

Summary
We report that neurofibromin, a tumor suppressor and Ras-GAP (GTPase Activating Protein), is also an estrogen receptor-α (ER) transcriptional co-repressor through leucine/isoleucine-rich motifs that are functionally independent of GAP activity. GAP activity, in turn, does not impact ER binding. Consequently, neurofibromin-depletion causes estradiol hypersensitivity and tamoxifen agonism, explaining the poor prognosis associated with neurofibromin-loss in endocrine therapy-treated ER+ breast cancer. Neurofibromin-deficient ER+ breast cancer cells initially retain sensitivity to selective estrogen receptor degraders (SERDs). However, Ras activation does play a role in acquired SERD resistance, which can be reversed upon MEK inhibitor addition, and SERD/MEK inhibitor combinations induce tumor regression. Thus, neurofibromin is a dual repressor for both Ras and ER signaling, and co-targeting may treat neurofibromin-deficient ER+ breast tumors.
Graphical Abstract
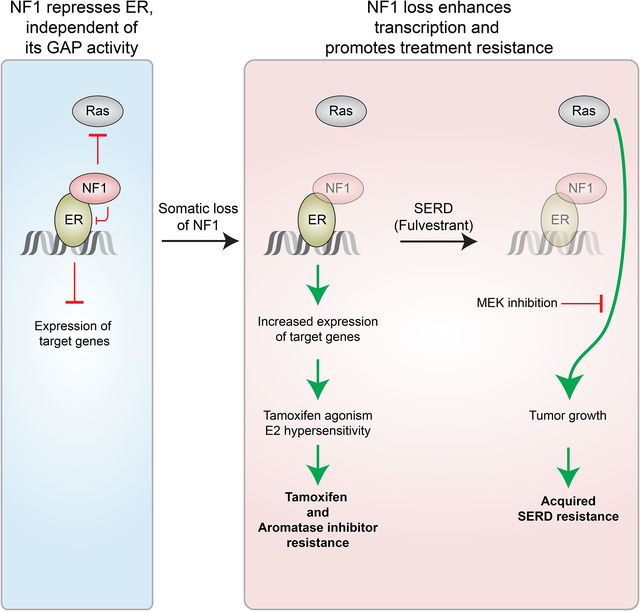
In Brief (eTOC blurb)
Zheng et al. find that the Ras-GAP NF1 is also a transcriptional co-repressor of estrogen receptor α (ER). NF1 loss leads to estradiol hypersensitivity and tamoxifen agonism. A selective ER degrader and MEK inhibitor combination induces tumor regression in mouse models of NF1-deficient ER+ breast cancer.
Introduction
Germline mutations in the NF1 (Neurofibromatosis type 1) gene are responsible for neurofibromatosis type 1, the most common inherited disorder that predisposes individuals to both benign and malignant tumors of the nervous system (Ratner and Miller, 2015), as well as an increased risk for breast cancer (Madanikia et al., 2012; Salemis et al., 2010; Sharif et al., 2007). Analysis of TCGA data has shown that NF1 is mutated in a wide range of common cancers (e.g., melanoma, lymphoma, and cancers of the lung, breast, and colon) (Yap et al., 2014). Thus, NF1-deficiency underlies the formation and/or progression of a large number of cancers.
NF1 encodes neurofibromin, an established GTPase-Activating-Protein (GAP), that attenuates Ras signaling (Maertens and Cichowski, 2014). The human genome contains up to 14 Ras GAPs that share no significant sequence homology beyond the GAP domain. However, across species there is extensive evolutionary sequence conservation outside of the relatively small GAP domain (e.g., 83% overall identity in amino acid sequence between human and salmon neurofibromins), suggesting that neurofibromin has other yet undisclosed functional domains.
Approximately 80% of breast cancers are positive for estrogen receptor-α (ER+), a ligand-dependent transcription factor that is activated by estradiol (E2) (Feng and O’Malley, 2014). The E2-liganded ER recruits co-activators (such as steroid receptor co-activators, SRC-1–3) to estrogen-responsive elements (EREs) in ER-regulated genes. Tamoxifen, a selective estrogen receptor modulator (SERM), is predominantly antagonistic in breast cancer cells, hence its therapeutic effect. When tamoxifen binds to ER, co-activators are displaced by co-repressors in the ER-ERE complex (Shang et al., 2000). Established ER co-repressors bind ER via their leucine/isoleucine-rich motifs, and substitutions of L or I with an A can disrupt binding (Hu and Lazar, 1999). Interactions are also mediated by electrostatic interactions (Heldring et al., 2007; Shiau et al., 1998).
Besides tamoxifen, ER+ breast cancer can be treated with estrogen deprivation through ovarian suppression and aromatase inhibition (AI), and ER can be directly targeted by selective estrogen receptor degraders (SERDs), such as fulvestrant, which induce ER protein turnover. However, relapse is common, and the majority of breast cancer deaths occur after a diagnosis of an ER+ primary cancer.
Results
NF1 loss correlates with poor patient outcome in ER+ breast cancer.
While NF1 mutation frequencies are low in primary ER+ breast cancer (2% in TCGA (TCGA, 2012) as shown in Figure 1A, and 4% in our patient cohort (Griffith et al., 2018)), NF1 mutation frequency is higher in metastatic ER+ breast cancer patients (n=535) (Lanman et al., 2015; Zill et al., 2018) when circulating cell-free tumor DNA (ctDNA) was sequenced (18%, Figure 1A and Table S1). Similar enrichment of NF1 mutations in metastatic ER+ breast cancer has been reported by others (Bertucci et al., 2019; Meric-Bernstam et al., 2014; Pearson et al., 2019; Razavi et al., 2018; Sokol et al., 2019; Yates et al., 2017). These observations suggest that somatic NF1 events are an important class of mutations driving breast cancer progression.
Figure 1. NF1 loss promotes tamoxifen agonism and E2 hypersensitivity leading to poor patient outcome in ER+ breast cancer.
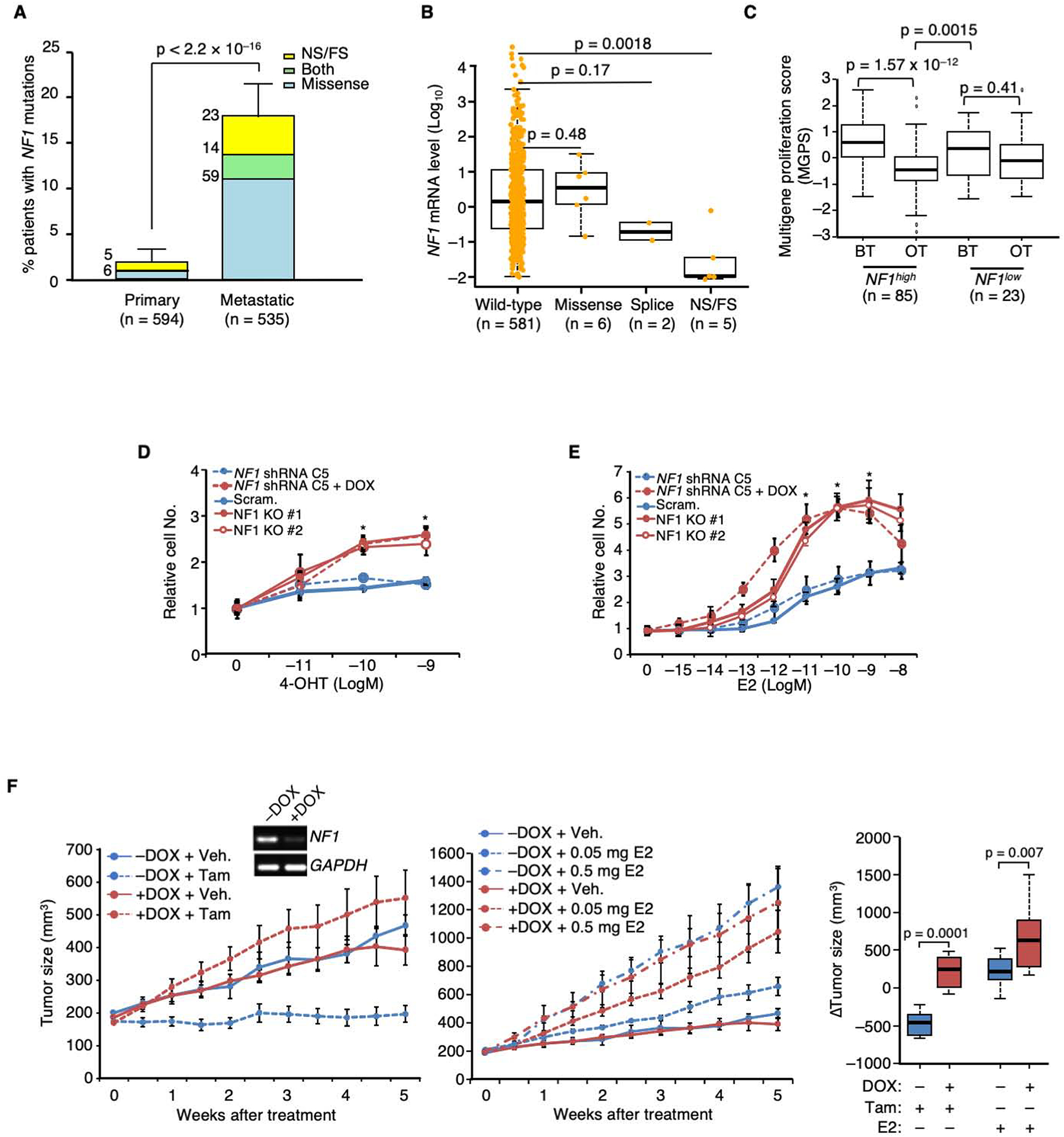
A. The percentages of ER+ primary vs. metastatic breast cancers carrying NF1 mutations were analyzed by Fisher Exact test. NS and FS stand for nonsense and frameshift, respectively. The number of patients carrying a particular type of NF1 mutation is shown at the upper left side of each column. B. Boxplot analysis of NF1 mRNA levels in ER+ breast tumors carrying different NF1 mutations in the RNA-seq database of TCGA. P value by Wilcoxon rank sum test. The line in the middle of the box is the median. The box edges are the 25th and 75th percentiles and the whiskers denote 1.5 times the inter-quartile range. C. Patient samples were stratified by NF1 mRNA levels according to TCGA definitions of high vs. low expression (mean – 1.5 × SD). The boxplot analysis was similarly performed as in (B) to compare multigene proliferation score (MGPS) in tumors before treatment (BT) and on treatment (OT) with AI. The differences in MGPS before and during treatment in each NF1 group were analyzed by the Wilcoxon signed-rank test. The differences in MGPS as a result of treatment between the two NF1 groups were further analyzed by Wilcoxon rank sum test. D. DOX-inducible gene silencing using NF1 shRNA clone C5 and CRISPR-mediated NF1-KO were performed in MCF-7 cells. These cells were seeded in E2-deprived medium, to which 4-OHT was added, and cultured for 6 days. Cell numbers relative to vehicle control are plotted. Experiments were conducted as biological triplicates (n = 3 experiments), except for MCF-7 cells carrying NF1 shRNA C5 (n = 8 experiments). E. Cell growth in response to E2 was similarly analyzed as in (D). n = 3 experiments, except for MCF-7 cells carrying NF1 shRNA C5 (n = 8 experiments). F. MCF-7 cells carrying DOX-inducible NF1 shRNA were transplanted into the mammary fat pads of ovariectomized nude mice, supplemented by an E2-capsule. When tumors appeared, the original E2-capsule was removed, and the resulting mice were randomized, DOX or vehicle treated. Each set was then treated by either tamoxifen (5 mg/mouse, left), or E2 (at two doses, middle). For NF1+ (−DOX) tumors, n=10, 12, 12, and 8 mice per group for treatment of vehicle, 0.05 mg E2, 0.5 mg E2, and tamoxifen; for NF1KD (+DOX) tumors, n = 10, 13, 11, and 8 mice per group. The inset shows NF1-silencing validation by qPCR 2 weeks post-DOX addition. Data are reported as mean±SEM. On the far right, “Δ tumor volumes” between vehicle and either tamoxifen or E2 treated were analyzed by box plot as in (C). *p<0.05, **p<0.01 by pair-wise two-tailed Student’s t-test, unless otherwise indicated.
We have previously analyzed somatic mutations in primary tumors accrued from ER+ breast cancer patients treated with adjuvant tamoxifen monotherapy and found that NF1 nonsense and frameshift mutations were associated with a markedly higher risk of breast cancer recurrence and death (Griffith et al., 2018). No GAP-inactivating missense mutations were found. When examining the COSMIC database, a C to T nonsense mutation at R2450 (R2450*) is the most frequent non-silent NF1 mutation, found 28 times (Figure S1A). Nonsense/frameshift mutations can trigger mRNA degradation through nonsense-mediated decay. Consistent with this mechanism, greatly reduced full-length NF1 mRNA levels were observed in both TCGA (Figure 1B) and METABRIC (Curtis et al., 2012) (Figure S1B) samples when nonsense/frameshift mutations were present. Similar reduction of NF1 mRNA in tumors carrying NF1 mutations have also been reported in another patient cohort (Pearson et al., 2019). ER+ breast cancer is also treated with aromatase inhibition (AI); therefore, we examined gene expression data from the ACOSOG Z1031 clinical trial, where patients were treated with an AI before surgery (Ellis et al., 2011). These data demonstrated that while a multi-gene proliferation score (MGPS) (Ellis et al., 2017) decreased after AI treatment in tumors with higher NF1 mRNA levels, reduced suppression of proliferation (high MGPS) was observed in tumors with low NF1 mRNA levels, indicating AI resistance (Figure 1C and Table S2). Finally, we found that NF1 proteins missing a small portion of the C-terminus caused by the NF1-R2450* mutation or by two other recurrent nonsense/frameshift mutations, NF1-R2258* and NF1-Y2285* (Figure S1A), could not be efficiently expressed unless a proteasome inhibitor was added (Figure S1C), supporting the possibility that some C-terminally truncated NF1 proteins may be unstable.
Collectively, these findings raise the possibility that depletion of neurofibromin is a common consequence of nonsense/frameshift mutations and most associated with poor outcomes. Importantly sequencing studies have found few recurrent inactivating mutations in the NF1 GAP domain, leading us to speculate that NF1 protein may harbor additional negative regulatory functions relevant to breast cancer pathogenesis.
NF1-depletion promotes tamoxifen agonism and E2 hypersensitivity in ER+ breast cancer cells.
To investigate the consequences of NF1-depletion on ER+ breast cancer, MCF-7 ER+ breast cancer cells were engineered to harbor lentiviruses expressing one of two doxycycline (DOX)-inducible shRNA clones (C5 and C6). Upon DOX addition (+DOX), NF1 protein levels were reduced by ~70% as detected by a monoclonal antibody (mAb) we raised (Figure S1 D and E). Increased ERK activating phosphorylation (pERK1/2) in DOX-treated cells vs. vehicle treated or scrambled shRNA controls indicated the anticipated reduction in GAP activity (Figure S1E). An NF1 shRNA-resistant expression construct reversed these effects (Figure S1E).
Remarkably, when NF1 expression was suppressed (+DOX) in MCF-7 cells as well as in two more ER+ breast cancer cell lines ZR-75B and T47D (Figure S1F), in vitro growth was consistently stimulated by 4-hydroxy-tamoxifen (4-OHT) under E2-deprived conditions (charcoal-stripped serum) (Figure 1D and Figure S1G), in comparison to the non-silenced control (−DOX) or the scrambled shRNA +DOX control, indicating an NF1 loss mediated transition from 4-OHT antagonism to agonism. Furthermore, NF1-silenced cells proliferated at lower concentrations of E2 than controls, and higher E2 concentrations paradoxically inhibited cell growth indicating E2 hypersensitivity (Figure 1E and Figure S1H). Two pools of NF1 “knock-out” (KO) MCF-7 cells were independently created by CRISPR-Cas9 (Figure S1D) with results similar to the shRNA data (Figure 1 D and E). MDA-MB-231 cells are ER−, and NF1 expression is barely detectable (Ogata et al., 2001) due to a frameshift mutation (NF1-T467fs) (Neve et al., 2006). These ER− “NF1low” cells were unresponsive to 4-OHT or E2, indicating a requirement for ER for the endocrine effects of NF1 perturbation (Figure S1 G and H). E2 hypersensitivity and 4-OHT agonism were reproduced in vivo using an MCF-7-based xenograft mouse model — tamoxifen stimulated the growth of NF1-silenced tumors (Figure 1F left), and these tumors grew better than control tumors at a lower dose of E2 (0.05 mg dose) (Figure 1F, right).
NF1-depletion globally enhances ER transcriptional activity.
Since the abnormal E2 and tamoxifen responses observed in NF1-depleted cells are ER-dependent, we investigated whether NF1-depletion affects ER-dependent transcription. First, the mRNA levels of established ER-target genes were examined by qPCR in MCF-7 cells, and the expression levels of GREB1 and pS2/TFF1 in the presence of E2 were elevated when NF1 was depleted by DOX-inducible shRNA (Figure S2A) or CRISPR-mediated KO (Figure S2B). This increase was not due to an increase in ER protein levels and could be reversed by the shRNA-refractory NF1 construct (“NF1-rescue”, Figure S2A), indicating the specificity of the shRNA targeting. Gene expression was also stimulated by 4-OHT in MCF-7 cells depleted for NF1 by shRNA (Figure S2C) or by CRISPR (Figure S2B), consistent with conversion to agonism. E2 and 4-OHT-stimulated gene expression were similarly observed in NF1-silenced ZR-75B cells (Figure S2C). ER-dependent transcription is promoted by co-activators SRC1–3, and these factors can be selectively degraded by bufalin (Wang et al., 2014). Our data showed that bufalin inhibited 4-OHT-induced enhanced gene expression in NF1KD MCF-7 cells (Figure S2D), indicating SRC1–3 is required for this activity. Finally, we examined tumor tissues from the MCF-7 xenograft model (Figure 1E) by qPCR and found that expression of two ER target genes GREB1 and TFF1 was greatly enhanced when NF1 was silenced (Figure S2E).
To examine the effects of NF1 on gene expression in a genome-wide fashion, RNA-seq experiments were performed in control (−DOX) and NF1 knock-down (+DOX, KD) MCF-7 cells with and without E2 stimulation. Overall, E2 altered expression from 540 genes in the control NF1+ cells and 955 genes in NF1KD cells (Figure 2A and Table S3). There was an overlap of 388 genes between the two gene sets (Table S3), indicating that expression changes of 72% (=388/540) of the observed E2-altered genes seen in NF1+ cells were also altered in NF1KD cells. These overlapping genes, referred to as the “common E2-regulon,” were analyzed by GSEA Hallmark Pathway analysis and found to be highly enriched with well-established E2-responsive genes (Figure 2A and Table S3). Furthermore, nearly all genes upregulated after E2-stimulation in NF1+ cells were more strongly induced in NF1KD cells, and the great majority of E2-repressed genes were also more strongly repressed, consistent with bidirectional ER-hyperactivity upon NF1-depletion (Zubairy and Oesterreich, 2005). The 415 (=955−540) E2-altered genes that were selectively observed in the NF1-depleted state (referred to as the “NF1KD-unique E2-regulon”) were also assessed by GSEA Hallmark pathway analysis (Table S3). Aside from a predominance of additional E2-regulated genes, a K-Ras-dependent gene expression signature was also observed (Figure 2A). Gene expression changes induced by 4-OHT were also examined by RNA-seq. While 4-OHT overall displays weak agonist activity as expected (Figure S2F, Table S3), the overlap of 4-OHT-altered gene expression between NF1+ and NF1KD cells was 65% and enriched with well-established E2-responsive genes (Figure S2G). Furthermore, genes that were induced by 4-OHT in NF1+ cells were more highly induced, while 4-OHT-repressed genes were more strongly repressed in NF1KD cells (Figure S2G).
Figure 2. NF1-depletion globally enhances ER transcriptional activity.
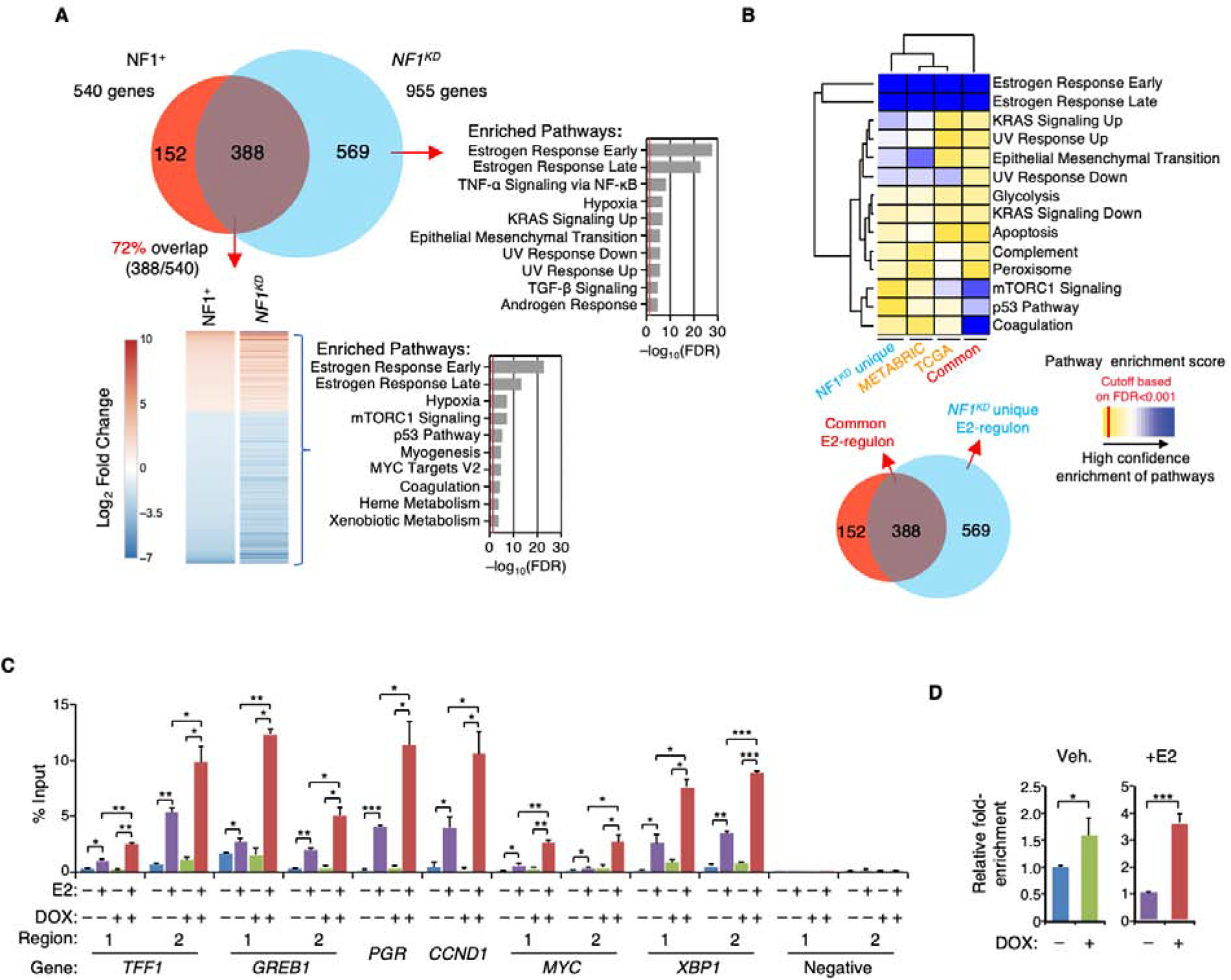
A. RNA-seq was performed on NF1+ or NF1KD MCF-7 cells treated with E2 or vehicle. A Venn diagram depicts the number of E2-mediated differentially expressed genes in NF1+ (red), NF1KD cells (blue), and those that overlap (“common E2-regulon,” purple). The common E2-regulon genes identified in NF1+ and NF1KD cells were ranked by (Log2) fold-change in gene expression, and enrichment for Hallmark Pathways by GSEA is shown to the right (red line marks an FDR cutoff at 0.05). GSEA analysis was also performed to examine Hallmark Pathways selectively enriched in NF1KD cells (“NF1KD unique E2-regulon”). B. Genes identified in (A) were examined in the TCGA and METABRIC ER+ breast cancer cases to identify those genes that are differentially expressed between tumors with wild-type NF1 and NF1 frameshift/nonsense mutations. The enriched Hallmark Pathways in the patient data are presented along with the results of the two E2-regulons identified in MCF-7 cells. C. ER was immunoprecipitated from cross-linked NF1+ or NF1KD MCF-7 cells treated by E2 or vehicle, and ChIP-qPCR was performed to measure ER occupancy at ten ERE sites in six genes. Two previous known ERE-negative regions (Carroll et al., 2005; Carroll et al., 2006) were assessed as negative controls. n = 2 ChIP experiments. D. ER ChIP-qPCR fold-enrichment values from all ERE sites examined in (C) were averaged and compared in NF1+ vs. NF1KD (+DOX) cells seeded with or without added E2 (n=10 sites). *p<0.05, **p<0.01, and ***p<0.001 by pair-wise two-tailed Student’s t-test.
To determine whether the E2-induced gene expression patterns shown in Figure 2A could be replicated in patient samples, differentially expressed genes according to NF1 status (with or without NF1 nonsense/frameshift mutation) in the METABRIC and TCGA ER+ data sets were identified, and pathway enrichments were similarly assessed (Table S4). The results were compared to the two MCF-7 E2-regulons described above. Supporting the conclusion that NF1-depletion dramatically affects the expression of E2-responsive genes in clinical ER+ specimens, E2-responsive pathway terms were most significantly modulated by NF1 mutation status, followed by the K-Ras signature (Figure 2B). A similar conclusion has been obtained by others after examining ER+ breast tumors with NF1 shallow deletion in METABRIC (Dischinger et al., 2018) and NF1 “truncating” mutations in TCGA (Pearson et al., 2019).
NF1-depletion increases ER recruitment to EREs.
In the presence of E2, NF1-depletion mostly affects expression of known E2-responsive genes. To assess whether this is due to direct ER binding to the EREs, we performed ChIP-qPCR to analyze ER recruitment to ten well-documented ERE sites in six genes, TFF1, GREB1, PGR, MYC, XBP1, and CCDN1. The data show that NF1-depletion enhances ER recruitment to all of these sites (Figure 2 C and D).
In the presence of 4-OHT, expression of 542 genes was mostly detected in NF1KD cells (Figure S2G). We examined one 4-OHT-dependent ER ChIP-seq data set (He et al., 2018) and found that 68% of these 542 genes (371/542) were also found in this ER ChIP-seq study (Table S3). All together, these data suggest that NF1-depletion mostly quantitatively enhances ER-dependent transcription, whether it is stimulated by E2 or 4-OHT, but NF1 loss does not substantially qualitatively alter the profile of genes that get transcribed under the experimental conditions tested.
Neurofibromin contains two leucine/isoleucine rich co-repressor motifs.
Tamoxifen agonism and E2 hypersensitivity, together with the RNA-seq and ER ChIP-seq data, suggested that neurofibromin may function as a ligand-dependent co-repressor for ER transcriptional activity. Consistent with this postulate, there are two consensus co-repressor motifs in neurofibromin, designated here as M1 and M2 (Figure 3A). Notably the ER binding motifs in neurofibromin family members appear to have co-evolved with emergence of estrogen-dependent reproduction. M1/2 are highly conserved in salmon, the most human-distant species examined to have an ER homolog (Rogers et al., 2000), but not in fly or yeast (Figure 3A), which do not. Furthermore, mutations affecting key residues in M1 and M2 can be found in cancer (COSMIC and METABRIC) and some of these mutations are recurrent. In particular, a somatic M1 I417M mutation was found in our patient cohort (Griffith et al., 2018), which coexisted with an NF1 nonsense mutation suggesting a biallelic event.
Figure 3. Neurofibromin binding to ER is selectively mediated by co-repressor motifs.
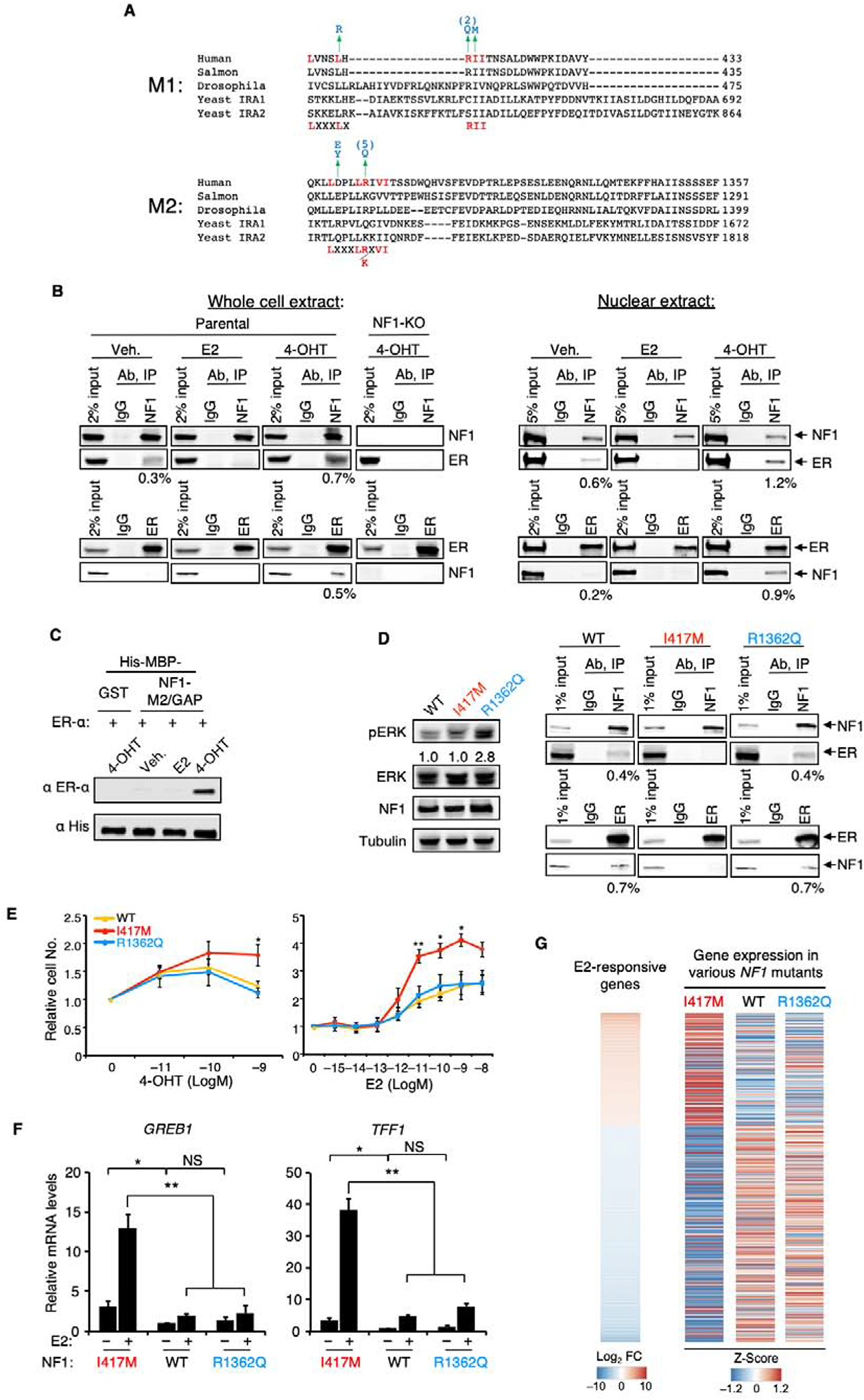
A. Protein alignment was created by ClustalW (MUSCLE). NF1 has two potential co-repressor motifs, M1 and M2 (potential consensus sequences shown at the bottom). Mutations found in cancers (COSMIC) or neurofibromatosis are colored blue, and the numbers in parentheses are the number of times found in the COSMIC database. B. Parental MCF-7 or the NF1-KO (clone #1) MCF-7 cells were grown in E2-deprived medium to which E2, 4-OHT, or vehicle was added. Whole cell or nuclear extracts were immunoprecipitated with NF1 or ER antibody, and the resulting samples were analyzed by immunoblotting. The number below the blot shows the percentage of co-immunoprecipitated protein. C. Recombinant purified ER-α preincubated with E2, 4-OHT, or vehicle was pulled-down by amylose beads containing His-MBP-tagged NF1-M2/GAP domain, or His-MBP-GST control, and the results were analyzed by immunoblotting. D. Left, whole cell lysates from parental MCF-7 cells (WT) or from a CRISPR “knock-in” mutant carrying either a I417M (colored red) or a R1362Q (colored blue) mutation grown in full-serum medium were examined by immunoblotting to measure the pERK/total ERK ratios (below). Right, reciprocal co-immunoprecipitation experiments in the presence of 4-OHT similar to those in (B) assessed ER binding to various NF1 proteins. E. Parental or NF1 mutant cells were seeded in E2-deprived medium and were examined for cell growth in response to E2 or 4-OHT (6 days later) n=3 experiments. F. GREB1 and TFF1 mRNA levels from parental or NF1 mutant cells treated with vehicle or E2 were measured by qPCR (n = 3 independent experiments). Expression levels were normalized to those of vehicle-treated wild-type cells. Data are reported as mean±SEM. *p<0.05, **p<0.01 by pair-wise two-tailed Student’s t-test. NS, not significant. G. mRNAs from cells seeded in charcoal-stripped serum were analyzed by RNA-seq. “E2-responsive genes” as defined in Figure 2A is shown on the left to which expression levels of genes (row Z-scores from “Log2 Transcripts per Million” values) in various strains were aligned.
The binding of ER to neurofibromin is selective, ligand-dependent, and direct.
Established ER co-repressors preferentially bind to the ER ligand-binding domain (LBD) in an interaction that is enhanced by 4-OHT but not by E2 (while the reverse is the case for co-activators) (Huang et al., 2002; Shiau et al., 1998). Ligand-dependent binding of neurofibromin to the ER LBD was detected in cells using a mammalian two-hybrid assay (Chang et al., 1999), mimicking a known co-repressor NCoR1 and opposite to the behavior of the coactivator SRC-1 (Figure S3 A and B). Reciprocal co-immunoprecipitation experiments in MCF-7 and ZR-75B cells were also performed to demonstrate that neurofibromin can co-immunoprecipitate ER using our mAb and vice versa (Figure S3C). NF1+ MCF-7 cells were used to illustrate the point that these interactions were enhanced by 4-OHT, but reduced by E2; in contrast, ER could not be pulled down in the NF1-KO line, indicating that our neurofibromin mAb is specific (Figure 3B). Finally, M2 is located in the GAP domain, which can be readily purified from Escherichia coli (Bollag et al., 1993). We thus determined that 4-OHT increased the efficiency of the interaction between purified NF1-M2/GAP domain and purified ER in a pull-down, confirming that the interaction between NF1 and ER is very likely direct (Figure 3C).
The human genome contains 14 potential Ras GAPs, but they do not share sequence homology beyond the GAP domain. For example, in p120GAP/RASA1, there is no identifiable M1; nor was there an M2 in a published GAP domain alignment study (Ballester et al., 1990) (Figure S3D). While ER co-immunoprecipitated NF1, ER did not co-immunoprecipitate p120/RASA1 (Figure S3C). Protein co-expression is a strong predictor for co-functionality (Wang et al., 2017a). Therefore, we assessed Gene Ontology molecular function terms of proteins whose levels positively correlated with those of neurofibromin from proteomics analysis of >100 breast cancer patient-derived samples (CPTAC) (Mertins et al., 2016). Whereas NF1 protein levels highly correlated with a number of transcription factor functionalities, and the “ligand-dependent nuclear receptor binding” term in particular, p120GAP, mostly negatively correlated with these factors (Figure S3E).
ER binding and GAP activity are two independent functions of neurofibromin.
To determine whether neurofibromin can directly interact with ER via M1/2 without a requirement for GAP activity or Ras pathway activation, we first took a pharmacological approach using Raf and MEK inhibitors, dabrafenib and trametinib (Robert et al., 2015). Here we found that Raf/MEK inhibition did not affect E2-dependent transcription measured by qPCR in either NF1+ or NF1-silenced MCF-7 cells (Figure S3F). Immunoblotting detected substantial loss of pERK levels by dabrafenib and trametinib, suggesting the Ras-Raf pathway was efficiently inhibited in these experiments (Figure S3F). Bufalin, which promotes degradation of SRC1–3 co-activators, was the positive control for this study. One of the best-known examples of a Ras-ER interaction is phosphorylation at serine 118 (pS118) in ER by ERK after growth factor stimulation (Kato et al., 1995). However, this event seems to be context (e.g., cell type and/or culture conditions)-dependent and can be catalyzed by other kinases (Anbalagan and Rowan, 2015). We could not detect a significant increase in ER-pS118 in NF1 KO MCF-7 cells, despite strong increase in activating phosphorylation in ERK1/2 (Figure S3G). Finally, we note that the average IC50 of trametinib in commonly used ER+ breast cancer cell lines under normal culture condition is 300 μM vs. 0.05 μM in melanoma cells (https://www.cancerrxgene.org). Thus, under standard culture conditions, Ras-Raf signaling is not critical for either ER-dependent gene expression or growth of E2-sensitive ER+ breast cancer cells.
To directly ascertain whether M1 and M2 are authentic ER-binding sites, residues I and V were substituted with A to create NF1-I417A-I418A and NF1-V1308A-V1309A. The I417M NF1 somatic M1 mutant found in an ER+ breast cancer was also generated. Conversely, two mutations found in metastatic breast cancer, NF1-R1362Q (Lanman et al., 2015) and NF1-K1444R (Li et al., 2013), were predicted (Wilkins et al., 2012) to inactivate GAP activity (Figure S3H). They were thus tested further to assess whether ER repression requires intact GAP activity. We confirmed that R1362Q and K1444R mutations greatly reduce the GAP activity of NF1, while the M1/2 mutations do not, by measuring pERK levels (Figure S3H).
Binding to ER (two-hybrid assay, Figure S3I) and repression of ER transcriptional activity (ERE-luciferase reporter assay (Hall and McDonnell, 1999), Figure S3J) were both greatly reduced by M1 and M2 mutations; in contrast, the two GAP mutants bound and suppressed ER activity as efficiently as wild-type NF1. One GAP and one M1 mutant were chosen for further study by expressing them in NF1-depleted cells to levels comparable to endogenous neurofibromin in control cells (Figure S3K). The enhanced GREB1 and TFF1 expression in NF1-silenced cells was completely repressed by wild-type NF1 and the GAP mutant, but only partially by the M1 mutant.
To ascertain whether ER-repression and GAP activity are two independent functions of NF1 without using ectopic overexpression, CRISPR-Cas9 mediated knock-in was used to create MCF-7 cells carrying homozygous NF1-I417M (M1) mutation or NF1-R1362Q (GAP) mutation (Figure S3L). The former cells had normal GAP activity, but their NF1-I417M cannot co-immunoprecipiate ER (Figure 3D). Similar to cells in which NF1 is depleted by shRNA or CRISPR, the NF1-I417M (ER binding) mutant cells showed abnormal endocrine responses because cell growth was stimulated by 4-OHT and low levels of E2; in contrast, the NF1-R1362Q (GAP) mutant behaved like wild-type cells (Figure 3E). We examined expression of two well-established ERE genes (TFF1 and GREB1) by qPCR (Figure 3F) and found that their expression after E2 stimulation was greatly enhanced by the I417M mutantion, which again behaved like full NF1-depletion. In contrast, the NF1-R1362Q mutatant behaved like wild-type cells showing only a modest increase in gene expression.
While conducting the ER ChIP experiments, we noted a trend that when NF1 was depleted, more ER was recruited to the chromatin even when the cells were seeded in the presence of vehicle alone (Figure 2D). Consistent with this, expression of GREB1 and TFF1 was significantly stronger in vehicle-treated NF1-I417M mutant cells than in wild-type and the NF1-R1362Q mutant cells (Figure 3F). To further examine these findings in a transcriptome-wide manner, cells were seeded in charcoal-stripped serum supplemented medium without adding ligand, and RNA-seq experiments were performed (Table S5 and Figure 3G). When expression levels of genes (row Z-scores from “Log2 Transcripts per Million” values) in various cell lines were aligned to the list of E2-responsive genes as identified previously (Figure 2A), it was evident that the majority of the E2-induced genes were more highly expressed and many E2-repressed genes were more strongly repressed in the NF1-I417M mutant (right panel, first column, Figure 3G). Essentially, the NF1-I417M mutation mimicked E2-stimulation. In contrast, the NF1-R1362Q mutant behaved like wild-type, showing no detectable transcriptional phenotype (middle and right columns in the right panel, Figure 3G). These results clearly illustrate that ER-repression and Ras-repression are two independent activites of NF1 that are mediated by distinct structural motifs.
Ligand-stimulated nuclear accumulation of neurofibromin.
For transcriptional regulation, neurofibromin must enter the nucleus. While neurofibromin is mainly cytoplasmic, nuclear neurofibromin has been previously reported in a variety of cell types including ER+ breast cancer cells (Beausoleil et al., 2004; Daston et al., 1992; Koliou et al., 2016; Kweh et al., 2009; Li et al., 2001; Nousiainen et al., 2006; Vandenbroucke et al., 2004). In one study, neurofibromin nuclear localization in neuronal cells was shown to be dependent on the Ran GTPase (Koliou et al., 2016). This mechanism may also operate in ER+ breast cancer, because neurofibromin co-immunoprecipitated with Ran in MCF-7 cells (Figure S3C). Leptomycin-B (LMB) blocks CRM1-dependent nuclear export. MCF-7, ZR-75B, and T47D cells were treated with LMB, and cell fractionation was then used to demonstrate that nuclear neurofibromin is increased after nuclear export blockade (Figure 4A). We further investigated whether NF1 nuclear levels are also affected by E2. Nuclear neurofibromin was decreased by E2 but increased by 4-OHT, indicating that ligand-regulated modulation of nuclear levels is an aspect of the co-repressor function of neurofibromin (Figure 4B). 4-OHT-enhanced ER and neurofibromin co-immunoprecipitation can be readily detected in nuclear extracts, and the percentages of co-immunoprecipitated proteins were higher in the nuclear extracts than in whole cell extracts (Figure 3B). To investigate neurofibromin subcellular localization by microscopy, an immunostaining protocol (Figure 4C) was developed using a set of cell lines with varying degrees of NF1 expression as controls (Figure S4), which included NF1-KO cells. Two cell lines were then chosen to confirm by microscopy the impact of LMB and ER ligands on nuclear neurofibromin levels (Figure 4 D and E). Overall these results suggest that while neurofibromin is mostly cytoplasmic at steady state, it is shuttled in and out of the nucleus, possibly by Ran and CRM-1, in a manner controlled by ER ligand exposure.
Figure 4. ER ligand-mediated nuclear accumulation of neurofibromin.
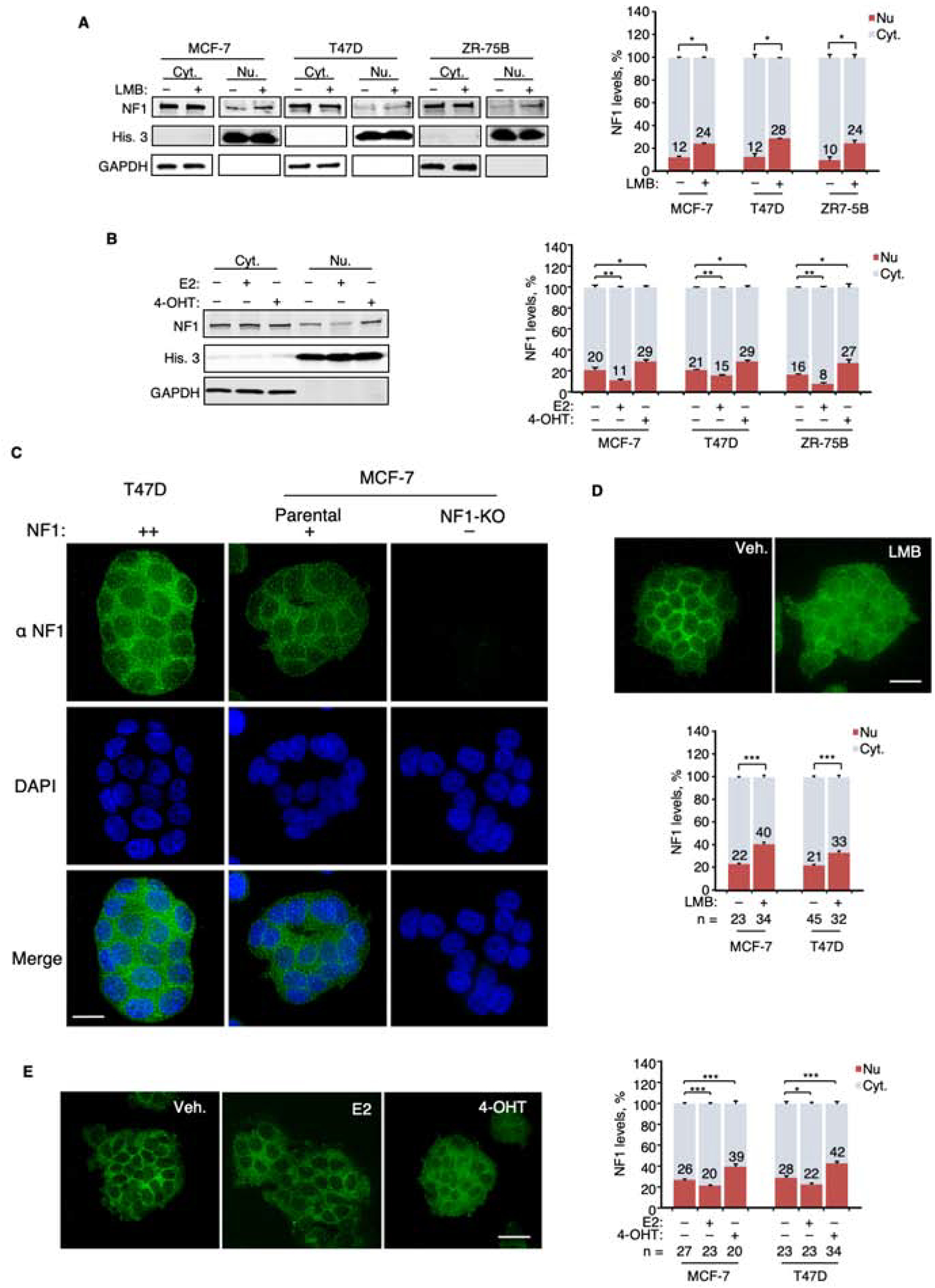
A. Left, whole cell lysates from indicated cell lines treated with LMB were separated into nuclear (Nu.) and cytoplasmic (Cyt.) fractions. NF1, histone-3 (His. 3, nuclear marker), and GAPDH (cytoplasmic marker) were examined by immunoblotting. Right, NF1 levels in the cytoplasmic fraction (light blue) and the nuclear fraction (red) were normalized to GAPDH and His.−3, respectively, and the sum of these two fractions is defined as total NF1 (100%). The numbers in the graph represent the percentages of NF1 determined to be nuclear. The pair-wise comparisons were between NF1 nuclear fractions. n = 2 experiments. B. Left, Cells after ligand stimulation were similarly analyzed as in (A) and an immunoblot of treated MCF-7 cells is shown as an example. n = 3 experiments. C. Immuno-fluorescence deconvolution microscopy was performed using our monoclonal NF1 antibody on cells with varying levels of NF1 protein. A single focal plane across the middle of the nucleus (marked by DAPI) is shown. D. Cells treated with LMB were examined by microscopy. NF1 nuclear fractions were quantified in the indicated number of cells. E. Cells after treatment with ER-ligands were similarly examined by microscopy and quantified. Scale bars = 20 μm. Data are reported as mean±SEM. *p<0.05, **p<0.01, ***p<0.001 by pair-wise two-tailed Student’s t-test.
See also Figure S4.
Ligand-dependent association of neurofibromin with the ER-ERE complex.
When E2 is present, E2-liganded ER recruits co-activators to the ERE; 4-OHT, in turn, displaces co-activators with co-repressors in the complex. These ligand-mediated actions can be recapitulated by a cell-free assay using HeLa cell nuclear extract (Foulds et al., 2013), which does not have endogenous ER. When purified recombinant ER, biotinylated ERE, and HeLa nuclear extract were combined, the E2-liganded ER-ERE complex could more efficiently pull down the co-activator SRC-1 by streptavidin beads, while NF1, like the co-repressor HDAC1, was more strongly recruited to the ER-ERE complex in the presence of 4-OHT (Figure 5A). To assess whether neurofibromin is recruited to endogenous EREs in a ligand-dependent manner, we performed ChIP-qPCR using two separate polyclonal neurofibromin antibodies, one of which, Ab-2, was generated by us (Figure S5A), and found that neurofibromin was more efficiently recruited to the EREs of GREB1 and TFF1 when 4-OHT, but not E2, was added (Figure S5B). NF1-KO cells were examined as the control to show antibody specificity and neurofibromin-dependency (Figure 5B). We then selected ten well-documented ERE sites in six genes, TFF1, GREB1, PGR, MYC, XBP1, and CCDN1, and performed three separate neurofibromin ChIP-qPCR experiments to show that neurofibromin recruitment to these sites was enhanced by 4-OHT; in contrast, no neurofibromin recruitment was detected in other regions (Carroll et al., 2005; Carroll et al., 2006) that do not contain an ERE (Figure 5C). Together, these results demonstrate that neurofibromin is an authentic ER co-repressor that localizes with ER on an ERE. These findings may explain the recent observation that NF1 loss (e.g., by loss of heterozygosity) and ESR1 activating LBD mutations are mutually exclusive (Sokol et al., 2019), because they both lead to enhanced ER transcriptional activity (Gates et al., 2018; Jeselsohn et al., 2018).
Figure 5. Ligand-dependent association of neurofibromin with the ER-ERE complex.
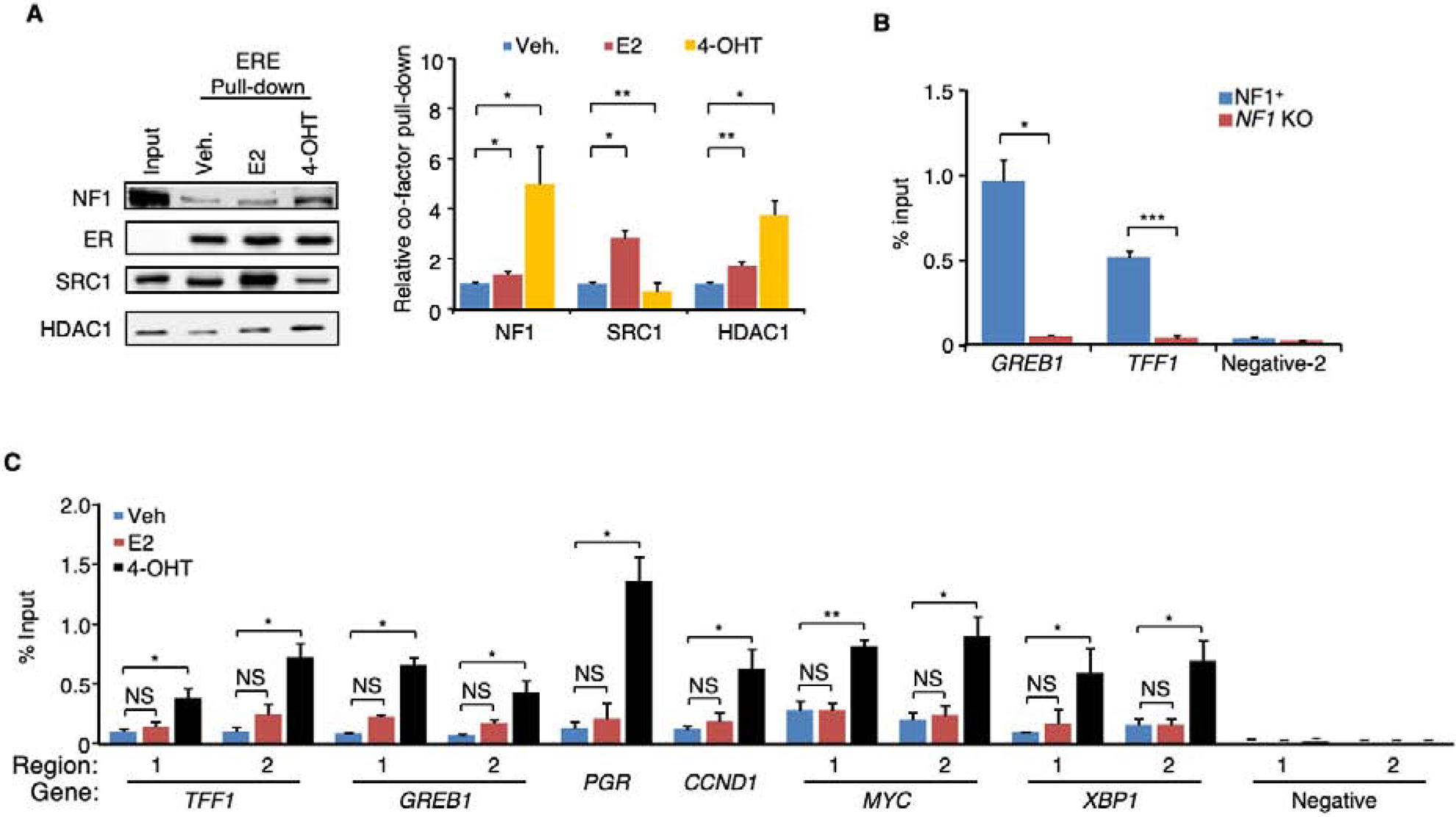
A. Ligands were added to the HeLa nuclear extract together with purified ER and biotinylated EREs immobilized onto streptavidin beads. After washing, the proteins bound to EREs with ER were analyzed by immunoblotting (left) and quantified (right). n = 4 experiments. B. ChIP-qPCR experiment was performed using NF1 antibody Ab-2 to assess NF1 occupancy at the EREs in GREB1 (Region 2, Figure 2C) or TFF1 (Region 1, Figure 2C) in parental or NF1-KO (clone 1) MCF-7 cells treated with 4-OHT. An ERE-negative region (site 2) (Carroll et al., 2006) was also analyzed as negative control. n = 2 experiments. C. NF1 occupancy in MCF-7 cells at ten ERE sites in six genes and two negative control regions was assessed by qPCR (n=3 separate ChIP experiments). Data are reported as mean±SEM. *p<0.05, **p<0.01, ***p<0.001 by pair-wise two-tailed Student’s t-test. NS, not significant.
See also Figure S5.
Co-targeting Ras and ER to treat NF1-deficient ER+ breast tumors.
The clinical and functional data presented thus far suggests that NF1-deficient ER+ breast tumors will not be effectively treated with tamoxifen or AI. However, when NF1 was depleted by either shRNA or CRISPR, the resulting MCF-7 cells were still sensitive to the SERD fulvestrant (Figure S6A). Similar observations were made in NF1KD ZR-75B and T47D cells (Figure S6B). However, in NF1-silenced MCF-7 cells fulvestrant produced an enhanced compensatory activation of the Ras-Raf-MEK-ERK pathway that may promote cell survival and/or drug resistance in spite of effective ER inhibition (Figure 6A). Thus, combinatorial targeting of both ER with fulvestant and the Ras-Raf pathways was assessed to determine if NF1-deficient ER+ breast cancer can be effectively treated with this strategy. In vitro, the combination of Raf and MEK inhibitors, dabrafenib and trametinib enhanced fulvestrant activity to inhibit cell growth by mostly increasing apoptosis in NF1KD MCF-7 and ZR-75B cells (Figure S6C). Immunoblots showed that all drugs inhibited the intended targets (Figure 6A). To develop a clinical protocol, we next investigated whether the dabrafenib and trametinib combination could be replaced by a single MEK inhibitor (MEKi). Selumetinib is a new generation MEKi that is less toxic than trametinib due to a shorter half-life is under consideration by the FDA for the treatment of plexiform neurofibromas in pediatric patients with neurofibromatosis (Dombi et al., 2016; Gross et al., 2018). Our data demonstrate that in vitro. fulvestrant plus selumetinib also strongly inhibited cell growth and induced apoptosis in NF1-silenced MCF-7, ZR-75B, and T47D cells whereas single agent exposure did not (Figure 6B). Immunoblotting was performed to confirm that the drugs effectively target ER and ERK activation (Figure 6A and Figure S6D).
Figure 6. Co-targeting Ras and ER to treat NF1-deficient ER+ breast cancer.
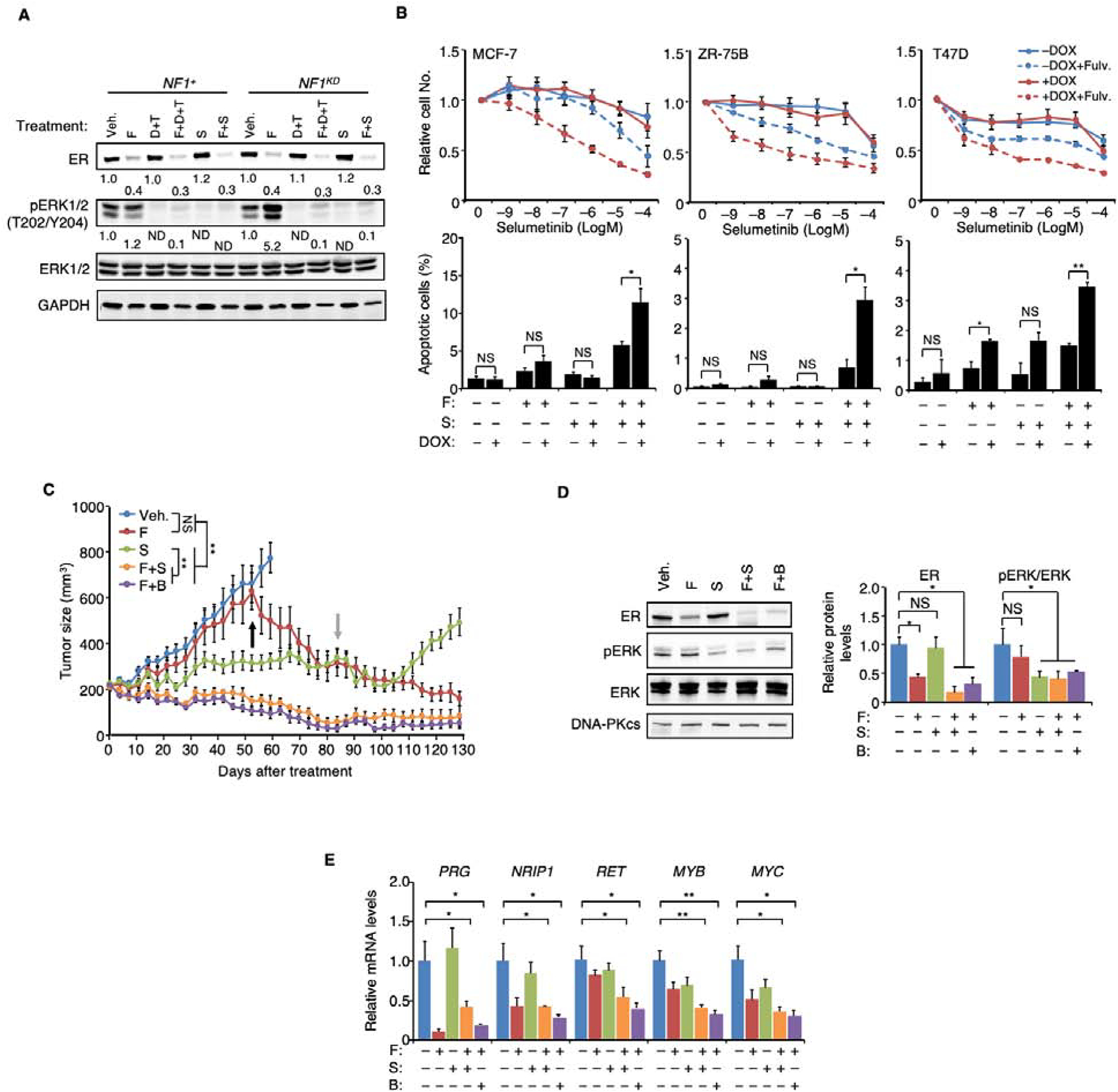
A. MCF-7 cells were seeded in 10−11 M E2 to which fulvestrant (F), dabrafenib (D), trametinib (T) or selumetinib (S) were subsequently added at 10−9, 10−6, 10−7 or 10−6 M, respectively. After 6 days, proteins were measured by immunoblotting, and phosphorylation levels (as defined by the levels of the phosphorylated form over total protein) relative to the vehicle-treated cells were set to 1. ND, not detectable. B. Top: cells were grown for 6 days in the presence of 10−11 M E2 and 10−9 M fulvestrant, to which increasing concentration of selumetinib was added. Cell numbers relative to the vehicle control are plotted. n = 3 experiments. Bottom: the cells were treated similarly except 10−6 M selumetinib was used, and apoptosis was measured 6 days post-treatment. n = 2 experiments. C. WHIM16 tumors were transplanted into cleared mammary fat pad of mice and later randomized to receive treatment (n=15 per treatment arm) when tumor volumes reached 200 mm3. Tumor volume comparison was performed at 52 days post-treatment by t-test between indicated treatment groups, and at the same time, binimetinib (B) was added to the fulvestrant-only arm (marked by the black arrow). After 84 days (grey arrow), treatments were withdrawn from all treatment groups expect the group receiving late binimetinib after initial fulvestrant monotherapy. D. WHIM16 tumors from each treatment arm at week-4 post-treatment in (C) were analyzed by immunoblot (one representative tumor shown on the left), and the results relative to those treated by the vehicle control were quantified on the right (n = 3 tumors). E. qPCR was performed to analyze expression levels of indicated genes from the same tumor samples as in (D). mRNA levels in vehicle-treated samples were set to 1. Data are reported as mean±SEM. *p<0.05, **p<0.01 by t-test. NS, not significant.
In vivo, while tumors from the MCF-7 NF1KD xenograft model (+DOX) initially responded to fulvestrant, at later time points these tumors acquired resistance (Figure S6E). Acquired fulvestrant resistance in NF1-depleted ER+ breast cancer cells has recently also been reported by others (Pearson et al., 2019). We further demonstrated that the fulvestrant-resistant tumor outgrowth in ER+ NF1low cells in vivo can be blocked by dabrafenib and trametinib (Figure S6E). In addition, RNA-seq (Li et al., 2013) and mass spectrometry data (Huang et al., 2017) suggested that a patient-derived xenograft (PDX), WHIM16 (Li et al., 2013), is NF1-deficient. Immunoblotting confirmed NF1 protein levels were barely detectable in WHIM16 (Figure S6F). This PDX line was derived from a patient who died after the development of resistance to multiple lines of endocrine therapies, including fulvestrant and AI.
An initial in vivo assessment of dabrafenib-trametinib in WHIM16 suggested activity for Ras-dependent kinase inhibition with fulvestrant over that observed for fulvestrant alone (Figure S6F), but longer term exposure was limited by weight loss and diarrhea. We therefore replaced the dabrafenib-trametinib combination with selumetinib-laced chow and conducted a four-arm study to compare single agent efficacy versus the combination. These data demonstrate that when selumetinib was combined with fulvestrant, efficient and long-term inhibition/regression of tumor was achieved (Figure 6C) without weight loss (Figure S6G) despite only 50% inhibition of ERK activity. In contrast, selumetinib alone slowed growth but did not induce tumor regression. Western blot confirmed ER degradation by fulvestrant and pERK inhibition by selumetinib (Figure 6D). Selumetinib did not reduce ER phosphorylation at S118 (Figure S6H), suggesting the MEKi efficacy cannot be easily explained on this basis. Binimetinib, another shorter half-life and better tolerated MEKi (Trojaniello et al., 2019), also efficiently inhibited tumor growth when combined with fulvestrant (Figure 6 C and D). Expression of several ERE-containing genes in treated tumors was examined by qPCR. Overall the lowest level of expression correlated with combination fulvestrant and MEKi exposure (Figure 6E). While fulvestrant was ineffective as a single agent, fulvestrant-treated tumors regressed with later addition of binimetinib (Black arrow, Figure 6C), mimicking the treatment of ER+ NF1low tumors progressing on fulvestrant monotherapy. Furthermore, while selumetinib alone can modestly inhibit tumor growth, growth soon resumed after the treatment was withdrawn; in contrast, no relapse was detected in the fulvestrant + MEKi groups after both agents were withdrawn (grey arrow, Figure 6C).
Discussion
This study presents comprehensive evidence that neurofibromin is a transcriptional co-repressor of ER in ER+ breast cancer, independent of its GAP activity. Consequently, when NF1 is depleted through somatic mutation, ER function is enhanced, leading to tamoxifen agonism, estradiol hypersensitivity, AI resistance, and poor outcome. These data suggest that the NF1-depleted ER+ breast cancers represent a distinct molecular subset of the disease that will require the development a new standard of care since tamoxifen is likely contraindicated and aromatase inhibition ineffective.
Our data suggest that in most cases, complete loss of the NF1 protein is responsible for enhanced ER transcriptional activity and endocrine therapy resistance. Such an NF1-null or NF1low state can be caused by nonsense and frame shift mutations due to nonsense mRNA decay and/or protein instability. Although nonsense and frame shift mutations are detectable by ctDNA sequencing on metastatic patients, NF1-depletion can also be caused by mechanisms not detectable by targeted sequencing. In a study of breast cancer by whole genome sequencing (Nik-Zainal et al., 2016), Nik-Zainal et al have described a wide range of structural variations affecting NF1 (tandem duplications, deletions, translocations, and inversions). Furthermore, in METABRIC database NF1 nonsense and frame shift mutations are also usually associated with shallow deletion (Griffith et al., 2018), suggesting biallelic loss of function events.
Human and salmon NF1 protein sequences share over 80% identity, which extends well beyond the GAP domain. However, defining a GAP-independent activity in neurofibromin has been difficult because of the broad range of cellular functions influenced by Ras. In hindsight, the commonly used ER+ breast cancer cells are ideal for investigating GAP-independent activities of neurofibromin because under standard culture conditions (e.g., in the absence of fulvestrant) growth and ER-dependent transcription operate independently of Ras. This is consistent with the observation that oncogenic mutations in RAS, RAF, MEK, and ERK almost never occur in primary ER+ breast cancer. However, Ras dependent cell survival becomes evident when the ER-dependent growth is inhibited by fulvestrant. Activation of Ras is made more efficient by NF1 loss, and NF1low cells appear to most addicted to this pathway for survival when ER activity is efficiently inhibited. In vivo acquired fulvestrant resistance modeled in the NF1-depleted MCF-7 tumors, was efficiently inhibited by dabrafenib + trametinib. WHIM16 represents metastatic ER+ breast cancer that already had acquired fulvestrant resistance when the tumor was accrued from the patient. Fulvestrant has little effect on tumor growth unless the MEK was co-inhibited.
As ER+ NF1low breast cancer acquires fulvestrant resistance, ER and MEK/ERK crosstalk likely occurs, but the finer details of this mechanism are beyond the scope of this paper. We investigated ER phosphorylation at S118 because this can be directly catalyzed by ERK. However, we could not detect a clear increase in pS118 levels in NF1-KO MCF-7 cells; conversely, in the presence of a MEKi, no decrease in pS118 can be seen in the WHIM16 tumors. Interpretation of WHIM16 experiments is complicated by the presence of fulvestrant, which greatly lowers total ER levels. There is a potential for selective stabilization of the S118-phosphorylated form of ER by other protein kinases, such as GSK3 (Grisouard et al., 2007).
In our experiments, estrogen deprivation therapy is mimicked by incubating cells in charcoal-stripped serum-containing media, as is customary in the field (Martin et al., 2017). We demonstrate that growth-simulation can be observed in NF1-depleted cells in vitro with as little as 10−12 M E2, which parallels E2 levels seen in AI-treated patients. We also present evidence from a neoadjuvant trial that AI-resistance can be seen in NF1low tumors defined at the mRNA level.
Our data suggest that the majority of the 569 NF1KD-unique genes are well-established E2-responsive genes. We speculate that the reason the expression of these genes was not readily detected unless NF1 is depleted is due to the low E2 concentration (10−11 M, see Methods) used to stimulate gene expression in this experiment. This E2 concentration was chosen because it best differentiates the growth of NF1+ from NF1KD cells, making it possible to more easily detect the differences in gene expression pattern we display. However, 10−11 M E2 is typical for the postmenopausal state and therefore may be too low to affect expression of some E2-responsive genes, until a co-repressor (NF1) is depleted. Of interest, the 569 genes also include genes responsible for EMT, a phenomenon that has also been seen in other resistance settings, i.e. the presence of ER-mutants (Jeselsohn et al., 2018) and ER-fusions (Lei et al., 2018).
The expression of the rest of the “NF1 knock-down only” genes that do not harbor an ERE is harder to decipher, and many of these genes appear in more than one functional category. Ras does not directly regulate gene expression, so gene expression influenced by Ras activity is likely to be context-dependent. Of the twelve “K-RAS signaling up genes”, three (ID2, SERPINA3, and PCP4) are also defined as “Estrogen response late,” so only the remaining 9 genes may be controlled by Ras activation.
In experimental systems, fulvestrant-resistance can be efficiently inhibited by further targeting MEK in both the MCF-7 and WHIM16 mouse models. These results strongly suggest that activation of the Ras-Raf pathway is the major driver for fulvestrant-resistance in NF1-deficient ER+ breast cancer. On this basis there is adequate clinical rationale to activate a clinical trial of a MEKi and a SERD specifically in patients with evidence for a loss of NF1 due to somatic mutation. Of note, WHIM16 regression was achieved with a well-tolerated dose of short half-life MEKi that inhibited ERK signaling by only 50%. This suggests that long term exposures to lower dose MEKi necessary for management of advanced breast cancer will be feasible, and the pediatric experience of neurofibromatosis-related plexiform neurofibroma treatment is relevant here (Gross et al., 2018). We note that a fulvestrant and selumetinib combination failed in a randomized Phase 2 trial (23 patients per arm) in unselected postmenopausal patients (Zaman et al., 2015). This study does not exclude a benefit in ER+ NF1low tumors, however, as it is likely that less than 5 patients harbored NF1low tumors in this study. Since the prevalence of ER+ advanced breast cancer in the USA exceeds 100,000 patients, the ctDNA based evidence suggest at least 7,000 to 10,000 of these individuals will have an NF1low tumor where a SERD/MEKi combination could be beneficial.
Further research may connect the ER hyperactivity induced by NF1 depletion to other paradoxical or unexplained observations in ER+ breast cancer, for example, regression of endocrine therapy-resistant ER+ breast cancer with estradiol treatment (Ellis et al., 2009). Additionally, the role of neurofibromin as an ER co-repressor may underlie the sexually dimorphic features of neurofibromatosis, including tumor growth and the preponderance of optic chiasm gliomas during female puberty (Diggs-Andrews et al., 2014).
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources or reagents should be directed to and will be fulfilled by the lead contact Dr. Eric C. Chang at echang1@bcm.edu. All materials generated by this study are available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
The human cell lines, growth media, and general methods of cell culture have been previously reported (Zheng et al., 2015), unless otherwise indicated. All lines were routinely tested for mycoplasma contamination. Estrogen deprivation was achieved by seeding cells in phenol red-free media (DMEM for ZR-75B, MDA-MB-231, and RPMI1640 for MCF-7 and T47D cells) containing 10% charcoal-stripped fetal bovine serum (Sigma-Aldrich) for two days prior to the experiment. Note that MDA-MB-175VII is an NF1-null cell line, apparently carrying biallelic frameshift mutation p.Y2285FS*5 (Neve et al., 2006).
The procedures for producing lentivirus, infecting breast cancer cells, and selecting for stable integrants were also conducted as previously described (Zheng et al., 2015). The cells stably expressing inducible shRNA were collected using FACS (Aria II, Becton Dickinson) two days after DOX treatment (2 μg/ml) by examining the presence of turboRFP, which is expressed together with the shRNA from the same bicistronic transcript. Typically, >85% of transduced cells were RFP+. For NF1 overexpression and CRISPR-mediated knock-out by two gRNA sequences, cells were transduced and selected in puromycin (1 μg/ml) and pooled.
Animals
All animal work was approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. For MCF-7 xenografts, prior to transplantation, 5–6 week old female nude mice (Envigo International) were ovariectomized and left to recover for two weeks before an estrogen “capsule” (Robinson and Jordan, 1989) (0.7 cm silicone tubing, Dow Corning) was inserted. These capsules contain 0.5 mg E2 mixed with silicone gel (Factor II) to keep the final weight of the filler at 2 mg. Three days later, one million human cancer cells were suspended in Growth Factor-Reduced Matrigel (BD Biosciences) and PBS and injected into the No. 9 mammary glands. When average tumor volume reached ≈200 mm3, the mice were randomized into different treatment groups, and the E2 capsule in the tumor-bearing mice was replaced with a fresh one, or with a tamoxifen pellet (5 mg/pellet, Innovative Research of America). DOX was added in the drinking water at 0.2 mg/ml to silence NF1 expression. For PDX studies, WHIM16 tumors were engrafted into cleared mammary fat pads of SCID/bg female mice (Envigo International) and allowed to grow without exogenous E2 supplementation until tumors reached ≈200 mm3. Mice were then randomized into different treatment groups. Fulvestrant was injected subcutaneously at 250 mg/kg weekly. Trametinib (1 mg/kg), dabrafenib (30 mg/kg), binimetinib (20 mg/kg), and selumetinib (20 mg/kg) were given daily as chow (Research Diets Inc.). Mouse body weight was measured twice weekly to monitor treatment toxicity.
METHOD DETAILS
Chemicals
Chemicals used are listed in Key Resource Table. Note that with the exception of the animal experiments, in all other experiments involving E2, the water-soluble version was used. Ethanol was used to dissolve 4-OHT, while DMSO was used to dissolve trametinib, dabrafenib, fulvestrant, and selumetinib. We note that drug concentrations were selected such that fulvestrant plus the selumetinib combination can more easily produce an effect on the cells that is greater than either group alone.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal NF1 (against aa2471-2839, 1:200) | This study | N/A |
| Rabbit polyclonal NF1 (against aa2471-2839) | This study | N/A |
| NF1 (against aa2760–2839) | Bethyl Laboratories | Cat#A300-140A; RRID:AB_2149790 |
| NF1 (against N-terminus, 1:500) | Cell Signaling Technology | Cat#14623; RRID:AB_2798543 |
| ER-α (1:500) | Cell Signaling Technology | Cat#8644; RRID:AB_2617128 |
| ER-α (HC-20) | Santa Cruz Biotechnology | Cat#sc-8002X; RRID:AB_627558 |
| ER-α (F-10, ChIP grade, 1:5000 for Western blots) | Santa Cruz Biotechnology | Cat# sc-8002X; RRID:AB_627558 |
| phospho-ER-α (Ser118, 1:500) | Millipore-Sigma | Cat#05–793; RRID:AB_310004 |
| ERK1/2 (1:500) | Cell Signaling Technology | Cat#9102; RRID:AB_330744 |
| Phospho-ERK1/2 (Thr202/Tyr204, 1:500) | Cell Signaling Technology | Cat#9101; RRID:AB_331646 |
| AKT (1:500) | Cell Signaling Technology | Cat#4691; RRID:AB_915783 |
| Phospho-AKT (Ser473, 1:500) | Cell Signaling Technology | Cat#4060; RRID:AB_2315049 |
| His-tag (1:1000) | Cell Signaling Technology | Cat#2365; RRID:AB_2115720 |
| Ran (1:1000) | Cell Signaling Technology | Cat#4462; RRID:AB_2284873 |
| Histone-3 (1:1000) | Cell Signaling Technology | Cat#4499; RRID:AB_10544537 |
| DNA-PKcs (1:1000) | Cell Signaling Technology | Cat#4602; RRID:AB_10692482 |
| K48-linkage specific polyubiquitin (1:1000) | Cell Signaling Technology | Cat#8081; RRID:AB_10859893 |
| α/β-tubulin (1:2000) | Cell Signaling Technology | Cat#2148; RRID:AB_2288042 |
| α-tubulin (1:10,000) | Santa Cruz Biotechnology | Cat#sc-5286; RRID:AB_628411 |
| p120/RasGAP (1:1000) | Santa Cruz Biotechnology | Cat#sc-63; RRID:AB_628206 |
| SRC-1 (1:1,000) | Santa Cruz Biotechnology | Cat#sc-6096; RRID:AB_661355 |
| HDAC1 (1:1,000) | Santa Cruz Biotechnology | Cat#sc-81598; RRID:AB_2118083 |
| GAPDH (1:10,000) | Santa Cruz Biotechnology | Cat#sc-32233; RRID:AB_627679 |
| Normal Mouse IgG | Santa Cruz Biotechnology | Cat# sc-2025; RRID:AB_628411 |
| Normal Rabbit IgG | Millipore-Sigma | Cat# 12-370; RRID:AB_145841 |
| Bacterial and Virus Strains | ||
| BL21 (DE3) | Agilent | Cat#200131 |
| Biological Samples | ||
| Patient-derived xenografts (PDX) WHIM16 | (Li et al., 2013) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 17β-estradiol (E2) | Sigma-Aldrich | Cat#E8875 |
| Cyclodextran-encapsulated “water-soluble” E2 | Sigma-Aldrich | Cat#E4389 |
| (Z)-4-hydroxytamoxifen (4-OHT) | Sigma-Aldrich | Cat#H7904 |
| Doxycycline (DOX) | Sigma-Aldrich | Cat#D9891 |
| Human epidermal growth factor (EGF) | Sigma-Aldrich | Cat#E4127 |
| Leptomycin B (LMB) | Sigma-Aldrich | Cat#L2913 |
| Bufalin | Sigma-Aldrich | Cat#B0261 |
| Phenylmethanesulfonylfluoride (PMSF) | Sigma-Aldrich | Cat#78830 |
| DMSO | Sigma-Aldrich | Cat#D8418 |
| Ethanol | Sigma-Aldrich | Cat#E7023 |
| MG132 | Millipore-Sigma | Cat#474790 |
| Polybrene | Millipore-Sigma | Cat#TR-1003-G |
| Trametinib | Selleck Chemicals, MedChem Express, or Novartis | Cat#S2673 or Cat#HY-10999A |
| Dabrafenib | Selleck Chemicals or MedChem Express | Cat#S2807 or Cat#HY-14660A |
| Fulvestrant | MedChem Express | Cat#HY-13636 |
| Binimetinib | MedChem Express | Cat#HY-15202 |
| Selumetinib | Selleck Chemicals or MedChem Express | Cat#S1008 or Cat#HY-50706 |
| Recombinant ERα protein | ThermoFisher Scientific | Cat#A15674 |
| Critical Commercial Assays | ||
| TruSeq RNA Library Prep Kit | Illumina | Cat# RS-122-2001 |
| KAPA Library Quantification Kit | Kapa Biosystems | Cat# KR0405 |
| Deposited Data | ||
| RNA-Seq | GEO | GSE142479 |
| Experimental Models: Cell Lines | ||
| MCF-7 | ATCC | Cat#HTB-22; RRID:CVCL_0031 |
| ZR-75B | A gift from Marc E. Lippman (Georgetown Univ., Med. Cntr.) | RRID:CVCL_5614 |
| T47D | ATCC | Cat#HTB-133; RRID:CVCL_0553 |
| Experimental Models: Organisms/Strains | ||
| Athymic nude mice | Envigo | N/A |
| SCID/bg | Envigo | N/A |
| Oligonucleotides | ||
| See Table S6. | This study | N/A |
| Recombinant DNA | ||
| pMD2.G | Addgene | Cat#12259 |
| pSPAX2 | Addgene | Cat#12260 |
| pINDUCER11-scrambled shRNA | This study | N/A |
| pINDUCER11-NF1 shRNA clone 5 | This study | N/A |
| pINDUCER11-NF1 shRNA clone 6 | This study | N/A |
| pLentiCRISPR v2-scramble | This study | N/A |
| pLentiCRISPR v2-NF1 clone 1 | GenScript | N/A |
| pLentiCRISPR v2-NF1 clone 2 | GenScript | N/A |
| pDONR225-NF1 | A gift from the RAS Initiative at the Frederick National Laboratory for Cancer Research at NCI | N/A |
| pCL-FLAG | Zheng et al., 2012 | N/A |
| pCL-FLAG-NF1 | This study | N/A |
| pCL-FLAG-NF1-R2258* | This study | N/A |
| pCL-FLAG-NF1-Y2285* | This study | N/A |
| pCL-FLAG-NF1-R2450* | This study | N/A |
| pCL-FLAG-NF1-R1362Q | This study | N/A |
| pCL-FLAG-NF1-K1444R | This study | N/A |
| pCL-FLAG-NF1-I417M | This study | N/A |
| pCL-FLAG-NF1-V1308A/I1309A | This study | N/A |
| pM-NF1 | This study | N/A |
| pM-NF1-R1362Q | This study | N/A |
| pM-NF1-K1444R | This study | N/A |
| pM-NF1-I417M | This study | N/A |
| pM-NF1-I417A/I418A | This study | N/A |
| pM-NF1-V1308A/I1309A | This study | N/A |
| pDEST566-GST | This study | N/A |
| pDEST566-NF1-M2/GAP | A gift from the RAS Initiative at the Frederick National Laboratory for Cancer Research at NCI | N/A |
| SpCas9(BB)-2A-GFP(PX458) | Addgene | Cat# 48138; RRID: Addgene_48138 |
| pGL4.70-RLuc | Promega | Cat#E688A |
| pGL2-ERE-Luc | A gift from Donald P. McDonnell (Duke Univ.) | Hall and McDonnell, 1999 |
| 5×Gal4-Luc | Promega | Cat#E2440 |
| pVP16-ERα | A gift from Donald P. McDonnell | Chang et al., 1999 |
| pVP16-SRC1 | A gift from Donald P. McDonnell | Chang et al., 1999 |
| pVP16-NCoR1 | A gift from Donald P. McDonnell | Chang et al., 1999 |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| SoftWorx v7.0 | GE Healthcare | N/A |
| FlowJo | FlowJo | N/A |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| RSEM v1.2.31 | Li and Dewey, 2011 | https://deweylab.github.io/RSEM/ |
| R 3.3.4 | R Core Team | N/A |
Plasmids
Scrambled and NF1-targeting shRNAs (clone C5 and C6) in pGIPZ were obtained from Dharmacon. All nucleotide sequences used in this study are described in Key Resource Table and Table S6. The shRNA clones were subcloned into pINDUCER11, with which cell lines stably expressing DOX-inducible scrambled or NF1 shRNA were later created as described (Zheng et al., 2015). Unless otherwise mentioned, data obtained from cell lines expressing NF1 shRNA C5 are shown in the figures, while the cells expressing C6 were also tested in most experiments. NF1 CRISPR-Cas9 knock-out (KO) constructs (clone #1 and #2 in pLentiCRISPR v2) were purchased from GenScript, and scrambled gRNA sequences were synthesized and then cloned into pLentiCRISPR v2. pDONR225-NF1 was a kind gift from the RAS Initiative at the Frederick National Laboratory for Cancer Research at NCI. The full-length NF1 coding sequence in this vector has been extensively modified to facilitate expression studies without changing the amino acid sequence. To analyze its expression, an “NF1 (NCI)” primer set was used for RT-PCR. This NF1 coding sequence is also refractory to both the C5 and C6 shRNA clones. NF1 mutants were created by site-directed mutagenesis (QuickChange II XL Site-Directed Mutagenesis Kit, Agilent Technologies) using pDONR225-NF1 as the template. These cDNAs were finally transferred into destination vectors pCL-FLAG and pM (Chang et al., 1999) to create pCL-FLAG-NF1, pM-NF1, pCL-FLAG-NF1-R2258*, pCL-FLAG-NF1-Y2285*, pCL-FLAG-NF1-R2450*, pCL-FLAG-NF1-R1362Q, pM-NF1-R1362Q, pCL-FLAG-NF1-K1444R, pM-NF1-K1444R, pCL-FLAG-NF1-I417M, pM-NF1-I417M, pM-NF1-I417A/I418A, pCL-FLAG-NF1-V1308A/I1309A, and pM-NF1-V1308A/I1309A. pDEST566-NF1-M2/GAP (Bollag et al., 1993), which expresses recombinant 6×His-MBP tag at the N-terminus of NF1-M2/GAP in E. coli, was also a gift from the NCI RAS Initiative. pDEST566-GST was built by transferring GST cDNA from pENTR-GST to pDEST566.
Generation of NF1 point mutation knock-in (KI) cells using CRISPR-Cas9
MCF-7 cells carrying homozygous mutations were generated by the Cell-Based Assay Screening Service core at Baylor College of Medicine (BCM). The general procedure is as described previously (Ran et al., 2013) and depicted in the right figure. Briefly, gRNA sequences were cloned into SpCas9(BB)-2A-GFP (PX458, a gift from Feng Zhang, Addgene plasmid #48138; http://n2t.net/addgene:48138; RRID: Addgene_48138). In addition to silent mutations that prevent Cas9 cleavage, each single-stranded oligodeoxynucleotide (ssODN) repair template also contains an engineered restriction site to facilitate identification of the KI allele. Cells were co-transfected with the CRISPR/gRNA plasmid and its corresponding ssODN template using the Neon transfection system (Thermo Fisher Scientific). The resulting GFP+ cells were plated by limiting dilution for single clone isolation. The genomic region surrounding each target site was PCR-amplified for validation by sequencing.
Assays for cell growth and apoptosis
To compare cell growth between NF1+ and NF1KD cells, cells stably expressing NF1 shRNA were first seeded in estrogen-deprived medium, typically in triplicate, with DOX added at the same time. Two days later (Day-0), the medium was replaced with a fresh medium containing either vehicle control or compound(s) under investigation, with medium change every three days thereafter. The number of viable cells was measured using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay kit (Promega) six days later (Day-6). Cell growth between parental and NF1 knock-out cells was similarly examined except no DOX was added. Apoptosis was measured by FITC-Annexin (Becton Dickinson) staining followed by FACS.
Generation of NF1 antibodies
A cDNA encoding amino acid residues 2471–2839 in NF1 was subcloned into pRP259 (Chang et al., 1994) to express this C-terminal fragment of NF1 (NF1C) as a GST-tagged protein in E. coli (BL21 DE3). Purified GST-NF1C was injected into rabbits (Thermo Fisher Scientific) or mice (Protein and Monoclonal Antibody Production Shared Resource at BCM) to generate polyclonal and monoclonal antibodies, respectively. The polyclonal antibody from one rabbit (NF1 Ab-2, Figure S5A) was later affinity-purified against the same immunizing antigen. One hybridoma clone (mAb-376) was expanded in vitro and IgG was purified from culture medium by protein-G Sepharose column chromatography.
Immunoprecipitation (IP)
Cells grown in E2-deprived medium were treated with E2 (10−11 M), 4-OHT (10−7 M), or the vehicle (ethanol) for 4 hrs before being isolated for whole cell or nuclear extracts. To conduct IP using whole cell extracts, cells were lysed in RIPA buffer supplemented with 1 mM PMSF and protease and phosphatase inhibitors (Roche Applied Science). The cell lysates were precleared with mouse IgG beads (Sigma, 3 hours at 4°C) before NF1 (mAb-376) or ER (F-10 monoclonal, Santa Cruz) antibody or the mouse IgG control (Santa Cruz) was added. Samples were incubated at 4°C overnight, followed by incubation with protein A/G agarose beads (Santa Cruz) for another 4 hours. All beads were finally washed three times with TBS (20 mM Tris-HCl (pH 7.5) and 150 mM NaCl). The bound proteins were eluted with 2× SDS sample buffer by boiling for 5 minutes and then examined by immunoblotting. To conduct IP using nuclear extracts, the same antibodies were pre-incubated with the protein-G Dynabeads™ (Thermo Fisher Scientific) overnight at 4°C before mixing with nuclear extracts which were prepared as described previously (Lanz et al., 2010). All beads were washed twice with NETN buffer (20 mM Tris-HCl (pH 7.5), 1 mM EDTA, 150 mM NaCl, and 0.5% NP-40), and once with PBS before elution and immunoblotting.
Immunoblotting
Cell lysates were generally prepared in RIPA buffer supplemented with protease and phosphatase inhibitors, and proteins were separated by SDS-PAGE before being transferred to nitrocellulose membranes. The primary antibodies used are listed in Key Resource Table. The fluorescein-conjugated secondary antibodies were from Li-COR Biosciences, and the protein levels were quantified by an Odyssey infrared imaging system (Li-COR Biosciences).
RNA-seq
Estrogen-deprived MCF-7 cells were treated with E2 (10−11 M), 4-OHT (10−7 M), or ethanol (vehicle) before RNA was extracted after 24 hrs using RNeasy Mini Kit (Qiagen) according to manufacturer’s directions. The E2 concentration used in this and the ChIP experiment was chosen because it can efficiently differentiate the growth between NF1+ and NF1-depleted cells (Figure 1E). Because the E2 concentration used was low, pilot qPCR experiments were conducted to determine that it takes 24 hrs after E2 stimulation to observe substantial difference in gene expression between these cells. MCF-7 CRISPR KI cells were simply grown in estrogen deprivation medium for 72 hrs before RNA extraction. The Genomic and RNA Profiling Core (GARP) at BCM constructed libraries with 250 ng of total RNA using the TruSeq RNA Library Prep Kit (Illumina). cDNA was generated from poly(A)-selected RNA. Libraries were quantified with the KAPA Library Quantification Kit (Kapa Biosystems) and were sequenced on the NextSeq 500 (Illumina) with paired-end 75bp reads and aligned to the hg19 (GRCh37) reference genome using RSEM v1.2.31 (Li and Dewey, 2011) and Bowtie 2 (Langmead and Salzberg, 2012).
Differential gene expression and fold change analysis was performed using EBseq (Leng et al., 2013) with FDR < 0.2 as a cutoff. Differentially expressed genes were subsequently used in Gene Set Enrichment Analysis (GSEA) for “Hallmark Pathways” in the Molecular Signatures Database (MSigDB) (Subramanian et al., 2005) and the top 10 enriched pathways were reported. For validation in clinical datasets, mRNA levels of the 1,107 genes identified from the MCF-7 cell line study were analyzed in TCGA and METABRIC ER+ breast cancer cohorts using cBioportal (Cerami et al., 2012; Gao et al., 2013). Genes in these cohorts that are differentially expressed between NF1-FS/NS and NF1-WT tumors were identified by t-test with p values cutoff at < 0.05. The differentially expressed genes in these clinical validation datasets were each similarly analyzed for “Hallmark” pathways enrichment as above (FDR<0.05). Clustering was performed using Hierarchical Clustering in R.
ChIP
The cells were seeded in E2-deprived medium. After 48 hrs, the cells were treated with E2 (10−11 M), 4-OHT (10−7 M), or the vehicle control (ethanol) for 45 min before ChIP, which was performed as described (Chen et al., 2008). Specifically, crosslinking was performed using 1% formaldehyde for 10 min at room temperature and quenched by glycine (final concentration of 125 mM). Cells were then scraped off the plate with ice-cold TBSE buffer (20 mM Tris-HCl, pH 7.5, 1.0 mM EDTA, 150 mM NaCl) and pelleted by centrifugation. Next, pelleted cells were resuspended in lysis buffer (10 mM Tris-HCl, pH 8.0, 0.25% Triton X-100, 10 mM EDTA, 100 mM NaCl) to release nuclei. The nuclei were further lysed in 1% SDS buffer (50 mM HEPESKOH, pH 7.5, 1% Triton X-100, 0.1% sodium deoxycholate, 1% SDS, 2 mM EDTA, 150 mM NaCl) and sonicated using a Branson Sonicator to shear genomic DNA to an average fragment size of 200 to 500 bp. ChIP was performed with a commercially available antibody against ER-α (HC-20 and F10 for ChIP-qPCR, and F10 for ChIP-seq, Santa Cruz), a rabbit polyclonal NF1 antibody (A300–140A from Bethyl Laboratories; Ab-1), a polyclonal NF1 antibody made by us (above; Ab-2), a rabbit IgG control (Millipore) or a mouse IgG control (Santa Cruz) added to the same amount of chromatin after SDS was diluted to 0.1%. The precipitated DNA was isolated by phenol-chloroform extraction. To assess ERE occupancy by qPCR, fold-enrichment of amplified ERE levels in the ChIPed samples vs. those in inputs were calculated.
qPCR
RNA was isolated using the RNeasy kit (Qiagen), and cDNAs were synthesized from 5 μg of total RNA using the SuperScript IV First-Strand Synthesis System (Thermo Fisher Scientific). Real-time PCR was performed with Fast SYBR Green Master Mix on a Light Cycle 96 (Roche Life Science) or CFX (Bio-Rad) Real-Time PCR system. The PCR primers used are described in Table S6. The relative amounts of PCR products generated from each primer set were determined on the basis of threshold cycle (Ct) using GAPDH as the loading control.
ERE-luciferase reporter assay
MCF-7 cells were seeded in E2-deprived medium for one day before being transfected with pGL2-ERE-Luc (Hall and McDonnell, 1999), which contains 3× vitellogenin EREs, for another day. The cells were then treated with 10−11 M E2 for 24 hrs. Cells were also co-transfected with pGL4.70-RLuc (Promega, Madison, WI, USA) to express Renilla luciferase as an internal transfection efficiency control. The firefly and Renilla luciferase levels were measured using the Dual-Luciferase Reporter Assay kit (Promega).
The mammalian two-hybrid assay
The mammalian two-hybrid assays measuring the binding between ER and its coregulators have been described (Chang et al., 1999). Briefly, HEK293 cells were co-transfected by pM vectors expressing various versions of NF1 fused to the Gal4 DNA binding domain (DBD), as well as pVP16 vectors expressing various ER fragments fused to the VP16 transcriptional activation domain. These cells were also co-transfected to carry the firefly luciferase reporter 5×Gal4-Luc3 and the pGL4.70-RLuc, whose Renilla luciferase activity was measured to control transfection efficiency. Fusion protein expression levels were also assessed by Western blot. Luciferase activities were measured two days later. To determine whether the NF1-ER interaction can be modulated by E2 and 4-OHT, cells were estrogen-starved before transfection. E2 (10−11 M), 4-OHT (10−7 M), or the vehicle control was added one day post-transfection. The luciferase activity was measured after another day.
ER and NF1-M2/GAP in vitro binding assay
E. coli BL21 (DE3) cells carrying either pDEST566-GST or pDEST566-NF1-M2/GAP were induced by 1 mM IPTG at 30°C for 4 hrs. The cell pellets were then resuspended in ice-cold MBP binding buffer (20 mM Tris-HCl (pH 7.4), 200 mM NaCl, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, and protease inhibitors) and sonicated. The cell lysates were incubated with amylose magnetic beads (New England Biolabs) for 4 hrs at 4°C and washed by the MBP binding buffer. After elution (2× SDS sample buffer), the concentrations of bound proteins on the beads were quantified using BSA as the standard. Purified recombinant ER-α (10 ng, Thermo Fisher Scientific) was pre-incubated with 100 nM E2, 5 μM 4-OHT, or ethanol for 1 hr at 4°C, and then mixed with 200 ng bead-bound NF1-M2/GAP or the GST control, which was pre-incubated with 1 mg/ml BSA in MBP binding buffer for 1 hr at 4°C to reduce non-specific binding. After 1 hr at 4°C, the bound proteins were eluted with 2× SDS sample buffer and examined by immunoblotting.
Cell fractionation
The experiments were conducted as previously described (Suzuki et al., 2010). Leptomycin B (LMB) at 40 nM or ethanol (vehicle) was given to the cells grown in regular growth medium for 6 hours. To assess the influence of ER ligands, cells were first starved in E2-deprived medium and then treated with E2 (10−11 M), 4-OHT (10−7 M), or the vehicle control for 16 hours.
Immunofluorescene deconvolution microscopy
Cells were seeded and treated by LMB or ER-ligands (above) in 12-well plates with an 18-mm No. 1.5 coverslip. The fixation (4% formaldehyde), permeabilization (200 mM glycine/1X PBS, 0.1% Triton X-100), and blocking (5% goat serum) were performed as previously described (Cheng et al., 2011). The sample was incubated with NF1 antibody mAb-376 at room temperature for 2.5 hrs. We found that it is important to optimize the dilution factor for the secondary antibody to reduce background. Alexa-Fluor™ 488 conjugated goat anti-mouse IgG secondary Ab (Thermo Fisher Scientific) was added after a 1:1,000 dilution in the blocking solution and incubation was performed in the dark for 1 hr. The samples were then mounted to slides with a DAPI-containing mounting medium (Vector Laboratories). Imaging was performed at the Integrated Microscopy Core at BCM on a DVLive epifluorescence image restoration microscope (GE Healthcare). The system is equipped with an Olympus PlanApo 60×/1.42 N.A. objective and a pco.EDGE sCMOS_5.5 camera with a resolution up to 2,560 ×2,160 pixels. The filter sets used were: DAPI and FITC. Z stacks (0.2 μm) covering the whole cell (~12 μm) were acquired before applying a conservative restorative algorithm for quantitative image deconvolution using SoftWorx v7.0. Image J was used to quantify NF1 levels. For each cell a single focal plane dissecting the middle of the nucleus was chosen, from which two ROIs were created: ROI-1 encircles the whole cell while ROI-2 encircles just the nucleus. The reported relative NF1 nuclear levels were defined as: ROI-2/ROI-1.
ERE pull-down using HeLa cell nuclear extract
These assays were conducted similarly to those previously described (Foulds et al., 2013). Briefly, 1 mg of HeLa S3 nuclear extract and 0.5 μg of purified recombinant ER were added to 4 μg of a 921 bp biotinylated 4× ERE DNA fragment immobilized onto 60 μl Dynabeads™ M280 (Thermo Fisher Scientific). Different ligands were included in these recruitment assays. After washing twice in NETN and once in DPBS, the retained proteins were detected by immunoblotting.
QUANTIFICATION AND STATISTICAL ANALYSIS
All reported data were typically presented as mean ± s.e.m. and pair-wise comparisons were analyzed by two-sided Student’s t-test unless otherwise mentioned. The n values are either separate biological replicates or numbers of cells or mice, as indicated. Human tumor databases were analyzed using the R statistical packages. We note that throughout this paper *, p<0.05; **, p<0.01; ***, p<0.001; NS, Not Significant (p≥0.05).
NF1 mutation frequency in patients
The extraction and general sequencing pipeline of cell-free DNA has been described previously (Lanman et al., 2015). Specifically, for NF1, most of its exons (except exons 1, 7, 15, 19, 22, 23, 29, 31, 51, and 56–61) were completely sequenced, and single nucleotide variants as well as small indel events were analyzed. M1 and M2 are encoded by exons 11 (25 amino acids) and 29 (35 amino acids). cBio (Cerami et al., 2012) was used for somatic mutation and clinical data mining. Mutations were visualized using the GeneVisR (Skidmore et al., 2016) Bioconductor package. The cohort of primary breast cancers was from TCGA (TCGA, 2012), while the metastatic cohort was described (Lanman et al., 2015).
Patient proliferation status after aromatase inhibitor (AI) treatments
Patients in the ACOSOG Z1031 trial (Ellis et al., 2011) were treated by AIs in a neoadjuvant setting, and their mRNAs before and after treatment were analyzed by Agilent microarray. A multigene proliferation score (MGPS) derived from mRNA expression of 772 genes was used to analyze proliferation status in the patient to assess treatment response (Ellis et al., 2017).
NF1 expression level analysis in patients
NF1 mutation types in TCGA (Ciriello et al., 2015) and METABRIC (Pereira et al., 2016) were mined from cBio (Cerami et al., 2012; Gao et al., 2013). NF1 mRNA levels in ER+ breast tumors from the METABRIC cohort (Curtis et al., 2012) together with survival information were obtained from Oncomine (Rhodes et al., 2007). High and low NF1 mRNA levels were as defined above. Univariate Cox regression was analyzed using the Survival package for R.
Protein levels correlation analysis
We have previously shown that protein co-expression is a strong predictor for co-functionality (Wang et al., 2017a). To identify proteins co-expressed with NF1, we used the LinkedOmics tool (http://www.linkedomics.org) (Vasaikar et al., 2018) to calculate the Pearson’s correlation coefficient between NF1 and all other proteins in the CPTAC (Clinical Proteomic Tumor Analysis Consortium) breast cancer mass spectrometry dataset, which contains 105 breast tumor samples (Mertins et al., 2016). We ranked the proteins based on correlation coefficient scores and then analyzed them using Gene Set Enrichment Analysis (GSEA) in WebGestalt (Wang et al., 2017b) to identify enriched Gene Ontology (GO) molecular functions. As a control, we similarly analyzed p120GAP/RASA1.
DATA AND SOFTWARE AVAILABILITY
RNA-seq data are deposited in Gene Expression Omnious (GEO) database, accession number GSE142479.
Supplementary Material
Table S1, related to Figure 1. Non-silent NF1 mutations identified by sequencing circulating tumor DNA in a metastatic ER+ breast cancer cohort.
Table S2, related to Figure 1. Multi Gene Proliferation Scores (MGPS) of samples in the Z1031 trial.
Table S3, related to Figure 2. E2 and 4-OHT modulated gene expression changes in the MCF-7 cell model.
Table S4, related to Figure 2. Hallmark Pathway enrichment validation in patient data in TCGA and METABRIC.
Table S5, related to Figure 3. Gene expression as analyzed by RNA-seq in MCF-7 cells carrying wild-type NF1 or an NF1 Knock-in mutation.
Table S6, related to STAR Methods. Oligonucleotides sequences used for this study.
Highlights.
NF1 is a GAP-independent estrogen receptor transcription co-repressor.
Somatic NF1 loss causes tamoxifen/aromatase inhibitor resistance in ER+ breast cancer.
A MEK inhibitor plus a SERD is effective in NF1− ER+ PDX and cell line models.
Significance.
Neurofibromin is a co-repressor of estrogen receptor (ER), therefore loss enhances ER transcriptional activity. Loss-of-function mutations in the NF1 gene promote tamoxifen agonism and estrogen-deprivation resistance. While ER+ NF1-depleted cells are initially responsive to fulvestrant, a selective ER degrader, enhanced Ras activity promotes acquired resistance. When Ras pathway activation is inhibited with a well-tolerated MEK inhibitor, fulvestrant resistance is reversed and full tumor regression ensues. Thus, neurofibromin-depletion represents a distinct breast cancer subset where the choice of endocrine treatment may be critical, and MEK inhibitor combinations require clinical investigation. A role as a dual repressor for both Ras and ER may also explain the sexually dimorphic characteristics of neurofibromatosis where tumorigenesis is promoted by female puberty.
Acknowledgements
We thank our colleagues for providing valuable resources and advice: Bert O’Malley, Anna Malovannaya, Jun Qin, Daniel Yan, Khushboo Shah, Jianhui Yao, Svasti Haricharan, Angela D. Wilkins, Dominic Esposito, Gary Chamness, Laura Smithson, David Gutmann, Ching-Yi Chang, and Donald McDonnell. This project was assisted by the following core facilities in the Dan L. Duncan Comprehensive Cancer Center: Cell-Based Assay Screening Service, Cytometry and Cell Sorting, Biostatistics and Informatics Shared Resource, Integrated Microscopy Core, Genomic and RNA Profiling Core, and the Protein and Monoclonal Antibody Production Shared Resource, which are all supported by a P30 Cancer Center Support Grant from NCI (CA125123). The Integrated Microscopy Core is also supported by grants from NIH (DK56338), CPRIT (RP150578 and RP170719) and the John S. Dunn Gulf Coast Consortium for Chemical Genomics. The Cell Sorting Core was also supported by P30AI036211 and S10RR024547. BC was supported by an ASCO Gianni Bonadonna Research Fellowship, and JTL was supported by T32GM008129 and T32CA203690 from NIH. CEF and YC were supported by the Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with BCM. ECC was supported by The Susan G. Komen Foundation (SAC150059), DOD (W81XWH-16-1-0538 and W81XWH-19-1-0527), Nancy Owen Memorial Foundation, NIH (R21CA185516 and P50CA186784), William and Ella Owens Foundation, and CPRIT (RP180844). M.J.E. is a CPRIT Scholar in cancer research supported by a CPRIT Established Investigator Recruitment Award RR140033 and is a McNair Scholar supported by the McNair Foundation. This research was also supported by NIH/NCI grants R01 CA095614 and U54CA233223 awarded to M.J.E. and by Susan G. Komen for the Cure Promise Grant PG12220321.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: Most authors declare no competing financial interests except: KCB and RBL are stockholders and employees of Guardant Health, Inc., CEF discloses an equity position in Coactigon, Inc., and MJE received consulting fees from Abbvie, Sermonix, Pfizer, AstraZeneca, Celgene, NanoString, Puma, and Novartis, and is an equity stockholder, consultant, and Board Director member of BioClassifier, and inventor on a patent for the Breast PAM50 assay.
References
- Anbalagan M, and Rowan BG (2015). Estrogen receptor alpha phosphorylation and its functional impact in human breast cancer. Mol Cell Endocrinol 418 Pt 3, 264–272. [DOI] [PubMed] [Google Scholar]
- Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, and Collins F (1990). The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 63, 851–859. [DOI] [PubMed] [Google Scholar]
- Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li J, Cohn MA, Cantley LC, and Gygi SP (2004). Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci U S A 101, 12130–12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berto M, Jean V, Zwart W, and Picard D (2018). ERalpha activity depends on interaction and target site corecruitment with phosphorylated CREB1. Life Sci Alliance 1, e201800055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertucci F, Ng CKY, Patsouris A, Droin N, Piscuoglio S, Carbuccia N, Soria JC, Dien AT, Adnani Y, Kamal M, et al. (2019). Genomic characterization of metastatic breast cancers. Nature 569, 560–564. [DOI] [PubMed] [Google Scholar]
- Bollag G, McCormick F, and Clark R (1993). Characterization of full-length neurofibromin: tubulin inhibits Ras GAP activity. EMBO J 12, 1923–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borromeo MD, Savage TK, Kollipara RK, He M, Augustyn A, Osborne JK, Girard L, Minna JD, Gazdar AF, Cobb MH, and Johnson JE (2016). ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell Rep 16, 1259–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, et al. (2005). Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122, 33–43. [DOI] [PubMed] [Google Scholar]
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, et al. (2006). Genome-wide analysis of estrogen receptor binding sites. Nat Genet 38, 1289–1297. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Norris JD, Gron H, Paige LA, Hamilton PT, Kenan DJ, Fowlkes D, and McDonnell DP (1999). Dissection of the LXXLL nuclear receptor-coactivator interaction motif using combinatorial peptide libraries: discovery of peptide antagonists of estrogen receptors alpha and beta. Mol Cell Biol 19, 8226–8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EC, Barr M, Wang Y, Jung V, Xu H, and Wigler HM (1994). Cooperative interaction of S. pombe proteins required for mating and morphogenesis. Cell 79, 131–141. [DOI] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. (2008). Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117. [DOI] [PubMed] [Google Scholar]
- Cheng CM, Li H, Gasman S, Huang J, Schiff R, and Chang EC (2011). Compartmentalized Ras proteins transform NIH 3T3 cells with different efficiencies. Mol Cell Biol 31, 983–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, et al. (2015). Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 163, 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. (2012). The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daston MM, Scrable H, Nordlund M, Sturbaum AK, Nissen LM, and Ratner N (1992). The protein product of the neurofibromatosis type 1 gene is expressed at highest abundance in neurons, Schwann cells, and oligodendrocytes. Neuron 8, 415–428. [DOI] [PubMed] [Google Scholar]
- de Cremoux P, Bieche I, Tran-Perennou C, Vignaud S, Boudou E, Asselain B, Lidereau R, Magdelenat H, Becette V, Sigal-Zafrani B, and Spyratos F (2004). Inter-laboratory quality control for hormone-dependent gene expression in human breast tumors using real-time reverse transcription-polymerase chain reaction. Endocr Relat Cancer 11, 489–495. [DOI] [PubMed] [Google Scholar]
- Diggs-Andrews KA, Brown JA, Gianino SM, Rubin JB, Wozniak DF, and Gutmann DH (2014). Sex Is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann Neurol 75, 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Li N, Dong B, Guo W, Wei H, Chen Q, Yuan H, Han Y, Chang H, Kan S, et al. (2019). Chromatin remodeling ATPase BRG1 and PTEN are synthetic lethal in prostate cancer. J Clin Invest 129, 759–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dischinger PS, Tovar EA, Essenburg CJ, Madaj ZB, Gardner EE, Callaghan ME, Turner AN, Challa AK, Kempston T, Eagleson B, et al. (2018). NF1 deficiency correlates with estrogen receptor signaling and diminished survival in breast cancer. NPJ Breast Cancer 4, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, Whitcomb P, Martin S, Aschbacher-Smith LE, Rizvi TA, et al. (2016). Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas. N Engl J Med 375, 2550–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis MJ, Gao F, Dehdashti F, Jeffe DB, Marcom PK, Carey LA, Dickler MN, Silverman P, Fleming GF, Kommareddy A, et al. (2009). Lower-dose vs high-dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer: a phase 2 randomized study. JAMA 302, 774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis MJ, Suman VJ, Hoog J, Goncalves R, Sanati S, Creighton CJ, DeSchryver K, Crouch E, Brink A, Watson M, et al. (2017). Ki67 Proliferation Index as a Tool for Chemotherapy Decisions During and After Neoadjuvant Aromatase Inhibitor Treatment of Breast Cancer: Results From the American College of Surgeons Oncology Group Z1031 Trial (Alliance). J Clin Oncol 35, 1061–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis MJ, Suman VJ, Hoog J, Lin L, Snider J, Prat A, Parker JS, Luo J, DeSchryver K, Allred DC, et al. (2011). Randomized phase II neoadjuvant comparison between letrozole, anastrozole, and exemestane for postmenopausal women with estrogen receptor-rich stage 2 to 3 breast cancer: clinical and biomarker outcomes and predictive value of the baseline PAM50-based intrinsic subtype--ACOSOG Z1031. J Clin Oncol 29, 2342–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, and O’Malley BW (2014). Nuclear receptor modulation–role of coregulators in selective estrogen receptor modulator (SERM) actions. Steroids 90, 39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulds CE, Feng Q, Ding C, Bailey S, Hunsaker TL, Malovannaya A, Hamilton RA, Gates LA, Zhang Z, Li C, et al. (2013). Proteomic analysis of coregulators bound to ERalpha on DNA and nucleosomes reveals coregulator dynamics. Mol Cell 51, 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates LA, Gu G, Chen Y, Rohira AD, Lei JT, Hamilton RA, Yu Y, Lonard DM, Wang J, Wang SP, et al. (2018). Proteomic profiling identifies key coactivators utilized by mutant ERalpha proteins as potential new therapeutic targets. Oncogene 37, 4581–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glont SE, Chernukhin I, and Carroll JS (2019). Comprehensive Genomic Analysis Reveals that the Pioneering Function of FOXA1 Is Independent of Hormonal Signaling. Cell Rep 26, 2558–2565 e2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith OL, Spies NC, Anurag M, Griffith M, Luo J, Tu D, Yeo B, Kunisaki J, Miller CA, Krysiak K, et al. (2018). The prognostic effects of somatic mutations in ER-positive breast cancer. Nature communications 9, 3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisouard J, Medunjanin S, Hermani A, Shukla A, and Mayer D (2007). Glycogen synthase kinase-3 protects estrogen receptor alpha from proteasomal degradation and is required for full transcriptional activity of the receptor. Mol Endocrinol 21, 2427–2439. [DOI] [PubMed] [Google Scholar]
- Gross AM, Wolters P, Baldwin A, Dombi E, Fisher MJ, Weiss BD, Kim AeRang, Blakeley JON, Whitcomb P, Holmblad M, et al. (2018). SPRINT: Phase II study of the MEK 1/2 inhibitor selumetinib (AZD6244, ARRY-142886) in children with neurofibromatosis type 1 (NF1) and inoperable plexiform neurofibromas (PN). J Clin Onco 36, 10503–10503. [Google Scholar]
- Hall JM, and McDonnell DP (1999). The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 140, 5566–5578. [DOI] [PubMed] [Google Scholar]
- Harrod A, Fulton J, Nguyen VTM, Periyasamy M, Ramos-Garcia L, Lai CF, Metodieva G, de Giorgio A, Williams RL, Santos DB, et al. (2017). Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 36, 2286–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Sinha I, Fan R, Haldosen LA, Yan F, Zhao C, and Dahlman-Wright K (2018). c-Jun/AP-1 overexpression reprograms ERalpha signaling related to tamoxifen response in ERalpha-positive breast cancer. Oncogene 37, 2586–2600. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldring N, Pawson T, McDonnell D, Treuter E, Gustafsson JA, and Pike AC (2007). Structural insights into corepressor recognition by antagonist-bound estrogen receptors. J Biol Chem 282, 10449–10455. [DOI] [PubMed] [Google Scholar]
- Hu X, and Lazar MA (1999). The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature 402, 93–96. [DOI] [PubMed] [Google Scholar]
- Huang HJ, Norris JD, and McDonnell DP (2002). Identification of a negative regulatory surface within estrogen receptor alpha provides evidence in support of a role for corepressors in regulating cellular responses to agonists and antagonists. Mol Endocrinol 16, 1778–1792. [DOI] [PubMed] [Google Scholar]
- Huang KL, Li S, Mertins P, Cao S, Gunawardena HP, Ruggles KV, Mani DR, Clauser KR, Tanioka M, Usary J, et al. (2017). Proteogenomic integration reveals therapeutic targets in breast cancer xenografts. Nature communications 8, 14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A, Xiao T, Li W, Qiu X, Buchwalter G, et al. (2018). Allele-Specific Chromatin Recruitment and Therapeutic Vulnerabilities of ESR1 Activating Mutations. Cancer Cell 33, 173–186 e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmakar S, Jin Y, and Nagaich AK (2013). Interaction of glucocorticoid receptor (GR) with estrogen receptor (ER) alpha and activator protein 1 (AP1) in dexamethasone-mediated interference of ERalpha activity. J Biol Chem 288, 24020–24034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, et al. (1995). Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 270, 1491–1494. [DOI] [PubMed] [Google Scholar]
- Kininis M, Isaacs GD, Core LJ, Hah N, and Kraus WL (2009). Postrecruitment regulation of RNA polymerase II directs rapid signaling responses at the promoters of estrogen target genes. Mol Cell Biol 29, 1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koliou X, Fedonidis C, Kalpachidou T, and Mangoura D (2016). Nuclear import mechanism of neurofibromin for localization on the spindle and function in chromosome congression. J Neurochem 136, 78–91. [DOI] [PubMed] [Google Scholar]
- Kweh F, Zheng M, Kurenova E, Wallace M, Golubovskaya V, and Cance WG (2009). Neurofibromin physically interacts with the N-terminal domain of focal adhesion kinase. Mol Carcinog 48, 1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanman RB, Mortimer SA, Zill OA, Sebisanovic D, Lopez R, Blau S, Collisson EA, Divers SG, Hoon DS, Kopetz ES, et al. (2015). Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS One 10, e0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz RB, Bulynko Y, Malovannaya A, Labhart P, Wang L, Li W, Qin J, Harper M, and O’Malley BW (2010). Global characterization of transcriptional impact of the SRC-3 coregulator. Mol Endocrinol 24, 859–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei JT, Shao J, Zhang J, Iglesia M, Chan DW, Cao J, Anurag M, Singh P, He X, Kosaka Y, et al. (2018). Functional Annotation of ESR1 Gene Fusions in Estrogen Receptor-Positive Breast Cancer. Cell reports 24, 1434–1444 e1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BM, Haag JD, Gould MN, Stewart RM, and Kendziorski C (2013). EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 29, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Cheng Y, Gutmann DA, and Mangoura D (2001). Differential localization of the neurofibromatosis 1 (NF1) gene product, neurofibromin, with the F-actin or microtubule cytoskeleton during differentiation of telencephalic neurons. Brain Res Dev Brain Res 130, 231–248. [DOI] [PubMed] [Google Scholar]
- Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, He X, Liu S, Hoog J, Lu C, et al. (2013). Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell reports 4, 1116–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Strom A, Vega VB, Kong SL, Yeo AL, Thomsen JS, Chan WC, Doray B, Bangarusamy DK, Ramasamy A, et al. (2004). Discovery of estrogen receptor alpha target genes and response elements in breast tumor cells. Genome Biol 5, R66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madanikia SA, Bergner A, Ye X, and Blakeley JO (2012). Increased risk of breast cancer in women with NF1. American journal of medical genetics Part A 158A, 3056–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens O, and Cichowski K (2014). An expanding role for RAS GTPase activating proteins (RAS GAPs) in cancer. Adv Biol Regul 55, 1–14. [DOI] [PubMed] [Google Scholar]
- Martin LA, Ribas R, Simigdala N, Schuster E, Pancholi S, Tenev T, Gellert P, Buluwela L, Harrod A, Thornhill A, et al. (2017). Discovery of naturally occurring ESR1 mutations in breast cancer cell lines modelling endocrine resistance. Nature communications 8, 1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meric-Bernstam F, Frampton GM, Ferrer-Lozano J, Yelensky R, Perez-Fidalgo JA, Wang Y, Palmer GA, Ross JS, Miller VA, Su X, et al. (2014). Concordance of genomic alterations between primary and recurrent breast cancer. Mol Cancer Ther 13, 1382–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, et al. (2016). Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. (2006). A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10, 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, et al. (2016). Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nousiainen M, Sillje HH, Sauer G, Nigg EA, and Korner R (2006). Phosphoproteome analysis of the human mitotic spindle. Proc Natl Acad Sci U S A 103, 5391–5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H, Sato H, Takatsuka J, and De Luca LM (2001). Human breast cancer MDA-MB-231 cells fail to express the neurofibromin protein, lack its type I mRNA isoform and show accumulation of P-MAPK and activated Ras. Cancer Lett 172, 159–164. [DOI] [PubMed] [Google Scholar]
- Paschou M, and Doxakis E (2012). Neurofibromin 1 is a miRNA target in neurons. PLoS One 7, e46773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson A, Proszek PZ, Pascual J, Fribbens C, Shamsher MK, Kingston B, O’Leary B, Herrera-Abreu MT, Cutts RJ, Garcia-Murillas I, et al. (2019). Inactivating NF1 mutations are enriched in advanced breast cancer and contribute to endocrine therapy resistance. Clin Cancer Res. [DOI] [PubMed] [Google Scholar]
- Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ, et al. (2016). The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nature communications 7, 11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner N, and Miller SJ (2015). A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer 15, 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, Cai Y, Bielski CM, Donoghue MTA, Jonsson P, et al. (2018). The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 34, 427–438 e426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, et al. (2007). Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia 9, 166–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, et al. (2015). Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372, 30–39. [DOI] [PubMed] [Google Scholar]
- Robinson SP, and Jordan VC (1989). Antiestrogenic action of toremifene on hormone-dependent, -independent, and heterogeneous breast tumor growth in the athymic mouse. Cancer Res 49, 1758–1762. [PubMed] [Google Scholar]
- Rogers SA, Llewellyn L, Wigham T, and Sweeney GE (2000). Cloning of the Atlantic salmon (Salmo salar) estrogen receptor-alpha gene. Comp Biochem Physiol B Biochem Mol Biol 125, 379–385. [DOI] [PubMed] [Google Scholar]
- Salemis NS, Nakos G, Sambaziotis D, and Gourgiotis S (2010). Breast cancer associated with type 1 neurofibromatosis. Breast Cancer 17, 306–309. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Hu X, DiRenzo J, Lazar MA, and Brown M (2000). Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103, 843–852. [DOI] [PubMed] [Google Scholar]
- Sharif S, Moran A, Huson SM, Iddenden R, Shenton A, Howard E, and Evans DG (2007). Women with neurofibromatosis 1 are at a moderately increased risk of developing breast cancer and should be considered for early screening. Journal of Medical Genetics 44, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, and Greene GL (1998). The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 95, 927–937. [DOI] [PubMed] [Google Scholar]
- Skidmore ZL, Wagner AH, Lesurf R, Campbell KM, Kunisaki J, Griffith OL, and Griffith M (2016). GenVisR: Genomic Visualizations in R. Bioinformatics 32, 3012–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol ES, Feng YX, Jin DX, Basudan A, Lee AV, Atkinson JM, Chen J, Stephens PJ, Frampton GM, Gupta PB, et al. (2019). Loss of function of NF1 is a mechanism of acquired resistance to endocrine therapy in lobular breast cancer. Ann Oncol 30, 115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Bose P, Leong-Quong RY, Fujita DJ, and Riabowol K (2010). REAP: A two minute cell fractionation method. BMC Res Notes 3, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCGA (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojaniello C, Festino L, Vanella V, and Ascierto PA (2019). Encorafenib in combination with binimetinib for unresectable or metastatic melanoma with BRAF mutations. Expert Rev Clin Pharmacol 12, 259–266. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke I, Van Oostveldt P, Coene E, De Paepe A, and Messiaen L (2004). Neurofibromin is actively transported to the nucleus. FEBS Lett 560, 98–102. [DOI] [PubMed] [Google Scholar]
- Vasaikar SV, Straub P, Wang J, and Zhang B (2018). LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res 46, D956–D963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liu C, Liu X, Wang M, Wu D, Gao J, Su P, Nakahata T, Zhou W, Xu Y, et al. (2018). MEIS1 Regulates Hemogenic Endothelial Generation, Megakaryopoiesis, and Thrombopoiesis in Human Pluripotent Stem Cells by Targeting TAL1 and FLI1. Stem Cell Reports 10, 447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ma Z, Carr SA, Mertins P, Zhang H, Zhang Z, Chan DW, Ellis MJ, Townsend RR, Smith RD, et al. (2017a). Proteome Profiling Outperforms Transcriptome Profiling for Coexpression Based Gene Function Prediction. Mol Cell Proteomics 16, 121–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Vasaikar S, Shi Z, Greer M, and Zhang B (2017b). WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 45, W130–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill TG, Wang J, Qi R, Matzuk AJ, Song X, Madoux F, et al. (2014). Bufalin is a potent small-molecule inhibitor of the steroid receptor coactivators SRC-3 and SRC-1. Cancer Res 74, 1506–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins AD, Bachman BJ, Erdin S, and Lichtarge O (2012). The use of evolutionary patterns in protein annotation. Current Opinion in Structural Biology 22, 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won Jeong K, Chodankar R, Purcell DJ, Bittencourt D, and Stallcup MR (2012). Gene-specific patterns of coregulator requirements by estrogen receptor-alpha in breast cancer cells. Mol Endocrinol 26, 955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap YS, McPherson JR, Ong CK, Rozen SG, Teh BT, Lee AS, and Callen DF (2014). The NF1 gene revisited - from bench to bedside. Oncotarget 5, 5873–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I, Alexandrov LB, Van Loo P, Haugland HK, Lilleng PK, et al. (2017). Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 32, 169–184 e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman K, Winterhalder R, Mamot C, Hasler-Strub U, Rochlitz C, Mueller A, Berset C, Wiliders H, Perey L, Rudolf CB, et al. (2015). Fulvestrant with or without selumetinib, a MEK 1/2 inhibitor, in breast cancer progressing after aromatase inhibitor therapy: a multicentre randomised placebo-controlled double-blind phase II trial, SAKK 21/08. Eur J Cancer 51, 1212–1220. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng ZY, Bay BH, Aw SE, and Lin VC (2005). A novel antiestrogenic mechanism in progesterone receptor-transfected breast cancer cells. J Biol Chem 280, 17480–17487. [DOI] [PubMed] [Google Scholar]
- Zheng ZY, Cheng CM, Fu XR, Chen LY, Xu L, Terrillon S, Wong ST, Bar-Sagi D, Songyang Z, and Chang EC (2012). CHMP6 and VPS4A mediate the recycling of Ras to the plasma membrane to promote growth factor signaling. Oncogene 31, 4630–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng ZY, Tian L, Bu W, Fan C, Gao X, Wang H, Liao YH, Li Y, Lewis MT, Edwards D, et al. (2015). Wild-type N-Ras, overexpressed in basal-like breast cancer, promotes tumor formation by inducing IL-8 secretion via JAK2 activation. Cell reports 12, 511–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zill OA, Banks KC, Fairclough SR, Mortimer SA, Vowles JV, Mokhtari R, Gandara DR, Mack PC, Odegaard JI, Nagy RJ, et al. (2018). The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin Cancer Res 24, 3528–3538. [DOI] [PubMed] [Google Scholar]
- Zubairy S, and Oesterreich S (2005). Estrogen-repressed genes – key mediators of estrogen action? Breast Cancer Res 7, 163–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, related to Figure 1. Non-silent NF1 mutations identified by sequencing circulating tumor DNA in a metastatic ER+ breast cancer cohort.
Table S2, related to Figure 1. Multi Gene Proliferation Scores (MGPS) of samples in the Z1031 trial.
Table S3, related to Figure 2. E2 and 4-OHT modulated gene expression changes in the MCF-7 cell model.
Table S4, related to Figure 2. Hallmark Pathway enrichment validation in patient data in TCGA and METABRIC.
Table S5, related to Figure 3. Gene expression as analyzed by RNA-seq in MCF-7 cells carrying wild-type NF1 or an NF1 Knock-in mutation.
Table S6, related to STAR Methods. Oligonucleotides sequences used for this study.
Data Availability Statement
RNA-seq data are deposited in Gene Expression Omnious (GEO) database, accession number GSE142479.