

Abstract
MurG is an essential bacterial glycosyltransferase that catalyses the GlcNAc-transformation of lipid I to lipid II during peptidoglycan biosynthesis. Park’s nucleotide has been a convenient biochemical tool to study the function of MraY and MurG, however, no fluorescent probe has been developed to differentiate individual processes in the biotransformation of Park’s nucleotide to lipid II via lipid I. Herein, we report a robust assay of MurG using either the membrane fraction of a M. smegmatis strain or a thermostable MraY and MurG of Hydrogenivirga sp. as enzyme sources, along with Park’s nucleotide or Park’s nucleotide-Nε-C6-dansylthiourea and UDP-GlcN-C6-FITC as acceptor and donor substrates. Identification of both the MraY and MurG products can be performed simultaneously by HPLC in dual UV mode. Conveniently, the generated lipid II fluorescent analogue can also be quantitated via UV-Vis spectrometry without separation of the unreacted lipid I derivative. The microplate-based assay reported here is amenable to high-throughput MurG screening. A preliminary screening of a collection of small molecules has demonstrated the robustness of the assays, and resulted in rediscovery of ristocetin A as a strong antimycobacterial MurG and MraY inhibitor.
Keywords: MurG translocase II, Lipid II, MraY translocase I, Lipid I, Park’s nucleotide, Fluorescence-based assay, ristocetin
Graphical Abstract
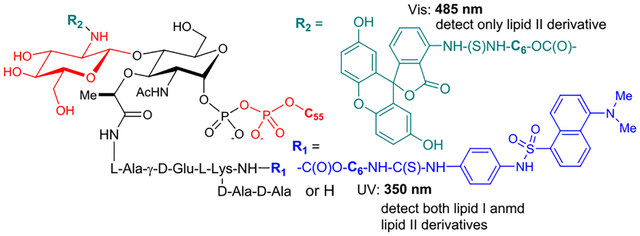
Glycosyltransferases (GTases) play an important role in the carbohydrate metabolisms in all living organisms.1,2 Many bacterial GTases are involved in cell wall biosynthesis transferring a carbohydrate unit from a nucleotide donor to a lipid-containing acceptor. Of this group, the membrane-associated and essential GTase, MurG (UDP-N-acetylglucosamine:N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase) catalyzes the rate-limiting step of lipid II (GlcNAc-MurNAc-(pentapeptide)-pyrophosphoryl prenol) synthesis by transferring GlcNAc from UDP-GlcNAc to lipid I (MurNAc-(pentapeptide)-pyrophosphoryl prenol). Lipid I synthesis is catalyzed by the transmembrane protein MraY/MurX (phospho-MurNAc-(pentapeptide) translocase) which transfers a MurNAc-pentapide from Park’s nucleotide to a phosphoprenol acceptor (Figure 1).3–5 Lipid II is then transferred across the cytoplasmic membrane to the outer leaflet where penicillin binding proteins (transpeptidases and transglycosylases) polymerize and cross-link lipid II to form peptidoglycan. We previously reported both chemoenzymatic and total chemical synthesis of Park’s nucleotide and its assay probes that allowed for the development of a convenient assay method for MraY/MurX.6–16 We have now extended our functional studies and inhibitor designs into MurG. Previously, the effect of inhibitor molecules on the biosynthesis of peptidoglycan has been monitored via radiolabeled precursors (e.g., UDP-GlcNAc, UDP-MurNAc-(pentapeptide), and prenyl-P) with cell-free particulate fractions.17 For MurG, enzyme inhibition can be monitored by the incorporation of a radiolabeled UDP-GlcNAc into lipid II using Park’s nucleotide. However, this requires subsequent separation of radiolabeled product from excess isotope-labeled substrates for quantitation. Moreover, the coupling assays with Park’s nucleotide and UDP-GlcNAc cause false-positive errors if molecules have MraY inhibitory activity. A number of assays were developed subsequent to this. A biotinylated lipid I analog was introduced and an avidin-derivatized resin was applied to remove excess [14C]UDP-GlcNAc (Men et al. 1998 and Branstorm et al. 1999).18–20 The Walker group developed a high-throughput screening (HTS) method to identify MurG UDP-GlcNAc antagonist utilizing fluorescence polarization (Helm et al. 2003).21 Conceptually unique UDP-GlcNAc probes having 2-(1H-indol-3-yl)acetamide were developed for MurG assay via Förster resonance energy transfer method (Li et al. 2004).22 The Wong group developed a MurG assay coupled with pyruvate kinase and lactic dehydrogenase, where MurG inhibitory activity is indirectly measured by the decrease of fluorescence of NADH (λex =340 nm, λem = 460 nm) (Liu et al. 2003).23 There are still a few MurG assays that can monitor inhibition of the lipid II transformation without relying on radioisotope(s). Lipid I or its analogs developed for MurG assays require total chemical synthesis.19,24–28 On the other hand, Park’s nucleotide can readily be obtained via enzymatic reactions from UDP-GlcNAc using MurA-F or from UDP-MurNAc using MurC-F chemo-enzymatically.6 Unlike lipid I, Park’s nucleotide is a water soluble molecule, and amenable to a medium scale synthesis and convenient purification methods. Therefore, the ideal method to study MurG function systematically would 1) avoid radioisotopes, 2) start with Park’s nucleotide, and 3) allow for the differentiation of MraY and MurG inhibitory activity. We have developed MurG assays using intact Park’s nucleotide (10) or Park’s nucleotide-Nε-C6-dansylthiourea fluorescent probe (1e), UDP-GlcN-C6-FITC (8), and exogenous C55-prenyl phosphate. Here we describe 1) the substrate tolerance of MurG, 2) novel MurG assay methods that do not require separation of the unreacted lipid I intermediate, and 3) characterization of MurG of Hydrogenivirga sp. (HyMurG) and Mycobacterium smegmatis (MsmegMurG) as convenient sources for HTS.
Figure 1.

Biosynthesis of lipid I and lipid II.
RESULTS AND DISCUSSION
Structures of Park’s nucleotide and lipid I probes recognized by MraY/MurX and MurG.
We have extensively studied the probing of Park’s nucleotide that can be recognized by translocase I (MraY and MurX).8 As summarized in Table 1, the lysine nitrogen (Nε-position) of Park’s nucleotide was modified with the sulfonyl chloride (-SO2Cl) or isothiocyanate (-N=C=S) of dansyl and fluorescein derivatives and all probes (1a-g) were effective in the formation of the corresponding lipid I-Nε-derivatives (50–60% yield via HPLC) using the crude membrane (P-60) prepared from a wild-type M. smegmatis strain (ATCC607). In our studies of lipid-acceptor, MurX and MraY showed tolerance in the length and E/Z-geometry of the β-double bond, but the α-double bond is required to be in the Z-configuration; P-60-catalyzed reaction of 1a-g with neryl phosphate (C10-P) and (2Z,6E)-farnesyl phosphate (C15-P) furnished the corresponding lipid I-Nε-derivatives in 60–80% yield (entries 3, 4, 8, 9, 13, 14, 18, and 19 in Table 1). On the other hand, (2Z)-phytyl phosphate did not provide the corresponding lipid I derivatives (entries 5, 10, 15, 20, 25, 30, and 36). It is worthwhile mentioning that C10- and C15-lipid I-Nε-derivatives are dissolved in the reaction buffer solutions, and these combinations have been applied to convenient MraY/MurX assays.8,9 On the other hand, none of the Park’s nucleotide probes (1a-d) modified with commercial reagents at the lysine Nε-position were effective in the formation of lipid II analogues with P-60 in the presence of UDP-GlcNAc (entries 1–20). With a C6-linker (6-aminohexanol) bridging to Park’s nucleotide, two types of dansyl fluorophores could be substrates for both MraY and MurG (entries 21,22, 26, and 27). In contrast, the lipid I-C6-FITC derivatives (e.g., 2g-C55) were not recognized by MurG (entries 32 and 33 in Table 1).
Table 1.
Park’s nucleotide and lipid I fluorescent probes effective in MurX- and MurG-catalysed reactions.

|
Unmodified lipid I and lipid II are difficult products to differentiate by reversed-phase chromatography. To establish an assay, we synthesized C55-lipid I-C6-dansyl (2e-C55) according to the synthetic scheme established previously with minor modifications (see, Scheme 1).7,27 We then converted the synthetic lipid I analogue, 2e-C55 to C55-lipid II-C6-dansyl (3e-C55) with P-60 of M. smegmatis. Purified Park’s nucleotide, lipid I, and lipid II derivatives (1e, 2e-C55, and 3e-C55) were used to establish HPLC-based assays for monitoring both MurX/MraY and MurG enzyme activities.8,9,27 We commenced HPLC studies with 2f-C55 and 3f-C55, establishing the best separation in retention times. The peak separation of 1 min was achieved via a gradient elution with 0.05 M NH4HCO3 and MeOH (15:85 to 0:100 over 30 min.). As shown in Figure 2B, separation of the peaks of lipid I and lipid II derivatives was better with 2e-C55 and 3e-C55; the difference between the retention time was over 3 min. Due to this observed chromatographic advantage, the Park’s nucleotide probe 1e was chosen for MurG assay development. MurG exhibited lower tolerance in the structure of the fluorescent probe at the lysine Nε-C6-linker and the donor substrate, prenyl phosphate, than those of MurX/MraY. The lipid I-C6-dansyl derivatives of neryl (C10) and (2Z,6E)-farnesyl phosphates (C15) were not converted to the corresponding lipid II analogues by using P-60 of M. smegmatis (entries 23, 24, and 25 in Table 1). We have continued exploring prenyl group mimetics that can be the substrates for both MurX/MraY and MurG, however, so far, natural forms of C55-P and C50-P are the only prenyl phosphates that fulfilled the biotransformation from 1e to 2e-C55 and 3e-C55. In the transformation of Figure 2A, over 50% of the lipid I derivative, 2e-C55 was generated within 1 h that was, in turn, consumed to <10% after 2 h, furnishing the lipid II derivative, 3e-C55 in 70% (42% overall yield based on consumption of 1e) (Figure 2C).
Scheme 1.

Chemical synthesis of C55-lipid I-Nε-C6-dansylthiourea, 2e-C55.
Figure 2.

A: Biotransformations of lipid I and lipid II derivatives, 2e-C55 and 3e-C55, from Park’s nucleotide Nε-C6-dansyl, 1e. B: HPLC chromatography of 1e, 2e-C55, and 3e-C55. C: Kinetics of transformation from 1e to 2e-C55, and 3e-C55 in A.
Convenient source of MraY and MurG.
We reported that MurX- and MraY-containing membrane fractions (P-60) obtained from wild-type M. tuberculosis, M. smegmatis, and E. coli strains could convert the Park’s nucleotide probes (1b and 1f in Table 1) to the corresponding lipid I analogues in 5–70% yields with 3 equivalents of C55- or C50-phosphate.8 This variation in yield conversion is dependent on the expression level of MurX/MraY. Mycobacterium spp. express MraY-type phosphotransferase much higher than Gram-negative and -positive bacteria.29 As demonstrated in Figure 3A, P-60 of a wild-type M. smegmatis strain could convert Park’s nucleotide Nε-C6-dansylthiourea (1e) to C55-lipid I-Nε-C6-dansylthiourea (2e-C55) in 60% yield and to C55-lipid II-Nε-C6-dansyl, 3e-C55 in 42% yield. To perform screening against MurG using crude membrane fractions, 1e should be converted to 2e-C55 in >90% yield in situ. We have been unable to successfully overexpress Mycobacterial MurX in E. coli. As an alternative, it has been demonstrated that recombinant proteins from M. thermoresistibile (Mtherm) are useful surrogates for production of problematic Mycobacterial proteins.30 We successfully expressed MthermMurX in E. coli and were able to purify it to a single homogenous species. As reported before,9 we have also routinely expressed and purified MraY of Hydrogenivirga spp. to study the catalytic mechanism and obtain insight into the binding mode of MraY/MurX inhibitors. Time-course experiments of prenylation of 1e with MraY/MurX from different sources of bacteria revealed that HyMraY (2.5 μM) yielded 2e-C55 in 95–100% yield within 1 h (Figure 3B). At the same concentration, MthermMurX furnished 2e-C55 in 70% yield, requiring a concentration of 5.0 μM to attain a similar level of conversion to that observed with HyMraY. Taking advantage of high-yielding 2e-C55 in a low concentration, we decided to apply the purified HyMraY to convert 1e to 2e-C55, and 2e-C55 generated in situ was used in the following MurG reactions (protocol A). Alternatively, the MurX activity can be terminated completely by addition of an MraY/MurX inhibitor, tunicamycin (50 μM). The MurG function of P-60 remains active after the addition of tunicamycin (protocol B). The latter protocol is particularly useful to study membrane fractions containing MurG where purification proves difficult (Figure 3C). Using MurG of a pathogen of research interest is ideal to discover selective antibacterial MurG inhibitors. Gamma-irradiated M. tuberculosis (NR-14819) obtained from BEI Resources has been a useful P-60 source for MtbMurG studies. However, it has proven unreliable as we often note a failure of the transformation from 1e to 3e-C55 due to an inactive P-60 membrane fraction from the obatined Mtb cells (Figure 2A). We turned to an M. smegmatis (ATCC607) strain that can serve as a surrogate of M. tuberculosis (H37Rv) to predict susceptibility of TB drugs under a slow growth condition.31 The IC50 levels of MraY inhibitors (e.g., tunicamycin, capuramycin, and muraymycins) obtained with MtbMurX were well-correlated with those with MsmegMurX. Importantly, M. smegmatis (ATCC607) can readily be cultured without an enrichment (growth rate: 48–72h at 37 °C to reach the OD value of 0.9). Thus, sufficient P-60 membrane fraction can be readily prepared from this M. smegmatis strain. In this study, it was determined that P-60 of M. smegmatis (ATCC607) is also a convenient surrogate for MtbMurG. We could expressed HyMurG in E. coli and successfully purified as its active form. Figure 3D summarises 3e-C55 yield-time curves for the transformation (2e-C55→3e-C55) using MurG enzymes from different sources. Under the conditions developed for MraY/MurG-catalyzed reactions (Figure 3A), 2e-C55 was converted to 3e-C55 in 80% and 100% yield with 2.5 μM and 5.0 μM concentration of HyMurG, respectively. P-60 of M. smegmatis required 10 μL (1 mg wet weight/μL) to convert 2e-C55 to 3e-C55 in 30–40% yield in a 50 μL scale. Conversion from 2e-C55 to 3e-C55 was dependent on P-60 concentration; MurG reaction with 30 μL of P-60 of M. smegmatis provided 3e-C55 in 70% yield. P-60 Membrane fractions prepared from wild-type S. aureus and E. coli were also examined. The same reaction with 30 μL of P-60 of E. coli provided 3e-C55 in less than 10% conversion of 3e-C55, and P-60 of S. aureus yielded 3e-C55 in 25%. These results suggested that P-60 of M. smegmatis is a convenient and reliable source to convert Park’s nucleotide to lipid II through lipid I and to identify antimycobacterial MurG inhibitors. Purified HyMurG is a robust enzyme, which is stable through repeated freezing and thawing cycles. Thus, we were interested in applying HyMurG as a convenient MurG source for discovering antibacterial MurG agents via HTS.
Figure 3.

Establishment of a MurG assay using Park’s nucleotide-Nε-C6-dansylthiourea, 1e.
Synthesis of C55-lipid I-Nε-C6-dansyl, 2e-C55, for kinetic studies.
We have previously reported chemical syntheses of Park’s nucleotide, lipid I, and lipid II.6,7,27 Synthesis of C55-lipid I-Nε-C6-dansylthioure (2e-C55) was adapted to the synthetic schemes developed for lipid I with minor modifications.7,9,27 Synthesis of 2e-C55 is summarized in Scheme 1. The common N-acetylmuramic acid (MurNAc) intermediate 4 was subjected to debenzylation of the anomeric position, and the generated free-alcohol was phosphorylated to form 5 in 85% overall yield with exclusive selectivity to the α-diasteromer by two step procedures of phosphite formation with dibenzyl N,N-diisopropylphosphoramidite and 5-(ethylthio)-1H-tetrazole followed by oxidation with aq. tBuO2H.9 Deprotection of the 2-(phenylsulfonyl)ethanol group of 5 was achieved by the treatment with DBU to furnish the free-carboxylic acid, which was coupled with the tetrapeptide, HCl•H-γ-D-Glu(OMe)-L-Lys(COCF3)-D-Ala-D-Ala-OMe, under EDCI, Glyceroacetonide-Oxima (GOx),32,33 NaHCO3 in DMF-H2O, furnishing the α-phosphoryl MurNAc-pentapeptide 6 in 90% overall yield. Hydrogenolitic debenzylations of 6 followed by the treatment with excess Et3N resulted in the corresponding monotriethylammonium phosphate, which was subjected to a carbonyldiimidazole (CDI)-promoted diphosphate-formation reaction with an ammonium salt of undecaprenyl phosphate (C55-P-NH4). The fully protected lipid I moeity was coverted to lipid I (7) by saponification with LiOH in THF-H2O. The crude lipid I was purified via reverse-phase HPLC (0.05 M NH4HCO3 : MeOH = 15 : 85 to 0 : 100 over 30 min, retention time : 25 min.) to furnish pure lipid I in 60% overall yield from 6. Lipid I could be stored in a 4 : 1 mixture of DMSO and 0.05M NaHCO3 at -20 °C for over 8 months without decomposition. An aliquot of lipid I was converted to lipid I-Nε-C6-dansylthiourea (2e-C55) in 80% overall yield in 3 steps from 7 including carbamate formation at the Nε-position with SuO-C(O)O-(CH2)6-NHCOCF3, deprotection of the CF3CO group, and thiourea formation with 4-(dansylamino)phenyl isothiocyanate. The structure of synthetic 2e-C55 was confirmed by 1H-NMR, LC-MS, and comparison of retention time with 2e-C55 synthesized from 1e using P-60 of M. smegmatis (Figure 2A).
Kinetic studies.
Kinetic studies provide insight into the catalytic mecahnism and help to optimize enymatic assay conditions. The kinetic parameters of MurG of M. smegmatis were investigated by varying concentrations of the substrates (2e-C55 and UDP-GlcNAc). The apparent Km for 2e-C55 was determined to be 40 ± 5.0 μM at 375 μM of UDP-GlcNAc, and the apparent Km for UDP-GlcNAc 35 ± 8.1 μM at 300 μM of 2e-C55. Many bacterial glycosyl transferases are believed to involve a ternary complex reaction mechanism.34,35 We further elaborated MurG kinetic studies to confirm its reaction mechanism whether MurG follows a ternary complex reaction mechanism using the new probe. The correlation between the concentrations of 2e-C55 or UDP-GlcNAc (x axis) and the reaction velocity (V) of 3e-C55 (y axis) at several fixed concentrations of UDP-GlcNAc or 2e-C55 was summarized in supporting information (SI) and the Km values for the enzymatic substrates are shown in Table 2. The Km values of UDP-GlcNAc were similar over the range of concentrations of 2e-C55. The Km values of 2e-C55 were also similar in a range between 93.8–375.0 μM of UDP-GlcNAc. The observations that different Km values were provided at lower concentrations of UDP-GlcNAc (below 62.5 μM); they may suggest that UDP-GlcNAc concentration affects MurG activity. These data indicate that 1) MurG catalyzes lipid II formation via a ternary complex mechanism in which lipid I and UDP-GlcNAc bind together to the enzyme, and 2) the lipid I analogue, 2e-C55, is an appropraite assay probe. Because detection limitation of the dansyl substrates (2e-C55 and 3e-C55) via UV-detector, kinetic studies were limited to higher than 12.5 μM of 2e-C55. Analogously, kinetic studies with HyMurG were performed. The Km value for 2e-C55 was 40 ± 7.0 μM at the concentration of 375 μM of UDP-GlcNAc; the Km (HyMurG) value was near equal to that obtained with MsmegMurG (Km: 40 ± 5.0 μM). The Vmax for the synthesis of lipid II analogue, 3e-C55, by P-60 of M. smegmatis was determined to be 0.98 μM/min and 2.0 μM/min for HyMurG. These kinetic parameters are not conclusive indexes to compare the catalytic effectiveness, however, these data support a faster-yielding 2e-C55 with HyMurG compared with the same reaction with P-60 of M. smegmatis (Figure 3C). Under the same reaction condition (buffer, detergent, pH, and temperature), the kcat values of HyMurG and HyMraY for 2e-C55 and 1e were determine to be 0.18 s−1 and 0.38 s−1, respectively, indicating that the turnover rate of MraY, which converts Park’s nucleotide to lipid I, is faster than that of MurG, which converts lipid I to lipid II. The Km and kcat values of EcoliMurG were reported to be 37–44 μM and 0.27–0.32 s−1 for the lipid I biotinylated analogue, respectively.19 Thus, these kinetic parameters indicated that affinity and catalytic turnover efficiency of MurG are similar among E. coli, Hydrogenivirga sp. and Mycobacterium spp.
Table 2.
Apparent Km values for C55-lipid I-Nε-C6-dansylthiourea (2e-C55) and UDP-GlcNAc at the different concentartions of the counterpart (UDP-GlcNAc or 2e-C55)a.
| C55-lipid I-Nε-C6-dansylthiourea (2e-C55) concentration (μM) |
Km of UDP-GlcNAc (μM) |
|---|---|
| 37.5 | 36 ± 10b |
| 75.0 | 37 ± 3.0 |
| 150 | 36 ± 5.0 |
| 225 | 36 ± 2.2 |
| 300 | 35 ± 8.1b |
| UDP-GlcNAc concentration (μM) |
Km of C55-lipid I-Nε-C6-dansylthiourea (2e-C55) (μM) |
| 46.9 | 13 ± 3.0b |
| 62.5 | 25 ± 3.0 |
| 93.8 | 36 ± 5.0 |
| 187.5 | 39 ± 3.0 |
| 375.0 | 40 ± 5.0b |
All reactions were performed in the presence of MgCl2 (50 μM)
Higher and lower concentrations were repeated three times.
Application of UDP-Glucosamine-C6-FITC (UDP-GlcN-C6-FITC) to MurG assay.
Previously, the intact UDP-GlcNAc and its radiolabeled substrates were the only nucleosides that have been applied in transformations with glycosyltransferases that utilize UDP-GlcNAc as the donor substrate. We have developed UDP-GlcN-C6-FITC probe (8) for assaying polyprenyl phosphate-GlcNAc-1-phosphate transferase (WecA),36 which catalyzes the conversion from UDP-GlcNAc to decaprenyl-P-P-GlcNAc. It was demonstrated, for the first time, that a UDP-GlcNAc-fluorescent probe can be a substrate for a glycosyltransferase. To facilitate the screening against MurG using coupled assays with Park’s nucleotide, tolerability of MurG against 8 was examined using P-60 of M. smegmatis and purified HyMurG. Gratifyingly, under the same condition developed in Figure 2, GlcN-C6-FITC addition to C55-lipid I-Nε-C6-dansyl (2e-C55) with 8 was catalyzed by P-60 of M. smegmatis and HyMurG to form C55-lipid II-Nε-C6-dansyl-FITC (9-C55) in 60–70% and 100% yield, respectively (Figure 4A). C55-lipid II-Nε-C6-dansyl-FITC (9-C55) can be detected by either 350 nm (for the dansyl group) or 485 nm (for the FITC group) or both wave lengths if a dual-wavelength UV detector is equipped with HPLC system (Figure 4B). The lipid I and II possessing different UV-visible absorbance have a significant advantage in undoubtedly distinguishing the enzymatic substrate and product in HPLC; regardless of chromatographic separation, only 9-C55 can be detected at 485 nm and both 2e-C55 and 9-C55 detected at 350 nm. Thus, MurG assays with 1e (or 2e-C55) and 8 will not provide false-positive or -negative results. The Km value for UDP-GlcN-C6-FITC probe (8) was 54 μM at the concentrations of 75 μM of C55-lipid I-Nε-C6-dansylthiourea (2e-C55); this was similar to the Km values obtained with lipid I (7) (Km: 49 μM). The Vmax values for C55-lipid II-Nε-C6-dansyl-FITC (9-C55) transformation by P-60 of M. smegmatis and HyMurG were determined to be 0.56 and 0.67 μM/min, respectively. In competition reactions in P-60 (M. smegmatis)-catalyzed lipid II synthesis with UDP-GlcN-C6-FITC probe (8) at 375 μM in the presence of UDP-GlcNAc (by varying concentration), 100%-disappearance of 9-C55 required >200 μM of UDP-GlcNAc (IC50 8.40 μM, Figure 5). These kinetic parameters (Km 54 μM for 8 and Km 44 μM for UDP-GlcNAc, and similar Vmax value of 0.5–0.6 μM/min) imply that 8 is an appropriate UDP-GlcNAc mimetic for MurG-catalyzed lipid II analogue formations. It is worthwhile mentioning that MraY/MurX followed by MurG-catalyzed lipid II synthesis from either Park’s nucleotide (10) or Park’s nucleotide-Nε-C6-dansylthiourea (1e) illustrated in Figure 4A is not a reversible process and polymerizations of C55-lipid II-FITC (11-C55) and C55-lipid II-Nε-C6-dansyl-FITC (9-C55) were not observed with P-60 of M. smegmatis.37 Thus, product yields for the lipid I and lipid II analogues are very high without addition of inhibitors of penicillin binding proteins.38
Figure 4.

A: Biotransformation of lipid II derivatives, 9-C55 from Park’s nucleotide-Nε-C6-dansylthiourea (1e). B: HPLC chromatography of 1e, 2e-C55, 9-C55, Park’s nucleotide (10), and C55-lipid II-FITC (11-C55).
Figure 5.

Competitive reaction of UDP-GlcN-C6-FITC (8) and UDP-GlcNAc in the biotransformation of 9-C55.
Development of UV/Vis spectroscopy-based assay for MurG.
The lipid II analogue, 9-C55 are readily extracted with n-BuOH and the contaminated UDP-GlcN-C6-FITC (8) in the organic phase can be removed by washing with a 1 : 1 mixture of saline and 0.2 M mannitol (an 8-washing solution). Because separation of lipid I and lipid II analogues are not necessary in this assay, the fluorescence in nBuOH extract of MurG reaction was monitored via ultraviolet-visible (UV-Vis) spectrometry (excitation of 485nm, emission of 528nm); the UV-Vis-based assay was performed at a sufficiently high concentration of Park’s nucleotide (10) or Park’s nucleotide-Nε-C6-dansylthiourea (1e) (45–75 μM) for UV-Vis spectrometry and enough concentrations of UDP-GlcN-C6-FITC (8) that fulfill the Km value (e.g., 135–375 μM). Progress of the MurG-catalyzed reaction of 2e-C55 was monitored for 3 h. As shown in Figure 6A, an increase in fluorescence signal was observed in a time-dependent manner that was well-correlated to the yield curve obtained via the HPLC method (Figure 6B). A UV-Vis-based MurG assay developed here was validated by demonstrating the inhibition of MurG activity by two antibiotics, ramoplanin A2 (12) and nikkomycin Z (13). Generation of 9-C55 was inhibited by both ramoplanin A2 and nikkomycin Z in a dose-dependent manner (Figure 7). The IC50 values of ramoplanin A2 against MsmegMurG and HyMurG were determined to be 25.5 and 22.4 μM by dose-response curves via UV-Vis spectrometry (Figure 7). These data obtained via a new assay method are close to the IC50 values (20–50 μM) reported by the other groups.39–41 In our preliminary screening of in-house library molecules, we found that nikkonmycin Z, an inhibitor of chitin synthases, shows a competitive inhibitory activity against MurG. The IC50 values of nikkonmycin Z against MsmegMurG and HyMurG were 9.5 and 8.7 μM, respectively. It was determined that DMSO did not inhibit the MurG assays at 5% (v/v) concentrations. However, inhibition of the reactions was started at 10% (v/v) of DMSO; approximately 50% of the enzyme activity was reduced at this concentration (see SI).
Figure 6.

Correlation of biotransformation of C55-lipid II-FITC determined via UV-Vis and HPLC.
Figure 7.

Dose-response curves of two inhibitors evaluated via a UV-Vis-based MurG assay.
A microplate-based assay for MurG.
The microplate-based assay were performed with Park’s nucleotide (10) under the condition established in Figure 4A (all substrate used are >Km concentrations). The microplate-based MurG assay via UV/Vis spectroscopy was validated by demonstrating screening of a collection of molecules including positive (ramoplanin A2, nikkomycin Z)-, negative (selective MraY and WecA inhibitors)- controls in triplicate with 96-well plates. In these screenings, purified HyMurG was applied. All compounds were screened at three different conditions (10, 50, and 100 μM). Each plate contained four control wells: the first one with the denatured MurG (heated at 100 °C for 2 min.), the second one without MurG, the third one addition of 30% (v/v) of DMSO and the fourth one with ramoplanin A2 (12) at 50 μM. Under the assay conditions, seven molecules including nikkomycin Z (13) were identified as MurG inhibitors. The identified MurG inhibitors were confirmed by the HPLC-based assays at 0.1, 1, 10, 50, and 100 μM concentrations, being created dose-response curves to obtain their IC50 value (Table 3). Two MurG inhibitors (ToXa-1 (15) and PyDT-1 (16) reported previously (Hu et al. 2004)42 displayed MurG inhibitory activity with the IC50 value of 2.2 and 2.7 μM, respectively.43 (Figure 8). As described above, nikkomycion Z (13) showed a completive inhibitor of MurG with the IC50 value of 8.6 μM. On the other hand, another chitin synthase inhibitor, polyoxin D (14) exhibited a weak MurG inhibitory activity (IC50 50.8 μM). Ristocetin A (17) was identified as a strong inhibitor of MurG (IC50 0.96 μM against HyMurG). Although moenomycin A (18), a transglycosylase inhibitor, was reported to inhibit MurG (IC50 10.6 μM) via a coupled assay using MurG-pyruvate kinase-lactic dehydrogenase (Liu et al. 2003),23 our assay method did not cause a false-positive result; moenomycin A did not inhibit MurG function at 50–100 μM. Vancomycin (19) is an antibiotic that has frequently applied as a positive-control in several coupling assays including the method developed by the Wong group.23 In our studies, it was demonstrated that vancomycin hampers the extraction of the lipid I and lipid II derivatives with n-BuOH, making pseud-inhibitory activity in a concentration independent manner (entry 9 in Table 3). The other molecules including MraY/MurX (tunicamycins (20), APPU (21),44,45 and capuramycin (22)),46,47 WecA (O-methylcapuramycin (23)48), triflumuron (24), and nisin did not inhibit the lipid II formation at 50–100 μM concentrations. Importantly, the inhibitory activities of the MurG inhibitors identified using HyMurG were well-correlated to those against MsmegMurG (Table 3).
Table 3.
MurG assay against a collection of positive- and negative-controls.
| entry | Compound | HthermMurG (MsmegMurG) inhibition (%)a | IC50(μM) HyMurGb |
IC50(μM) MsmegMurGb |
IC50 (μM) MsmegMraYb |
||
|---|---|---|---|---|---|---|---|
| 10 μM | 50 μM | 100 μM | |||||
| 1 | Ramoplanin A2 (12) | 0 (0)c | 100(100) | 100 (100) | 22.4 | 23.5 | >100 |
| 2 | Nikkomycin Z (13) | 33 (35) | 50 (45) | 95 (93) | 8.7 | 9.5 | >100 |
| 3 | Polyoxin D (14) | 34 (40) | 48 (48) | 50 (45) | 50.8 | 55.0 | >100 |
| 4 | ToXa-1 (15) | 51(50) | 65 (68) | 98(100) | 2.2 | 2.0 | >100 |
| 5 | PyDT-1 (16) | 46 (48) | 55 (60) | 98(100) | 2.7 | 3.5 | >100 |
| 7 | Ristocetin A (17) | 55 (58) | 70 (65) | 95 (93) | 0.96 | 1.4 | 0.81 |
| 8 | Moenomycin A (18) | 0(0) | 0(0) | 12(15) | >100 | >100 | >100 |
| 9 | Vancomycin (19) | 59 (60)c | 55 (67)c | 50 (58)c | ND | ND | ND |
| 10 | Tunicamycin (20) | 0(0) | 0(0) | 0(0) | >100 | >100 | 2.9 |
| 11 | APPU (21) | 0(0) | 0(0) | 0(0) | >100 | >100 | 0.085 |
| 13 | Capuramycin (22) | 0(0) | 0(0) | 0(0) | >100 | >100 | 0.13 |
| 14 | O-Methylcapuramycin (23) | 0(0) | 0(0) | 0(0)d | >100 | >100 | >100 |
| 15 | Triflumuron (24) | 0(0) | 0(0) | 0(0) | >100 | >100 | >100 |
| 16 | Nisin (not shown in Fig. 7) | 0(0) | 0(0) | 2(5) | >100 | >100 | >100 |
| 17 | DMSO | 0(0) | 0(0) | 0(0) | ND | ND | ND |
1) Reaction conditions: CHAPS (20 wt%): 1.25 μL, β-mercaptoethanol (50 mM): 5 μL, MgCl2 (0.5 M): 5μL, KCl (2 M): 5μL, Park’s nucleotide (10 for UV-Vis- based assays, 2 mM) or Park’s nucleotide-Nε-C6-dansyl (1e-C55, for HPLC-based assays, 2 mM): 1.88 μL, C55-P (4 mM): 2.81 μL (3 eqv), HyMraY (125 μM): 1 μL, after 1h, UDP-GluN-C6-FITC (8, 10 mM): 1.88 μL (5 eqv), inhibitor molecule (0.1–100 μM), HyMurG (8.3 μg/mL): 5 μL or P-60-M. smegmatis (1 mg/μL, 10–30 μL), for 1 h at 37 °C. Work-up: n-BuOH (150 μL), a 1:1 mixture of saline/0.2 M mannitol. Analyses: UV-Vis method
Each experiment was performed two-three times, and the average IC50 values were summarized. Analyses: HPLC-based method
The lipid I and lipid II derivatives formed complexes with vancomycin (19), causing pseudo-inhibitory activity in a concentration independent manner.
Figure 8.

Structures of positive- and negative-controls.
Antimycobacterial activity of ristocetin A.
The glycopeptide antibiotics such as vancomycin (17) display antibacterial activity by forming hydrogen bondings between the glycopeptide aglycones and the L-Lys-D-Ala-D-Ala segment of the peptidoglycan precursors (e.g., lipid II) located in Gram-positive bacterial cell membrane.49–53 While vancomycin and ristocetin A are structurally similar, the mode of action of ristocetin A is different from that of vancomycin;54 the interaction of ristocetin A with the L-Lys-D-Ala-D-Ala segment of lipid II is very weak as demonstrated by our MurG assay methods (Table 3). Kinetic studies of inhibition of HyMurG in the presence of ristocetin A (5 μM) revealed that ristocetin A competes for the UDP-GlcNAc binding-site (Figure 9). Ristocetin A is also a strong inhibitor of MurX (IC50 0.81 μM against MsmegMurX, see SI) via a mechanism of non-competitive inhibition against Park’s nucleotide and prenyl-P. Recognition and complexation of the L-Lys-D-Ala-D-Ala portion of lipid II have been the interest of ristocetin A and other glycopeptide antibiotics.55
Figure 9.

Michaelis-Menten plots for MurG-catalyzed formation of C55-lipid II-Nε-C6-dansylthiourea (3e-C55) in the presence and absence of ristocetin A.
To the best of our knowledge, a molecule that has dual inhibitory activities against MurG and MraY/MurX at such a low concentrations has never been reported.
Antimycobacterial activity of the MurG inhibitors identified in Table 3 were examined against M. smegmatis (ATCC607) and M. tuberculosis (H37Rv). Nikkomycin Z (13), polyoxin D (14), ToXa-1 (15), and PyDT-1 (16) did not inhibit growth of these bacteria even at 50 μg/mL concentration. A moderate MurG inhibitor, ramoplanin A2 (12) has antimycobacterial activity with the MIC level of 3.25–12.5 μg/mL. Ristocetin A (17) exhibited strong bactericidal activity against Mycobacterium spp. with MIC below 0.35 μg/mL. ToXa-1 exhibited cytotoxicity against Vero cell with the IC50 value of 12.5 μg/mL. All other MurG inhibitors identified in Table 3 did not show cytotoxicity against Vero cells at 100 μg/mL concentration (Table 4).
Table 4.
MIC of MurG inhibitors (12-17) against Mycobacterium spp.
| entry | Compound |
M. smegmatis (ATCC607)a MIC (μg/mL) |
M. tuberculosis (H37Rv)b MIC (μg/mL) |
Vero cellc (μg/mL) |
|---|---|---|---|---|
| 1 | Ramoplanin A2 (12) | 3.25 | 6.25–12.5 | >100 |
| 2 | Nikkomycin Z (13) | >50.0 | >50.0 | >100 |
| 3 | Polyoxin D (14) | >50.0 | >50.0 | >100 |
| 4 | ToXa-1 (15) | >50.0 | >50.0 | 12.5 |
| 5 | PyDT-1 (16) | >50.0 | >50.0 | >100 |
| 7 | Ristocetin A (17) | <0.39 | 0.5 | >100 |
| 8 | Rifampicin | 1.58 | 0.17 | >100 |
| 9 | Ethambutol | 0.39 | 0.78 | >100 |
| 10 | INH | 0.78 | 0.16 | >100 |
| 11 | Vancomycin (19) | 3.12 | 12.5 | >100 |
| 12 | Tunicamycin (20) | 6.25–12.5 | 12.5 | 1.56 |
M. smegmatis (ATCC607) was cultured with 7H9 containing 0.5% tween 80. The bacterial culture in a 96-well plate treated or non-treated with compounds was incubated for 3 days at 37 °C in a static incubator. Resazurin (0.01 %, 20 μL) was added to each well and incubated at 37 °C for 4h. The MIC values were determined according to NCCLS method (pink = growth, blue = no visible growth).
M. tuberculosis (H37Rv) was cultured with 7H9 containing OADC. The culture was incubated for 14 days.
Kidney epithelial cells extracted from an African green monkey (ATCC).
Structural comparison between MurG proteins of Mycobacterium spp. and Hydrogenivirga sp.
We demonstrated that MurG of Hydrogenivirga sp. is a convenient surrogate for MsmegMurG for screening of antimycobacterial MurG inhibitors. BLAST search [Altschul et al. 1990]56 of MurG enzymes of M. smegmatis (MC2 155), Hydrogenivirga sp. (128–5-R1–1), and E. coli (K-12) against M. tuberculosis (H37Rv) revealed that MurG between M. tuberculosis and M. smegmatis: 84% similarity / 76% identity, Hydrogenivirga sp. and M. tuberculosis: 42% similarity / 26% identity, and E. coli and M. tuberculosis: 53% similarity / 39% identity (see SI). Although moderate primary sequence similarity between Hydrogenivirga sp. and M. smegmatis or M. tuberculosis, we confirmed that inhibitory response of the inhibitor molecules against MurG is similar among those obtained from Mycobacterium spp. and Hydrogenivirga sp. MurG is tightly associated with peripheral membrane. Although MurG of E. coli was successfully crystallized and X-ray diffraction experiments were successfully carried out (Ha et al. 2000),57 our studies suggested that purification of MurG enzymes of Mycobacterium spp. remains a very challenging task. Thus, applying thermally stable HyMurG has significant advantage in context from reliable and practical enzyme sources for assay screenings. In order to understand correlation of MurG inhibitory activity across the bacterial species, we constructed a homology model of HyMurG based on EcMurG (PDB: 1F0K). Albeit a lower sequence identity (31%) between two MurG enzymes, no apparent difference in overall fold was observed (Figure 10A). Some structural deviations were observed in the loop regions, but all hydrophobic segments associated with the putative active site are highly conserved. Multiple sequence alignment of MurG homologs revealed high conservation in the active site (Figure 10B).
Figure 10.

Conservation of MurG homologs. A: Structural models of MurG with E. coli (PDB ID: 1F0K) aligned to a homology model of MurG from Hydrogenivirga sp. HyMurG colored in green and EcMurG in gray. Putative active site is magnified in the inset with important catalytic residues as sticks, numbering based on EcMurG. Homology model was created in SWISS-MODEL [Waterhouse et al. 201858 using EcMurG (PDB ID: 1F0K) chain B as a template. Figure was generated using PyMOL (Version 2.2.3, Schrödinger, LLC, Portland, OR, USA)59. B: Alignment of MurG sequences from EcMurG, HyMurG, and MtbMurG highlighting residues from A. Alignment made using T-Coffee and ESPript [Tommaso et al. 2011; Robert et al. 2014]60,61.
CONCLUSIONS
We have studied the fluorescent probe-conjugated substrates for MraY/MurX and MurG enzymes. To date, a very few number of Park’s nucleotide fluorescent probes have been demonstrated in their transformations to the corresponding lipid II derivatives with the purified enzymes. MraY/MurX- and MurG-catalyzed biotransformation with the Park’s nucleotide fluorescent probe, 1e, yields the lipid II-fluorescent, 3e-C55, in very high yield. The MurG assay protocols developed here do not require separation of the reaction products via specific biopolymer(s) that require extensive washing processes. In contrast, our assays developed herein take advantage of a strong hydrophobicity of the MraY/MurX and MurG products. A washing condition (a 1 : 1 mixture of saline and 0.2 M mannitol) can prevent a micelle formation of the lipid I and lipid II derivatives, retaining these products in the BuOH phase and solubilizing the donors, UDP-GlcNAc or UDP-GlcN-C6-FITC (8) in the aqueous phase. Importantly, the Park’s nucleotide fluorescent probe, 1e, can readily be synthesized from Park’s nucleotide (10). Conveniently, the intact Park’s nucleotide can be applied to the MurG assays with 8. The microplate-based MurG assays using 10 and 8 summarized in Figure 4A and Figure 6 show good correlations with the assays via HPLC, and could be applicable to HTS. The HPLC-based MurG assays summarized in Figure 4B can monitor both lipid I and lipid II derivative simultaneously at different wavelengths, thus, false results will not be generated in these assays. The membrane fraction (P-60) prepared from M. smegmatis (ATCC607) is a convenient surrogate of MtbMurX and MtbMurG. We experimentally proved that the purified MurG of Hydrogenivirga sp. can serve as a reliable and alternative source of MsmegMurG; IC50 values obtained with HyMurG are very close to those with P-60 of M. smegmatis. HyMraY and HyMurG can keep at -80 °C for over a year without loss of activity and tolerate to multiple freeze and thaw cycles. Therefore, combination of the unique donor/acceptor substrates (1e and 8) and enzyme sources (HyMraY and HyMurG) will provide robust MurG assay screenings. In preliminary screening of a collection of small molecules, ristocetin A (17) shows strong MurG inhibitory activity by competing with UDP-GlcNAc. Ristocetin A is also a strong MraY/MurX inhibitor, whereas, vancomycin (19) does not bind both MraY/MurX and MurG enzymes. Antimycobacterial activity of ristocetin A is stronger than that of vancomycin. Our studies imply that strong antimycobacterial activity of ristocetin A cannot be explained solely by the binding ability to the L-Lys-D-Ala-D-Ala portion of lipid II. Ristocetin A has about 3.0 times less binding affinity against the L-Lys-D-Ala-D-Ala mimetic than that of vancomycin.53 It has never been reported previously that a single molecule inhibits both MraY/MurX and MurG at low concentrations. Ramoplanin A2 with a larger molecular weight (Mw =2,554) is widely accepted as a MurG inhibitor with a membrane disrupting activity.41 Ramoplanin’s MurG inhibition is very weak, thus, a strong antibacterial MurG inhibitor will be a useful lab tool as well as a lead compound for developing new MurG inhibitors. We have been attempting to generate ristocetin A resistant mutants of M. smegmatis (ATCC 607) to obtain insights into the mode of antimycobacterial activity of ristocetin A. Appropriate chemical modifications of ristocetin A are known to attenuate thrombocyte aggregation,62 making ristocetin A analogues as new TB drug leads to combat MDR strains. SAR of ristocetin A against drug resistant Mtb and platelet aggregation activity and screening date for a large library molecule will be reported elsewhere.
EXPERIMENTAL SECTION
All experimental detail are provided in Supporting Information
Cloning, expression, and purification
Expression and purification of HyMraY: The gene mraY of Hydrogenivirga sp.128–5-R1–1 was cloned with an N-terminal His6 tag into a pET22b vector. The plasmid was transformed and expressed in E. coli NiCo21(DE3) pLEMO competent cells. The proteins were purified using a nickel-affinity, cation exchange, and size exclusion chromatography. The final storage buffer was 20 mM HEPES pH 7.5, 100 mM NaCl, 10% glycerol, 5 mM β-mercaptoethanol, 0.15% decyl β-D-maltopyranoside.
Expression and purification of HyMurG: The gene murG of Hydrogenivirga sp.128–5-R1–1 was cloned with an N-terminal His6 tag into a pET33b vector. The plasmid was transformed and expressed in E. coli NiCo21(DE3) competent cells. The proteins were purified using a cobalt-affinity and size exclusion chromatography. The final storage buffer was 20 mM Tris pH 7.5, 150 mM NaCl, 10 % glycerol, 0.15 % decyl β-D-maltopyranoside, and 5 mM β-mercaptoethanol.
Preparation of P-60 membrane fraction from M. smegmatis (ATCC607): The cells were harvested by centrifugation followed by washing with saline and buffer A (50 mM potassium phosphate, 5 mM MgCl2, 5 mM DTT, 10% glycerol, pH 7.2). The washed cell pellets were suspended in buffer A and disrupted by sonication on ice-bath. The resulting suspension was centrifuged at 4,700 xg for 15 min at 4 °C. The supernatant was centrifuged at 25,000 xg for 20 min at 4 °C. The supernatant was subjected to ultracentrifugation at 100,000 xg for 1 h at 4 °C. The supernatant was discarded and the pellet containing the membrane was suspended in Tris buffer (pH 7.5, 1 mg/1 μL).
Enzymatic assay procedures
Protocol A (in Figure 3): Park’s nucleotide-Nε-C6-dansylthiourea (1e) (2 mM stock solution, 1.88 μL), CHAPS (20%, 1.25 μL), β-mercaptoethanol (50 mM, 5 μL), MgCl2 (0.5 M, 5 μL), KCl (2 M, 5 μL), and C55-phosphate dissolved in NaHCO3 (50 mM) : DMSO (1 : 4) (4 mM, 2.81 μL) were placed in a 1.5 mL Eppendorf tube. To a reaction mixture, HyMraY (4.18 mg/mL, 1 μL) was added (total volume of reaction mixture: 50 μL adjust with Tris buffer (50 mM, pH = 8.0)). The reaction mixture was incubated for 1 h at 37 °C. To a reaction mixture, inhibitor molecule (0 – 100 μg/mL in Tris buffer), UDP-GlcNAc (10 mM stock solution, 1.88 μL), and P-60 (1 mg/μL, 30 μL) or HyMurG (5.2 mg/mL, 5 μL) were added. The reaction mixture was incubated for 1 h at 37 °C, and quenched with water saturated n-butanol (150 μL). Two phases were mixed via vortex for 2 min and centrifuged at 25,000 xg for 10 min. The upper n-butanol phase was assayed via reverse-phase HPLC. The n-butanol phase (30 μL) was injected into HPLC (solvent: a gradient elution of CH3OH/0.05 M aq. NH4HCO3 = 70 : 30 to 100 : 0 over 30 min; UV: 350 nm; flow rate: 1.0 mL/min; column: Luna 5μm C8, 100 A, 250 × 4.60 mm), and the area of the peak for C55-lipid II-Nε-C6-dansyl-FITC was quantified to obtain the IC50 value. The IC50 values were calculated from plots of the percentage product inhibition versus the inhibitor concentration.
Protocol B (in Figure 3): Park’s nucleotide-Nε-C6-dansylthiourea (1e) (2 mM stock solution, 1.88 μL), CHAPS (20%, 1.25 μL), β-mercaptoethanol (50 mM, 5 μL), MgCl2 (0.5 M, 5 μL), KCl (2 M, 5 μL), and C55-phosphate dissolved in NaHCO3 (50 mM) : DMSO (1 : 4) (4 mM, 2.81 μL) were placed in a 1.5 mL Eppendorf tube. To a stirred reaction mixture, P-60 (1 mg/μL, 30 μL) was added (total volume of reaction mixture: 60 μL adjust with Tris buffer (50 mM, pH = 8.0)). The reaction mixture was incubated for 1 h at 37 °C. To a reaction mixture, tunicamycin (10 mg/mL stock solution, 0.25 μL) was added, and inhibitor molecule (0 – 100 μg/mL in Tris buffer) and UDP-GlcNAc (10 mM stock solution, 1.88 μL) were added. The reaction mixture was incubated for 1 h at 37 °C, and quenched with water saturated n-butanol (150μL). Two phases were mixed via vortex for 2 min and centrifuged at 25,000 xg for 10 min. The upper n-butanol phase was assayed via reverse-phase HPLC. See, Protocol A for the analyses.
UV/VIS spectroscopy-based assay (non-microplate MurG assay): Park’s nucleotide-Nε-C6-dansylthiourea (1e) (2 mM stock solution, 1.88 μL), CHAPS (20%, 1.25 μL), β-mercaptoethanol (50 mM, 5 μL), MgCl2 (0.5 M, 5 μL), KCl (2 M, 5 μL), and C55-phosphate dissolved in NaHCO3 (50 mM) : DMSO (1 : 4) (4 mM, 2.81 μL) were placed in a 1.5 mL Eppendorf tube. To a stirred reaction mixture, HyMraY (4.18 mg/mL, 1 μL) was added (total volume of reaction mixture: 50 μL adjust with Tris buffer (50 mM, pH = 8.0)). The reaction mixture was incubated for 1 h at 37 °C. To a reaction mixture, inhibitor molecule (0 – 100 μg/mL in Tris buffer), UDP-GlcN-C6-FITC (10 mM stock solution, 1.88 μL), and P-60 (1 mg/μL, 30 μL) or HyMurG (5.2 mg/mL, 5 μL) were added. The reaction mixture was incubated for 1 h at 37 °C, and quenched with water saturated n-butanol (150μL). Two phases were mixed via vortex for 2 min and centrifuged at 25,000 xg for 10 min. The n-butanol phase was washed with a 1 : 1 mixture of saline and 0.2 M mannitol (50 μL, thrice) and the washed n-butanol phase (20 μL) was transferred to a 384 well black plate and fluorescence was measured at an excitation of 485 nm and emission of 528 nm. The IC50 values were calculated from plots of the percentage product inhibition versus the inhibitor concentration.
Microplate MurG assay: The assay was performed in 96-well plates. The reaction mixture contained park’s nucleotide-Nε-C6-dansylthiourea (1e) (2 mM stock solution, 1.88 μL), CHAPS (20%, 1.25 μL), β-mercaptoethanol (50 mM, 5 μL), MgCl2 (0.5 M, 5 μL), KCl (2 M, 5 μL), C55-phosphate dissolved in NaHCO3 (50 mM) : DMSO (1 : 4) (4 mM, 2.81 μL), and HyMraY (4.18 mg/mL, 1 μL) (total volume of reaction mixture: 50 μL adjust with Tris buffer (50 mM, pH = 8.0)). The reaction mixture was incubated for 1 h at 37 °C. To a reaction mixture, inhibitor molecule (0 – 100 μg/mL in Tris buffer), UDP-GlcN-C6-FITC (10 mM stock solution, 1.88 μL), and HyMurG (5.2 mg/mL, 5 μL) were added. The reaction mixture was incubated for additional1 h at 37 °C. The reaction was quenched by adding water-saturated n-butanol (150 μL) and thoroughly mixed 20 times using a multichannel pipette. The upper phase was transferred to another well and washed with a 1 : 1 mixture of saline and 0.2 M mannitol (50 μL). Two phases were thoroughly mixed and the upper phase was washed with a 1 : 1 mixture of saline and 0.2 M mannitol (50 μL) (repeated twice). The upper phase (20 μL) was transferred to a 384 well black plate and fluorescence was measured at an excitation of 485 nm and emission of 528 nm.
Synthesis and characterization of representative molecules
Park’s nucleotide-Nε-C6-dansylthiourea (1e): To a solution of Park’s nucleotide (10) (6.3 mg, 5.5 μmol) and SuO-C(O)O-(CH2)6-NHCOCF3 (5.8 mg, 0.017 mmol) in MeCN (0.5 mL) was added Et3N (3.9 μL, 0.028 mmol). After being stirred for 12 h at r.t., the solution was concentrated under reduced pressure and the resulting product was dried under high vacuum. To a solution of the crude product in THF (0.5 mL) was added 0.2 mL of aq. LiOH (2.3 mg, 0.055 mmol). After being stirred for 3 h at r.t., the reaction mixture was filtered. The crude product was purified by reverse phase HPLC [column: HYPERSIL GOLD™ (175 Å, 12 μm, 150 × 20 mm), solvents: 0 : 100 CH3CN : 0.05 M aq. NH4HCO3 for 5 min then 5 : 95 CH3CN : 0.05 M aq. NH4HCO3 for 10 min then 10 : 90 CH3CN : 0.05 M aq. NH4HCO3 for 10 min, flow rate: 4.0 mL/min, UV: 254nm]. To a solution of the product and NaHCO3 (4.6 mg, 0.055 mmol) in a 4 : 1 mixture of THF and H2O (0.5 mL) was added 4-(dansylamino)phenyl isothiocyanate (10.5 mg, 0.028 mmol). After being stirred for 4 h at r.t., the reaction mixture was filtered. The filtrate was purified by reverse phase HPLC [column: Phenomenex Luna (100 Å, 10 μm, C18, 250 × 10 mm), solvents: 10 : 90 CH3CN : 0.05 M aq. NH4HCO3 for 5 min then 20 : 80 CH3CN : 0.05 M aq. NH4HCO3 for 10 min then 30 : 70 CH3CN : 0.05 M aq. NH4HCO3 for 10 min, flow rate: 3.0 mL/min, UV: 350nm] to afford 1e (6.4 mg, 70% overall, retention time: 24.7 min): 1H NMR (400 MHz, Deuterium Oxide) δ 8.55 (d, J = 8.2 Hz, 1H), 8.30 (d, J = 8.2 Hz, 1H), 8.15 (d, J = 7.4 Hz, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.32 (d, J = 7.2 Hz, 1H), 7.07 (s, 1H), 6.87 (s, 4H), 5.93 (ddd, J = 3.8, 1.6, 0.9 Hz, 1H), 5.89 (d, J = 8.0 Hz, 1H), 5.41 (dd, J = 7.7, 3.2 Hz, 1H), 4.30 (d, J = 3.3 Hz, 3H), 4.28 – 4.01 (m, 10H), 3.92 – 3.87 (m, 1H), 3.82 – 3.77 (m, 2H), 3.74 – 3.70 (m, 1H), 3.61 – 3.57 (m, 1H), 3.39 – 3.32 (m, 2H), 2.98 – 2.90 (m, 2H), 2.80 (s, 6H), 2.24 – 2.18 (m, 4H), 2.14 – 2.09 (m, 3H), 1.95 (s, 3H), 1.80 – 1.76 (m, 1H), 1.62 – 1.43 (m, 4H), 1.37 (d, J = 7.5 Hz, 3H), 1.34 (d, J = 7.1 Hz, 3H), 1.28 (d, J = 7.0 Hz, 3H), 1.26 (d, J = 6.9 Hz, 3H), 1.17 – 1.11 (m, 4H), 0.86 – 0.78 (m, 4H); HRMS (ESI+) m/z calcd for C66H96N13O30P2S2 [M + H] 1676.5303, found: 1676.5322.
Lipid I (7). Synthesis of 5: To a solution of 4 (0.33 g, 0.46 mmol) in a 9:5:1 mixture of MeOH, formic acid and H2O (15 mL) was added Pd-C (0.65 g). The reaction solutionn was stirred under hydrogen atmosphere (400 psi) for 17 h. The reaction mixture was filtrated and the residue was concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (EtOAc/MeOH = 90/10) to afford the free-alcohol (0.26 g, 91%). To a solution of the anomeric-free alcohol (0.21 g, 0.34 mmol) and 5-(ethylthio)-1H-tetrazole (0.13 g, 1.0 mmol) in CH2Cl2 (3.4 mL) was added dibenzyl N,N-diisopropylphosphoramidite (0.29 mL, 0.86 mmol) at 0 °C. After 2 h at 0 °C, the reaction was quenched with sat. NaHCO3 solution and the mixture was separated. The aqueous layer was extracted with CHCl3 and the combined organic layer was dried over Na2SO4, and evaporated. 70% aq. tert-butyl hydroperoxide (0.48 mL, 3.4 mmol) was added to a solution of the residue and NaHCO3 (58 mg, 0.69 mmol) in THF (3.4 mL) at 0 °C. After 30 min. at r.t., the reaction was quenched with aq. Na2S2O3 and the mixture was extracted with CHCl3 and the combined organic layer was dried over Na2SO4, and evaporated. The residue was purified by silica gel column chromatography (EtOAc/MeOH = 90/10) to afford 5 (0.28 g, 93% for 2 steps). 1H NMR (400 MHz, Chloroform-d) δ 7.92 (dd, J = 8.4, 1.3 Hz, 2H), 7.68 (tt, J = 7.4, 1.3 Hz, 1H), 7.61 – 7.56 (m, 2H), 7.39 – 7.32 (m, 10H), 6.73 (d, J = 7.0 Hz, 1H), 6.12 (d, J = 9.0 Hz, 1H), 5.62 (dd, J = 5.8, 3.2 Hz, 1H), 5.14 – 4.97 (m, 6H), 4.45 (td, J = 6.2, 1.3 Hz, 2H), 4.33 (ddt, J = 10.6, 9.0, 3.1 Hz, 1H), 4.19 (t, J = 7.2 Hz, 1H), 4.10 (dd, J = 13.1, 4.6 Hz, 1H), 3.92 (d, J = 13.8 Hz, 1H), 3.89 (d, J = 9.8 Hz, 1H), 3.50 – 3.41 (m, 3H), 2.07 (s, 3H), 2.01 (s, 3H), 1.75 (s, 3H), 1.30 (d, J = 6.9 Hz, 3H), 1.29 (d, J = 7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 171.93, 171.51, 170.63, 170.60, 169.12, 139.05, 134.11 (2C), 129.47 (2C), 129.04 (2C), 128.80 (4C), 128.16 (2C), 128.08 (4C), 96.85, 96.78, 78.37, 70.15, 70.03, 69.98, 68.65, 61.45, 58.07, 54.84, 52.92, 52.85, 47.92, 22.97, 20.79, 20.65, 18.62, 17.00; 31P NMR (162 MHz, CDCl3) δ -2.39; HRMS (ESI+) m/z calcd for C40H49N2NaO16PS [M + Na] 899.2438, found: 899.2412. Synthesis of 6: To a solution of 6 (78 mg, 0.089 mmol) in CH2Cl2 (0.45 mL) was added DBU (15 μL, 0.098 mmol). After stirring the solution for 1 h at r.t., the reaction was quenched with 1 M aq. HCl and the mixture was separated. The aqueous layer was extracted with EtOAc and the combined organic layer was dried over Na2SO4. After concentration under reduced pressure, GOx (41 mg, 0.18 mmol) and EDCI (34 mg, 0.18 mmol) were added to a solution of the crude product, tetrapeptide (0.10 g, 0.18 mmol) and NaHCO3 (38 mg, 0.45 mmol) in 24:1 solution of DMF and H2O (1.0 mL). After stirring the solution for 2 h at r.t., the reaction was added 9:1 solution of chloroform and methanol (5 mL). The solution was washed with aq. NH4Cl and aq. NaHCO3, and the organic layer was dried over Na2SO4. Concentration under reduced pressure followed by purification by silica gel column chromatography (EtOAc/MeOH/Et3N = 93/7/0.5 – 90/10/0.5) afforded 99 mg (90% for 2 steps) of 7. 1H NMR (400 MHz, Chloroform-d) δ 7.75 (t, J = 5.8 Hz, 1H), 7.60 (d, J = 7.7 Hz, 1H), 7.38 – 7.30 (m, 10H), 7.15 (d, J = 5.8 Hz, 1H), 7.10 (d, J = 8.5 Hz, 1H), 6.88 (d, J = 8.7 Hz, 1H), 5.64 (dd, J = 5.9, 3.2 Hz, 1H), 5.06 (ddd, J = 9.2, 8.4, 3.0 Hz, 6H), 4.54 – 4.30 (m, 7H), 4.24 (quin, J = 6.9 Hz, 1H), 4.15 – 4.09 (m, 2H), 3.91 (d, J = 10.6 Hz, 2H), 3.70 (s, 3H), 3.68 (s, 3H), 3.69 – 3.66 (m, 1H), 3.59 (t, J = 9.9 Hz, 1H), 3.31 (q, J = 6.5 Hz, 2H), 3.10 (q, J = 7.3 Hz, 1H), 2.38 – 2.13 (m, 4H), 2.07 (s, 3H), 1.99 (s, 3H), 1.78 (s, 3H), 1.58 (tt, J = 13.5, 6.2 Hz, 2H), 1.43 (d, J = 7.0 Hz, 3H), 1.38 (d, J = 7.4 Hz, 3H), 1.37 (d, J = 7.3 Hz, 3H), 1.29 (d, J = 6.6 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 173.32, 173.24, 172.73, 172.60, 172.18, 172.02, 171.94, 171.05, 170.57, 169.26, 157.45 (q, J = 36.8 Hz), 129.00 (2C), 128.76 (4C), 128.63, 128.56, 128.09 (4C), 70.02, 69.98, 68.63, 61.46, 53.82, 53.09, 53.01, 52.48, 52.29, 50.76, 50.27, 49.12, 48.03, 45.85, 39.42, 31.40, 31.16, 29.66, 28.16, 27.58, 22.78, 22.36, 20.79, 20.61, 18.54, 17.82, 17.64, 17.22, 8.57; 31P NMR (162 MHz, CDCl3) δ -2.72; HRMS (ESI+) m/z calcd for C53H74F3N7O21P [M + H] 1232.4628, found: 1232.4646. Synthesis of 7: To a solution of 6 (9.2 mg, 7.5 μmol) in MeOH (10 mL) was added 10% Pd-C (18 mg). After being stirred the reaction mixture under hydrogen atmosphere (using double-fold balloons) for 1 h, Et3N (0.5 mL) was added to the mixture. After 1 h, the catalyst was filtered through a pad of Celite. The filtrate was concentrated under reduced pressure and the resulting product was dried under high vacuum. To a solution of the crude product in a 3:1 mixture of THF and DMF (0.5 mL) was added CDI (3.6 mg, 0.023 mmol). After being stirred for 3 h at r.t., MeOH (20 μL) was added to the reaction mixture. After 30 min, the solution was evaporated, concentrated under high vacuum, and the resulting product was dried. To a solution of the crude product in a 3:1 mixture of THF and DMF (0.5 mL) was added C55-P-NH4 (5.3 mg, 6.0 μmol). After 48 h at r.t., the reaction mixture was filtered and concentrated. To a solution of the crude product in THF (0.5 mL) was added aq. LiOH (3.2 mg, 0.075 mmol). After 3 h at r.t., the reaction mixture was filtered. The filtrate was purified by reverse phase HPLC [column: Phenomenex Luna (100 Å, 10 μm, C18, 250 × 10 mm), solvents: a gradient elution of 85 : 15 to 100 : 0 MeOH : 0.05 M aqueous NH4HCO3 over 30 min, flow rate: 3.0 mL/min, UV: 220nm] to afford 7 (7.5 mg, 60% overall, retention time: 24.3 min). 1H NMR (400 MHz, Methanol-d4) δ 5.57 – 5.51 (m, 1H), 5.48 – 5.42 (m, 1H), 5.18 – 5.06 (m, 11H), 4.55 – 4.48 (m, 2H), 4.41 – 4.10 (m, 6H), 4.03 – 3.96 (m, 1H), 3.88 (q, J = 10.3 Hz, 2H), 3.74 (dd, J = 12.3, 5.1 Hz, 1H), 3.53 (t, J = 9.5 Hz, 1H), 2.99 – 2.88 (m, 2H), 2.79 (s, 1H), 2.31 (s, 3H), 2.14 – 2.03 (m, 31H), 1.99 (q, J = 7.8, 7.1 Hz, 8H), 1.90 – 1.78 (m, 2H), 1.74 (s, 3H), 1.68 (s, 21H), 1.63 – 1.58 (m, 10H), 1.47 (d, J = 7.2 Hz, 3H), 1.41 (t, J = 6.7 Hz, 6H), 1.36 (d, J = 7.2 Hz, 3H), 1.30 (d, J = 7.6 Hz, 8H), 0.89 (d, J = 8.1 Hz, 2H); HRMS (EI) calcd for C86H144N7O21P2 ([M + H]+): 1672.9891, found: 1672.9908.
UDP-GlcN-C6-FITC (8): The title compound was synthesized according to the procedure reported previously: [20] 1H NMR (400 MHz, Deuterium Oxide) δ 7.90 (d, J = 8.1 Hz, 1H), 7.71 – 7.63 (m, 1H), 7.61 – 7.51 (m, 1H), 7.37 – 7.30 (m, 3H), 7.30 – 7.20 (m, 4H), 5.99 – 5.85 (m, 2H), 5.52 (d, J = 6.3 Hz, 1H), 4.35 – 4.27 (m, 3H), 4.26 – 4.13 (m, 2H), 4.12 – 4.00 (m, 2H), 3.92 – 3.84 (m, 1H), 3.80 (dd, J = 16.2, 3.2 Hz, 1H), 3.76 – 3.69 (m, 2H), 3.62 – 3.56 (m, 2H), 3.54 – 3.47 (m, 1H), 3.45 – 3.37 (m, 1H), 1.70 – 1.57 (m, 4H), 1.46 – 1.33 (m, 4H); HRMS (ESI+) m/z calcd for C44H51N5NaO23P2S [M + H] 1134.2069, found: 1134.2084.
Kinetic studies: For the determination of apparent Km values, the substrates were added at various concentrations. Each reaction was applied the procedure described in Protocol A, but 2e-C55 was used instead of 1e. HyMraY was excluded. The apparent Km values were obtained by a nonlinear regression method using GraphPad Prism 7.04.
Supplementary Material
ACKNOWLEDGMENTS
The National Institutes of Health is greatly acknowledged for financial support of this work (Grant GM114611). MK thanks UTRF (University of Tennessee Health Science Center) for generous financial support (Innovation award R079700292). We thank Miss Kendal G. Crawley (UTHSC Research Scholar Program), Miss Shakiba Eslamimehr, and Mr. Stewart J. Clayton (UTHSC) for preparing membrane fractions from the bacteria and for cytotoxicity assays. NMR and HR-MS data were obtained on instruments supported by the NIH Shared Instrumentation Grant. The following reagent was obtained through BEI Resources, NIAID, NIH: Mycobacterium tuberculosis, strain H37Rv and gamma-irradiated Mycobacterium tuberculosis, NR-14819.
Funding Sources
GM114611 (NIGMS/NIH)
ABBREVIATIONS
- GT
glycosyltransferase
- MurG
UDP-N-acetylglucosamine:N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase
- Park’s nucleotide
UDP-MurNAc-pentapeptide
- Lipid I
MurNAc(pentapeptide)-pyrophosphoryl prenol
- Lipid II
GlcNAc-MurNAc(pentapeptide)-pyrophosphoryl prenol
- GlcNAc
N-acetylglucosamine
- GlcN
glucosamine
- MurNAc
N-acetylmuramic acid
- UDP
uridine diphosphate
- FITC
fluorescein isothiocyanate
- UV-Vis
ultraviolet-visible
- HPLC
high performance liquid chromatography
- NADH
nicotinamide adenine dinucleotide phosphate
- MurA
UDP-N-acetylglucosamine enolpyruvyl transferase
- MurB
UDP-N-acetylenolpyruvoylglucosamine reductase
- MurC
UDP-N-acetylmuramyl-L-alanine ligase
- MurD
UDP-N-acetylmuramoyl-L-alanine:D-glutamate ligase
- MurE
UDP-N-acetylmuramoyl-dipeptide-L-lysine ligase
- MurF
UDP-N-acetylmuramoyl-tripeptide-D-alanyl-D-alanine ligase
- MraY/MurX
polyprenyl phosphate-GlcNAc-1-phosphate transferase
- Hy
Hydrogenivirga sp.
- M. smeg
Mycobacterium smegmatis
- HTS
high-throughput screening
- P-60
membrane fraction containing ~60 KDa protein
- Mtherm
Mycobacterium thermoresistibile
- TB
tuberculosis
- WecA
polyprenyl phosphate-GlcNAc-1-phosphate transferase
- DPAGT1
dolichyl-phosphate GlcNAc-1-phosphotransferase 1
- tBu
tertiary-butyl
- nBu
normal-butanol
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- GOx
glyceroacetonide-Oxyma, Su, N-hydroxysuccinimide
- EDCI
1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
- Ac
acetyl
- Bn
benzyl
- Ph
phenyl
- DMSO
dimethyl sulfoxide
- OD
optical density
- SAR
structure-activity relationship
- Km
Michaelis constant
- kcat
turnover number
- Vmax
maximal velocity
- Vero
kidney epithelial cells extracted from an African green monkey
- PDB
Protein Data Bank
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Assay data, copies of NMR spectra, HPLC chromatogram of new compounds, and assay procedures.
Conflict of Interest
The authors declare no competing financial interest.
REFERENCES AND NOTES
- (1).Schmid J, Heider D, Wendel NJ, Sperl N, Sieber V (2016) Bacterial glycosyltransferases: Challenges and opportunities of a highly diverse enzyme class toward tailoring natural products. Front. Microbiol 7, 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Unligil UM, Rini JM (2000) Glycosyltransferase structure and mechanism. Curr. Opin. Struct. Biol 10, 510–517. [DOI] [PubMed] [Google Scholar]
- (3).van Heijenoort J (2007) Lipid intermediates in the biosynthesis of bacterial peptidoglycan. Microbiol. Mol. Biol. Reviews : MMBR. 71, 620–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bouhss A, Trunkfield AE, Bugg TDH, Mengin-Lecreulx D (2008) The biosynthesis of peptidoglycan lipid-linked intermediates. FEMS Microbiol. Reviews. 32, 208–233. [DOI] [PubMed] [Google Scholar]
- (5).Ha S, Gross B, Walker S (2001) E. Coli MurG: a paradigm for a superfamily of glycosyltransferases. Curr. Drug Targets: Infect. Disord 1, 201–213. [DOI] [PubMed] [Google Scholar]
- (6).Kurosu M, Mahapatra S, Narayanasamy P, Crick DC (2007) Chemoenzymatic synthesis of Park’s nucleotide: toward the development of high-throughput screening for MraY inhibitors. Tetradedron Lett 48, 799–803. [Google Scholar]
- (7).Li K, Kurosu M (2008) Synthetic Studies on Mycobacterium tuberculosis Specific Fluorescent Park’s Nucleotide Probe. Heterocycles 76, 455–469. [Google Scholar]
- (8).Siricilla S, Mitachi K, Skorupinska-Tudek K, Swiezewska E, Kurosu M (2014) Biosynthesis of a water-soluble lipid I analogue and a convenient assay for translocase I. Anal. Biochem 46, 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mitachi K, Siricilla S, Klaić L, Clemons WM, Kurosu M (2015) Chemoenzymatic syntheses of water-soluble lipid I fluorescent probes. Tetrahedron Lett 56, 3441–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
References
Synthesis of Park’s nucleotide or its derivatives from other groups (10–16):
- (10).Hitchcock SA, Eid CN, Aikins JA, Zia-Ebrahimi M, Blaszczak LC (1998) The first total synthesis of bacterial cell wall precursor UDP-N-acetylmuramyl-pentapeptide (Park Nucleotide). J. Am. Chem. Soc 120, 1916–1917. [Google Scholar]
- (11).Eid CN, Nesler MJ, Zia-Ebrahimi M, Wu C-YE, Yao R, Cox K, Richardson J (1998) Synthesis of a radioiodinated Park nucleotide analog: a new tool for antibacterial screen development. J. Lab. Comp. Radiopharm 41, 705–716. [Google Scholar]
- (12).Liu H, Sadamoto R, Sears PS, Wong C-H (2001) An efficient chemoenzymatic strategy for the synthesis of wild-type and vancomycin-resistant bacterial cell-wall precursors: UDP-N-acetylmuramyl-peptides. J. Am. Chem. Soc 123, 9916–9917. [DOI] [PubMed] [Google Scholar]
- (13).Ueda T, Feng F, Sadamoto R, Niikura K, Monde K, Nishimura S (2004) Synthesis of 4-fluorinated UDP-MurNAc pentapeptide as an inhibitor of bacterial growth. Org. Lett 6, 1753–1756. [DOI] [PubMed] [Google Scholar]
- (14).Wohnig S, Spork AP, Koppermann S, Mieskes G, Gisch N, Jahn R, Ducho C (2016) Total synthesis of dansylated Park’s nucleotide for high-throughput MraY assays. Chem. Eur. J 22, 17813–17819. [DOI] [PubMed] [Google Scholar]
- (15).Chen K-T, Chen P-T, Lin C-K, Huang L-Y, Hu C-M, Chang Y-F, Hsu H-T, Cheng T-R, Wu Y-T; Cheng W-C (2016) Structural investigation of Park’s nucleotide on bacterial translocase MraY: Discovery of unexpected MraY inhibitors. Sci. Rep 6, 31579–31590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Katsuyama A, Sato K, Yakushiji F, Matsumaru T, Ichikawa S (2018) Solid-phase modular synthesis of park nucleotide and lipids I and II analogues. Chem. Pharm. Bull 66, 84–95. [DOI] [PubMed] [Google Scholar]
- (17).Chandrakala B, Elias BC, Mehra U, Umapathy NS, Dwarakanath P, Balganesh TS, deSousa SM (2001) Novel scintillation proximity assay for measuring membrane-associated steps of peptidoglycan biosynthesis in Escherichia coli. Antimicrob. Agents Chemother 45, 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Men H, Park P, Ge M, Walker S (1998) Substrate synthesis and activity assay for MurG. J. Am. Chem. Soc 120, 2484–2485. [Google Scholar]
- (19).Ha S, Chang E, Lo M-C, Men H, Park P, Ge M, Walker S (1999) The kinetic characterization of Escherichia coli MurG using synthetic substrate analogues. J. Am. Chem. Soc 121, 8415–8426. [Google Scholar]
- (20).Branstrom AA, Midha S, Longley CB, Han K, Baizman ER, Axelrod HR (2000) Assay for identification of inhibitors for bacterial MraY translocase or MurG transferase. Anal. Biochem 280, 315–319. [DOI] [PubMed] [Google Scholar]
- (21).Helm JS, Hu Y, Chen L, Gross B, Walker S (2003) Identification of active-site inhibitors of MurG using a generalizable, high-throughput glycosyltransferase screen. J. Am. Chem. Soc 125, 11168–11169. [DOI] [PubMed] [Google Scholar]
- (22).Li J-J, Bugg TDH (2004) A fluorescent analogue of UDP-N-acetylglucosamine: application for FRET assay of peptidoglycan translocase II (MurG). Chem. Commun 182–183. [DOI] [PubMed] [Google Scholar]
- (23).Liu H, Ritter TK, Sadamoto R, Sears PS, Wu M, Wong C-H (2003) Acceptor specificity and inhibition of the bacterial cell-wall glycosyltransferase MurG. ChemBioChem 4, 603–609. [DOI] [PubMed] [Google Scholar]
- (24).Schwartz B, Markwalder JA, Wang Y (2001) Lipid II: Total synthesis of the bacterial cell wall precursor and utilization as a substrate for glycosyltransfer and transpeptidation by penicillin binding protein (PBP) 1b of Escherichia coli. J. Am. Chem. Soc 123, 11638–11643. [DOI] [PubMed] [Google Scholar]
- (25).Van Nieuwenhze MS, Mauldin SC, Zia-Ebrahimi M, Aikins JA, Blaszczak LC (2001) The total synthesis of lipid I. J. Am. Chem. Soc 123, 6983–6988. [DOI] [PubMed] [Google Scholar]
- (26).Van Nieuwenhze MS, Mauldin SC, Zia-Ebrahimi M, Winger BE, Hornback WJ, Saha SL, Aikins JA, Blaszczak LC (2002) The first total synthesis of lipid II: The final monomeric intermediate in bacterial cell wall biosynthesis. J. Am. Chem. Soc 124, 3656–3660. [DOI] [PubMed] [Google Scholar]
- (27).Mitachi K, Mohan P, Siricilla S, Kurosu M (2014) One-pot protection-glycosylation reactions for synthesis of lipid II analogues. Chem. Eur. J 20, 4554–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Huang L-Y, Huang S-H, Chang Y-C, Cheng W-C, Cheng T-JR, Wong C-H (2014) Enzymatic synthesis of lipid II and analogues. Angew. Chem. Int. Ed 53, 8060–8065. [DOI] [PubMed] [Google Scholar]
- (29).Liu Y, Breukink E (2016) The membrane steps of bacterial cell wall synthesis as antibiotic targets. Antibiotics (Basel) 5, 28/1–28/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Edwards TE, Liao R, Phan I, Myler PJ, Grundner C (2012) Mycobacterium thermoresistibile as a source of thermostable orthologs of Mycobacterium tuberculosis proteins. Protein Sci 21, 1093–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lelovic N, Mitachi K, Lemieux MR, Ji Y, Kurosu M manuscript in preparation.
- (32).Wang Q, Wang Y, Kurosu M (2012) A new Oxyma derivative for nonracemizable amide-forming reactions in water. Org. Lett 14, 3372–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wang Y, Aleiwi BA, Wang Q, Kurosu M (2012) Selective esterifications of primary alcohols in a water-containing solvent. Org. Lett 14, 4910–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Gloster TM (2014) Advances in understanding glycosyltransferases from a structural perspective. Curr. Opin. Struct. Biol 28, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Grzegorzewicz AM; Ma F; Jones V; Crick D; Liav A; McNeil MR (2008) Microbiology 154, 3724–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Mitachi K, Siricilla S, Yang D, Kong Y, Skorupinska-Tudek K, Swiezewska E, Franzblau SG, Kurosu M (2016) Fluorescence-based assay for polyprenyl phosphate-GlcNAc-1-phosphate transferase (WecA) and identification of novel antimycobacterial WecA inhibitors. Anal. Biochem 512, 78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Stachyra T, Dini C, Ferrari P, Bouhss A, van Heijenoort J, Mengin-Lecreulx D, Blanot D, Biton J, Le Beller D (2004) Fluorescence detection-based functional assay for high-throughput screening for MraY. Antimicrob. Agents Chemother 48, 897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Chandrakala B, Shandil RK, Mehra U, Ravishankar S, Kaur P, Usha V, Joe B, deSousa SM (2004) High-throughput screen for inhibitors of transglycosylase and/or transpeptidase activities of Escherichia coli penicillin binding protein 1b. Antimicrob. Agents Chemother 48, 30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Somner EA, Reynolds PE (1990) Inhibition of peptidoglycan biosynthesis by ramoplanin. Antibicrob. Agents Chemother 34, 413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).McCafferty DG, Cudic P, Frankel BA, Barkallah S, Kruger RG, Wenkai L (2002) Chemistry and biology of the ramoplanin family of peptide antibiotics. Biopolymers 66, 261–284. [DOI] [PubMed] [Google Scholar]
- (41).Hu Y, Helm JS, Chen L, Ye X-Y, Walker S (2003) Ramoplanin inhibits bacterial transglycosylases by binding as a dimer to lipid II. J. Am. Chem. Soc 125, 8736–8737. [DOI] [PubMed] [Google Scholar]
- (42).Hu Y, Helm JS, Chen L, Ginsberg C, Gross B, Kraybill B, Tiyanont K, Fang X, Wu T, Walker S (2004) Identification of selective inhibitors for the glycosyltransferase MurG via high-throughput screening. Chem. Biol 11, 703–711. [DOI] [PubMed] [Google Scholar]
- (43).A promising antibacterial MurG inhibitor. Mann PA, Muller A, Xiao L, Pereira PM, Yang C, Ho LS, Wang H, Trzeciak J, Schneeweis J, dos Santos MM, Murgolo N S. Xinwei; Gill C, Balibar CJ, Labroli M, Su J, Flattery A, Sherborne B, Maier R, Tan CM, Black T, Onder K, Kargman S, Monsma FJ, Pinho MG, Schneider T, Roemer T (2013) Murgocil is a highly bioactive Staphylococcal-specific inhibitor of the peptidoglycan glycosyltransferase enzyme MurG. ACS Chem. Biol 8, 2442–2451. [DOI] [PubMed] [Google Scholar]
- (44).Mitachi K, Kurosu SM, Eslamimehr S, Lemieux MR, Ishizaki Y, Clemons WM, Kurosu M (2019) Semisynthesis of an anticancer DPAGT1 inhibitor from a muraymycin biosynthetic intermediate. Org. Lett 21, 876–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Mitachi K, Yun H-G, Kurosu SM, Eslamimehr S, Lemieux MR, Klaić L, Clemons WM, Kurosu M (2018) Novel FR-900493 analogues that inhibit the outgrowth of Clostridium difficile spores. ACS Omega, 3, 1726–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Kurosu M, Li K, Crick DC (2009) Concise synthesis of capuramycin. Org. Lett 11, 2393–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Wang Y, Siricilla S, Aleiwi BA, Kurosu M (2013) Improved synthesis of capuramycin and its analogues. Chem. Eur. J 19, 13847–13858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Siricilla S, Mitachi K, Wan B, Franzblau SG, Kurosu M (2015) Discovery of a capuramycin analog that kills nonreplicating Mycobacterium tuberculosis and its synergistic effects with translocase I inhibitors. J. Antibiot 68, 271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Groves P, Searle MS, Chicarelli-Robinson I, Williams DH, Dudley H (1994) Recognition of the cell-wall binding site of the vancomycin-group antibiotics by unnatural structural motifs: 1H NMR studies of the effects of ligand binding on antibiotic dimerization. J. Chem. Soc. Perkin Trans. 1: Org. Bio-Org. Chem 6, 659–665. [Google Scholar]
- (50).Rao J, Lahiri J, Weis RM, Whitesides GM (2000) Design, synthesis, and characterization of a high-affinity trivalent system derived from vancomycin and L-Lys-D-Ala-D-Ala. J. Am. Chem. Soc 122, 2698–2710. [Google Scholar]
- (51).McComas CC, Crowley BM, Boger DL (2003) Partitioning the loss in vancomycin binding affinity for D-Ala-D-Lac into lost H-bond and repulsive lone pair contributions. J. Am. Chem. Soc 125, 9314–9315. [DOI] [PubMed] [Google Scholar]
- (52).Crane CM, Boger DL (2009) Synthesis and evaluation of vancomycin aglycon analogues that bear modifications in the N-terminal D-leucyl amino acid. J. Med. Chem 52, 1471–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Jia Z, O’Mara ML, Zuegg J, Cooper MA, Mark AE (2013) Vancomycin: ligand recognition, dimerization and super-complex formation. FEBS J 280, 1294–1307. [DOI] [PubMed] [Google Scholar]
- (54).Nahoum V, Spector S, Loll PJ (2009) Structure of ristocetin A in complex with a bacterial cell-wall mimetic. Acta Cryst. D 65, 832–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Cheng M, Ziora ZM, Hansford KA, Blaskovich MA, Butler MS, Cooper MA (2014) Anti-cooperative ligand binding and dimerisation in the glycopeptide antibiotic dalbavancin. Org. Biomol. Chem 12, 2568–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J. Mol. Biol 215, 403–410. [DOI] [PubMed] [Google Scholar]
- (57).Ha S, Walker D, Shi Y, Walker S (2000) The 1.9 A crystal structure of Escherichia coli MurG, a membrane-associated glycosyltransferase involved in peptidoglycan biosynthesis. Protein Sci 9, 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, deBeer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).PyMOL: An Open-Source Molecular Graphics Tool. DeLano WL, see. https://www.ccp4.ac.uk/newsletters/newsletter40/11_pymol.pdf.
- (60).Robert X, Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42, W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Tommaso PD Moretti S, Xenarios I, Orobitg M, Montanyola A, Chang J-M, Taly J-F, Notredame C (2011) T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res 39, W13–W17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Sztaricskai F, Pintér G, Röth E, Herczegh P, Kardos S, Rozgonyi F, Boda Z (2007) N-Glycosylthioureido aglyco-ristocetins without platelet aggregation activity. J. Antibiot 60, 529–533. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.