using immunofluorescence assays (IFAs) with antibodies against the early gametocyte marker Pfs16, which is expressed from stage I of gametocyte development onward (5). The proportion of Pfs16-positive cells was determined 3 days after induction, when asexual parasites are at the schizont stage and parasites undergoing sexual development are at gametocyte stage I and already express Pfs16. In some experiments, cultures were treated with the cGMP-dependent protein kinase (PKG) inhibitor ML10 to block schizont rupture and reinvasion (25), thus preventing amplification of nonconverting parasites. In all cases, ~90% of parasites were Pfs16 positive in rapamycin-induced E5ind cultures, whereas no Pfs16-positive parasites were observed in DMSO-treated control cultures (Fig. 2C). The majority of Pfs16-negative parasites in ML10-treated induced cultures were mononucleated, suggesting that parasites that failed to convert upon induction were predominantly dead or growth arrested. Only ~2% of parasites in these cultures were at the multinucleated schizont stage, the stage expected for healthy replicating parasites 3 days after induction (Fig. 2C).
Conditional activation of pfap2-g rescues gametocyte production in a gametocyte nonproducer line
To test the inducible system in a gametocyte nonproducer line, we used the 3D7 subclone 1.2B, derived from the 3D7-A stock. In the 3D7-A line and its subclones, including 1.2B, pfap2-g is expressed at very low levels and consequently gametocytes are not produced (6, 26). Next-generation sequencing of the 1.2B genome, conducted as part of an ongoing comparative chromatin immunoprecipitation sequencing (ChIP-seq) study, revealed a nonsense mutation in the gdv1 gene that introduces a premature STOP codon (Q578*), resulting in a truncated GDV1 protein lacking the last 21 amino acids. The mutation was validated by Sanger sequencing (fig. S3). Given that GDV1 is an upstream regulator of pfap2-g activation (14), the GDV1 truncation in the 1.2B line likely underlies its inability to form gametocytes.
We edited the 1.2B genome in an analogous way as we did with the E5 line to generate the 1.2Bind line. Induction with rapamycin resulted in ~70% sexual conversion, determined by either light microscopy or Pfs16 IFA, whereas no gametocytes were observed in control cultures (Figs. 1C and 2, A to C). This result shows that the conditional pfap2-g activation system can overcome a deficiency in a factor operating upstream of pfap2-g, indicating that PfAP2-G activation is sufficient to trigger sexual conversion. It also supports the idea that GDV1 is needed to reverse the silencing of pfap2-g (14) but is dispensable for normal gametocyte development thereafter.
Transcriptional changes at the pfap2-g locus upon induction
Transcriptional analysis of E5ind and 1.2Bind 24 hours after rapamycin treatment revealed activation of pfap2-g expression and loss of hdhfr expression, as expected (Fig. 2, D and E). However, pfap2-g transcript levels were >25-fold lower than the levels of endogenous cam transcripts (Fig. 2F) and rather resembled the pfap2-g levels in wild-type parasites (5). Furthermore, pfap2-g transcript levels decreased severely in maturing E5ind gametocytes (day 4 or 7 gametocytes), similar to the pfap2-g expression pattern in wild-type parasites (5, 27) but in contrast to endogenous cam transcripts (Fig. 2, D and F). Three features of the system can nonexclusively explain these observations: First, there are important differences between the sequence of the 5′cam used here (and widely used in malaria research to drive the expression of drug resistance markers) (28) and the endogenous cam upstream region, which can determine different promoter activity. Second, after rapamycin-induced recombination pfap2-g expression is under the control of two promoters in tandem, the intact endogenous pfap2-g promoter and the 5′cam promoter. Although the 5′cam promoter is located in a more proximal position (Fig. 1A), the distal pfap2-g promoter and the associated PfAP2-G autoregulatory positive feedback loop (6, 7, 29) may also contribute to determining pfap2-g expression. Transcripts containing sequences of the proximal part of the pfap2-g promoter or the 5′cam promoter occurred at similarly high levels (comparable to endogenous cam transcript levels), which may either reflect transcription from the pfap2-g promoter or the bidirectional activity of the 5′cam promoter (fig. S4A) (28). However, transcripts containing the pfap2-g coding sequence were far less abundant than transcripts containing promoter sequences, revealing a complex landscape suggestive of an important role for posttranscriptional mechanisms, or truncation of transcription immediately after the loxP site. Third, the pfap2-g locus heterochromatin environment may influence the activity of the 5′cam promoter. Regardless of the underlying mechanism, the roughly physiological pfap2-g transcripts levels and temporal dynamics in the inducible lines are a clear advantage of our system, as it resembles natural sexual conversion more faithfully than if the gene was markedly overexpressed.
To further explore the influence of reversible epigenetic states on pfap2-g activation, we maintained E5ind and 1.2Bind cultures without WR99210 pressure for 5 weeks and then induced conversion with rapamycin. In both parasite lines, conversion rates were clearly lower than in cultures under constant drug pressure (fig. S4B). Selecting back the cultures with WR99210 recovered conversion rates (upon induction) to the original levels (fig. S4C), consistent with conversion levels depending on reversible epigenetic states. These results suggest that in the absence of selection, heterochromatin restricts transcription at the pfap2-g locus, even after rapamycin-induced recombination. In contrast, drug pressure selects for parasites that have the 5′cam-hdhfr chimera in an active state, which determines pfap2-g expression upon recombination. Heterochromatin expansion and influence of the epigenetic state of one gene on its neighbors has been previously reported at other P. falciparum loci (30, 31).
The few parasites that continue asexual growth after induction have heterochromatin at the endogenous pfap2-g promoter
In ~1 week, we could establish stably growing populations from the few parasites that did not convert upon induction and continued proliferating, which we termed E5ind+Rapa_prol and 1.2Bind+Rapa_prol. Diagnostic PCR analysis of genomic DNAs revealed that DiCre-mediated recombination had proceeded correctly in these parasites (Fig. 3A). Unexpectedly, new gametocytes were formed at each growth cycle in E5ind+Rapa_prol but not in 1.2Bind+Rapa_prol, with sexual conversion rates and pfap2-g transcript levels that resembled those in the respective wild-type E5 and 1.2B parental lines (Fig. 3B). This observation supports the idea that, despite the constitutive 5′cam promoter being immediately upstream of the pfap2-g coding sequence, the probability of pfap2-g activation is affected by the heterochromatin environment at the pfap2-g locus.
Fig. 3. Characterization of parasites that continue asexual growth after rapamycin-induced recombination.
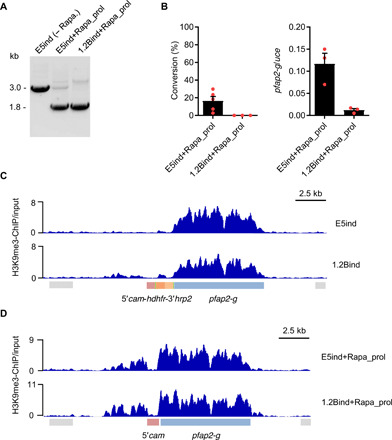
(A) Diagnostic PCR to assess recombination at the pfap2-g locus in parasites that continue proliferating after rapamycin induction. Genomic DNA was extracted 2 to 3 weeks after induction. (B) Sexual conversion rates (left) and analysis of pfap2-g relative transcript levels (right) in parasites that continue proliferating after rapamycin induction, determined 2 to 3 weeks after induction. Transcript levels are normalized against ubiquitin-conjugating enzyme (uce). Data are presented as the average and SEM of three biological replicates from independent inductions (except for conversion rates in E5ind+Rapa_prol, N = 5). (C and D) ChIP-Seq profiles of normalized H3K9me3 signal relative to input at the pfap2-g locus. The E5ind and 1.2Bind lines were analyzed before induction (C) or in parasites that continue proliferating after induction (D).
To further investigate this possibility, we performed H3K9me3 ChIP-seq analysis to characterize the distribution of heterochromatin in the E5ind+Rapa_prol and 1.2Bind+Rapa_prol lines and also in the E5ind and 1.2Bind lines before rapamycin induction. While in wild-type asexual parasites heterochromatin spans the full coding sequence and about 3.5 kb of the upstream region (32), in the noninduced E5ind and 1.2Bind lines, H3K9me3 was present at the pfap2-g coding sequence but almost absent from the endogenous pfap2-g upstream region and the inserted construct (Fig. 3C). However, in parasites that failed to convert into gametocytes upon rapamycin-induced recombination (E5ind+Rapa_prol and 1.2Bind+Rapa_prol lines), both the endogenous pfap2-g upstream region and coding sequence were heterochromatic and only the 5′cam sequence was devoid of H3K9me3 (Fig. 3D). This result indicates that when E5ind and 1.2Bind cultures are maintained under WR99210 pressure, in the majority of parasites, the endogenous pfap2-g promoter remains in a euchromatic state that enables them to readily express pfap2-g and convert into gametocytes upon rapamycin-induced recombination. In a small subpopulation of parasites, which was larger in 1.2Bind (Fig. 3C), this promoter is in a heterochromatic conformation that prevents expression of pfap2-g even after recombination places the 5′cam promoter adjacent to the gene. Since these parasites continue proliferating, they are selected when the culture is maintained for several days after induction and exhibit a sexual conversion rate that likely depends on GDV1-mediated disruption of heterochromatin at the pfap2-g promoter (not occurring in 1.2B because GDV1 is truncated).
Characterization of mature-induced gametocytes
Both E5ind and 1.2Bind produced male and female mature gametocytes, with apparently normal morphology and at similar rates to the NF54 control line (fig. S5, A to D). However, because of defective exflagellation of mature male gametocytes and consequently severely reduced ookinete formation, neither inducible line produced oocysts upon mosquito infection (Fig. 4, A and B, and fig. S5, E and F). The exflagellation defect in E5ind is not attributable to the conditional activation system, as the parental E5 line also failed to exflagellate (fig. S5G). IFA analysis during gametocyte activation suggests that deficient DNA replication underlies the failure of E5ind and 1.2Bind male gametocytes to exflagellate and infect mosquitoes (Fig. 4, C and D, and fig. S5H). The specific molecular defect in the E5 and 1.2B linages has not been identified. While this defect does not prevent gametocyte development or activation (including roundup and egress), it remains formally possible that it results in currently unknown alterations at stages preceding gamete formation.
Fig. 4. Characterization of E5ind and 1.2Bind mature gametocytes.
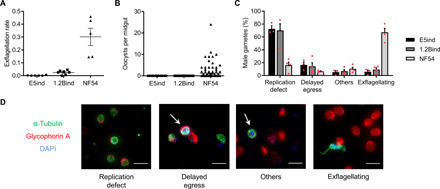
(A) Exflagellation rate (percentage of exflagellation normalized by gametocytemia) of mature gametocytes of the different parasite lines. Data are presented as the average and SEM of five to seven independent experiments. (B) Number of oocysts per mosquito midgut 9 days after blood feeding. Data are presented as average and SEM of the pooled values from three independent feeding experiments (E5, 117 midguts; 1.2B, 155 midguts; NF54, 137 midguts). (C) Distribution of male gamete types 25 min after activation. Male gametes were classified into four categories after IFA with anti–α-tubulin (stains microtubules, including the flagella) and anti–glycophorin A (an erythrocyte membrane marker) antibodies and 4′,6-diamidino-2-phenylindole (DAPI) staining of nuclei. Replication defect: Parasites that rounded up but had not replicated the genome (single nucleus). Delayed egress: Parasites that replicated the genome and formed axonemes but were still inside the red blood cells. Others: Other defects such as parasites lacking properly formed axonemes. Exflagellating: Parasites that developed correctly. Data are presented as the average and SEM of three or four independent experiments (E5ind, 389 male gametes; 1.2Bind, 769 male gametes; NF54, 406 male gametes). (D) Representative images of the categories described in (C). Scale bars, 10 μm.
Transcriptomic profiling of sexually committed schizonts and sexual rings
Our inducible sexual conversion system yields high amounts of synchronous parasites at the initial stages of sexual development, with a level of purity that was not achieved by previous approaches. This provides a unique opportunity to describe the largely uncharacterized sexually committed schizont and sexual ring stages. Here, we characterized the initial transcriptional changes upon pfap2-g activation using time course genome-wide transcriptomic analysis.
Overall, we identified 379 genes with altered expression between rapamycin-treated and control E5ind cultures [log2(fold change) of >1 in two independent biological replicates], of which 77 showed a log2(fold change) of >3 (Fig. 5, A and B, fig. S6A, and data file S1). Gene families such as surfin or etramp were enriched in up-regulated genes, whereas large families involved in antigenic variation were generally enriched in down-regulated genes (fig. S6B and data file S2). The majority of transcriptional differences were observed at the latest time point analyzed, which corresponds to the late sexual ring stage (~15 to 20 hpi of the cycle after induction) (Fig. 5C). At the early sexual ring stage (~5 to 10 hpi), the most up-regulated gene was the early sexual marker gexp02 (Fig. 5, A and B) (22). Genes highly up-regulated upon pfap2-g activation included well-established early gametocyte markers such as pfs16, pfg27/25, and pfg14-744 and many genes up-regulated early during P. falciparum sexual development identified by recent genome-wide studies (Fig. 5A) (6, 7, 10, 14, 17, 19, 33, 34). Many of the most up-regulated genes (71% of genes with log2[fold increase] >3) were bound by PfAP2-G according to a recent ChIP-seq study (Fig. 5A) (29), whereas only 7% of the rest of genes in the genome were bound (P = 2.2 × 10−16 using Fisher’s exact test). This result indicates that the majority of highly up-regulated genes are direct targets of PfAP2-G. In contrast, very few of the genes down-regulated upon pfap2-g activation are bound by PfAP2-G (5% of genes with a log2[fold-decrease] >3) or had been reported to change during sexual development (Fig. 5A). Down-regulated genes included genes with known functions associated with asexual development, such as mesa, hrpIII, pfemp3, pf332, and kahrp, and revealed many potential previously unidentified asexual parasite markers. The high level of purity of our sexual and control preparations (~90% sexual and 100% asexual, respectively) enables the identification of genes down-regulated during sexual commitment and development. This was not possible using previous approaches in which many asexual parasites were still present in the preparations enriched in sexual parasites. Only a study using fluorescence-activated cell sorting–sorted early sexual parasites (17) identified a substantial fraction of the most down-regulated genes in our analysis, which are genes that show decreased expression at the late sexual ring stage (Fig. 5A).
Fig. 5. Transcriptomic analysis of the alterations that ensue upon pfap2-g activation.
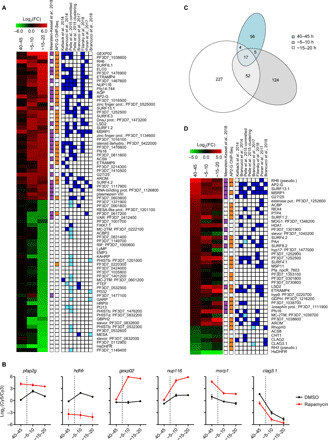
(A) Expression profile of genes with a log2 of the expression fold change (FC) between induced and control cultures of >3 in two independent biological replicates (at any of the time points analyzed). Values are the average of the log2(FC) in the two replicates. Genes are ordered by FC value. Samples were collected at 40 to 45 hpi of the cycle of induction or ~5 to 10 and ~15 to 20 hpi of the next cycle. PlasmoDB IDs are provided for genes that do not have a unique annotation. Genes that were identified as gametocyte markers in an integrative analysis (purple), direct targets of PfAP2-G as determined by ChIP-seq (orange), and genes up-regulated (dark blue) or down-regulated (light blue) in gametocytes in previous studies are indicated. (B) Time course relative expression levels (normalized log2 ratio of Cy5-labeled samples signal versus Cy3-labeled reference pool signal) of selected genes in DMSO (control) and rapamycin-treated cultures. The dotted lines indicate the time of schizont rupture and reinvasion. (C) Venn diagram showing the number of genes with altered expression at each time point [log2(FC) > 1]. (D) Expression FC only for genes showing a log2(FC) > 1 at 40 to 45 hpi.
Next, we focused specifically on genes showing altered expression at the committed schizont stage (40 to 45 hpi of the induction cycle). Thirty-six genes were up-regulated in committed versus noncommitted schizonts (log2 [fold-change] >1 in two biological replicates), including several previously reported commitment markers and PfAP2-G targets, whereas only six genes were down-regulated in addition to the hdhfr marker (Fig. 5, B and D). Of note, three of these six genes (clag3.1, clag2, and rhoph3) encode components of the RhopH complex, which participates in erythrocyte invasion and solute transport (35–37).
A recent report showed that, in sexually committed schizonts, PfAP2-G binds the promoter of many invasion-related genes, which suggests that it may contribute to their regulation (29). We found that the expression of the majority of invasion genes bound by PfAP2-G was not altered between induced and control cultures (fig. S6C). We identified a very small number of differentially expressed invasion genes: Excluding pseudogenes, only msrp1 and, to a lesser extent, msp11 were up-regulated, whereas only components of the RhopH complex were down-regulated. Both up- and down-regulated invasion genes and pseudogenes are bound by PfAP2-G (fig. S6C), in contrast to genes down-regulated at the sexual ring stage that are generally not bound by PfAP2-G (Fig. 5A). Of note, the up-regulated pseudogene rh6 is in close proximity to msrp1, which is suggestive of regional activation events possibly associated with chromosome topology (38).
The RhopH complex and solute transport are down-regulated during sexual development
To determine whether the expression of RhopH complex genes is reduced in wild-type committed cells, we analyzed their transcript levels in the NF54 line under basal conditions (sexual conversion rate, ~10%) and after stimulating conversion by choline depletion (~50% conversion) (22). Consistent with the results for the E5ind line, the expression of clag3.1 and rhoph3 was lower in stimulated cultures, whereas rhoph2 and rap1 transcript levels did not change (Fig. 6A). The modest magnitude of the differences observed in these experiments was expected given that choline-depleted cultures still contain many asexual parasites. Western blot analysis of E5ind schizont pellets and culture supernatants with antibodies against CLAG3 (cytoadherence linked asexual protein 3) revealed a clear reduction in CLAG3 levels in induced compared to control cultures (Fig. 6B), confirming that the lower transcript levels in committed schizonts translate into lower protein levels.
Fig. 6. Functional characterization of sexually committed schizonts and early sexual stages.
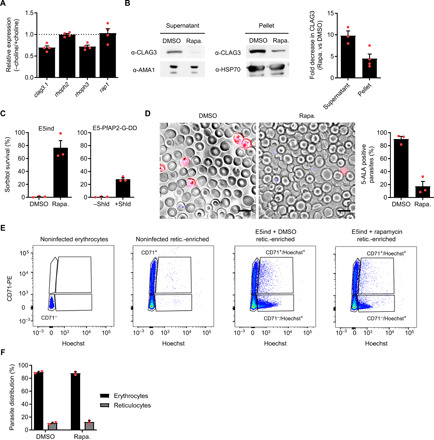
(A) Expression of RhopH complex genes and rap1 (control) in committed schizonts of the NF54 line. Values are the FC between inducing (−choline) and noninducing (+choline) conditions. Transcript levels were normalized against rama. In all panels, data are presented as the average and SEM of independent biological replicates. N = 4. (B) Western blot analysis of E5ind culture supernatant or mature schizont pellets with antibodies against CLAG3 and the loading controls AMA1 or HSP70. The bar chart shows the fold decrease in normalized band intensity between induced and noninduced cultures. N = 3 (supernatant) or 4 (pellet). (C) Sorbitol resistance assay performed ~30 to 40 hpi at the cycle after induction. Values are the percentage of pigmented parasites surviving sorbitol treatment. Shield 1 (Shld) stabilizes PfAP2-G in E5-PfAP2-G-DD cultures. N = 3. (D) Uptake of 5-ALA in induced and noninduced E5ind cultures. Representative fluorescence microscopy images and quantification of the proportion of positive parasites are shown. N = 3. Scale bars, 10 μm. (E) Flow cytometry analysis of invasion preference by committed and noncommitted merozoites. Reticulocytes were identified by CD71 signal and parasites by Hoechst signal. Representative dot plots are shown. PE, phycoerythrin. (F) Distribution of parasites between erythrocytes and reticulocytes after invasion by committed and noncommitted merozoites. N = 2.
The RhopH complex is initially expressed in schizonts. After reinvasion and maturation to the trophozoite stage, it participates in the formation of the plasmodial surface anion channel (PSAC) (36), which determines the permeability of infected erythrocytes to multiple solutes, including sorbitol. To determine whether lower expression of RhopH components in sexually committed schizonts is associated with reduced PSAC activity, we performed sorbitol lysis experiments with E5ind cultures at the cycle following rapamycin induction. A 10-min treatment with 5% sorbitol produced osmotic lysis of all mature parasites (trophozoites and schizonts) in control E5ind cultures, whereas in induced cultures, the majority of parasites (stage I gametocytes) survived. The level of sorbitol survival also correlated with the level of sexual conversion in the independent parasite line E5-PfAP2-G-DD, which has a destabilization domain appended to PfAP2-G to modulate sexual commitment (Fig. 6C) (6). These results are consistent with previous reports showing that gametocytes are more resistant to sorbitol lysis than late stage asexual parasites (39). We also characterized PSAC function by assessing the uptake of 5-aminolevulinic acid (5-ALA), which requires functional PSAC (37). As expected, the proportion of parasites incorporating the compound was much lower in induced than in control cultures (Fig. 6D). Together, these results confirm that the expression of some RhopH complex components is reduced in sexually committed schizonts, resulting in lower PSAC activity in sexual parasites.
Sexually committed merozoites do not have a preference for reticulocyte invasion
The high purity of the E5ind committed schizont preparations enables the phenotypic characterization of parasites at this stage. Differential ligand expression in committed schizonts and merozoites was previously proposed as a mechanism underlying homing of sexual parasites to the bone marrow or enhancing invasion of reticulocytes, which are abundant in this tissue (29, 40). To test whether committed merozoites preferentially invade reticulocytes, we compared the relative invasion of reticulocytes and erythrocytes by Percoll-purified schizonts from induced and control cultures. As a source of reticulocytes and erythrocytes, we used reticulocyte-enriched cord blood. Flow cytometry analysis revealed a similar distribution of new rings in reticulocytes (CD71-positive) and erythrocytes (CD71-negative) between induced and control cultures (Fig. 6, E and F). While we cannot completely exclude the possibility that reticulocytes from other sources may be preferentially invaded by sexually committed merozoites, this result suggests that committed merozoites do not have a specific tropism for reticulocytes.
DISCUSSION
Investigations of sexually committed schizonts and sexual rings have been hampered by the relatively low abundance of these stages in mixed cultures and the difficulty in achieving efficient separation from their asexual counterparts. Here, we describe a conditional activation system for pfap2-g that can be used to induce massive synchronous sexual conversion. The high efficiency of the system, with asexually growing parasites almost absent after induction, enables the characterization of committed schizonts and early sexual stages without any further purification step. We demonstrate the utility of the system by providing a detailed characterization of the transcriptome of these stages and showing its suitability for phenotypic characterization. The system can also be used to characterize early sexual parasites at any other level (e.g., drug susceptibility screens, nascent transcript measurements, and epigenomic, metabolomic, or proteomic analysis). We also used the system to show that pfap2-g activation is sufficient to trigger productive sexual conversion, even in parasite lines with defects in GDV1. The same approach used here to generate the E5ind and 1.2Bind lines could be applied in the future on the background of a parasite line competent for mosquito infection (e.g., NF54). However, while obtaining parasite preparations highly enriched in the initial stages of sexual development was not possible until now, efficient methods to obtain pure preparations of mature gametocytes in large amounts are already available (41, 42). Thus, we consider that our system would have limited utility for the study of mature gametocytes.
Previous systems for inducible sexual conversion in P. falciparum achieved sexual conversion rates of 30 to 60% (5, 6, 10, 14, 19), as the expression of the master regulator pfap2-g was not directly controlled. With a consistent sexual conversion rate of ~90% in the E5ind line and the majority of the remaining ~10% parasites failing to develop rather than growing asexually, the system presented here stands out as the method that yields highest purity P. falciparum early sexual parasite preparations. In induced parasites, the 5′cam promoter is adjacent to the pfap2-g coding sequence, resulting in activation of the gene in the vast majority of parasites. However, the temporal dynamics of expression and transcript levels of pfap2-g resemble endogenous pfap2-g expression in wild-type parasites rather than expression of the endogenous cam gene. While we were unable to fully clarify the underlying mechanism for this unexpected observation, physiological pfap2-g expression is a beneficial feature of the system that favors the correct formation of gametocytes and dispensed the need to screen multiple promoters. Conditional activation of sexual conversion has also been described in P. berghei. Using an approach similar to the one described here, based on conditional activation of ap2-g upon DiCre-mediated promoter flipping, conversion of the majority of parasites was achieved (15).
Together with single-cell transcriptomic approaches, purification of sexual parasites, and comparative analysis of gametocyte producing and nonproducing lines (7, 17, 27, 33, 43, 44), previously reported inducible systems were instrumental to gain insight into the transcriptome of the initial stages of sexual development. However, the high conversion rate obtained with our inducible system provides important advantages for an accurate transcriptomic profiling of these stages. While the majority of genes up-regulated in committed or early sexual parasites had been previously identified by other approaches, the high level of purity of our committed schizont and sexual ring preparations also enabled the identification of down-regulated genes. In previous studies, down-regulation was probably masked by transcripts arising from abundant asexual parasites present in the bulk population. Our approach also provides insight into the temporal order of events in the sexual commitment regulatory cascade: Genes proposed to be altered upstream of PfAP2-G activation, such as the ISWI and SNF2L helicases or GDV1 (7, 14), were not up-regulated upon PfAP2-G activation, supporting the idea that they operate upstream of PfAP2-G. Our results also clearly demonstrate that, while expression of some genes involved in sexual commitment and development may occur independently of PfAP2-G (18, 45), activation of this transcription factor is sufficient to drive the full process. However, characterization of the transcriptional changes occurring immediately upon PfAP2-G activation with high temporal resolution was not possible because of the intrinsic time required for rapamycin-induced recombination with the DiCre system (24).
We show that a relatively small number of genes have altered expression at the committed schizont stage, and a broad remodeling of the transcriptome does not occur until the sexual ring stage. Very few genes involved in erythrocyte invasion showed altered expression in sexually committed versus noncommitted schizonts. Apart from pseudogenes, msrp1 and genes encoding components of the RhopH complex were the only clearly up- and down-regulated invasion-related genes, respectively. Up-regulation of msrp1 in committed schizonts, of still unknown functional significance, has been consistently observed in several studies (7, 10, 14, 17, 19, 29, 33). In contrast, the down-regulation of genes encoding components of the RhopH complex was not previously identified. A previous study reported up-regulation of some components of the complex (33). While the reasons for the discrepancy are unclear, our observations are consistent with the known reduction in PSAC function and sorbitol sensitivity of gametocytes (39), which we confirmed. Our results suggest that committed schizonts express RhopH components at reduced levels to adjust the permeability of sexually developing parasites upon reinvasion, possibly to adapt to lower nutrient needs or to prevent the entry of toxic compounds (46). Of note, genes that are up-regulated and genes that are down-regulated in committed schizonts (but not at other stages) are bound by PfAP2-G. Interactions between PfAP2-G and another ApiAP2 transcription factor, PfAP2-I (47), have been proposed to underlie the regulation of invasion genes in committed schizonts (29). Complex interactions between the two factors may determine the transcriptional outcome of PfAP2-G binding, ranging from activation for the majority of its sexual ring and gametocyte-stage targets to no transcriptional change for many invasion-related genes, and even reduced expression for genes involved in solute transport such as rhoph3 and clag genes.
The insertion of a constitutive promoter to drive pfap2-g expression also provided insight into the regulation of pfap2-g itself. In cultures maintained in the absence of drug pressure, sexual conversion rates were lower, and this was reverted upon reselection with the drug. This is suggestive of reversible heterochromatin expansion from neighbor regions influencing the activation of pfap2-g in the inducible system. Furthermore, in uninduced E5ind and 1.2Bind cultures under constant WR99210 pressure, heterochromatin is present at the pfap2-g coding sequence but not at the promoter, roughly matching the heterochromatin distribution in gametocytes (32). In these parasites, the gene is readily activated upon rapamycin-induced recombination, indicating that heterochromatin at the pfap2-g coding sequence is compatible with active expression of the gene. In contrast, we found that the few parasites that escaped conversion after rapamycin-induced recombination had the endogenous pfap2-g promoter in a heterochromatic state, which was associated with restricted activation of the gene and led to sexual conversion rates that reflected those observed in the respective parental lines. Together, these observations indicate that, in the inducible lines, the constitutive promoter adjacent to the gene, the endogenous promoter and the locus chromatin environment all contribute to pfap2-g regulation. We propose a model for endogenous pfap2-g regulation whereby the coding sequence is constitutively heterochromatic at all stages of the life cycle, including sexually committed and early sexual stages in which the gene is actively expressed. This may serve as a permanent “reservoir” from which heterochromatin spreads to the promoter region at stages at which the gene needs to be epigenetically silenced, including asexual blood stages and mosquito stages (32, 48). In this scenario, activation of pfap2-g expression in committed parasites requires dismantling heterochromatin only at the promoter region.
A possible additional layer of regulation of pfap2-g expression in gametocytes of the inducible lines, operating at the posttranscriptional level, is transcript stability. Of note, altering the activity of the exonuclease PfRNase II resulted in up-regulation of PfAP2-G, among other sexual markers (49). This raises the intriguing possibility that this exonuclease, involved in var gene regulation, also regulates pfap2-g expression. In support of this idea, a recent transcriptomic time course analysis of gametocyte development revealed that the expression of the pfrnase II gene peaks at stage I of gametocyte development, coinciding with the drop in pfap2-g transcripts (27).
In summary, we have developed inducible P. falciparum transgenic lines that show the highest level of sexual conversion reported so far. Using these lines, we provide new insight into the regulation of pfap2-g and the transcriptomic and functional changes that occur during the initial steps of sexual development. These inducible lines will be instrumental to gain a deeper understanding of the formation of sexual stages, which is expected to facilitate the design of new tools to block malaria transmission.
MATERIALS AND METHODS
Parasite cultures and induction of sexual conversion
The 3D7 subclones E5 (derived from the 3D7-B stock) and 1.2B (derived from the 3D7-A stock), the E5-PfAP2-G-DD line, and the NF54 line have been previously described and characterized (6, 26, 41). Parasites were cultured in B+ erythrocytes at 3% hematocrit under standard conditions, with RPMI 1640–based culture medium. For 3D7-derived cultures, we used media supplemented with 0.5% Albumax II (Invitrogen). NF54 cultures were grown in media supplemented with 0.25% Albumax II and 5% human serum (41). To produce gametocytes for mosquito infection, a 4% hematocrit was used. Cultures were regularly synchronized by sorbitol lysis to eliminate late asexual stages (trophozoites and schizonts). For some experiments, cultures were tightly synchronized to a 5-hour age window by purification of schizont stages using Percoll gradients (63% Percoll) followed by sorbitol lysis 5 hours later. The PKG inhibitor ML10 was used at a concentration of 80 nM to inhibit merozoite egress (25). AquaShield-1 (Cheminpharma) was used at a concentration of 0.5 μM to stabilize PfAP2-G in the E5-PfAP2-G-DD line.
To induce sexual conversion, E5ind or 1.2Bind cultures were sorbitol-synchronized and, after synchronization, maintained without WR99210 (Jacobus Pharmaceutical Co., USA). Typically at ~20 hours after synchronization, cultures were treated with 10 nM rapamycin (Sigma-Aldrich, R0395) or DMSO solvent (control) for 1 hour (24), which was followed by one wash with incomplete culture media (culture media without Albumax II) before placing back in culture. After reinvasion, cells were grown in serum-supplemented media, as it improved gametocyte development. Sexual conversion rates were measured by treating cultures at the ring stage after reinvasion (day 0) with 50 mM N-acetylglucosamine (Sigma-Aldrich, A3286) for 5 days to eliminate asexual parasites. The sexual conversion rate was calculated as the gametocytemia at day 5 relative to the initial rings parasitemia at day 0 (5). Parasitemia and gametocytemia were measured by light microscopy quantification of Giemsa-stained smears.
To stimulate sexual conversion in the NF54 line, we used the choline depletion method (14, 19). Cultures were regularly maintained in RPMI 1640-based culture medium with 0.5% Albumax and 2 mM choline (Sigma-Aldrich, C7527), and choline was removed at the ring stage to stimulate conversion, as previously described (22).
Plasmids
The pL7-Ind-ap2g plasmid was derived from the pL6-egfp-yfcu plasmid (20). The yfcu cassette was removed using restriction sites Not I and Sac II. Next, a loxP site was cloned into a BamH I site between the 5′cam and the hdhfr gene using two annealed and phosphorylated oligonucleotides (p1 and p2) containing the loxP sequence. The second loxP site was cloned together with the pfap2-g homology region 2 (HR2) between the 3′ hrp2 and HR2. The loxP sequence was added to primer p3, which was used together with p4 to amplify the HR2 (positions −73 to +311 bp relative to the pfap2-g start codon). The pfap2-g HR1 (positions −449 to −160 bp) was amplified with primers p5 and p6. HR1 and HR2 were cloned into Spe I/Afl II and EcoR I/Nco I sites, respectively. Last, to clone the guide, the plasmid was digested with BtgZ I, and two annealed oligonucleotides (p7 and p8) containing the guide sequence (located at −139 to −158 bp) were cloned into this site using the In-Fusion HD Cloning Kit (Clontech).
Plasmid pHH1-cambsd-DiCre was based on plasmids pHH1_SERA5del3DC (21) and E140-0 (50). The bsd coding sequence was amplified with primers p9 and p10 and cloned into E140-0 using BamH I and Xho I restriction sites, replacing the hdhfr coding sequence and removing unnecessary plasmid elements. After generating Afl II and Spe I restriction sites upstream of the bsd cassette with a PCR-amplified (primers p11 and p12) fragment of the plasmid recloned into Hind III and EcoR I sites, the DiCre cassette obtained by Afl II and Spe I digestion of pHH1SERA5del3DC was cloned into the new Afl II and Spe I sites.
Plasmid pHHI-DiCre-lisp1, which contains the DiCre expression cassette flanked by lisp1 HRs, was derived from pHH1-cambsd-DiCre. First, the HR2 (positions +5891 to +6235 bp relative to the lisp1 start codon) was PCR-amplified using primers p13 and p14 and cloned into Spe I and Not I sites in pHHI-cambsd-DiCre. The HR1 (positions +5169 to +5523 bp) was amplified using primers p15 and p16 and cloned into an Afl II site using the In-Fusion kit system. To generate plasmid pDC2-Cas9-hDHFRyFCU-lisp1, a guide covering positions +5526 to +5545 bp from the lisp1 start codon was prepared by annealing oligonucleotides p17 and p18 and cloning into a Bbs I site of plasmid pDC2-Cas9-hDHFRyFCU (24) using the In-Fusion system.
Guides were designed using the EuPaDGT Web-based tool. PCR amplifications from P. falciparum genomic DNA were performed using LA Taq DNA Polymerase (Takara). To clone the plasmids, we used Escherichia coli DH5α or ultracompetent MAX Efficiency DH5α (Invitrogen) for difficult cloning. Oligonucleotides were from Integrated DNA Technologies. All primers and oligonucleotides are described in table S1.
Generation of transgenic lines
All transfections were performed by electroporation of cultures at the ring stage. For the inducible line with episomal expression of DiCre, E5 cultures were transfected with 60 μg of pUF1-Cas9-ydhodh (20) and 12 μg of pL7-Ind-ap2g linearized using a Sca I site (located in the plasmid backbone). Cultures were permanently maintained under 10 nM WR99210 pressure. After confirming that the pfap2-g locus was correctly edited, cultures were transfected with 100 μg of the pHHI-cambsd-DiCre plasmid and selected continuously with blasticidin S (2.5 μg/ml) (Thermo Fisher Scientific, R21001). For approaches involving stable integration of the DiCre expression cassette, we used a three-plasmid strategy. Sixty micrograms of pDC2-Cas9-hDHFRyFCU-lisp1, 12 μg of pHHI-DiCre-lisp1, and 12 μg of pL7-Ind-ap2g, the latter two linearized using a Sca I site (located in the plasmid backbone), were cotransfected and selected with 10 nM WR99210 for 4 days as previously described (24). Correct edition of the two loci using this strategy was achieved only for the E5 line. For the 1.2B line, we had to use a sequential editing strategy: Cultures were first transfected with 60 μg of pDC2-Cas9-hDHFRyFCU-lisp1 and 12 μg of linearized pHHI-DiCre-lisp1 plasmids and selected with 10 nM WR99210 for 4 days. After subcloning and treatment with 1 μM 5-fluorocytosine (clinical grade Ancotil, Mylan N.V.) for 2 weeks to eliminate parasites that maintain episomal copies of the pDC2-Cas9-hDHFRyFCU-lisp1 plasmid, a second round of transfection was performed with 60 μg of pUF1-Cas9-ydhodh and 12 μg of linearized pL7-Ind-ap2g plasmids, followed by continuous selection with 10 nM WR99210.
Diagnostic PCR to assess the correct integration of the transgenes was performed using the LA Taq DNA Polymerase (Takara). The primers used for diagnostic PCR are described in table S1, and their relative positions are shown in Fig. 1.
RNA extraction and transcriptional analysis by reverse transcriptase quantitative PCR
For the majority of samples, RNA was extracted using the TRIzol method, deoxyribonuclease-treated and purified with a protocol optimized for low amounts of RNA (51), and reverse-transcribed using the AMV Reverse Transcription Kit (Promega) with a mixture of oligo (dT) and random primers. Transcript abundance was measured by real-time quantitative PCR (qPCR) using the standard curve method in a 7900HT Fast Real-Time PCR System and the Power SYBR Green Master Mix (both from Applied Biosystems). For samples from stage V gametocytes, RNA collected in TRIzol was extracted with the Direct-zol RNA MiniPrep Kit (Zymo Research), complementary DNA (cDNA) synthesis performed using the iScript cDNA Synthesis Kit (Bio-Rad), and qPCR analysis of male and female markers (52) was performed using the standard curve method in a LightCycler 480 Instrument II (Roche) with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad). For the standard curve, we used genomic DNA from the same parasite line being analyzed. Transcript levels of the ubiquitin-conjugating enzyme (uce; ID: PF3D7_0812600), serine-tRNA ligase (serrs; ID: PF3D7_0717700), or rhoptry-associated membrane antigen (rama; ID: PF3D7_0707300) were used for normalization as indicated. All primers used for qPCR analysis are described in table S1. For the analysis of pfap2-g transcript levels in Figs. 2 and 3, we used primers p23 and p24.
IFAs and analysis of 5-ALA uptake
For the analysis of asexual parasites and immature gametocytes, IFA analysis was performed on paraformaldehyde (PFA)–fixed smears essentially as described (5). For the analysis of stage V gametocytes or activated gametes, samples were fixed with 4% PFA for 15 min and incubated overnight on top of glass coverslips previously coated with poly-l-lysine (Merck Millipore), followed by permeabilization, blocking, and antibody incubations. To identify ookinetes in mosquito midguts (24 hours after blood feeding), air-dried blood drops from single midguts were PFA-fixed and analyzed by IFA. The primary antibodies used were mouse anti-Pfs16 (1:400 to 1:2000; 32F717:B02, a gift from R. Sauerwein, Radboud University), mouse anti–α-tubulin (1:700; DM1A; Sigma-Aldrich, T6199), rabbit anti–glycophorin A (1:200; EPR8200; Abcam, ab129024), rabbit anti-Pfg377 (1:1000; batch 6809; a gift from L. Ranford-Cartwright, University of Glasgow), and mouse anti-Pfs25 clone 4B7 (1:3000; MRA-315, BEI Resources) conjugated to Cy3 (GE Healthcare) (41). The secondary antibodies were goat anti-mouse immunoglobulin G (IgG)–Alexa Fluor 488 (1:500 to 1:1000; Thermo Fisher Scientific, A11029) and donkey anti-rabbit IgG–Alexa Fluor 594 (1:500; Thermo Fisher Scientific, R37119). Preparations were visualized with an Olympus IX51 epifluorescence microscope, and images were acquired with an Olympus DP72 camera using cellSens Standard 1.11 software or, for experiments with gametes, a Nikon Ti-E widefield microscope with ×60 to 100 objective lens and captured in 0.3-μm slice z-stack images, which were converted to maximum intensity projections in NIS Elements v4.20. All further image processing was performed using ImageJ.
5-ALA uptake was determined as previously described (37, 46). Synchronized E5ind cultures were treated with rapamycin or DMSO and, after reinvasion, when cultures were at the ring stage, 200 μM 5-ALA (Sigma-Aldrich, A3785) was added. Inside of infected erythrocytes, 5-ALA is converted to fluorescent protoporphyrin IX. Uptake was determined at ~30 to 40 hpi after staining nuclei with Hoechst (2 μg/ml) for 10 min at 37°C in an eight-well chamber slide for live cell fluorescence microscopy. Images were acquired using an Olympus IX51 epifluorescence microscope and analyzed using ImageJ.
Chromatin immunoprecipitation sequencing
Chromatin extraction from cultures at the late trophozoite/schizont stage was performed as previously described (30), with minor modifications. Briefly, after the cross-linking and washing steps, the MAGnify Chromatin Immunoprecipitation System (Life Technologies) was used. Samples were sonicated using an M220 sonicator (Covaris) at 10% duty factor, 200 cycles per burst, 140 W of peak incident power for 10 min. Immunoprecipitations were performed overnight at 4°C with 4 μg of chromatin and 8 μg of antibodies against H3K9me3 (Diagenode, C15410193) previously coupled to protein A/G magnetic beads provided in the kit. Washing, decross-linking, and elution were performed following the MAGnify ChIP System recommendations but avoiding high temperatures that may result in denaturation of extremely AT-rich intergenic regions: De-crosslinking, proteinase K treatment, and elution were performed at 45°C (for 2 hours, overnight, and 1.5 hours, respectively).
Libraries for Illumina sequencing were prepared from 5 ng of immunoprecipitated DNA using a protocol adapted to a genome with an extremely high AT richness (53). Briefly, after end repair and addition of 3′ A-overhangs, NEBNext Multiplex Oligos for Illumina (NEB, E7335 and E7500) were ligated. Purification steps were performed with Agencourt AMPure XP beads (Beckman Coulter). Libraries were amplified using the KAPA HiFi PCR Kit (Kapa Biosystems) in KAPA HiFi Fidelity Buffer (5×) with the following conditions: 95°C for 3 min, nine cycles at 98°C for 20 s and 62°C for 2.5 min, and 62°C for 5 min. Amplified libraries were purified using 0.9× AMPure XP beads to remove adapter dimers. The library size was analyzed in a 4200 TapeStation System (Agilent Technologies). We obtained 12 to 26 million 125 bp paired-end reads per sample using a HiSeq2500 System (Illumina).
Transcriptomic analysis using microarrays
Time course transcriptomic analysis was performed using tightly synchronized cultures induced at 25 to 30 hpi. Samples for transcriptomic analysis were collected 15, 28, and 38 hours after induction (corresponding to 40 to 45 hpi schizonts of the first cycle and ~5 to 10 or ~15 to 20 hpi rings of the next cycle, respectively). RNA was purified using the TRIzol method, and cDNA was synthesized, purified, and labeled essentially as described (26, 54). Samples were analyzed on two-color long oligonucleotide-based custom Agilent microarrays. The microarray design was based on Agilent design AMADID #037237 (54), modified by adding new probes for genes lacking unique probes and for some noncoding RNAs and reporter genes (new designs: AMADID-084561 and AMADID-085763; data file S3). All samples, labeled with Cy5, were hybridized against a reference pool labeled with Cy3, consisting of a mixture of cDNA from rings, trophozoites, and schizonts of E5 and E5ind in equal amounts. Microarray hybridization and image acquisition were performed as described using a Microarray Scanner (no. G2505C, Agilent Technologies) located in a low ozone hood (54).
Bioinformatic analysis
For the analysis of ChIP-seq data, we first removed repetitive k-mers from the reads using BBDuK (v36.99) with parameters ktrim = r, k = 22, and mink = 6. After this initial filtering, reads where aligned to modified 3D7 reference genomes reflecting the alterations introduced by CRISPR-Cas9 genome editing and rapamycin-induced recombination (i.e., integration of the DiCre expression cassette into the lisp1 locus, integration of the conditional activation cassette upstream of the pfap2-g coding sequence, and recombination between loxP sites in rapamycin-treated samples). Reads alignment was performed using Bowtie (v1.2) with parameters --very-sensitive and --local, trimming 4 bp from both the 5′ and 3′ ends and restricting fragment size to 50 to 200 bp. After aligning, duplicate reads where removed using PicardTools MarkDuplicates (v2.9.4). For each sample, per-base coverage was calculated using BEDtools (v2.27.1) and divided by the number of million reads (RPM). For each base, H3K9me3 enrichment was calculated as ChIP (RPM)/input (RPM) with a pseudocount of 0.1 to avoid division by 0. Data were visualized using IGV (Integrative Genomics Viewer; v2.4.10).
Initial processing of microarray data, including normalization, was performed using Feature Extraction software (Agilent) with default options. The next steps of the analysis were performed using Bioconductor in an R environment (R version 3.5.3). For each individual microarray, we calculated Cy3 and Cy5 background signals as the median of the 100 lowest signal probes for each channel. Probes with both Cy3 and Cy5 signals below three times the array background were excluded. Gene level log2(Cy5/Cy3) values and statistical estimation of parasite age were computed as described (26). Genes with missing values or genes that in all samples had expression values within the lowest 20th percentile of intensity (Cy5 channel) were excluded from downstream analysis, including identification of differentially expressed genes, because differences in genes expressed at near-background levels are of low confidence. Heatmaps were generated using TMEV 4.9. Gene set enrichment analysis (GSEA) with gene lists corresponding to gene families (data file S2) was performed using GSEA preranked.
Western blot
Sample preparation for Western blot analysis was performed essentially as previously described (50). Briefly, Percoll-purified schizonts were placed in culture at a 0.3% hematocrit, and, after 13 to 20 hours, culture supernatants were harvested and mixed with an equal volume of 2× SDS–polyacrylamide gel electrophoresis (PAGE) protein loading buffer. Schizont extracts were prepared by resuspending Percoll-purified schizonts into 20 pellet volumes of phosphate-buffered saline (PBS) and adding an equal volume of 2× SDS-PAGE protein loading buffer. Samples were heated for 5 min at 95°C before storing at −80°C. For SDS-PAGE, β-mercaptoethanol was added to a 4% final concentration, and, after boiling again for 5 min at 95°C, proteins were resolved in 4 to 8% SDS-PAGE, transferred to nitrocellulose membranes, incubated with antibodies, and washed following standard procedures. The primary antibodies used were mouse anti-CLAG3 polyclonal antibodies (36) at 1:2000 (167#2, recognize both CLAG3.1 and CLAG3.2; a gift from S. A. Desai, NIAID-NIH, USA), rabbit anti-3D7 AMA1 polyclonal antibodies at 1:2000 (a gift from R. F. Anders, La Trobe University, Australia), and rabbit anti-PfHSP70 polyclonal antibodies at 1:10,000 (StressMarq Biosciences, SPC-186C, lot no. 1007). As secondary antibody, we used anti-mouse-horseradish peroxidase (HRP) (Sigma-Aldrich, A9044) or anti-rabbit-HRP (Sigma-Aldrich, A6154) at 1:5000. HRP signal was detected using the Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific), and membranes were imaged in a LAS4000 system. Band quantification was performed using ImageJ.
Reticulocyte invasion assay
Human cord blood samples were obtained from the Barcelona Blood and Tissue Bank (www.bancsang.net/), following approval for the protocol and informed consent by the Clinical Research Ethics Committee of Vall d’Hebron University Hospital [PR(CS)236/2017]. Human reticulocyte preparations were performed as previously described (55), with some modifications. Briefly, samples were centrifuged at 1000g for 15 min (acceleration = 6, deceleration = 2), and plasma was removed. Cell pellets were washed with incomplete culture media at 500g for 10 min and resuspended at 50% hematocrit with incomplete culture media. Five milliliters of resuspended blood was carefully loaded on 6 ml of 70% Percoll (GE Healthcare) and centrifuged at 1200g for 15 min at 20°C (acceleration = 4, deceleration = 0). Reticulocytes concentrated in the Percoll interface were collected and washed three times with incomplete culture media (centrifugation at 400g for 5 min). To deplete leukocytes, reticulocyte-enriched pellets were resuspended to a final volume of 900 μl with incomplete culture media and incubated with 100 μl of CD45 MicroBeads (Miltenyi Biotec, 130-045-801) for 18 min at 4°C, mixing every 3 min. The cells were washed with incomplete culture media and passed through LS Columns (Miltenyi Biotec, 130-042-401) placed on a magnetic stand. Eluents, containing reticulocyte-enriched fractions, were washed twice with incomplete culture media, and reticulocyte enrichment was quantified by incubating for 15 min at room temperature with anti–CD71–phycoerythrin (PE) (Miltenyi Biotec, clone AC102, 130-099-219) at a final dilution of 1:400. Reticulocyte quantification was performed using a BD LSRFortessa flow cytometer.
E5ind schizonts treated with rapamycin or DMSO were purified in 70% Percoll gradients and mixed with reticulocyte-enriched or control regular erythrocyte preparations to achieve a ~1% parasitemia. After establishing 3% hematocrit cultures, parasites were allowed to reinvade overnight and analyzed the following day by flow cytometry. Samples were washed in PBS and incubated for 15 min at room temperature with anti–CD71-PE antibodies. After washing with PBS, parasite nuclei were stained with Hoechst (2 μg/ml) for 20 min. Samples were washed in PBS again before analysis in a BD LSRFortessa Cytometer. A total of 200,000 events were recorded per sample. After gating erythrocytes, forward scatter height (FSC-H) versus forward scatter area (FSC-A) plots were used to define the singlets population. Unstained samples and uninfected erythrocyte controls were used to define the thresholds for positivity.
Gamete activation assays
To evaluate exflagellation, a 30-μl sample from day 12 E5ind and 1.2Bind mature gametocyte cultures was mixed with an equal volume of ookinete medium [100 μM xanthurenic acid, sodium bicarbonate (2 g/liter), and hypoxanthine (50 mg/liter) in RPMI 1640–Hepes (pH 7.4)] with a 1:1500 dilution of anti–Pfs25-Cy3 antibody. As a control, we used NF54 mature gametocytes induced as previously described (41). Part of the preparation was immediately transferred to a hemocytometer and incubated at room temperature. After 15 min, exflagellation centers and erythrocyte density were counted using bright-field microscopy (10× magnification) to calculate the percentage of exflagellation (number of exflagellating centers relative to the number of erythrocytes) (56). To compare exflagellation between experiments, we determined exflagellation rates by normalizing the percentage of exflagellation by the gametocytemia.
To determine the number of egressing females, the rest of the preparation was incubated overnight at 26°C in the darkness to allow for maximal expression of Pfs25 on the macrogamete surface (56). Female numbers were counted on FastRead slides (Immune Systems) and normalized by gametocytemia. For IFA analysis of activated male gametes, 200 μl of stage V gametocyte cultures was mixed with 200 μl of ookinete medium and incubated at room temperature for 25 min before fixing with 4% PFA.
Mosquito infection experiments
E5ind, 1.2Bind, and NF54 mature gametocyte cultures (days 12 to 16) were provided to 3- to 7-day-old Anopheles stephensi mosquitoes that were starved the day before the feed. Cultures for mosquito feeding were always manipulated at 37°C to prevent premature activation of the gametocytes. Fifty to 150 μl of prewarmed fresh human erythrocytes was used as a cushion to add 10 to 40 ml of gametocyte culture on top. The mixture was centrifuged at 500g for 5 min at 38°C, the supernatant was removed, and the pellet was mixed with an equal volume of prewarmed human serum. Five hundred microliters of the mixture was transferred into three-dimensionally printed feeders attached to a 38°C circulating water bath (Grant Instruments) (57), with Parafilm placed at the bottom of the feeders to have a thin membrane through which the mosquitoes can feed. Mosquitoes were allowed to feed for 20 to 30 min and then maintained at 26°C and 80% humidity. Twenty-four hours after feeding, unfed mosquitoes were removed. To determine the presence of ookinetes, mosquitoes were dissected to obtain the midguts (also 24 hours after feeding), the blood bolus was smeared onto a glass slide and fixed with 4% PFA, and ookinetes/females were detected using the Pfs25 antibody. Nine days after the feed, mosquito midguts were stained in 0.1% mercurochrome in PBS for 15 min, fixed in 4% PFA, and stained with 4′,6-diamidino-2-phenylindole (DAPI). Infection intensity was assessed by counting the number of oocysts per midgut with a 20× objective.
Supplementary Material
Acknowledgments
We thank H. A. del Portillo (ICREA at ISGlobal) for helpful scientific discussions; G. A. Josling and M. Llinás (Pennsylvania State University) for providing the list of PfAP2-G ChIP-seq targets before publication; M. Llinás for useful comments on the manuscript; R. Bartfai (Radboud University) for technical advice for ChIP-seq; H. Portugaliza and C. Bancells (ISGlobal) for providing NF54 cDNAs and for useful discussion; A. Churchyard, S. Yahiya, and F. Dahalan (Imperial College) for technical assistance with gamete activation and mosquito feeding assays; C. Collins and M. Blackman (The Francis Crick Institute) for providing plasmid pHH1_SERA5del3DC; J. J. López-Rubio (University of Montpellier) for plasmid pL6-egfp-yfcu; E. Knuepfer (The Francis Crick Institute) for plasmid pDC2-Cas9-hDHFRyFCU; S. Osborne (LifeArc) and D. A. Baker (LSHTM) for the compound ML10; R. W. Sauerwein (Radboud University) for the anti-Pfs16 antibody; L. Ranford-Cartwright (University of Glasgow) for the anti-Pfg377 antibody; S. A. Desai (NIAID-NIH) for the anti-CLAG3 antibody; and R. F. Anders (La Trobe University) for the anti-AMA1 antibody. Funding: This work was supported by a grant from the Spanish Ministry of Economy and Competitiveness (MINECO)/Agencia Estatal de Investigación (AEI) (SAF2016-76190-R to A.C.), cofunded by the European Regional Development Fund (ERDF, European Union), and also received funding from “la Caixa” Banking Foundation under the project code HR18-00267 awarded to A.C. O.L.-B. was supported by a FPU fellowship from the Spanish Ministry of Education, Culture and Sports (FPU014/02456) and an EMBO Short-Term Fellowship (no. 7732). L.M.-T. was supported by a fellowship from the Spanish Ministry of Economy and Competitiveness (BES-2017-081079). H.T. was supported by Secretaria d’Universitats i Recerca del Departament d’Economia i Creixement, Generalitat de Catalunya. C.F.-B. was partially supported by a grant from MINECO (SAF2016-80655-R). Work in the Baum laboratory was supported through funding from Wellcome (Investigator Award 100993/Z/13/Z). This research is part of ISGlobal’s Program on the Molecular Mechanisms of Malaria, which is partially supported by the Fundación Ramón Areces. We acknowledge support from the Spanish Ministry of Science and Innovation through the “Centro de Excelencia Severo Ochoa 2019-2023” Program (CEX2018-000806-S) and support from the Generalitat de Catalunya through the CERCA Program. Author contributions: O.L.-B. performed the experiments. L.M.-T. and O.L.-B. performed the bioinformatics analysis. K.W. performed experiments, provided training, and, together with J.B., provided supervision for experiments with mature gametocytes and mosquito infections. H.T. and C.F.-B. provided reticulocyte-enriched cord blood and supervision for reticulocyte invasion experiments. O.L.-B. and A.C. conceived the project, designed the experiments, and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The ChIP-seq and microarray data have been deposited in the GEO database with accession number GSE135006. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Materials and protocols are available from the corresponding author on reasonable request. The scripts used for the analysis of microarray and ChIP-seq data will be made available upon reasonable request, without any restrictions.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/24/eaaz5057/DC1
REFERENCES AND NOTES
- 1.Josling G. A., Williamson K. C., Llinás M., Regulation of sexual commitment and gametocytogenesis in malaria parasites. Annu. Rev. Microbiol. 72, 501–519 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meibalan E., Marti M., Biology of Malaria Transmission. Cold Spring Harb. Perspect. Med. 7, a025452 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The malERA Refresh Consultative Panel on Basic Science and Enabling Technologies , malERA: An updated research agenda for basic science and enabling technologies in malaria elimination and eradication. PLOS Med. 14, e1002451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruce M. C., Alano P., Duthie S., Carter R., Commitment of the malaria parasite Plasmodium falciparum to sexual and asexual development. Parasitology 100, 191–200 (1990). [DOI] [PubMed] [Google Scholar]
- 5.Bancells C., Llorà-Batlle O., Poran A., Nötzel C., Rovira-Graells N., Elemento O., Kafsack B. F. C., Cortés A., Revisiting the initial steps of sexual development in the malaria parasite Plasmodium falciparum. Nat. Microbiol. 4, 144–154 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kafsack B. F., Rovira-Graells N., Clark T. G., Bancells C., Crowley V. M., Campino S. G., Williams A. E., Drought L. G., Kwiatkowski D. P., Baker D. A., Cortés A., Llinás M., A transcriptional switch underlies commitment to sexual development in malaria parasites. Nature 507, 248–252 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poran A., Nötzel C., Aly O., Mencia-Trinchant N., Harris C. T., Guzman M. L., Hassane D. C., Elemento O., Kafsack B. F. C., Single-cell RNA sequencing reveals a signature of sexual commitment in malaria parasites. Nature 551, 95–99 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sinha A., Hughes K. R., Modrzynska K. K., Otto T. D., Pfander C., Dickens N. J., Religa A. A., Bushell E., Graham A. L., Cameron R., Kafsack B. F., Williams A. E., Llinás M., Berriman M., Billker O., Waters A. P., A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature 507, 253–257 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeninga M. D., Quinn J. E., Petter M., ApiAP2 transcription factors in apicomplexan parasites. Pathogens 8, E47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brancucci N. M., Bertschi N. L., Zhu L., Niederwieser I., Chin W. H., Wampfler R., Freymond C., Rottmann M., Felger I., Bozdech Z., Voss T. S., Heterochromatin protein 1 secures survival and transmission of malaria parasites. Cell Host Microbe 16, 165–176 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Lopez-Rubio J.-J., Mancio-Silva L., Scherf A., Genome-wide analysis of heterochromatin associates clonally variant gene regulation with perinuclear repressive centers in malaria parasites. Cell Host Microbe 5, 179–190 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Flueck C., Bartfai R., Volz J., Niederwieser I., Salcedo-Amaya A. M., Alako B. T., Ehlgen F., Ralph S. A., Cowman A. F., Bozdech Z., Stunnenberg H. G., Voss T. S., Plasmodium falciparum heterochromatin protein 1 marks genomic loci linked to phenotypic variation of exported virulence factors. PLOS Pathog. 5, e1000569 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eksi S., Morahan B. J., Haile Y., Furuya T., Jiang H., Ali O., Xu H., Kiattibutr K., Suri A., Czesny B., Adeyemo A., Myers T. G., Sattabongkot J., Su X.-z., Williamson K. C., Plasmodium falciparum gametocyte development 1 (Pfgdv1) and gametocytogenesis early gene identification and commitment to sexual development. PLOS Pathog. 8, e1002964 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filarsky M., Fraschka S. A., Niederwieser I., Brancucci N. M. B., Carrington E., Carrió E., Moes S., Jenoe P., Bártfai R., Voss T. S., GDV1 induces sexual commitment of malaria parasites by antagonizing HP1-dependent gene silencing. Science 359, 1259–1263 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kent R. S., Modrzynska K. K., Cameron R., Philip N., Billker O., Waters A. P., Inducible developmental reprogramming redefines commitment to sexual development in the malaria parasite Plasmodium berghei. Nat. Microbiol. 3, 1206–1213 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker D. A., Malaria gametocytogenesis. Mol. Biochem. Parasitol. 172, 57–65 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pelle K. G., Oh K., Buchholz K., Narasimhan V., Joice R., Milner D. A., Brancucci N. M., Ma S., Voss T. S., Ketman K., Seydel K. B., Taylor T. E., Barteneva N. S., Huttenhower C., Marti M., Transcriptional profiling defines dynamics of parasite tissue sequestration during malaria infection. Genome Med. 7, 19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tibúrcio M., Dixon M. W., Looker O., Younis S. Y., Tilley L., Alano P., Specific expression and export of the Plasmodium falciparum Gametocyte EXported Protein-5 marks the gametocyte ring stage. Malar. J. 14, 334 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brancucci N. M. B., Gerdt J. P., Wang C., De Niz M., Philip N., Adapa S. R., Zhang M., Hitz E., Niederwieser I., Boltryk S. D., Laffitte M. C., Clark M. A., Grüring C., Ravel D., Blancke Soares A., Demas A., Bopp S., Rubio-Ruiz B., Conejo-Garcia A., Wirth D. F., Gendaszewska-Darmach E., Duraisingh M. T., Adams J. H., Voss T. S., Waters A. P., Jiang R. H. Y., Clardy J., Marti M., Lysophosphatidylcholine regulates sexual stage differentiation in the human malaria parasite Plasmodium falciparum. Cell 171, 1532–1544.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghorbal M., Gorman M., Macpherson C. R., Martins R. M., Scherf A., Lopez-Rubio J.-J., Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat. Biotechnol. 32, 819–821 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Collins C. R., Das S., Wong E. H., Andenmatten N., Stallmach R., Hackett F., Herman J.-P., Müller S., Meissner M., Blackman M. J., Robust inducible Cre recombinase activity in the human malaria parasite Plasmodium falciparum enables efficient gene deletion within a single asexual erythrocytic growth cycle. Mol. Microbiol. 88, 687–701 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Portugaliza H. P., Llorà-Batlle O., Rosanas-Urgell A., Cortés A., Reporter lines based on the gexp02 promoter enable early quantification of sexual conversion rates in the malaria parasite Plasmodium falciparum. Sci. Rep. 9, 14595 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishino T., Boisson B., Orito Y., Lacroix C., Bischoff E., Loussert C., Janse C., Ménard R., Yuda M., Baldacci P., LISP1 is important for the egress of Plasmodium berghei parasites from liver cells. Cell. Microbiol. 11, 1329–1339 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knuepfer E., Napiorkowska M., van Ooij C., Holder A. A., Generating conditional gene knockouts in Plasmodium – a toolkit to produce stable DiCre recombinase-expressing parasite lines using CRISPR/Cas9. Sci. Rep. 7, 3881 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baker D. A., Stewart L. B., Large J. M., Bowyer P. W., Ansell K. H., Jiménez-Díaz M. B., El Bakkouri M., Birchall K., Dechering K. J., Bouloc N. S., Coombs P. J., Whalley D., Harding D. J., Smiljanic-Hurley E., Wheldon M. C., Walker E. M., Dessens J. T., Lafuente M. J., Sanz L. M., Gamo F.-J., Ferrer S. B., Hui R., Bousema T., Angulo-Barturén I., Merritt A. T., Croft S. L., Gutteridge W. E., Kettleborough C. A., Osborne S. A., A potent series targeting the malarial cGMP-dependent protein kinase clears infection and blocks transmission. Nat. Commun. 8, 430 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rovira-Graells N., Gupta A. P., Planet E., Crowley V. M., Mok S., Ribas de Pouplana L., Preiser P. R., Bozdech Z., Cortés A., Transcriptional variation in the malaria parasite Plasmodium falciparum. Genome Res. 22, 925–938 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Biljon R., van Wyk R., Painter H. J., Orchard L., Reader J., Niemand J., Llinás M., Birkholtz L.-M., Hierarchical transcriptional control regulates Plasmodium falciparum sexual differentiation. BMC Genomics 20, 920 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crabb B. S., Triglia T., Waterkeyn J. G., Cowman A. F., Stable transgene expression in Plasmodium falciparum. Mol. Biochem. Parasitol. 90, 131–144 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Josling G. A., Russell T. J., Venezia J., Orchard L., van Biljon R., Painter H. J., Llinás M., Dissecting the role of PfAP2-G in malaria gametocytogenesis. Nat. Commun. 11, 1503 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rovira-Graells N., Crowley V. M., Bancells C., Mira-Martínez S., Ribas de Pouplana L., Cortés A., Deciphering the principles that govern mutually exclusive expression of Plasmodium falciparum clag3 genes. Nucleic Acids Res. 43, 8243–8257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Voss T. S., Healer J., Marty A. J., Duffy M. F., Thompson J. K., Beeson J. G., Reeder J. C., Crabb B. S., Cowman A. F., A var gene promoter controls allelic exclusion of virulence genes in Plasmodium falciparum malaria. Nature 439, 1004–1008 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Fraschka S. A., Filarsky M., Hoo R., Niederwieser I., Yam X. Y., Brancucci N. M. B., Mohring F., Mushunje A. T., Huang X., Christensen P. R., Nosten F., Bozdech Z., Russell B., Moon R. W., Marti M., Preiser P. R., Bártfai R., Voss T. S., Comparative heterochromatin profiling reveals conserved and unique epigenome signatures linked to adaptation and development of malaria parasites. Cell Host Microbe 23, 407–420.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brancucci N. M. B., De Niz M., Straub T. J., Ravel D., Sollelis L., Birren B. W., Voss T. S., Neafsey D. E., Marti M., Probing Plasmodium falciparum sexual commitment at the single-cell level. Wellcome Open Res. 3, 70 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meerstein-Kessel L., van der Lee R., Stone W., Lanke K., Baker D. A., Alano P., Silvestrini F., Janse C. J., Khan S. M., van de Vegte-Bolmer M., Graumans W., Siebelink-Stoter R., Kooij T. W. A., Marti M., Drakeley C., Campo J. J., van Dam T. J. P., Sauerwein R., Bousema T., Huynen M. A., Probabilistic data integration identifies reliable gametocyte-specific proteins and transcripts in malaria parasites. Sci. Rep. 8, 410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ito D., Schureck M. A., Desai S. A., An essential dual-function complex mediates erythrocyte invasion and channel-mediated nutrient uptake in malaria parasites. eLife 6, e23485 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguitragool W., Bokhari A. A., Pillai A. D., Rayavara K., Sharma P., Turpin B., Aravind L., Desai S. A., Malaria parasite clag3 genes determine channel-mediated nutrient uptake by infected red blood cells. Cell 145, 665–677 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sherling E. S., Knuepfer E., Brzostowski J. A., Miller L. H., Blackman M. J., van Ooij C., The Plasmodium falciparum rhoptry protein RhopH3 plays essential roles in host cell invasion and nutrient uptake. eLife 6, e23239 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szabo Q., Bantignies F., Cavalli G., Principles of genome folding into topologically associating domains. Sci. Adv. 5, eaaw1668 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saul A., Graves P., Edser L., Refractoriness of erythrocytes infected with Plasmodium falciparum gametocytes to lysis by sorbitol. Int. J. Parasitol. 20, 1095–1097 (1990). [DOI] [PubMed] [Google Scholar]
- 40.Nilsson S. K., Childs L. M., Buckee C., Marti M., Targeting human transmission biology for malaria elimination. PLOS Pathog. 11, e1004871 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delves M. J., Straschil U., Ruecker A., Miguel-Blanco C., Marques S., Baum J., Sinden R. E., Routine in vitro culture of P. falciparum gametocytes to evaluate novel transmission-blocking interventions. Nat. Protoc. 11, 1668–1680 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Duffy S., Loganathan S., Holleran J. P., Avery V. M., Large-scale production of Plasmodium falciparum gametocytes for malaria drug discovery. Nat. Protoc. 11, 976–992 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Silvestrini F., Bozdech Z., Lanfrancotti A., Di Giulio E., Bultrini E., Picci L., deRisi J. L., Pizzi E., Alano P., Genome-wide identification of genes upregulated at the onset of gametocytogenesis in Plasmodium falciparum. Mol. Biochem. Parasitol. 143, 100–110 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Silvestrini F., Lasonder E., Olivieri A., Camarda G., van Schaijk B., Sanchez M., Younis Younis S., Sauerwein R., Alano P., Protein export marks the early phase of gametocytogenesis of the human malaria parasite Plasmodium falciparum. Mol. Cell. Proteomics 9, 1437–1448 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Painter H. J., Carrasquilla M., Llinás M., Capturing in vivo RNA transcriptional dynamics from the malaria parasite Plasmodium falciparum. Genome Res. 27, 1074–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mira-Martínez S., Pickford A. K., Rovira-Graells N., Guetens P., Tintó-Font E., Cortés A., Rosanas-Urgell A., Identification of antimalarial compounds that require CLAG3 for their uptake by Plasmodium falciparum-infected erythrocytes. Antimicrosb. Agents Chemother. 63, e00052-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Santos J. M., Josling G., Ross P., Joshi P., Orchard L., Campbell T., Schieler A., Cristea I. M., Llinás M., Red blood cell invasion by the malaria parasite is coordinated by the PfAP2-I transcription factor. Cell Host Microbe 21, 731–741.e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gómez-Díaz E., Yerbanga R. S., Lefèvre T., Cohuet A., Rowley M. J., Ouedraogo J. B., Corces V. G., Epigenetic regulation of Plasmodium falciparum clonally variant gene expression during development in Anopheles gambiae. Sci. Rep. 7, 40655 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Q., Siegel T. N., Martins R. M., Wang F., Cao J., Gao Q., Cheng X., Jiang L., Hon C.-C., Scheidig-Benatar C., Sakamoto H., Turner L., Jensen A. T., Claes A., Guizetti J., Malmquist N. A., Scherf A., Exonuclease-mediated degradation of nascent RNA silences genes linked to severe malaria. Nature 513, 431–435 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Cortés A., Carret C., Kaneko O., Yim Lim B. Y., Ivens A., Holder A. A., Epigenetic silencing of Plasmodium falciparum genes linked to erythrocyte invasion. PLOS Pathog. 3, e107 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mira-Martínez S., van Schuppen E., Amambua-Ngwa A., Bottieau E., Affara M., Van Esbroeck M., Vlieghe E., Guetens P., Rovira-Graells N., Gómez-Pérez G. P., Alonso P. L., D'Alessandro U., Rosanas-Urgell A., Cortés A., Expression of the Plasmodium falciparum clonally variant clag3 genes in human infections. J. Infect. Dis. 215, 938–945 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Santolamazza F., Avellino P., Siciliano G., Yao F. A., Lombardo F., Ouédraogo J. B., Modiano D., Alano P., Mangano V. D., Detection of Plasmodium falciparum male and female gametocytes and determination of parasite sex ratio in human endemic populations by novel, cheap and robust RTqPCR assays. Malar. J. 16, 468 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kensche P. R., Hoeijmakers W. A., Toenhake C. G., Bras M., Chappell L., Berriman M., Bártfai R., The nucleosome landscape of Plasmodium falciparum reveals chromatin architecture and dynamics of regulatory sequences. Nucleic Acids Res. 44, 2110–2124 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Painter H. J., Altenhofen L. M., Kafsack B. F., Llinás M., Whole-genome analysis of Plasmodium spp. utilizing a new agilent technologies DNA microarray platform. Methods Mol. Biol. 923, 213–219 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Díaz-Varela M., de Menezes-Neto A., Perez-Zsolt D., Gámez-Valero A., Seguí-Barber J., Izquierdo-Useros N., Martinez-Picado J., Fernández-Becerra C., Del Portillo H. A., Proteomics study of human cord blood reticulocyte-derived exosomes. Sci. Rep. 8, 14046 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Delves M. J., Ruecker A., Straschil U., Lelièvre J., Marques S., López-Barragán M. J., Herreros E., Sinden R. E., Male and female Plasmodium falciparum mature gametocytes show different responses to antimalarial drugs. Antimicrob. Agents Chemother. 57, 3268–3274 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Witmer K., Sherrard-Smith E., Straschil U., Tunnicliff M., Baum J., Delves M., An inexpensive open source 3D-printed membrane feeder for human malaria transmission studies. Malar. J. 17, 282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/24/eaaz5057/DC1