

Abstract
Wolfram Syndrome 1 (WFS1) protein is an endoplasmic reticulum (ER) factor whose deficiency results in juvenile-onset diabetes secondary to cellular dysfunction and apoptosis. The mechanisms guiding β-cell outcomes secondary to WFS1 function, however, remain unclear. Here, we show that WFS1 preserves normal β-cell physiology by promoting insulin biosynthesis and negatively regulating ER stress. Depletion of Wfs1 in vivo and in vitro causes functional defects in glucose-stimulated insulin secretion and insulin content, triggering Chop-mediated apoptotic pathways. Genetic proof of concept studies coupled with RNA-seq reveal that increasing WFS1 confers a functional and a survival advantage to β-cells under ER stress by increasing insulin gene expression and downregulating the Chop-Trib3 axis, thereby activating Akt pathways. Remarkably, WFS1 and INS levels are reduced in type 2 diabetic (T2DM) islets, suggesting that WFS1 may contribute to T2DM β-cell pathology. Taken together, this work reveals essential pathways regulated by WFS1 to control β-cell survival and function primarily through preservation of ER homeostasis.
Summary Urano
WFS1 is a causative gene for Wolfram syndrome, a rare neurodegenerative disorder characterized by juvenile-onset diabetes mellitus and optic nerve atrophy. Genetic proof of concept studies coupled with RNA-seq reveal that increasing WFS1 confers a survival advantage to cells under ER stress by activating Akt pathways and preserving ER homeostasis. This work reveals essential pathways regulated by WFS1 and therapeutic targets for Wolfram syndrome.
Introduction
Diabetes mellitus (DM) encompasses a set of metabolic disorders of glucose homeostasis characterized by a deficiency in insulin production or secretion. While the etiology of this deficiency varies by disorder, it inevitably involves pancreatic β-cell dysfunction that usually culminates in cell death1, 2. Accumulating evidence underscores endoplasmic reticulum (ER) dysfunction as a key factor in diabetic pathophysiology, particularly in type 2 diabetes mellitus (T2DM), due to the importance of ER homeostasis to insulin production and secretion3, 4. Still, there remains a gap in our understanding of the key molecules that mediate ER homeostasis and the mechanisms by which they preserve β-cell health.
The Wolfram syndrome 1 (WFS1) gene was identified as the major causative locus for Wolfram syndrome, a rare neurodegenerative disorder characterized by juvenile-onset diabetes mellitus, optic nerve atrophy, diabetes insipidus, and deafness5, 6. WFS1 encodes an ER transmembrane protein in which common variants are associated with T2DM susceptibility and over one hundred recessive mutations are linked to the genetic form of diabetes associated with Wolfram syndrome5, 7. A recent study also identified a mutation in WFS1 causative for autosomal dominant diabetes, further implicating WFS1 in DM pathology8. Various reports suggest that WFS1 may play a pivotal role in maintaining ER health through modulation of ER stress and calcium homeostasis9–11. Evidently, WFS1 is a vital component of normal β-cell physiology that when altered causes systemic disruption. Yet, we still do not fully understand the mechanisms or targets of WFS1 action in β-cells, particularly the downstream effectors that mediate WFS1’s pro-survival effects.
Here we describe loss-of-function and gain-of-function cell and mouse models of WFS1 that have enabled us to elucidate molecular pathways regulated by WFS1 in pancreatic β cells. Our results reveal essential pathways regulated by WFS1 which control β cell survival and function. Activation of such pathways has therapeutic implications for Wolfram syndrome and, more broadly, diabetes.
Materials and Methods
Reagents
Tunicamycin and thapsigargin (Sigma) were used at the concentrations specified in the figure legends. For chemical ER stress experiments involving inducible overexpression or knockdown of Wfs1, RPMI 1640 media (ThermoFisher Scientific) was supplemented with tetracycline-free fetal bovine serum (Clontech) and β-mercaptoethanol (Sigma). For glucotoxic ER stress experiments, RPMI 1640 no glucose media (ThermoFisher Scientific, cat no 11879020) was supplemented with D-glucose (Gibo) as specified in figure legends.
Human islets
De-identified normal and type-2 diabetic donor human islets were obtained from Prodo Laboratories. Informed consent was obtained from all subjects. Experiments were approved by Washington University’s Human Research Protection Office and conducted in accordance with the National Institutes of Health guidelines. All human islets used in this study were cultured in CMRL 1066 medium (Corning, 15–110-CV) supplemented with 10% FBS (Gibco), 1% non-essential amino acids (Gibco), 1% sodium pyruvate (Corning) and 1% penicillin/streptomycin (Gibco) at 37°C in 5% CO2 incubators to recover overnight prior to experiments. Further information regarding islet donors can be found in Supplementary Table S4.
Mice, in vivo physiology and pancreatic insulin content
129S6 whole body Wfs1-knockout mice were a kind gift from Dr. Sulev Kõks 12. These mice were generated using a Wfs1 targeting construct that replaced amino acids 360 – 890 of the Wfs1 protein with an in‐frame NLS-LacZ-Neo cassette. Genotypes were ascertained by multiplex PCR as previously described12. All animal experiments were performed according to procedures approved by the Institutional Animal Care and Use Committee at the Washington University School of Medicine (A-3381–01). In vivo glucose tolerance tests and insulin tolerance tests were performed according to standard procedures of the NIH-sponsored National Mouse Metabolic Phenotyping Centers (http://www.mmpc.org). Blood glucose was measured by glucometer (Arkray). Total pancreatic insulin was extracted from minced pancreata in ice-cold acid ethanol incubated at −20°C for 72-hours. Pancreatic and serum insulin content was measured by rat/mouse insulin ELISA kit (EMD Millipore).
β-cell morphometry
Pancreata from WT and Wfs1 KO mice were weighed, then fixed in zinc-formaldehyde and paraffin-embedded for sectioning. Morphometric analysis of pancreata from these mice was preformed as previously reported13. Cross-sectional areas calculated using ImageJ. The β cell mass for each specimen was quantified by multiplying the fraction of the cross-sectional area of pancreatic tissue positive for insulin staining by the pancreatic weight. All staining and subsequent morphometric analyses were conducted by an operator blinded to the genotypes of the specimens.
Immunofluorescence
Pancreata from WT and Wfs1 KO mice were fixed in 4% PFA and paraffin-embedded for sectioning. After rehydration, sections were permeabilized in 0.3% Triton-X prior to blocking in 2% BSA. Primary antibodies were treated overnight at 4℃. Incubation with secondary antibodies was for 1 h at RT. Slides were mounted with Vectashield with DAPI (Vector Laboratories). The antibodies used for the staining are listed in Supplementary Table S5. For calculating the percentage of Ki67 positive β-cells, Ki67 positive cells were counted among more than 200 insulin positive cells in each sample
Cell lines and insulin secretion assays
Tet-inducible INS1 832/13 cell lines were cultured using published media formulations, with modifications to glucose concentration as specified in the figure legends14. To generate β-cells with tet-inducible knockdown of endogenous Wfs1, INS-1 832/13 pTetR Tet-On were transduced with lentivirus expressing shRNA directed against rat Wfs1. To generate β-cells with tet-inducible overexpression of WFS1, INS-1 832/13 pTetR Tet-On were transduced with lentivirus expressing FLAG-tagged human WFS1. For knockdown and overexpression experiments, cells were cultured in 2 μg/ml doxycycline for 48 hours prior to any experimental manipulation. For rescue experiments, cells were transfected per manufacter’s instructions using the 4D-Nucleofector system (Lonza) or transduced with lentivirus expressing WT WFS1. Glucose-stimulated insulin secretion assays were performed as previously described15.
RNA Sequencing and Analysis
Samples were prepared according to library kit manufacturer’s protocol, indexed, pooled, and sequenced on an Illumina HiSeq. Basecalls and demultiplexing were performed with Illumina’s bcl2fastq software and a custom python demultiplexing program with a maximum of one mismatch in the indexing read. RNA-seq reads were aligned to the Ensembl release 76 top-level assembly with STAR version 2.0.4b16. Gene counts were derived from the number of uniquely aligned unambiguous reads by Subread:featureCount version 1.4.517. Isoform expression of known Ensembl transcripts were estimated with Sailfish version 0.6.1318. Sequencing performance was assessed for total number of aligned reads, total number of uniquely aligned reads, and features detected. The ribosomal fraction, known junction saturation, and read distribution over known gene models were quantified with RSeQC version 2.319.
All gene counts were imported into the R/Bioconductor package EdgeR and TMM normalization size factors were calculated to adjust for samples for differences in library size20. Ribosomal genes and genes not expressed in the smallest group size minus one sample greater than one count-per-million were excluded from further analysis. The TMM size factors and matrix of counts were imported into the R/Bioconductor package Limma 21. Weighted likelihoods based on the observed mean-variance relationship of every gene and sample were calculated for all samples with the voomWithQualityWeights 22. The performance of all genes was assessed with plots of residual standard deviation of every gene to their average log-count with a robustly fitted trend line of the residuals. Differential expression analysis was performed to analyze for differences between conditions and results were filtered for only those genes with Benjamini-Hochberg false-discovery rate adjusted p-values less than or equal to 0.05.
For each contrast extracted with Limma, global perturbations in known Gene Ontology (GO) terms were detected using R/Bioconductor package GAGE to test for changes in expression of the reported log 2 fold-changes reported by Limma in each term versus the background log 2 fold-changes of all genes found outside the respective term23. Differentially expressed genes in thapsigargin and tunicamycin stress conditions are listed in Supplementary Tables 3 and 4.
Intracellular cytosolic calcium
Confocal microscopy was performed on a Zeiss LSM 880 using a C-Apochromat 40× Water NA 1.2 objective lens. The cell permeable calcium sensor Fluo-4 AM (Invitrogen) was added to cells at a concentration of 4 μM in 1 ml of RPMI for 15–20 minutes. Cells were washed to remove excess Fluo-4AM prior to imaging. Experiments were performed in 37 °C KRBH with increasing glucose doses (0mM, 2.8mM and 16.7mM). The incubator stage provided the physiological conditions of 37 °C and 5% CO2. Images were acquired as a time series of 2 minutes for each condition before and after the addition of increasing glucose doses. Because Fluo-4 AM is a single-wavelength calcium sensor, the data shown are related to the variation of fluorescence intensity of the stimulated cells (ΔF) relative to the basal condition (F0). The signal from at least 20 cells was averaged for each condition to avoid differences due to different Fluo-4 concentrations in the cells. Photostability experiments were performed prior to each imaging session to confirm best acquisition conditions with minimal photobleaching. All data analysis and image processing were carried out with ImageJ.
Real-time polymerase chain reaction
Total RNA was isolated using RNeasy Kits (Qiagen) and cDNA libraries were generated using high-capacity cDNA reverse transcription kits (Applied Biosystem). Relative amounts of each transcript were calculated by the ΔΔCt method and normalized to either 18S rRNA expression for INS1 cells or to human β-actin for human islets. Quantitative PCR was performed with Applied Biosystems ViiA7 using SYBR green dye. Primers used for qPCR are listed in Supplementary Table S5.
Immunoblot analysis
Cells were lysed on ice for 10 minutes in freshly prepared Mammalian Protein Extraction Reagent (Thermo Scientific) supplemented with 1X Protease Complete, EDTA-free (Sigma) and 1X PhosSTOP (Sigma) before centrifugation at 15,000 rpm for 12-min at 4°C. Protein lysates were prepared using 4× Laemmli sample buffer (BioRad) heated in boiling water for 5-min. Immunoblot analysis was conducted with the antibodies listed in Supplementary Table S5. Immunoblot analysis of Akt phosphorylation was performed sequentially using the same PVDF membrane, first probing for phosphorylated Akt (Ser473), then total Akt. PVDF membranes were stripped for 8-min using Restore PLUS buffer (Thermo Scientific) and blocked with 5% BSA between subsequent probes. Tubulin was used a loading control for all immunoblot experiments. Images were digitally captured using a Bio-Rad ChemiDoc MP and analyzed with ImageJ.
Measurement of apoptosis
Caspase 3/7 activity was monitored using the Caspase-Glo® 3/7 Assay kit (Promega) and luminescence was measured using an Infinite M1000 plate reader (Tecan). Data was analyzed relative to untreated control cells.
Statistical analysis
Statistical significance was calculated by two-tailed unpaired Student’s T-test, with a p-value less than 0.05 considered significant. All values are shown as means ± SEM.
Results
WFS1 is critical for maintenance of β-cell mass and function
Diabetes is a cardinal phenotype of Wolfram syndrome that has been recapitulated in C57BL/6J mice on a delayed time scale relative to human disease24, 25. Recent reports of 129S6 whole body Wfs1 knockout (Wfs1 KO) mice suggest that this strain may more faithfully model human disease phenotypes12. To characterize the progression of diabetic pathology in 129S6 Wfs1 KO mice, we performed a detailed analysis of glucose tolerance in this model between 4.5- and 16-weeks of age. Wfs1 KO mice developed a progressive glucose tolerance impairment between 4.5- and 6.5-weeks old that persisted through 16-weeks of age (Figure 1, A–C). During this period, insulin tolerance was not impaired. Instead, insulin secretion was blunted in Wfs1 KO mice at both 6.5- and 16-weeks of age (Figure 1D). To determine whether this secretory defect was associated with pancreatic insulin deficiency, we assessed total pancreatic insulin content in Wfs1 KO mice and littermate controls. At 6.5-weeks old, the age of onset of glucose intolerance, Wfs1 KO mice had considerably less total pancreatic insulin content than wildtype (WT) mice (6.23±1.18 μg insulin/ mg wet pancreas in Wfs1 KO compared to 20.53± 1.34 μg insulin/mg wet pancreas in WT) (Figure 1E). A similar disparity in total pancreatic insulin content was apparent at 16-weeks of age (11.36±1.15 μg insulin/mg wet pancreas in Wfs1 KO compared to 35.99±4.69 μg insulin/mg wet pancreas in WT). To further interrogate these insulin deficits in Wfs1 KO mice, we examined pancreatic tissue of 6.5- and 16-week old mice. WT islets retained their characteristic core insulin staining surrounded by peripheral glucagon staining at both ages. Wfs1 KO islets, in contrast, displayed a disrupted islet architecture with central glucagon staining from the onset of glucose intolerance (Figure 1F). This observation prompted us to quantify the ratio of α-cell area to β-cell area in WT versus KO islets. As suspected, this ratio was 2-fold higher in KO islets than WT islets at both 6.5- and 16-weeks of age (Figure 1G). Moreover, the β-cell mass of Wfs1 KO mice at 6.5-weeks of age (0.53±0.07 mg) was considerably lower than that of WT littermate controls (0.96±0.13 mg), as determined by blinded morphometric analysis (Figure 1H). This difference in β-cell mass between Wfs1 KO (0.27±0.08 mg) and WT mice (0.87±0.28 mg) persisted through 16-weeks of age, with Wfs1 KO mice exhibiting a more precipitous drop in β-cell mass with time. Wfs1 KO β-cells also displayed reduced Ki-67 staining, a marker for cellular proliferation, at 6.5-weeks of age relative to WT β-cells (2.74±0.64% in Wfs1 KO compared to 5.88±1.05% in WT), consistent with published data reporting impaired cell cycle progression in the context of Wfs1 deficiency (Supplementary Figure S1)26. Although not statistically significant, a similar trend persisted at 16-weeks of age (1.76±0.50% in Wfs1 KO compared to 2.96±0.43% in WT). Collectively, these findings suggest that WFS1 plays a critical role in the maintenance of β-cell mass and β-cell function, particularly in the context of insulin production and secretion. These data also suggest a potential role for WFS1 in preserving proper islet composition.
Figure 1. Glucose tolerance and β-cell morphometry in whole body WFS1–/– 129S6 mice.
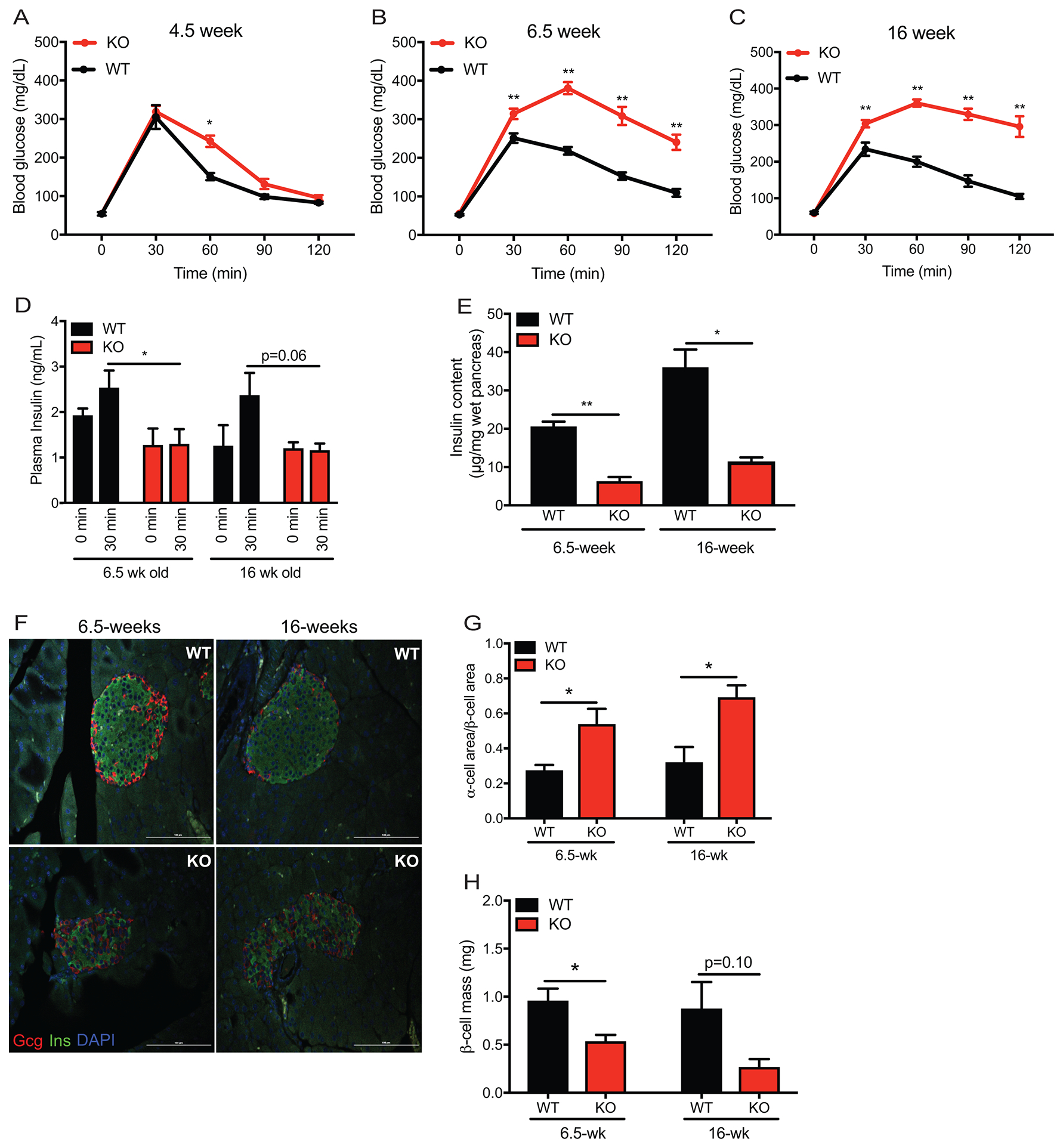
Intraperitoneal glucose tolerance test of: (A) WFS1 KO (n=12) and WT control (n=6) mice at 4.5 weeks old, (B) WFS1 KO (n=21) and WT control (n = 19) mice at 6.5-weeks old and (C) WFS1 KO (n=7) and WT control (n=7) mice at 16-weeks old (*p<0.05, **p< 0.01). (D) Insulin levels 30 min after injection of glucose (2g/kg) in 6.5-week old and 16-week old mice (n=4 WFS1 KO mice at each age and n = 4 WT mice at each age, *p<0.05). (E) Total pancreatic insulin content in 6.5-week-old and 16-week-old mice (n = 4 WFS1 KO mice at each age and n = 4 WT mice at each age, *p<0.05, **p< 0.01) (F) Immunofluorescence of islets from control and WFS1 KO mice at 6.5 and 16-weeks of age. (G) Beta cell mass in WT and WFS1 KO mice at 6.5 and 16-weeks of age mice (n = 3 WFS1 KO mice at each age and n = 3 WT mice at each age). Black bars represent data from WT mice and red bars indicate data from Wfs1 KO mice. (H) Quantification of alpha cell area to beta cell area in WT and WFS1 KO mice at 6.5 and 16-weeks of age mice (n=3 Wfs1 KO mice and n = 3 WT mice at each age, *p<0.05). Data are represented as mean ± SEM from at least three mice per experiment. Statistical significance was determined by unpaired t-test.
WFS1 promotes β-cell viability and function through suppression of pro-apoptotic pathways and upregulation of insulin biosynthesis
Given the degree of β-cell dysfunction caused by WFS1 depletion in humans and mice, we hypothesized that overexpressing WFS1 in β-cells could confer β-cells with a functional and/or survival benefit, particularly under stress. To test this hypothesis, we established a tet-inducible system that could express Flag-tagged WFS1 at levels 4-fold higher than endogenous Wfs1 in INS1 832/13 cells. To agnostically interrogate the gene networks affected by WFS1 in the context of ER stress, we performed RNA sequencing on INS1 cells overexpressing WFS1 under tunicamycin or thapsigargin conditions. First, a list of genes differentially expressed between control and Wfs1-overexpressing cells was generated for each stress condition. These lists were then compared against each other to identify genes differentially expressed across both stressors in the context of WFS1 overexpression (Figure 2A). Consistent with our hypothesis, pathways associated with the negative regulation of apoptosis and the positive regulation of hormone, peptide and insulin secretion were among the top 25 biological processes most affected by WFS1 overexpression under ER stress (Figure 2, B and C). These results support our in vivo data suggesting a critical role for WFS1 in preserving β-cell function and survival. Our model was further reinforced by the presence of Ins1, Ins2 and Nkx6.1 within the list of genes upregulated under both tunicamycin and thapsigargin-mediated ER stress (Supplementary Table S1). This subset of genes was particularly prominent to us because of their importance in defining β-cell maturity. Interestingly, validation by quantitative PCR revealed that Ins1 and Ins2 were significantly upregulated by WFS1 overexpression in both the presence and absence of ER stress (Figure 2, D and E). Correspondingly, Nkx2.2, Nkx6.1 and Pdx1 were differentially expressed under thapsigargin stress in a similar pattern to rat insulin gene expression under WFS1 overexpression (Figure 2, F–H and Supplementary Table 3). These data, coupled to the disrupted islet architecture and β-cell dysfunction observed in Wfs1 KO mice, suggest a role for WFS1 in maintaining β-cell function through the preservation of insulin biosynthesis and the positive regulation of β-cell maturity factors, particularly under ER stress.
Figure 2. WFS1 positively regulates insulin biosynthesis and β-cell maturity markers.
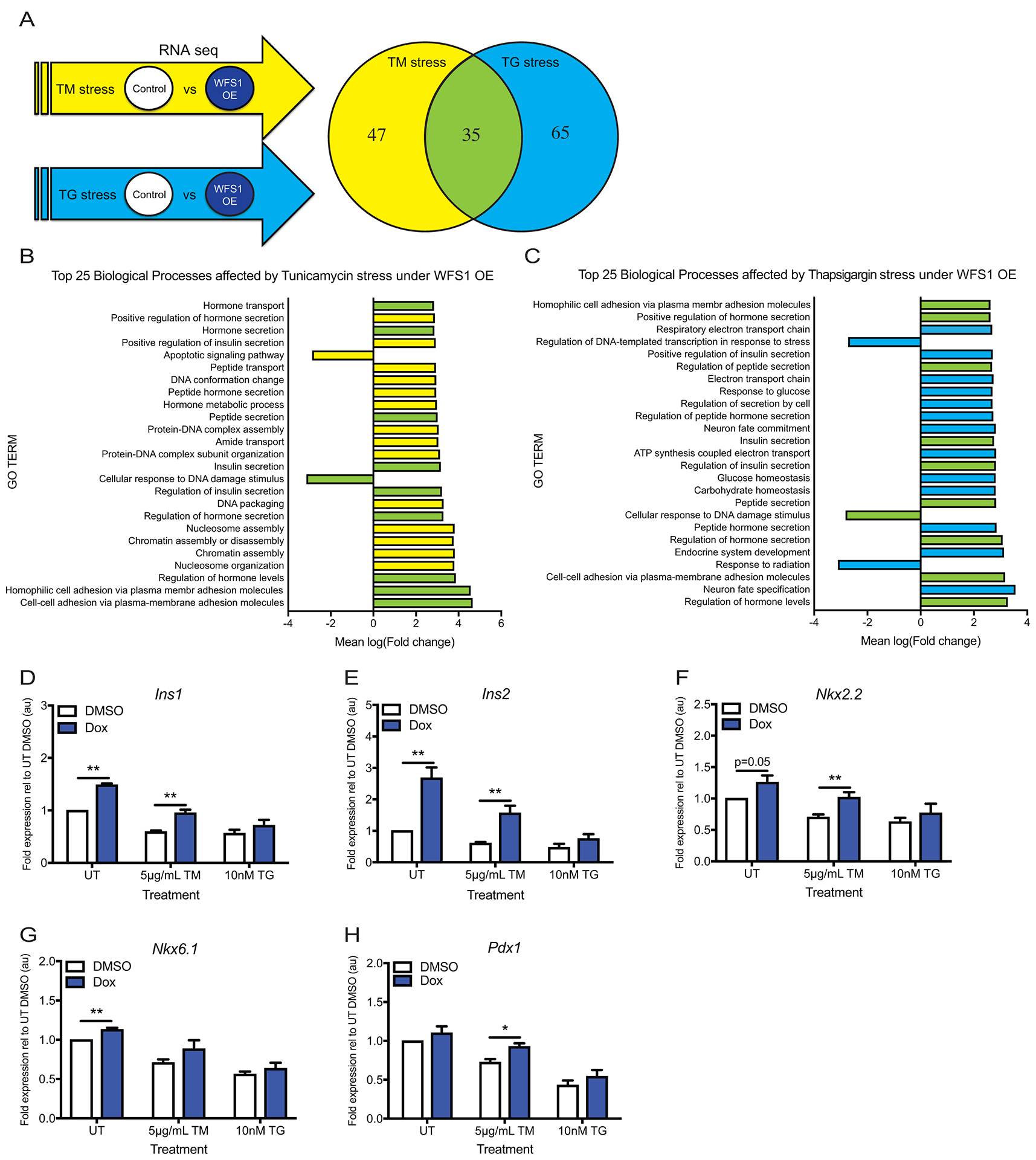
(A) Experimental strategy for identifying gene networks affected by WFS1 overexpression in INS1 cells via RNA sequencing. INS-1 832/13 with inducible pTetR Tet On (TO) WFS1 overexpression were treated with DMSO or doxycycline for 48 hours, followed by 16 hours of 5μg/ml tunicamycin or 10nM thapsigargin. Venn diagram represents number of genes with q-value ≤ 0.05 in each stress condition. (B) Top 25 biological processes affected by WFS1 overexpression under tunicamycin-mediated ER stress. Yellow bars indicate biological processes affected only by tunicamycin-mediated stress. Green bars indicate biological processes affected by both tunicamycin- and thapsigargin-mediated stress. (C) Top 25 biological processes affected by WFS1 overexpression under thapsigargin-mediated ER stress. Blue bars indicate biological processes affected only by thapsigargin-mediated stress. Green bars indicate biological processes affected by both tunicamycin- and thapsigargin-mediated stress. (D-E) Relative expression of Ins1 and Ins2 by quantitative PCR in cells treated as indicated in (A) (n=4 for all conditions, **p< 0.01). (F-H) Relative expression of Nkx2.2, Nkx6.1 and Pdx1 by quantitative PCR in cells treated as indicated in (A) (n=4 for all conditions, *p<0.05, **p< 0.01). Data are represented as mean ± SEM from at least four independent experiments or three independent mice. Statistical significance was determined by unpaired t-test.
WFS1 negatively regulates the Chop-Trib3 axis to promote Akt activation
To parse out the pro-apoptotic pathways suppressed by WFS1 overxpression across tunicamycin and thapsigargin-mediated ER stress, we examined the genes comprising the GO term for cellular response to DNA damage stimulus in our RNA seq study. Remarkably, Chop was reduced in WFS1-overexpressing cells across both ER stress conditions (Supplementary Table S1). Since Chop is an ER stress inducible molecule downstream of Atf6 and published reports suggest that WFS1 modulates Atf6 degradation, we hypothesized that upregulating WFS1 would downregulate apoptosis through the suppression of Chop-mediated cell death pathways10. To test this hypothesis, we challenged WFS1-overexpressing and control INS1 cells with tunicamycin and thapsigargin. Under no stress, WFS1 overexpression alone reduced caspase 3/7 activity by 20% (Figure 3A). Under tunicamycin challenge, WFS1 overexpressing cells exhibited 24% less caspase 3/7 activity than WT cells. Although not statistically significant, WFS1 overexpression also reduced caspase 3/7 activity under thapsigargin-mediated ER stress by 14%. To determine if WFS1’s protective effect on INS1 cells stemmed in part from targeted downregulation of Chop, we monitored Chop expression under ER stress. Chop was downregulated at the transcriptional and the translational level under no treatment and under tunicamycin-mediated stress (Figure 3, B and D), the two conditions in which WFS1 overexpression reduced apoptosis, as corroborated by caspase 3 cleavage (Figure 3, D and F). To further assess the effects of WFS1 overexpression on this Chop-mediated pro-apoptotic axis, we measured expression of Trib3, a pro-apoptotic downstream effector of Chop. As anticipated, Trib3 was significantly downregulated in conditions where Chop was reduced by WFS1 overxpression (Figure 3C). To determine if WFS1 modulates this pathway under more physiological forms of ER stress, we examined caspase 3/7 activity and Chop-Trib3 expression under high glucose (25mM glucose) conditions. As with chemical ER stress, WFS1 overexpression decreased caspase 3/7 activity by 34% under high glucose (Figure 3G). This decrease in pro-apoptotic caspase activity again corresponded with reduced Chop and Trib3 expression, further supporting a role for WFS1 in the negative regulation of these molecules (Figure 3, H – L).
Figure 3. WFS1 overexpression suppresses ER stress-mediated cell death.
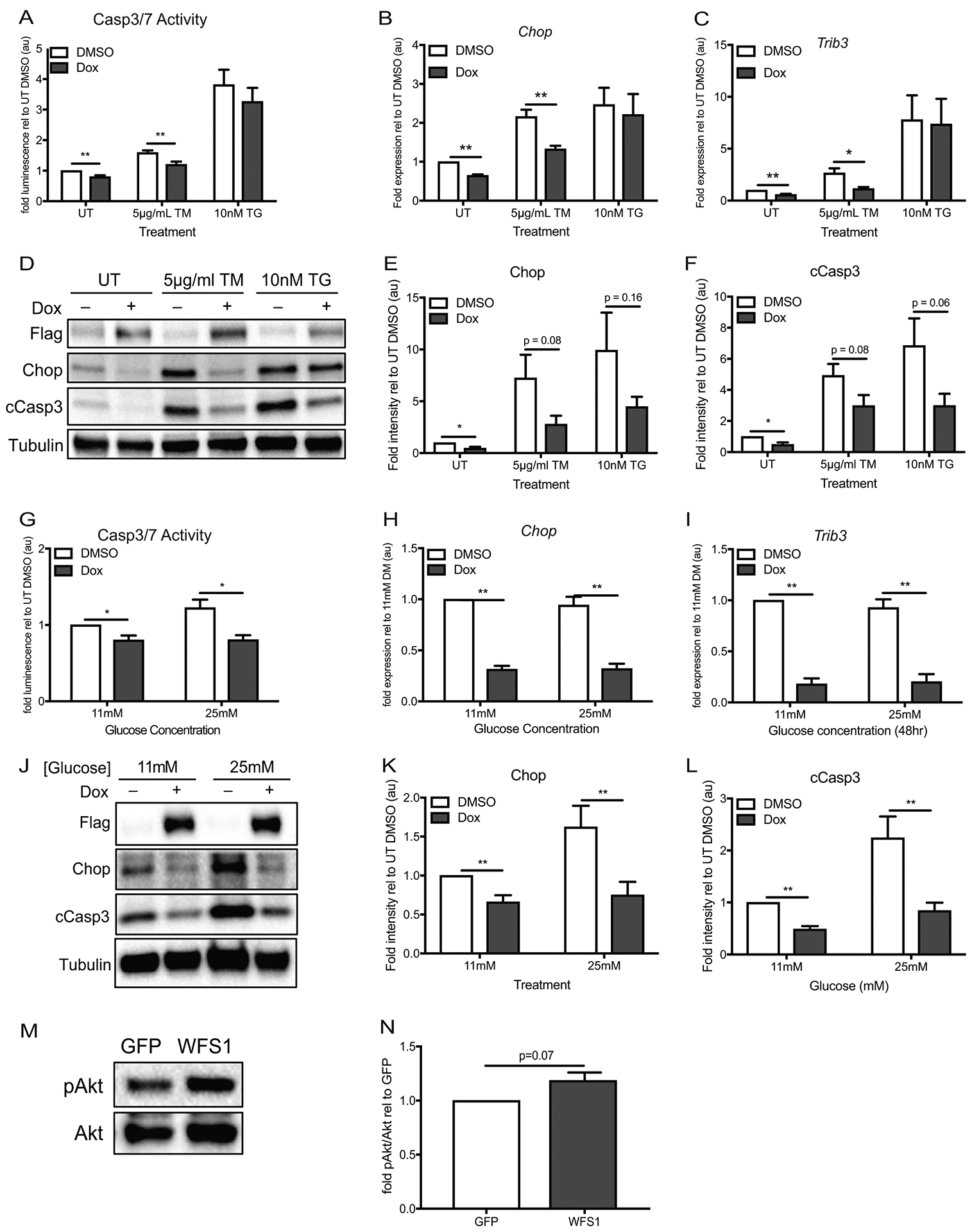
(A) INS-1 832/13 with inducible pTetR Tet On (TO) Flag-tagged WFS1 overexpression were treated with DMSO or doxycycline for 48 hours, followed by 16 hours of no treatment (UT) or chemical ER stress by specified doses of tunicamycin (TM) or thapsigargin (TG) before measurement of caspase 3/7 activity (n=8 for all conditions, **p< 0.01). (B-C) Relative expression of Chop and Trib3 by quantitative PCR in cells treated as (a) (n=4 for all conditions, *p<0.05, **p< 0.01). (D) Immunoblot analysis of Chop and cleaved caspase 3 expression in cells treated as (A). (E-F) Quantification of Chop and cCasp3 protein expression relative to untreated control cells—UT DMSO (n=8 for all conditions, *p<0.05). (G) INS-1 832/13 with inducible pTetR TO Flag-tagged WFS1 overexpression were treated with DMSO or doxycycline for 48 hours in 11mM glucose or 25mM glucose media prior to measurement of caspase 3/7 activity (n=4 for all conditions, *p< 0.05). (H-I) Relative expression of Chop and Trib3 by quantitative PCR in cells treated as (G) (n=4 for all conditions, **p< 0.01). (J) Immunoblot analysis of Chop and cleaved caspase 3 expression in cells treated as (G). (K-L) Quantification of Chop and cCasp3 protein expression relative to untreated control cells—11mM glucose DMSO (n=8 for Chop quantification, n=9 for cCasp3 quantification, **p< 0.01). (M) Immunoblot analysis of Wfs1, p(473)-Akt and total Akt expression in WT INS1 832/13 cells transduced with GFP or WFS1 lentivirus (n=3). (N) Quantification of p(473)-Akt/Akt in WFS1 overexpression (WFS1) cells relative to WT control (GFP) cells (n=3). White bars represent data from control cells and blue bars indicate data from WFS1 overexpression cells. Data are represented as mean ± SEM from at least four independent experiments. Statistical significance was determined by unpaired t-test.
Prompted by the anti-apoptotic effects of WFS1 upregulation in β-cells, we explored the role of WFS1 on β-cell survival pathways. Our data indicate that WFS1 mitigates apoptosis through the negative regulation of the Chop-Trib3 axis. Since Trib3 is a well-documented negative regulator of Akt activation, we hypothesized that overexpressing WFS1 in INS1 cells would enhance Akt (Ser473) phosphorylation27. Indeed, Akt (Ser473) phosphorylation increased upon WFS1 overexpression in INS1 cells (Figure 3, M and N), suggesting that WFS1 not only suppresses β-cell death, but also enhances β-cell survival through Akt activation.
To corroborate our model of WFS1’s role in β-cell viability and function, we evaluated a tet-inducible Wfs1 knockdown loss-of-function model in INS1 832/13 cells. To determine if this in vitro system modeled our in vivo observations, we assessed glucose stimulated insulin secretion in Wfs1-depleted and control cells. Consistent with our Wfs1 KO animal data, Wfs1 knockdown cells secreted less insulin than control cells (Figure 4A). Insulin content was also reduced in Wfs1-depleted cells (Figure 4B). Since insulin secretion is triggered by an increase in intracellular calcium and WFS1 has been implicated in maintaining calcium homeostasis, we assessed the effect of Wfs1 depletion on glucose-stimulated calcium mobilization11, 24, 28, 29. More specifically, we monitored changes in cytoplasmic calcium upon exposure to increasing glucose concentrations. Control cells exhibited a step-wise increase in cytoplasmic calcium in response to rising glucose concentrations. Wfs1-depleted cells exhibited no response to the same glucose stimuli (Figure 4C). Given the importance of calcium balance to β-cell function and viability, we also examined the effects of Wfs1 knockdown on ER stress-mediated cell death. Interestingly, knockdown of Wfs1 alone increased caspase 3/7 activity 5-fold in INS1 cells (Figure 4D). When challenged with tunicamycin and thapsigargin, Wfs1 knockdown cells exhibited 3-fold and 2-fold higher levels of caspase 3/7 activity, respectively, compared to WT cells under the same conditions. As we hypothesized, both Chop and cleaved caspase 3 were increased in Wfs1-depleted cells, suggesting a role for Wfs1 in the negative regulation of Chop-mediated apoptosis (Figure 4, E–G). To assess this pathway in a more physiologic context, we examined caspase 3/7 activity and Chop expression in Wfs1-knockdown and control cells under high glucose. As observed under chemical ER stress, high glucose elicited an increase in caspase 3/7 activity in Wfs1-depleted cells relative to WT cells (Figure 4H). Moreover, Wfs1 knockdown increased Chop and cleaved caspase 3 expression under high glucose (Figure 4, I – K). To evaluate the effect of Wfs1-depletion and subsequent Chop induction on Akt activation, we measured Akt (Ser473) phosphorylation relative to total Akt expression in Wfs1 knockdown cells. In keeping with our hypothesis, Akt (Ser473) phosphorylation was reduced by 30% in Wfs1 knockdown cells (Figure 4, L and M). Taken together, these data support a model wherein Chop acts a key mediator of β-cell death in the context of Wfs1 deficiency by simultaneously suppressing Akt activation and promoting pro-apoptotic pathways. These data thus corroborate the survival benefits observed with the upregulation of WFS1.
Figure 4. WFS1 knockdown promotes β-cell dysfunction, ER stress and β-cell death.
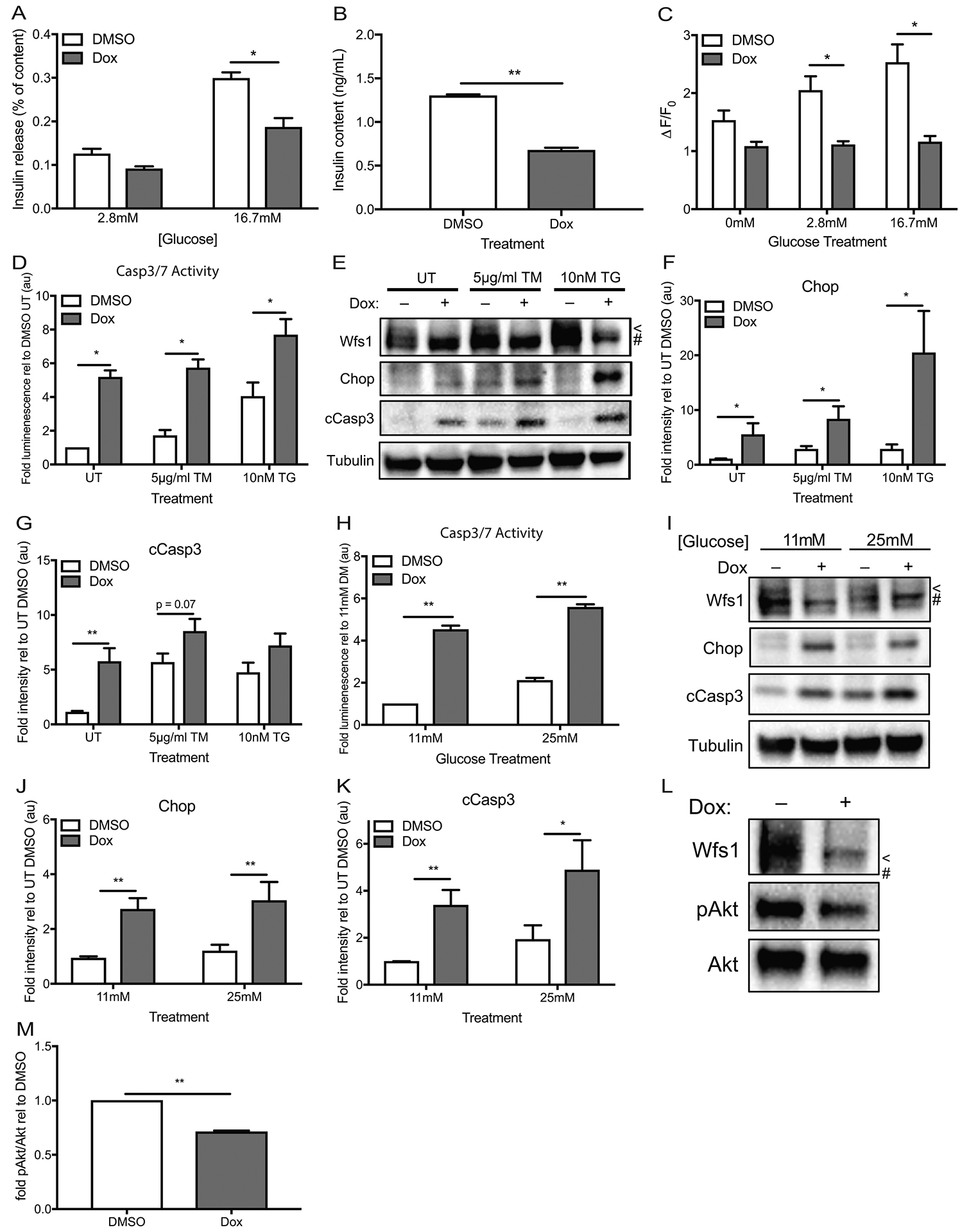
(A) INS-1 832/13 with inducible pTetR Tet-On (TO) shWfs1 were treated with DMSO or doxycycline for 48 hours, then subjected to glucose stimulated insulin secretion by sequential exposure to 2.8mM glucose then 16.7mM glucose (n=4, *p< 0.05). (B) Insulin content was measured from the cells used in (a) (n=4, **p< 0.01). (C) INS-1 832/13 with inducible pTetR TO shWfs1 were treated with DMSO or doxycycline for 48 hours, then loaded with Fluo-4 AM and imaged over 2 minutes in 0mM glucose KRBH, 2.8mM glucose KRBH and 16.7mM glucose KRBH. The change in fluorescence intensity at each glucose condition was averaged over 2 minutes of image acquisition and is expressed relative to baseline fluorescence to account for uneven concentration of dye loading in each cell. ΔF/F0 was averaged over 20 cells per glucose concentration (n=3, *p<0.05). (D) INS-1 832/13 with inducible pTetR TO shWfs1 were treated with DMSO or doxycycline for 48 hours, followed by 16 hours of no treatment (UT) or chemical ER stress by specified doses of tunicamycin (TM) or thapsigargin (TG) before measurement of caspase 3/7 activity (n=8 for all conditions, **p< 0.01). (E) Immunoblot analysis of Wfs1, Chop and cleaved caspase 3 expression in cells treated as (d). (F-G) Quantification of Chop and cCasp3 protein expression relative to untreated control cells—UT DMSO (n=9 for all conditions, *p<0.05, **p<0.01). (H) INS-1 832/13 with inducible pTetR TO shWfs1 were treated with DMSO or doxycycline for 48 hours in 11mM glucose or 25mM glucose media prior to measurement of caspase 3/7 activity (n=5 for all conditions, *p< 0.05). (I) Immunoblot analysis of Wfs1, Chop and cleaved caspase 3 expression in cells treated as (h). (J-K) Quantification of Chop and cCasp3 protein expression relative to untreated control cells—UT DMSO (n=11 for all conditions, *p<0.05, **p<0.01). (L) Immunoblot analysis of Wfs1, Ser743 Akt phosphorylation (pAkt) and total Akt expression in INS-1 832/13 with inducible pTetR Tet On (TO) shWfs1 treated for 48 hours with DMSO or doxycycline. (M) Quantification of pAkt/Akt in Wfs1 knockdown (Dox) cells relative to control (DMSO) cells (n=7, **p<0.01). White bars represent data from control cells and red bars indicate data from Wfs1 knockdown cells. For immunoblots, < indicates WFS1 band, # indicates a background band. Data are represented as mean ± SEM from at least four independent experiments. Statistical significance was determined by unpaired t-tests.
WFS1 and insulin are reduced in T2DM islets
Wfs1 is a critical gene for maintaining β-cell health as evidenced by our loss-of-function and gain-of-function models. In mice, Wfs1-depletion alters islet architecture and elicits profound insulin loss in association with β-cell dysfunction. This raised the possibility that Wfs1 might play a role in more complex metabolic disorders like T2DM. To evaluate the potential contribution of WFS1 to T2DM pathology in islets, we assessed WFS1 expression in T2DM human donor islets relative to normoglycemic donor islets. Interestingly, islets from T2DM donors had significantly lower levels of WFS1 and insulin gene expression than islets from normoglycemic donors (Figure 5, A and B). Conversely, T2DM donor islets had slightly elevated TXNIP levels and a more robust increase in IL-1β expression than normoglycemic islets (Figure 5, C and D). In the context of our in vivo and in vitro WFS1 loss of function data, these results from humans islets suggest that WFS1 downregulation may contribute to the β-cell dysfunction and β-cell death associated with T2DM pathology3, 30–32.
Figure 5. WFS1 expression is decreased in type-2 diabetic human donor islets.
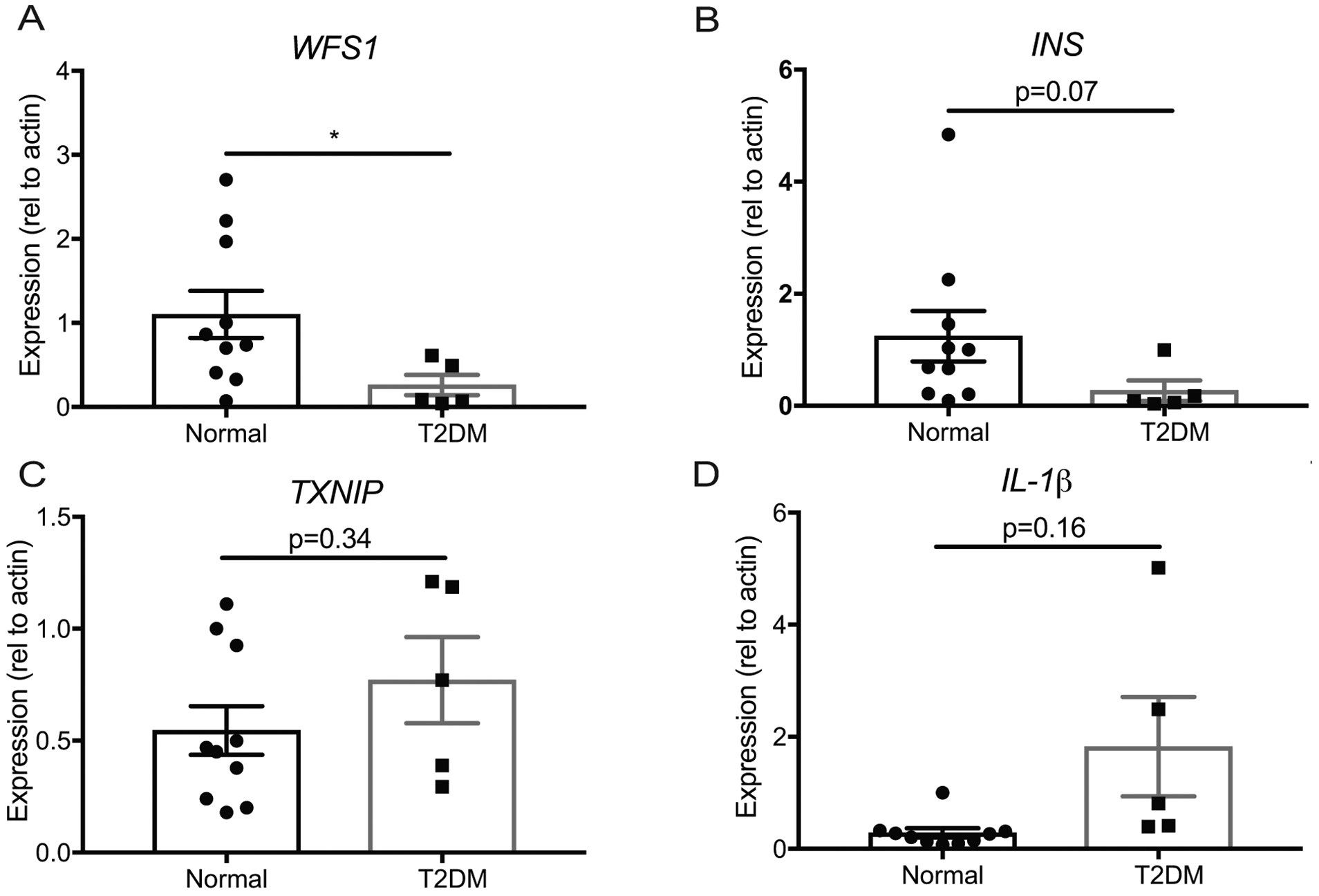
Relative expression of (A) WFS1, (B) INS, (C) TXNIP and (D) IL-1β by quantitative PCR in normal (n=10) and type-2 diabetic (T2DM) (n=5) donor islets (*p<0.05). (e) Proposed model of Wfs1 role in β-cell viability and function. Data are represented as mean ± SEM from at least five individual donors. Statistical significance was determined by unpaired t-test.
DISCUSSION
In this study, we demonstrate a role for WFS1 in preserving β-cell function and viability in physiologic and pathophysiologic states through the positive regulation of insulin biosynthesis and the negative regulation of ER stress-inducible pro-apoptotic molecules. We identify new targets of WFS1 action downstream of ATF6 through the negative regulation of the CHOP-TRIB3 axis, thereby establishing a link between WFS1 and AKT activation. Further, we propose a role for WFS1 in islets as a molecular determinant of proper β-cell function through the preservation of islet composition and key β-cell maturity factors.
Consistent with previous reports of diabetic pathology in C57BL/6J Wfs1 KO mice, we observed a progressive decline in glucose tolerance with concurrent β-cell loss and pancreatic insulin insufficiency in 129S6 Wfs1 KO mice. Unexpectedly, however, our data showed that Wfs1 KO in the 129S6 strain led to an earlier onset of diabetic pathology (at 6.5-weeks) than previously reported in whole body (at 17-weeks) and conditional (at 12-weeks) C57BL/6J KO models, or even in previous reports of this Wfs1 KO model24, 25, 33, 34. While the contribution of NLS-LacZ-Neo cassette expression under the Wfs1 promoter cannot be fully parsed out, our data suggests a potential role for genetic background in mediating disease severity in the context of WFS1 depletion that may resonate with the clinical variability observed in patients35–37. Notably, our study narrows the window of pathological progression in 129S6 Wfs1 KO mice to the 2-week period preceding 6.5-weeks of age, providing a discrete window for future studies to evaluate the events precipitating β-cell decline.
The aberrant islet architecture we observed at 6.5-weeks in 129S6 Wfs1 KO mice parallels published C57BL/6J data from 36-week old whole body Wfs1 KO mice and 24-week old β-cell-specific Wfs1 KO mice, as well as 24-week old mixed strain [(129S6xB6)xB6]F2 whole body Wfs1 KO mice24, 25. Counter to the hyperglycemia reported in C57BL/6J Wfs1 KO models at those ages, however, 129S6 Wfs1 KO mice are normoglycemic at 6.5-weeks old. This suggests a potential role for WFS1 in maintaining proper islet composition independent of the β-cell loss expected from chronic hyperglycemia. Perhaps WFS1 loss-of-function not only increases β-cell death, but also affects β-cell maturity by causing downregulation of key β-cell identity markers, leading to the anomalous α-cell/β-cell ratios we observed in Wfs1 KO islets. Further studies aimed at ascertaining WFS1’s role in maintaining β-cell maturity are warranted.
While insulin production is critical for maintaining glucose homeostasis, insulin biosynthesis is also a major source of β-cell ER stress38, 39. It is likely for this reason that insulin gene expression is strongly downregulated under ER stress40, 41. Our data supports this phenomenon, as genetic depletion of Wfs1 not only reduced insulin content in β-cells, but also induced β-cell death, especially under chemical or glucotoxic ER stress. However, our data also showed that WFS1 overexpression increased insulin gene expression to varying degrees, even under different forms of ER stress. While the degree of ER stress reduction and insulin upregulation varied by each specific chemical stressor, the trends in Chop-Trib3 and caspase 3/7 activity were consistent across both glucotoxic and chemical ER stress.
Taken together, these data posit a model where WFS1 hosts dual and opposing roles in mitigating ER stress while also promoting insulin synthesis. This may, in part, be due to WFS1’s role in the negative regulation of key unfolded protein response (UPR) factors such as ATF6, which when overactive dampen insulin expression10, 41. Still, WFS1 likely has an independent role in promoting insulin biosynthesis under homeostatic conditions given the apparent increase in insulin gene expression under conditions devoid of ER stress. This independent pathway may well depend on WFS1’s positive regulation of β-cell maturity factors, such as Pdx1, Nkx2.2 and Nkx6.1, which also promote insulin production42–46. Indeed, this may also help explain the consistent disruption in islet architecture observed across Wfs1 KO mouse models that requires further address in the literature.
Recent reports have described the complex relationship between ER stress, glucose homeostasis and β-cell mass47–50. One study proposed that a mild ER load promotes an adaptive response via UPR signaling, primarily through ATF6, that results in β-cell proliferation49. Another study proposed that decreasing insulin production decreases ER stress, thus promoting β-cell proliferation through AKT activation48. In our model, WFS1 relieves ER stress and also burdens the ER with increased insulin bioynthesis. We therefore propose a model where WFS1 acts as fulcrum in β-cells between the dichotomous reality of insulin production and ER stress management to maintain a crucial cell population for organismal glucose homeostasis. Under physiologic conditions, WFS1 promotes insulin biosynthesis to meet glucose demand, but also modulates pro-apoptotic branches of the ER stress response to sustain an adaptive outcome through AKT activation. Under pathologic conditions, as in the case of WFS1 mutation or depletion, the absence of WFS1 leads to unregulated ER stress that culminates in β-cell death at least in part due to repression of AKT survival pathways. In this manner, WFS1 may lie at the intersection of the observations made by aforementioned reports, by serving as a molecular arbiter of β-cell survival that negotiates the outcome of increasing insulin demand in the face of UPR signaling.
ER stress is a potent contributor to β-cell pathology in T2DM, while Wolfram syndrome is arguably a prototype of ER stress disease51–54. Moreover, T2DM is associated with β-cell death and dysfunction similar to our models of WFS1 depletion55. Accordingly, we questioned whether WFS1 played a role in the complex milieu of T2DM pathology. Intriguingly, we found WFS1 and insulin gene expression decreased in T2DM donor islets. These findings are consistent with our in vivo and in vitro knockout and knockdown studies of WFS1, suggesting that perhaps the downregulation of WFS1 may be contributing to the β-cell pathology of T2DM. Considering reports of WFS1 variants that confer risk of T2DM, it is possible that these variants affect WFS1 mRNA or protein stability7, 56. In light of our findings regarding the effects of WFS1 expression on β-cell viability and function, further studies should examine the effects of T2DM-associated WFS1 variants on the pathways highlighted by our studies.
This study provides novel insights into the cellular function of WFS1 in β-cells through the identification of key downstream effectors under physiologic and pathologic states. Our findings demonstrate the importance of WFS1 as a negative regulator of ER stress, but, more importantly, highlight a previously unreported role for WFS1 in promoting insulin biosynthesis and β-cell health through downregulation of the CHOP-TRIB3 axis and activation of AKT survival pathways. The striking effect of Wfs1 depletion on islet composition coupled to the upregulation of key markers of β-cell maturity by WFS1 overexpression suggests a role for WFS1 in maintaining β-cell mass through an intricate balance of insulin production and ER stress management. Given the positive effects of WFS1 upregulation on overall β-cell health, particularly in the context of metabolic stress, our findings provide motivation for novel therapeutic strategies targeting WFS1 for stabilization or upregulation as a treatment for diabetic disorders.
Supplementary Material
Acknowledgments
We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analysis. The Center is partially supported by NCI Cancer Center Support Grant P30CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant UL1TR000448 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH. The authors would like to acknowledge Cris Brown, Akari Takesato, and Lucas Peng for their technical assistance. This work was partly supported by the grants from the National Institutes of Health/NIDDK (DK112921, DK020579) and National Institutes of Health/NCATS (TR002065) and philanthropic supports from the Unravel Wolfram Syndrome Fund, the Silberman Fund, the Stowe Fund, the Ellie White Foundation for Rare Genetic Disorders, the Eye Hope Foundation, and the Snow Foundation to F. Urano. D. Abreu was supported by the NIH training grant (F30DK111070).
Footnotes
Disclosure/Conflict of Interest
The authors declare no competing interests relevant to this article.
References
- 1.Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 2005;54 Suppl 2:S97–107. [DOI] [PubMed] [Google Scholar]
- 2.Donath MY, Ehses JA, Maedler K, Schumann DM, Ellingsgaard H, Eppler E, et al. Mechanisms of beta-cell death in type 2 diabetes. Diabetes 2005;54 Suppl 2:S108–113. [DOI] [PubMed] [Google Scholar]
- 3.Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem 2012;81:767–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harding HP, Ron D. Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes 2002;51 Suppl 3:S455–461. [DOI] [PubMed] [Google Scholar]
- 5.Inoue H, Tanizawa Y, Wasson J, Behn P, Kalidas K, Bernal-Mizrachi E, et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet 1998;20(2):143–148. [DOI] [PubMed] [Google Scholar]
- 6.Urano F. Wolfram Syndrome: Diagnosis, Management, and Treatment. Curr Diab Rep 2016;16(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandhu MS, Weedon MN, Fawcett KA, Wasson J, Debenham SL, Daly A, et al. Common variants in WFS1 confer risk of type 2 diabetes. Nat Genet 2007;39(8):951–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonnycastle LL, Chines PS, Hara T, Huyghe JR, Swift AJ, Heikinheimo P, et al. Autosomal dominant diabetes arising from a Wolfram syndrome 1 mutation. Diabetes 2013;62(11):3943–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fonseca SG, Fukuma M, Lipson KL, Nguyen LX, Allen JR, Oka Y, et al. WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic beta-cells. J Biol Chem 2005;280(47):39609–39615. [DOI] [PubMed] [Google Scholar]
- 10.Fonseca SG, Ishigaki S, Oslowski CM, Lu S, Lipson KL, Ghosh R, et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J Clin Invest 2010;120(3):744–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu S, Kanekura K, Hara T, Mahadevan J, Spears LD, Oslowski CM, et al. A calcium-dependent protease as a potential therapeutic target for Wolfram syndrome. Proc Natl Acad Sci U S A 2014;111(49):E5292–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luuk H, Koks S, Plaas M, Hannibal J, Rehfeld JF, Vasar E. Distribution of Wfs1 protein in the central nervous system of the mouse and its relation to clinical symptoms of the Wolfram syndrome. J Comp Neurol 2008;509(6):642–660. [DOI] [PubMed] [Google Scholar]
- 13.Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic beta cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab 2014;19(5):872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 2000;49(3):424–430. [DOI] [PubMed] [Google Scholar]
- 15.Clark AL, Kanekura K, Lavagnino Z, Spears LD, Abreu D, Mahadevan J, et al. Targeting Cellular Calcium Homeostasis to Prevent Cytokine-Mediated Beta Cell Death. Sci Rep 2017;7(1):5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30(7):923–930. [DOI] [PubMed] [Google Scholar]
- 18.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 2017;14(4):417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 2012;28(16):2184–2185. [DOI] [PubMed] [Google Scholar]
- 20.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26(1):139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu R, Holik AZ, Su S, Jansz N, Chen K, Leong HS, et al. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res 2015;43(15):e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 2009;10:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishihara H, Takeda S, Tamura A, Takahashi R, Yamaguchi S, Takei D, et al. Disruption of the WFS1 gene in mice causes progressive beta-cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum Mol Genet 2004;13(11):1159–1170. [DOI] [PubMed] [Google Scholar]
- 25.Riggs AC, Bernal-Mizrachi E, Ohsugi M, Wasson J, Fatrai S, Welling C, et al. Mice conditionally lacking the Wolfram gene in pancreatic islet beta cells exhibit diabetes as a result of enhanced endoplasmic reticulum stress and apoptosis. Diabetologia 2005;48(11):2313–2321. [DOI] [PubMed] [Google Scholar]
- 26.Yamada T, Ishihara H, Tamura A, Takahashi R, Yamaguchi S, Takei D, et al. WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic beta-cells. Hum Mol Genet 2006;15(10):1600–1609. [DOI] [PubMed] [Google Scholar]
- 27.Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 2003;300(5625):1574–1577. [DOI] [PubMed] [Google Scholar]
- 28.Takei D, Ishihara H, Yamaguchi S, Yamada T, Tamura A, Katagiri H, et al. WFS1 protein modulates the free Ca(2+) concentration in the endoplasmic reticulum. FEBS Lett 2006;580(24):5635–5640. [DOI] [PubMed] [Google Scholar]
- 29.Hara T, Mahadevan J, Kanekura K, Hara M, Lu S, Urano F. Calcium efflux from the endoplasmic reticulum leads to beta-cell death. Endocrinology 2014;155(3):758–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003;52(1):102–110. [DOI] [PubMed] [Google Scholar]
- 31.Evans-Molina C, Hatanaka M, Mirmira RG. Lost in translation: endoplasmic reticulum stress and the decline of beta-cell health in diabetes mellitus. Diabetes Obes Metab 2013;15 Suppl 3:159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henquin JC, Ibrahim MM, Rahier J. Insulin, glucagon and somatostatin stores in the pancreas of subjects with type-2 diabetes and their lean and obese non-diabetic controls. Sci Rep 2017;7(1):11015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noormets K, Koks S, Muldmaa M, Mauring L, Vasar E, Tillmann V. Sex differences in the development of diabetes in mice with deleted wolframin (Wfs1) gene. Exp Clin Endocrinol Diabetes 2011;119(5):271–275. [DOI] [PubMed] [Google Scholar]
- 34.Ivask M, Hugill A, Koks S. RNA-sequencing of WFS1-deficient pancreatic islets. Physiol Rep 2016;4(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Heredia ML, Cleries R, Nunes V. Genotypic classification of patients with Wolfram syndrome: insights into the natural history of the disease and correlation with phenotype. Genet Med 2013;15(7):497–506. [DOI] [PubMed] [Google Scholar]
- 36.Chaussenot A, Bannwarth S, Rouzier C, Vialettes B, Mkadem SA, Chabrol B, et al. Neurologic features and genotype-phenotype correlation in Wolfram syndrome. Ann Neurol 2011;69(3):501–508. [DOI] [PubMed] [Google Scholar]
- 37.Cryns K, Sivakumaran TA, Van den Ouweland JM, Pennings RJ, Cremers CW, Flothmann K, et al. Mutational spectrum of the WFS1 gene in Wolfram syndrome, nonsyndromic hearing impairment, diabetes mellitus, and psychiatric disease. Hum Mutat 2003;22(4):275–287. [DOI] [PubMed] [Google Scholar]
- 38.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev 2008;29(3):317–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fonseca SG, Gromada J, Urano F. Endoplasmic reticulum stress and pancreatic beta-cell death. Trends Endocrinol Metab 2011;22(7):266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lipson KL, Fonseca SG, Ishigaki S, Nguyen LX, Foss E, Bortell R, et al. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab 2006;4(3):245–254. [DOI] [PubMed] [Google Scholar]
- 41.Seo HY, Kim YD, Lee KM, Min AK, Kim MK, Kim HS, et al. Endoplasmic reticulum stress-induced activation of activating transcription factor 6 decreases insulin gene expression via up-regulation of orphan nuclear receptor small heterodimer partner. Endocrinology 2008;149(8):3832–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cissell MA, Zhao L, Sussel L, Henderson E, Stein R. Transcription factor occupancy of the insulin gene in vivo. Evidence for direct regulation by Nkx2.2. J Biol Chem 2003;278(2):751–756. [DOI] [PubMed] [Google Scholar]
- 43.Gutierrez GD, Bender AS, Cirulli V, Mastracci TL, Kelly SM, Tsirigos A, et al. Pancreatic beta cell identity requires continual repression of non-beta cell programs. J Clin Invest 2017;127(1):244–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Le Lay J, Stein R. Involvement of PDX-1 in activation of human insulin gene transcription. J Endocrinol 2006;188(2):287–294. [DOI] [PubMed] [Google Scholar]
- 45.Schaffer AE, Taylor BL, Benthuysen JR, Liu J, Thorel F, Yuan W, et al. Nkx6.1 controls a gene regulatory network required for establishing and maintaining pancreatic Beta cell identity. PLoS Genet 2013;9(1):e1003274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shang L, Hua H, Foo K, Martinez H, Watanabe K, Zimmer M, et al. beta-cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes 2014;63(3):923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest 2008;118(10):3378–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szabat M, Page MM, Panzhinskiy E, Skovso S, Mojibian M, Fernandez-Tajes J, et al. Reduced Insulin Production Relieves Endoplasmic Reticulum Stress and Induces beta Cell Proliferation. Cell Metab 2016;23(1):179–193. [DOI] [PubMed] [Google Scholar]
- 49.Sharma RB, O’Donnell AC, Stamateris RE, Ha B, McCloskey KM, Reynolds PR, et al. Insulin demand regulates beta cell number via the unfolded protein response. J Clin Invest 2015;125(10):3831–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Porat S, Weinberg-Corem N, Tornovsky-Babaey S, Schyr-Ben-Haroush R, Hija A, Stolovich-Rain M, et al. Control of pancreatic beta cell regeneration by glucose metabolism. Cell Metab 2011;13(4):440–449. [DOI] [PubMed] [Google Scholar]
- 51.Marchetti P, Bugliani M, Lupi R, Marselli L, Masini M, Boggi U, et al. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 2007;50(12):2486–2494. [DOI] [PubMed] [Google Scholar]
- 52.Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, et al. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007;50(4):752–763. [DOI] [PubMed] [Google Scholar]
- 53.Eizirik DL, Cnop M. ER stress in pancreatic beta cells: the thin red line between adaptation and failure. Sci Signal 2010;3(110):pe7. [DOI] [PubMed] [Google Scholar]
- 54.Urano F Wolfram syndrome iPS cells: the first human cell model of endoplasmic reticulum disease. Diabetes 2014;63(3):844–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Papa FR. Endoplasmic reticulum stress, pancreatic beta-cell degeneration, and diabetes. Cold Spring Harb Perspect Med 2012;2(9):a007666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stitzel ML, Sethupathy P, Pearson DS, Chines PS, Song L, Erdos MR, et al. Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci. Cell Metab 2010;12(5):443–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.