

Abstract
We investigated the role of nitric oxide synthase (NOS) in mediating blood-brain barrier (BBB) disruption and peripheral immune cell infiltration in the cerebellum following blast exposure. Repetitive, but not single blast exposure, induced delayed-onset BBB disruption (72 hours post-blast) in cerebellum. The NOS inhibitor N(G)-nitro-L-arginine methyl ester (L-NAME) administered after blast blocked BBB disruption and prevented CD4+ T-cell infiltration into cerebellum. L-NAME also blocked blast-induced increases in intercellular adhesion molecule-1 (ICAM-1), a molecule that plays a critical role in regulating blood-to-brain immune cell trafficking. Blocking NOS-mediated BBB dysfunction during this acute/subacute post-blast interval (24–71 hours after the last blast) also prevented sensorimotor impairment on a rotarod task 30 days later, long after L-NAME cleared the body. In postmortem brains from Veterans/military Servicemembers with blast-related TBI, we found marked Purkinje cell dendritic arbor structural abnormalities, which were comparable to neuropathologic findings in the blast-exposed mice. Taken collectively, these results indicate that blast provokes delayed-onset of NOS-dependent pathogenic cascades that can later emerge as behavioral dysfunction. These results also further implicate the cerebellum as a brain region vulnerable to blast-induced mTBI.
Subject terms: Neuroscience, Blood-brain barrier, Diseases of the nervous system, Motor control, Neuroimmunology, Somatosensory system, Diseases, Trauma, Neurology, Brain injuries
Introduction
Blast-induced mild traumatic brain injury (mTBI) is prevalent among military personnel and Veterans who have served in Operation Enduring Freedom/Operation Iraqi Freedom/Operation New Dawn (OEF/OIF/OND). Between 2000–2018, more than 380,000 OEF/OIF/OND Veterans have been diagnosed with TBI, which is considered the signature injury of these wars1.
The risk of dementia more than doubles among military Veterans with TBI, but the mechanisms mediating this increased risk remain poorly understood2. Neurovascular dysfunction, including blood-brain barrier (BBB) disruption, may be an important initiating mechanism by which mTBI sets in motion pathogenic cascades leading to chronic neurodegenerative pathophysiology3–19. BBB disruption has been examined in a number of pre-clinical models of blast mTBI10,11,20–25. We and others have shown that blast exposure causes transient BBB disruption8,10,26, suggesting that blast also stimulates short-term neurovascular repair processes that interact with more protracted, co-occurring, pathological cascades that undermine BBB integrity. In this regard, blast has been shown to provoke delayed-onset BBB disruption, days after an initial BBB opening was restored to normal8. This delayed-onset BBB dysintegrity occurs in specific brain regions and is prominent in the hippocampus and the cerebellum8,10,27.
Multiple clinical studies show the cerebellum is vulnerable to the effects of blast mTBI11,28–32. Moreover, biophysical simulations suggest a specific vulnerability of the human cerebellum to blast exposure33,34. Pre-clinical models of blast-induced mTBI have revealed persistent cerebellar white matter injury11,35,36. The cerebellum is classically appreciated for its role in sensorimotor integration, which is affected by blast and other forms of neurotrauma11,37–41. However, it is increasingly clear that the cerebellum also plays important roles in modulating higher order cognitive and behavioral functions that are prominently affected in individuals with blast-related mTBI30,42,43. The translational significance of the cerebellum, coupled with its vulnerability to blast-induced mTBI, prompted us to focus on better understanding the mechanisms of BBB disruption in the cerebellum.
In this study, we sought to determine whether cerebellar blast-induced BBB disruption is NOS-dependent and to address whether delayed onset BBB disruption and co-occurring leukocyte infiltration are associated with cerebellar pathophysiology and sensorimotor function.
Results
Repetitive blast exposure increases BBB permeability to radiolabeled albumin
Using established methods8,10,11,14,44–46, male C57BL/6 mice were exposed to one or three blast overpressures (BOPs) using a pneumatic shock tube delivering a peak static pressure of 20.19 psi (±0.29), positive phase duration of 5.86 ms (±0.065), and impulse of 0.038 psi*ms (±0.0016) (Fig. 1).
Figure 1.
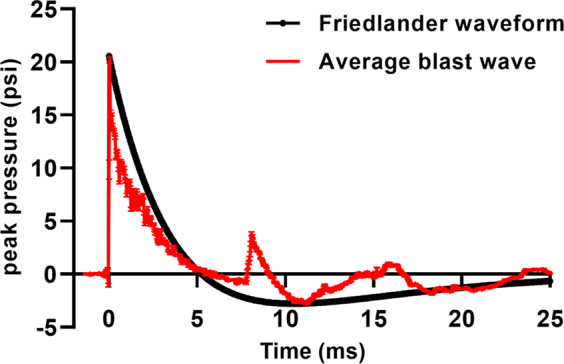
Blast overpressures generated with a well-established pneumatic shock tube simulate a Friedlander waveform expected from high explosives. Red trace denotes the mean waveform of 24 blasts sampled throughout the experiments comprising this report. Black trace shows an estimated Friedlander waveform expected from detonation of approximately 21 kg TNT detonated at a distance of 8 m in the open field. Error bars denote SEM.
Under normal conditions, only very low levels of blood-borne albumin cross the intact BBB into the CNS47–49. We have previously reported that repetitive (2X), but not single (1X) blast causes delayed-onset (72 h) BBB permeability to albumin in whole brain8. For the experiments in this report, we used a 3X blast paradigm (one per day for three days) because it better corresponds with commonly used preclinical exposure regimens and is in keeping with our previous translational mTBI findings11. The results in Fig. 2 confirmed that in whole brain 3X, but not 1X blast, significantly increased delayed-onset 99mTc-albumin BBB disruption (Fig. 2; F(2,25) = 6.94; p ≤ 0.01). We have previously shown that this same 3X exposure regimen caused acute BBB disruption, which was closely associated with later emerging chronic neuron loss in the cerebellum11. Thus, for this study we focused attention on the effects of 3X blast exposure on BBB disruption in the cerebellum.
Figure 2.
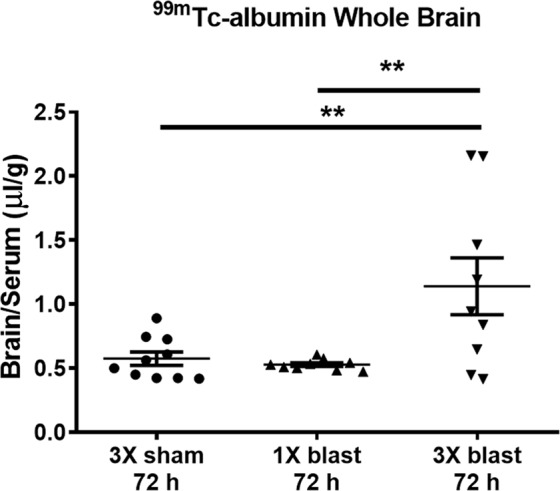
Repetitive blast exposure increases BBB permeability to radiolabeled albumin. A significant increase (**p ≤ 0.01) in brain/serum (uμl/g) ratios of 99mTc-albumin was observed in whole brain at 72 h after repetitive mTBI (3X blast; n = 12) compared to 1X blast (n = 12), and sham control mice (n = 12). One-way ANOVA post-hoc Newman-Keuls. Values represent means ± SEM and are expressed as microliters per gram of brain tissue. Brain/serum ratios were calculated by dividing the cpm per brain by the cpm per microliter in the corresponding serum and then by the weight of the brain.
Nitric oxide inhibition blocks albumin permeability in the cerebellum following repetitive blast
Nitric oxide (NO) signaling is known to regulate BBB permeability50–52. In keeping with this, we have reported that nitric oxide synthase (NOS) inhibition attenuates single and double blast-induced delayed-onset BBB disruption8. To address whether 3X blast-induced BBB disruption in the cerebellum is similarly regulated by NOS, we measured uptake of blood-borne 99mTc-albumin 72 h after the final exposure in mice injected with the pan-specific NOS inhibitor, N(G)-nitro-L-arginine methyl ester (L-NAME). We found that 3X blast significantly increased delayed-onset BBB disruption in cerebellum (Fig. 3a; F(2,30) = 4.23; p ≤ 0.05), which was significantly ameliorated by L-NAME treatment (Fig. 3a; p ≤ 0.05). In contrast to the cerebellum, 3X blast under these specific experimental conditions did not increase BBB permeability to blood-borne radiolabeled albumin in forebrain structures that excluded the hindbrain (i.e., posterior midbrain, pons, medulla, and cerebellum) (Fig. 3b; F(2, 30) = 0.073; n.s.).
Figure 3.

Nitric oxide synthase inhibition blocks blast-induced delayed-onset BBB disruption in cerebellum. (a) 3X blast significantly increased brain/serum ratios of 99mTc-albumin in the cerebellum (*p ≤ 0.05), which was significantly attenuated by L-NAME administration (10 mg/kg; ip at 48, 54, and 71 h after the last blast) (*p ≤ 0.05). (b) No differences were observed in brain/serum ratios of 99mTc-albumin in the forebrain after repetitive mTBI (p > 0.05). One-way ANOVA post-hoc Newman-Keuls. Values represent means ± SEM. Timeline portrays the mTBI exposure, L-NAME treatment paradigm, and measurement of BBB permeability.
Nitric oxide synthase inhibition blocks blast-induced CD4+ T-cell infiltration in the cerebellum
The results above further support the idea that the cerebellum is particularly vulnerable to blast-induced BBB dysfunction and that delayed-onset BBB disruption is mediated (at least in part) by NOS-dependent signaling cascades. In addition, there is evidence that NOS activity underlies T-cell infiltration into the CNS53,54, corresponds with BBB breakdown, and occurs within a temporal window consistent with the delayed-onset BBB disruption we observed following blast8,55,56. This prompted us to ask: (i) whether blast increases immune cell infiltration in cerebellum, (ii) whether this occurs in a NOS-dependent fashion, and (iii) if blast-induced immune cell infiltration follows the same brain region-specific pattern (i.e., cerebellum versus forebrain) as BBB disruption.
To test these questions, we employed flow cytometry to quantify CD4+ T-cell (CD45+/CD3+/CD8−) and CD8+ T-cell (CD45+/CD3+/CD4−) infiltration into cerebellum 72 h after 3X blast exposure (see Methods, Fig. 4, and Supplemental Fig. 1). Blast significantly increased CD4+ infiltration into the cerebellum (Fig. 4a; F(2,24) = 5.56; p ≤ 0.01; scatter plots Fig. 4c-e). Neuman-Keuls post-hoc analyses adjusting for multiple comparisons confirmed that L-NAME treatment blocked blast-induced CD4+ T-cell infiltration (blast + vehicle versus blast + L-NAME, p ≤ 0.027), reducing infiltration to levels comparable to sham + L-NAME controls. An additional a priori planned comparison Helmert analysis further confirmed that CD4+ T-cell infiltration into the cerebellum was significantly greater in the blast + vehicle treated group than in the blast + L-NAME and sham control groups (p ≤ 0.004); and that the blast + L-NAME group was statistically equivalent to shams (n.s.). In contrast to cerebellum, blast exposure did not increase CD4+ T-cell infiltration into the forebrain of the same animals (Fig. 4f; F(2,24) = 0.331, n.s.; scatter plots Fig. 4h-j). Blast exposure did not affect infiltration of CD8+ T-cells into either the cerebellum (Fig. 4b; F(2,24) = 0.16, n.s.) or forebrain (Fig. 4g; F(2,24) = 0.69, n.s.). We also measured peripheral monocyte (CD11b+, CD45 high) infiltration, which was not significantly affected by blast exposure in either the cerebellum (F(2,24) = 0.081, n.s.) or forebrain (F(2,24) = 0.902, n.s.).
Figure 4.

Nitric oxide inhibition attenuates CD4+ T-cell infiltration in the cerebellum following repetitive blast. (a) Flow cytometry revealed that repetitive TBI significantly increased CD4+ counts in the cerebellum (**p ≤ 0.01), which was significantly attenuated by L-NAME administration (*p ≤ 0.05). (b) No differences were measured in cerebellar CD8+ counts after repetitive TBI (p > 0.05). (c) Scatter plots for cerebellum of shams, (d) blast vehicle-treated, and (e) blast L-NAME-treated mice. (f) No differences in CD4+ counts (p > 0.05), or (g) CD8+ counts were measured in forebrain after repetitive mTBI (p > 0.05). (h) Scatter plots for forebrain of shams, (i) blast + vehicle-treated, and (j) blast + L-NAME-treated mice. One-way ANOVA post-hoc Newman-Keuls. Values represent means ± SEM.
These results demonstrate that repetitive blast exposure induces brain region-specific, NOS-dependent, CD4+ T-cell infiltration that corresponds to the differential effects of blast on BBB integrity in the cerebellum versus forebrain regions. These results, coupled with reports that NOS signaling regulates expression of Intercellular Adhesion Molecule-1 (ICAM-1)66, which is known to play a critical role in T-cell transit across the BBB57,58, raised the question whether blast increases cerebellar ICAM-1 expression in a NOS-dependent fashion.
Repetitive blast exposure specifically increases cerebellar ICAM-1, but not VCAM-1 expression in a NOS-dependent fashion
Brain endothelial cell-expressed ICAM-1 plays a critical role in mediating T-cell infiltration into the brain57. Thus, we investigated whether blast-induced changes in ICAM-1 could play a role mediating the T-cell results above. Western blot analysis revealed a significant difference in ICAM-1 protein expression in the cerebellum at 72 h after 3X TBI (Fig. 5; F(2,15) = 7.66; p ≤ 0.01). Moreover, L-NAME administration after blast significantly attenuated ICAM-1 protein expression (Fig. 5a; p ≤ 0.05). VCAM-1 is another endothelial membrane protein involved in T-cell tethering59. In contrast to the ICAM-1 findings, blast did not alter cerebellar VCAM-1 levels, neither was it affected by L-NAME treatment. We found no significant differences in VCAM-1 protein expression after repetitive blast (Fig. 5b; F(2,15) = 0.93; p > 0.05).
Figure 5.

Repetitive blast significantly increased ICAM-1, but not VCAM-1 expression in cerebellum. (a) Western blot analysis revealed that repetitive mTBI significantly increased ICAM-1 expression in cerebellum at 72 h (**p ≤ 0.01), and that L-NAME significantly attenuated this effect (*p ≤ 0.05). (b) No significant differences were observed in VCAM-1 expression in cerebellum at 72 h after repetitive mTBI (p > 0.05). One-way ANOVA post-hoc Newman-Keuls. Values represent means ± SEM.
The increase in ICAM-1 expression corresponds with the observed T-cell infiltration in the cerebellum. Complementing this finding and in keeping with the functional BBB results in Figs. 2 and 3, we found that L-NAME attenuated blast-induced ICAM-1 co-localized with the endothelial cell marker, glucose transporter 1 (GLUT1)60 on microvessels in the cerebellum (Supplementary Fig. 4; F(2,9) = 10.93; p ≤ 0.01). Taken together, these data suggest that repetitive TBI increases cerebellar ICAM-1 expression in a NOS-dependent fashion to regulate endothelial T-cell entry into the CNS.
L-NAME prevents blast-induced sensorimotor impairment
In cerebella of mice exposed to repetitive TBI, L-NAME prevented BBB disruption (Fig. 3a) and CD4+ T-cell infiltration 72 h post-blast (Fig. 4a). To test whether this intervention during the acute/subacute post-blast interval prevents sensorimotor performance deficits, we examined rotarod performance 30 days later. Sensorimotor performance was measured by latency to fall (Fig. 6). Sham and 3X blasted mice received L-NAME or vehicle injections 48, 54, and 71 h after the last blast. We first confirmed that there were no significant effects of L-NAME treatment among sham controls (F(1,19) = 3.05, n.s.). Thus, vehicle- and L-NAME-treated shams were pooled for comparisons to 3X blast vehicle- and 3X blast L-NAME-treated animals. A two-way mixed (between-within) repeated measures ANOVA revealed a significant overall difference between the sham, blast-vehicle, and blast-L-NAME groups (F(2,34) = 3.31; p ≤ 0.045). Further post-hoc paired comparison tests confirmed that blast L-NAME-treated mice performed significantly better than blast vehicle-treated (p ≤ 0.02) and sham controls (p ≤ 0.03). Because L-NAME improved sensorimotor function in mice exposed to TBI, we investigated whether L-NAME treatment influenced the degree of blast-induced neuropathology.
Figure 6.
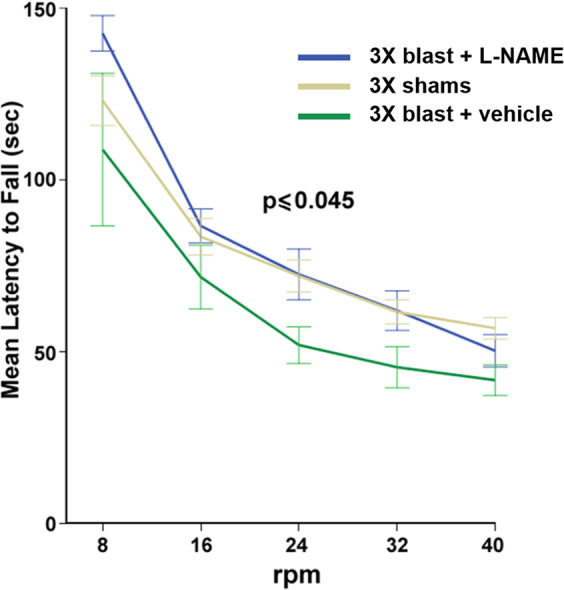
L-NAME improves sensorimotor impairment in mice exposed to repetitive blast. 3X blast and sham mice received L-NAME mice. In shams, L-NAME treatment had no significant effect on performance and were thus pooled (sham-vehicle n = 10, and sham L-NAME-treated n = 11). There was a significant overall difference (p ≤ 0.045) between shams, blast vehicle-treated (n = 14), and blast L-NAME-treated (n = 14) mice. Mice exposed to repetitive mTBI one month prior exhibited impaired sensorimotor performance on a rotarod task (p ≤ 0.05) compared to sham controls (pooled mice, mice administered L-NAME). Post-hoc analysis revealed blast L-NAME-treated mice performed significantly better than blast vehicle-treated mice (p ≤ 0.02). Two-way ANOVA Helmert’s test. Values represent means ± SEM.
Repetitive blast causes dystrophic Purkinje cell arbor EAAT4 expression
Using the same blast exposure regimen employed in this report, we have previously found significant Purkinje cell loss occurring in patchy domains consistent with anatomical sub-domains of transient BBB disruption11. Following up on this, we examined the effects L-NAME treatment on Purkinje cell arbor structure following 3X blast exposure. To accomplish this, we performed confocal microscopy on cerebella obtained 30 days post-3X blast and immunostained them for EAAT4. EAAT4 is a neuronal glutamate transporter expressed exclusively in the plasma membranes of mature Purkinje cell bodies and their dense dendritic arbors that ramify throughout the cerebellar molecular layer61,62.
Secondary only fluorescent-labeled antibodies were included to validate and support EAAT4 (Fig. 7a), and calbindin (Fig. 7b) morphological specificity (merged Fig. 7c; zoomed Fig. 7d). EAAT4 immunostaining in sham control mice revealed normal-appearing, dense Purkinje cell arbors (Fig. 7e). Normal Purkinje cell morphology was also observed in sham control mice, as evidenced by calbindin immunostaining (Fig. 7f) in lobule IX (merged Fig. 7g; zoomed Fig. 7h). One month after 3X blast exposure, reduced EAAT4 (Fig. 7i) and calbindin (Fig. 7j) immunoreactivity was observed in lobule IX (merged Fig. 7k; zoomed Fig. 7l). This is in keeping with our previous reports of persistent Purkinje cell body pathology, particularly in lobule IX11. Of note, L-NAME administration did not rescue the blast-induced decreases in EAAT4 (Fig. 7m) or calbindin immunoreactivity (Fig. 7n) in lobule IX (merged Fig. 7o; zoomed Fig. 7p).
Figure 7.

Reduced EAAT4 expression on cerebellar Purkinje cells was observed in mice exposed to repetitive blast injury. (a) Shows representative images of secondary only controls (no primary antibodies) for Alexa 488, (b) Alexa Cy3, (c) merged, and (d) zoomed (40×). (e) Shows representative immunofluorescent images of EAAT4 (green), (f) calbindin (red), (g) merged, and (h) zoomed images in lobule IX of the cerebellum in control mice at one month after 3X sham and L-NAME administration. (i) Shows representative images of EAAT4, (j) calbindin, (k) merged, and (l) zoomed images at one month after repetitive mTBI. (m) Shows representative images of EAAT4, (n) calbindin, (o) merged, and (p) zoomed images at one month after repetitive mTBI with L-NAME. A significant decrease in (q) EAAT4 (*p ≤ 0.05), and (r) calbindin (*p ≤ 0.05) immunofluorescence was observed in lobule IX of the cerebellum of mice exposed to both repetitive mTBI plus vehicle (*p ≤ 0.05), and repetitive mTBI plus L-NAME (*p ≤ 0.05). One-way ANOVA post hoc Newman-Keuls. Values represent means ± SEM. Arrowheads highlight EAAT4+/calbindin+ Purkinje cell bodies. Scale bars = 30 µm, 20 µm (zoomed).
Quantification revealed that repetitive blast mTBI significantly reduced the expression of EAAT4 (Fig. 7q; F(2,9) = 5.25; p ≤ 0.05), and calbindin (Fig. 7r; F(2,9) = 6.93; p ≤ 0.05). These results confirm that repetitive TBI causes persistent cerebellar pathology, which may be associated with persistent sensorimotor impairment. Although L-NAME prevented blast-induced sensorimotor deficits, this treatment failed to prevent overt blast-induced cerebellar Purkinje cell neuropathology. This suggests NOS inhibition may possess beneficial effects on persistent functional outcomes after repetitive TBI that are not reflected by gross Purkinje cell loss under these experimental conditions. Nonetheless, this does not rule out the possibility that properly timed NOS inhibition could still be beneficial, or at least delay development of persistent neuropathology in the cerebellum.
Cerebella from Veterans with blast-related TBI display Purkinje cell dysmorphology
We and others have shown a number of functional and structural neuroimaging abnormalities in Veterans with blast-related mTBI11,28,32,63–68. However, currently there is little neuropathological information regarding blast-related mTBI in the cerebellum in humans. In order to gain initial insights into the possible translational significance of the cerebellar pathology in blast exposed mice, we examined cerebella from a rare set of four postmortem brains from US military Servicemembers and Veterans with blast-related mTBI and two comparable non-TBI veteran controls.
Control1 was a male Navy officer with no known TBIs who died at 43 due to cardiac arrest (likely myocardial infarction). Medical history included hypertension, hypercholesterolemia, and chronic obstructive pulmonary disease. There were no significant neuropathological abnormalities, including no evidence of tauopathy or neurodegeneration. Control2 was a male Army veteran who died at age 57 due to cardiac arrest. He had no lifetime history of TBI. However, he reported falling down the stairs and bumping his head twice at the ages of 4 and 7 years without loss of consciousness or requiring any urgent care or emergency room evaluation. Medical/psychiatric diagnoses included headaches and remote history of a “mental breakdown.” TBI1 was a male who died at age 31 of undetermined causes. He had known exposure to high explosives and had a history of PTSD and reported memory and concentration impairment, headaches, anxiety, and depression. Neuropatholgical findings were interface astroglial scarring (IAS), and two tau foci. TBI2 was a 46-year-old male Navy SEAL who died from a self-inflicted gunshot wound. Military blast exposure included multiple improvised explosive devices (IEDs) and other explosive ordnance. Contact sports history consisted of high school, college, semi-professional football, and mixed martial arts. Medical/psychiatric history included diagnoses of PTSD, alcohol use disorder, hearing loss, and chronic pain. Neuropathologic findings were CTE (Stage 1) and IAS. TBI3 was a male Navy SEAL who died at age 35 by drowning. Military blast exposure included multiple IEDs and other explosive ordnance. There was no history of participation in contact sports. Medical/psychiatric diagnoses included PTSD, alcohol/substance use disorder, chronic pain, paranoid ideation, and manic episodes. Neuropathological findings were IAS without tau pathology. TBI4 was a male Army veteran who died at age 46 following complications of elective back surgery. This case had previously participated in a study of blast mTBI and had extensive pre-mortem clinical characterization. Military blast exposure consisted of >50 blast mTBIs due to IEDs and other explosive ordnance with acute symptoms consistent with VA/DoD criteria for mTBI and no history of contact sports or non-blast TBIs. Medical/psychiatric diagnoses included PTSD, alcohol use disorder, migraine headaches, chronic back pain, hearing loss, and tinnitus. Neuropathologic diagnoses included CTE (Stage 1) and meningitis.
In keeping with the mouse neuropathologic findings (Fig. 8) and our previous results in 3X blast exposed mice also examined 30 days post treatment11, we observed patchy domains of cerebellar Purkinje cell loss (Fig. 8) in the cases with blast-related mTBI (red calbindin staining). Accompanying this, we observed striking patchy domains of EAAT4+ arbor loss in the molecular layer (green). In some instances, these arbors were not lost (evidenced by the calbindin immunostaining), yet still displayed a marked decrease in EAAT4, a molecule that is critical in regulating glutamate uptake and glutamatergic signaling in these cells. Overall, these results support the translational significance of the neuropathology we and others have reported in blast-exposed animals11,14,69,70, and suggest that the vast network of highly arborized Purkinje cells, which are the sole output of the cerebellum, may be chronically disrupted by blast-related mTBI.
Figure 8.

Dystrophic EAAT4 expression in cerebellar Purkinje cells in human with blast-related mTBI. Confocal microscopy shows representative immunofluorescent images of EAAT4 (green), and calbindin (red) in the cerebella of Veteran/military Servicemember controls with no blast TBI (a–c and d–f, Controls1–2, respectively described in Results), and in the cerebellum of Veteran/military Servicemembers with blast-related mTBI (g–i, j–l, m–o, and p–r, TBIs1–4, respectively as described in Results) Scale bars = 50 µm.
Discussion
We have found that repetitive mTBI causes delayed-onset BBB disruption in the cerebellum and was markedly more robust than in forebrain structures of blast-exposed mice. Biphasic BBB disruption has been observed in other rodent models of neurotrauma71,72. This dynamic property of BBB injury is potentially important because latent BBB disruption has been proposed as a target for post-injury TBI intervention8. In this report, we found that the cerebellum was more susceptible to BBB dysfunction than the forebrain. This finding is well in keeping with our previous results showing that the cerebellum is vulnerable to blast-induced BBB disruption8, subsequent Purkinje cell loss11, and is also in keeping with pioneering findings in other neurotrauma animal models that show cerebellum is prone to injury even when the initiating insult is focused elsewhere in the brain40,41.
Previously it has been thought that the CNS and the peripheral immune system are functionally independent of one another. This view has more recently undergone important revisions based on studies that demonstrated active interactions between immune cells in the periphery and in the CNS73. In this study, CD4+ T-cell infiltration was observed in the cerebellum. Leukocyte infiltration has been considered a functional surrogate for BBB dysfunction, which has been observed in the injured CNS after more severe impact TBI74. Recently there has been a report that blast can induce CCL2+ monocytes to cross the BBB9. The results herein demonstrate that CD4+ T-cells infiltrate the cerebellum following repetitive blast-induced mTBI.
A growing body of evidence indicates that neurotrauma can provoke peripheral immune cells to infiltrate the brain56. While there is gross histological evidence indicating that leukocytes enter the CNS after blast exposure75,76, the effects of blast on immune cell brain infiltration have thus far received limited attention and there has been no information regarding potential mechanisms or functional consequences. One mechanism by which leukocytes and, in particular, T-cells infiltrate the brain involve changes in ICAM-1 expression57. We found that ICAM-1 expression was both increased in cerebellum after blast mTBI and was reduced by NOS inhibition. Increased expression of ICAM-1 has also been observed in human endothelial cells exposed to inflammatory cytokines77. In addition, NOS signaling modulates ICAM-1 expression and function53. Excessive inflammation can cause microvascular hyper-permeability through NOS-dependent mechanisms78. Interestingly however, inhibition of constitutive NOS signaling promotes leukocyte adhesion79. Blast exposed mice exhibit excessive NOS signaling80 and NOS-dependent BBB dysfunction8. The data in this report shed new light on the NOS-dependent mechanisms of delayed leukocyte infiltration associated with BBB dysfunction.
In keeping with our previous work on delayed-onset BBB disruption8, these results speak further to the potential significance of the acute/subacute post-injury interval in shaping long term outcomes. Even though L-NAME and it’s active NOS inhibiting metabolite L-NG-nitro arginine (LNOARG) have short half-lives on the order of 20 minutes to 22 hours, respectively81, we found that L-NAME treatment from 48–71 hours post-blast prevented impaired sensorimotor performance tested 30 days after the last blast exposure.
Changes in NOS signaling have been observed in other pre-clinical brain injury models82,83 and have been implicated in disease progression after TBI84,85. A current clinical trial using inhaled nitric oxide is underway as a means to improve TBI outcome (NCT03260569 clinicaltrails.gov). The potential relationships between peripheral NO administration, the timing of such treatment post injury, and other factors make it very difficult to draw precise correspondences between the findings in this report, Nonetheless, this effort in the clinical arena lends additional support to the idea NO/NOS signaling pathways are relevant targets for further investigation in treating mTBI. NOS inhibition has also been shown to improve sensorimotor function38 and reduce neuronal cell death86 in preclinical models of TBI. Our results suggest that acute/subacute NOS inhibition does not block the full spectrum of cerebellar neuropathology. It is possible that this specific intervention may only delay the sequelae of blast mTBI. Nonetheless, it is possible that long-term strategies to modulate NOS function could offer additional neuroprotection; or at least even further delay onset and progression of mTBI-related neurotrauma.
Currently there is very little information from postmortem human cerebellum after blast exposure, primarily because available tissue from such cases is extremely rare. In the cerebella from four Veterans and military Servicemembers with histories of blast-related TBI, we found marked dystrophic Purkinje cell arbor morphology and loss of EAAT4 expression, which strengthens the potential translational significance of the neuropathologic findings we and others have observed in animal models of blast mTBI.
The vulnerability of the cerebellum to mTBI is not well understood. It is likely due to a number of interacting factors that include: (i) high metabolic demand due to the very large number of neurons (69 billion of the 86 billion total neurons in adult human brain are in cerebellum); (ii) a distinct microvascular structure, and (iii) its anatomical location with respect to the skull87–89. In considering these factors, BBB disruption and/or immune cell infiltration may still represent key interacting pathologic processes that drive persistent cerebellar dysfunction and neuropathology.
Veterans with mild TBI exhibit aberrant cerebellar function30,31,90,91. BBB dysfunction in the cerebellum could set in motion chronic low-grade inflammation associated with NOS signaling that could eventually result in long-term neurodegeneration. Overall, our results argue that NOS plays an important role in mediating cerebellar responses to mild blast injury and suggest that properly timed interventions blocking NO signaling delay pathophysiological progression. Not only do these complex interactions play important roles in mediating delayed BBB dysfunction, but they may also play a critical role in mTBI neurodegenerative progression.
Methods
Animals
Male C57BL/6 (n = 148) mice (Jackson Laboratories) 8–12 weeks of age were studied. Mice had ad libitum access to food, water, and were group housed. Animals were randomly assigned to sham, drug, or TBI groups. All studies were approved by the Veterans Affairs Puget Sound Health Care System’s Institutional Animal Care and Use Committee. Experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and reported in compliance with the ARRIVE guidelines.
Blast exposure
Mice were acclimatized to the animal facility for at least one week prior to experimentation. For blast exposure, animals were first anesthetized with 5% isoflurane, followed by maintenance with 2% isoflurane (1 L/min oxygen). Mice were mounted in a restraint harness in the shock tube with their ventral body surface facing the oncoming shock wave in a pneumatic shock tube as previously described10,11,44. Sham control animals were mounted in the restraint harness and held under anesthesia for the same amount of time as the blast-exposed mice. Mice that survived blast exposure (146/148) all displayed normal ambulation, visually guided grasping responses when elevated by the tail near a foothold, and grooming behavior within 1 hour after exposure.
Radiolabeled tracer preparation
Following established procedures50, albumin (Sigma, St. Louis MO) was labeled with 99mTc (GE Healthcare, Piscataway, NJ). A mixture of 240 mg/ml stannous tartrate and 1 mg/ml albumin was adjusted to pH 3.0 with HCl. One millicurie of 99mTc-NaOH4 was added to this mixture and allowed to incubate for 20 min. The 99mTc-albumin was purified on a column of G-10 Sephadex (GE Healthcare) in 0.1 ml fractions of phosphate buffer (0.25 M). Radioactivity in the purified 99mTc-albumin peak was more than 90% acid precipitable in an equal volume of 1% bovine serum albumin (BSA) and trichloroacetic acid (30%). 5 × 106 cpm/mouse of purified 99mTc-albumin fraction was prepared in a final volume (0.2 ml/mouse) of lactated Ringer’s solution containing 1% BSA.
Radiolabeled tracer injections
Following established procedures50, at 72 h after the final blast/sham treatment, mice were anesthetized with urethane (4 g/kg; 0.2 ml; ip); jugular veins were exposed, and injected with 99mTc-albumin (5 × 106 Counts per minute (cpm)) in 0.2 ml of lactated Ringer’s solution with 1% BSA for 10 min. Blood was collected from a cut in the descending abdominal aorta. Vascular space of the brain was then washed free of blood by opening the thorax, clamping the descending thoracic aorta, severing both jugular veins, and perfusing 20 ml of lactated Ringer’s solution through the left ventricle of the heart in less than 1 min. After washout, brain was removed. The cerebellum and forebrain structures (remaining brain regions excluding posterior midbrain, pons, medulla, and cerebellum) were dissected and individually weighed. Brains with visible blood after washout were excluded from analysis (n = 2). Serum was obtained by centrifuging the carotid artery blood for 10 min at 3,200 × g at 4 °C. Levels of 99mTc radioactivity in the serum and brain regions were determined in a gamma counter. Brain tissue radioactivity was calculated by dividing the cpm in the brain region by the weight of the brain region to yield cpm/g. Serum radioactivity was calculated by dividing the cpm in the serum by the microliters of serum counted to yield cpm/microliter. The brain tissue radioactivity was then divided by the corresponding serum radioactivity and the results given in units of microliters/gram of brain tissue.
N(G)-nitro-L-arginine methyl ester treatment
For all studies in this report, L-NAME treatment indicates that the mice received three intraperitoneal injections of N(G)-nitro-L-arginine methyl ester (L-NAME; Sigma; 10 mg/kg; 100 µl 7% NaHCO3; ip) at 48, 54, and 71 h after the last blast. For these experiments, shams received identical L-NAME injections along with their respective blast cohorts. We employed this 3X dosing paradigm based on the plasma half-life of L-NAME81. Vehicle controls received three 100 µl injections of 7% NaHCO3.
Rotarod
A rotarod apparatus (San Diego Instruments, San Diego, CA) was used to measure motor coordination and balance. Five trials are used, each with a different speed of rotation (8, 16, 24, 32, and 40 rpm). For each trial the rod was accelerated to its final speed over 120 s, remained at the final speed for an additional 30 s, and finally decreased back to 0 rpm over 30 s. 2 min intertrial intervals were used. The latency to fall of the rotarod was recorded for each trial.
Confocal microscopy
Using previously established methods8, brains were post-fixed in 4% paraformaldehyde in PBS at 4° C for ten 99mTc-albumin half-lives (~60 h). Hemisections were equilibrated in 30% sucrose in PBS for 24 h at 4 °C and embedded in OCT (Tissue-Tek, Torrance, CA). Antigen retrieval was performed with 50 mM sodium citrate (pH 9.0) and then heating at 80 °C for 30 min. Immunostaining was performed as previously described14. Floating tissue sections were cover slipped with a drop of Prolong Gold Antifade Reagent. The following antibodies were used: rabbit polyclonal anti-EAAT4 (Abcam, Cambridge, MA; 1:1,000), mouse monoclonal calbindin (Abcam; 1:1,000), rabbit polyclonal anti-GLUT1 (Millipore, Billerica, MA; 1:500), and goat polyclonal anti-ICAM-1 (Novus Biologics, Centennial, CO; 1:1,000). Corresponding secondary antibodies labeled with Alexa 488, and Cy3 were applied for 2 h (Jackson Immunoresearch, West Grove, PA; 1:1,000). Confocal microscopy was performed with a Leica TCS SP5 II microscope. Microscopic images were acquired with the Leica Application Suite and processed using image adjustments limited only to linear contrast and brightness adjustments applied identically to data from blast- and sham-treated animals in each experiment.
Flow cytometry
At 72 h after last blast or after time-matched sham procedures, mice were anesthetized with urethane (4 g/kg; 0.2 ml; ip), and transcardially perfused with ice-cold PBS. Brains were extracted and dissected into cerebellum and forebrain. Brain regions were mashed on ice in PBS. Homogenates were centrifuged at 300 × g for 5 min at 4o C. Pellets were resuspended in enzyme dissociation buffer (Tumor Dissociation kit, Miltenyi Biotec, Auburn, CA), triturated with a 22-gauge needle, and rocked at 37 oC. Digested brain suspension were then passed through a 70 μm SmartStrainer and rinsed thoroughly with 5 ml HBSS/FBS/EDTA. The collected suspension was centrifuged at 300 × g. Pellets were resuspended in 5 ml of 40% Percoll (GE Healthcare, Pittsburgh, PA, USA) and centrifuged at 400 × g for 20 min at 25 oC. The myelin layer (top) and Percoll supernatant were aspirated and the pellets were washed with 5 ml HBSS/FBS/EDTA and centrifuged at 300 × g for 10 min at 25 oC. Supernatant was aspirated and pellet was resuspended in 100 μl PBS and transferred to 1.5 ml centrifuge tubes.
Aliquots of each cell suspension were pooled for use as controls for staining. One aliquot from the pool was reserved as an unstained control; a second was heat-treated for 10 min at 55 °C. All cell suspensions, including the pool and the heat-killed aliquot (but not the unstained control) were incubated with an equal volume of viability dye (Ghost Dye510, Tonbo Biosciences, San Diego, CA) diluted 1:500 in PBS for 30 min at 4 °C. Labeling was terminated by the addition of 5 volumes HBSS/FBS/EDTA, and centrifuging at 300 × g for 5 min. Cells were resuspended in HBSS/FBS/EDTA containing 5 μg/ml mouse Fc Block (ThermoFisher Grand Island, NY) for 15 min on ice before staining with a panel of fluorophore-conjugated primary antibodies (Supplementary Table 1). Cells were incubated with antibodies on ice for 30 min, covered, then washed and resuspended in 250 μl 1% paraformaldehyde in PBS, and stored at 4 °C until analyzed. Data were acquired on an LSRFortessa SORP using FACSDiva 8.0 (BD Biosciences, San Jose, CA, USA) and analyzed using FlowJo (Tree Star, San Carlos, CA, USA). A standardized gating strategy was used for infiltrating T-cells (Supplementary Fig. 1). Fluorescence-minus-one (FMO) controls were used to determine antibody positivity, and compensation controls were performed by adding 1 drop of UltraComp eBeads (ThermoFisher Invitrogen).
Immunoblotting
Using previously established methods8, brains were flash frozen in liquid nitrogen and stored at −80 °C. Dissected tissue was sonicated in radioimmunoprecipitation assay (RIPA) buffer with protease/phosphatase inhibitors (Thermo; Rockford, IL 1:100), centrifuged for 15 min at 12,000 × g, supernatant collected, and protein determined with a bicinchoninic acid (BCA) assay. Protein lysates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (20 µg total protein/lane) under reducing conditions, transferred to nitrocellulose, and probed with goat polyclonal anti-ICAM-1 (Novus Biologics, 1:1,000), rabbit monoclonal anti-VCAM-1 (Abcam, 1:2000), and rabbit monoclonal β-actin (Cell signaling, Danvers, MA; 1:10,000) as loading controls. Blots were probed with anti-goat and anti-rabbit horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch; 1:5,000) and visualized by enhanced chemiluminescence (GE Healthcare, Piscataway, NJ). Optical densities of the immunoreactive protein bands were quantified using Image-Quant with background subtraction and normalization to loading control (β-actin). Full immunoblots can be found in Supplementary Fig. 2 (ICAM-1), and Supplementary Fig. 3 (VCAM-1).
Human tissue
Collection of all brain specimens and case characteristics information were conducted in accordance with approved University of Washington and Uniformed Services University Institutional Review Board (IRB) procedures allowing for use of de-identified tissue samples. Tissue samples were obtained from autopsies of blast-exposed Veterans and age, sex-matched, non-exposed controls. Brain sections were stained using a commercially available kit (Opal Manual IHC Kit, Akoya Bioscience). The following antibodies were applied overnight at 4 °C: rabbit polyclonal anti-EAAT4 (Abcam; 1:1,000), and mouse monoclonal calbindin (Millipore; 1:1,000). Heat-mediated antigen retrieval was performed in AR6 buffer. Stained slides were mounted with ProLong Diamond antifade mountant (ThermoFisher). Confocal microscopy was performed as described above.
Statistical analysis
Statistical analyses used GraphPad Prism 8.0 8 (GraphPad Software, Inc., La Jolla, CA) and SPSS software (IBM Corp, Armonk, NY). Error bars represent standard error of the mean (SEM). One-way analysis of variance (ANOVA) were followed by Newman-Keuls, post-hoc tests. Two-way ANOVAs were followed by a Helmert test92,93.
Notification
The opinions expressed herein are those of the authors and are not necessarily representative of those of the Uniformed Services University, the United States Department of Defense or the United States Army, Navy, or Air Force.
Supplementary information
Acknowledgements
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the VA Biomedical Laboratory Research and Development Service from the U.S. Department of Veterans Affairs I01BX002311(DGC); Veterans Affairs Rehabilitation Research and Development Service #B77421 (ERP); University of Washington Friends of Alzheimer’s Research (DGC, ERP); VA Puget Sound Health Care System Seed Grant (JSM); and NIH National Institute on Aging projects R01AG046619 (WAB) and T32AG052354 (AFL); Alzheimer’s Disease Research Center P50AG005136 (CDK, DAM); Nancy and Buster Alvord Endowment (CDK): VA Biomedical Laboratory Research and Development Service from the U.S. Department of Veterans Affairs IK2 BX003258-01A1 (AGS).
Author contributions
Study design (AFL, AGS, JSM, WAB, DGC.); conducted experiments (AFL, AGS, JSM, MJH, MY); analyzed data (AFL, AGS, MJH, MY, DGC); wrote manuscript (AFL, AGS, JSM, MJH, MY, WAB, DGC); carried out clinical (ERP) and neuropathological evaluations of postmortem specimens (CDK, DPP, DAM); provided critical scientific input in manuscript preparation and interpreting results (AFL, AGS, MJH, ERP, JSM, MAR, WAB, CDK, DPP, DAM, DGC).
Data availability
Authors will provide published data and relevant protocols upon request to the Principal Investigator by phone or email.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-66113-7.
References
- 1.AFHSB. DoD Worldwide Numbers for TBI https:/dvbic.dcoe.mil/dod-worldwide-numbers-tbi (2019).
- 2.Barnes DE, et al. Association of Mild Traumatic Brain Injury With and Without Loss of Consciousness With Dementia in US Military Veterans. JAMA Neurol. 2018;75:1055–1061. doi: 10.1001/jamaneurol.2018.0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janelidze S, et al. Increased blood-brain barrier permeability is associated with dementia and diabetes but not amyloid pathology or APOE genotype. Neurobiol. aging. 2017;51:104–112. doi: 10.1016/j.neurobiolaging.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nation DA, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019;25:270–276. doi: 10.1038/s41591-018-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta. 2016;1862:887–900. doi: 10.1016/j.bbadis.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elder GA, et al. Vascular and inflammatory factors in the pathophysiology of blast-induced brain injury. Front. Neurol. 2015;6:48. doi: 10.3389/fneur.2015.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glushakova OY, Johnson D, Hayes RL. Delayed increases in microvascular pathology after experimental traumatic brain injury are associated with prolonged inflammation, blood-brain barrier disruption, and progressive white matter damage. J. neurotrauma. 2014;31:1180–1193. doi: 10.1089/neu.2013.3080. [DOI] [PubMed] [Google Scholar]
- 8.Logsdon AF, et al. Blast exposure elicits blood-brain barrier disruption and repair mediated by tight junction integrity and nitric oxide dependent processes. Sci. Rep. 2018;8:11344. doi: 10.1038/s41598-018-29341-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuriakose M, Rama Rao KV, Younger D, Chandra N. Temporal and Spatial Effects of Blast Overpressure on Blood-Brain Barrier Permeability in Traumatic Brain Injury. Sci. Rep. 2018;8:8681. doi: 10.1038/s41598-018-26813-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huber BR, et al. Blast exposure causes dynamic microglial/macrophage responses and microdomains of brain microvessel dysfunction. Neuroscience. 2016;319:206–220. doi: 10.1016/j.neuroscience.2016.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meabon JS, et al. Repetitive blast exposure in mice and combat veterans causes persistent cerebellar dysfunction. Sci. Transl. Med. 2016;8:321ra326. doi: 10.1126/scitranslmed.aaa9585. [DOI] [PubMed] [Google Scholar]
- 12.Gama Sosa MA, et al. Low-level blast exposure disrupts gliovascular and neurovascular connections and induces a chronic vascular pathology in rat brain. Acta neuropathologica Commun. 2019;7:6. doi: 10.1186/s40478-018-0647-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abutarboush, R. et al. Exposure to Blast Overpressure Impairs Cerebral Microvascular Responses and Alters Vascular and Astrocytic Structure. Journal of neurotrauma, 10.1089/neu.2019.6423 (2019). [DOI] [PMC free article] [PubMed]
- 14.Huber BR, et al. Blast exposure causes early and persistent aberrant phospho- and cleaved-tau expression in a murine model of mild blast-induced traumatic brain injury. J. Alzheimers Dis. 2013;37:309–323. doi: 10.3233/JAD-130182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kondo A, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature. 2015;523:431–436. doi: 10.1038/nature14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldstein LE, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 2012;4:134ra160. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mez J, et al. Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football. Jama. 2017;318:360–370. doi: 10.1001/jama.2017.8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lucke-Wold BP, et al. Linking traumatic brain injury to chronic traumatic encephalopathy: identification of potential mechanisms leading to neurofibrillary tangle development. J. neurotrauma. 2014;31:1129–1138. doi: 10.1089/neu.2013.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Omalu, B. I. et al. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 57, 128-134; discussion 128–134 (2005). [DOI] [PubMed]
- 20.Shetty AK, Mishra V, Kodali M, Hattiangady B. Blood brain barrier dysfunction and delayed neurological deficits in mild traumatic brain injury induced by blast shock waves. Front. Cell. Neurosci. 2014;8:232. doi: 10.3389/fncel.2014.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Logsdon AF, et al. Altering endoplasmic reticulum stress in a model of blast-induced traumatic brain injury controls cellular fate and ameliorates neuropsychiatric symptoms. Front. Cell. Neurosci. 2014;8:421. doi: 10.3389/fncel.2014.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lucke-Wold BP, et al. Bryostatin-1 Restores Blood Brain Barrier Integrity following Blast-Induced Traumatic Brain Injury. Mol. Neurobiol. 2015;52:1119–1134. doi: 10.1007/s12035-014-8902-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hue CD, et al. Time Course and Size of Blood-Brain Barrier Opening in a Mouse Model of Blast-Induced Traumatic Brain Injury. J. neurotrauma. 2016;33:1202–1211. doi: 10.1089/neu.2015.4067. [DOI] [PubMed] [Google Scholar]
- 24.Garman RH, et al. Blast exposure in rats with body shielding is characterized primarily by diffuse axonal injury. J. neurotrauma. 2011;28:947–959. doi: 10.1089/neu.2010.1540. [DOI] [PubMed] [Google Scholar]
- 25.Yeoh S, Bell ED, Monson KL. Distribution of blood-brain barrier disruption in primary blast injury. Ann. Biomed. Eng. 2013;41:2206–2214. doi: 10.1007/s10439-013-0805-7. [DOI] [PubMed] [Google Scholar]
- 26.Readnower RD, et al. Increase in blood-brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. J. Neurosci. Res. 2010;88:3530–3539. doi: 10.1002/jnr.22510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hall AA, et al. Repeated Low Intensity Blast Exposure Is Associated with Damaged Endothelial Glycocalyx and Downstream Behavioral Deficits. Front. Behav. Neurosci. 2017;11:104. doi: 10.3389/fnbeh.2017.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peskind ER, et al. Cerebrocerebellar hypometabolism associated with repetitive blast exposure mild traumatic brain injury in 12 Iraq war Veterans with persistent post-concussive symptoms. Neuroimage. 2011;54(Suppl 1):S76–82. doi: 10.1016/j.neuroimage.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warden DL, et al. Case report of a soldier with primary blast brain injury. Neuroimage. 2009;47(Suppl 2):T152–153. doi: 10.1016/j.neuroimage.2009.01.060. [DOI] [PubMed] [Google Scholar]
- 30.Petrie EC, et al. Neuroimaging, behavioral, and psychological sequelae of repetitive combined blast/impact mild traumatic brain injury in Iraq and Afghanistan war veterans. J. neurotrauma. 2014;31:425–436. doi: 10.1089/neu.2013.2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jorge RE, et al. White matter abnormalities in veterans with mild traumatic brain injury. Am. J. Psychiatry. 2012;169:1284–1291. doi: 10.1176/appi.ajp.2012.12050600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mac Donald CL, et al. Detection of blast-related traumatic brain injury in U.S. military personnel. N. Engl. J. Med. 2011;364:2091–2100. doi: 10.1056/NEJMoa1008069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taylor PA, Ford CC. Simulation of blast-induced early-time intracranial wave physics leading to traumatic brain injury. J. Biomech. Eng. 2009;131:061007. doi: 10.1115/1.3118765. [DOI] [PubMed] [Google Scholar]
- 34.Sundaramurthy A, et al. Blast-induced biomechanical loading of the rat: an experimental and anatomically accurate computational blast injury model. J. neurotrauma. 2012;29:2352–2364. doi: 10.1089/neu.2012.2413. [DOI] [PubMed] [Google Scholar]
- 35.Bauman RA, et al. An introductory characterization of a combat-casualty-care relevant swine model of closed head injury resulting from exposure to explosive blast. J. neurotrauma. 2009;26:841–860. doi: 10.1089/neu.2009-0898. [DOI] [PubMed] [Google Scholar]
- 36.Badea A, et al. Repeated mild blast exposure in young adult rats results in dynamic and persistent microstructural changes in the brain. Neuroimage Clin. 2018;18:60–73. doi: 10.1016/j.nicl.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peterson TC, Maass WR, Anderson JR, Anderson GD, Hoane MR. A behavioral and histological comparison of fluid percussion injury and controlled cortical impact injury to the rat sensorimotor cortex. Behav. Brain Res. 2015;294:254–263. doi: 10.1016/j.bbr.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wada K, Chatzipanteli K, Busto R, Dietrich WD. Effects of L-NAME and 7-NI on NOS catalytic activity and behavioral outcome after traumatic brain injury in the rat. J. neurotrauma. 1999;16:203–212. doi: 10.1089/neu.1999.16.203. [DOI] [PubMed] [Google Scholar]
- 39.Bhowmick, S., D’Mello, V., Ponery, N. & Abdul-Muneer, P. M. Neurodegeneration and Sensorimotor Deficits in the Mouse Model of Traumatic Brain Injury. Brain Sci8, 10.3390/brainsci8010011 (2018). [DOI] [PMC free article] [PubMed]
- 40.Potts MB, Adwanikar H, Noble-Haeusslein LJ. Models of traumatic cerebellar injury. Cerebellum. 2009;8:211–221. doi: 10.1007/s12311-009-0114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fukuda K, et al. Purkinje cell vulnerability to mild traumatic brain injury. J. neurotrauma. 1996;13:255–266. doi: 10.1089/neu.1996.13.255. [DOI] [PubMed] [Google Scholar]
- 42.Stoodley CJ. The cerebellum and cognition: evidence from functional imaging studies. Cerebellum. 2012;11:352–365. doi: 10.1007/s12311-011-0260-7. [DOI] [PubMed] [Google Scholar]
- 43.Koziol LF, et al. Consensus paper: the cerebellum’s role in movement and cognition. Cerebellum. 2014;13:151–177. doi: 10.1007/s12311-013-0511-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schindler AG, et al. Blast-related disinhibition and risk seeking in mice and combat Veterans: Potential role for dysfunctional phasic dopamine release. Neurobiol. Dis. 2017;106:23–34. doi: 10.1016/j.nbd.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 45.Cao J, et al. ApoE4-associated phospholipid dysregulation contributes to development of Tau hyper-phosphorylation after traumatic brain injury. Sci. Rep. 2017;7:11372. doi: 10.1038/s41598-017-11654-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Del Razo MJ, et al. Computational and in vitro studies of blast-induced blood-brain barrier disruption. Soc. Ind. Appl. Mathematics: J. Sci. Computing. 2016;38:B347–374. [Google Scholar]
- 47.Levin VA, Fenstermacher JD, Patlak CS. Sucrose and inulin space measurements of cerebral cortex in four mammalian species. Am. J. Physiol. 1970;219:1528–1533. doi: 10.1152/ajplegacy.1970.219.5.1528. [DOI] [PubMed] [Google Scholar]
- 48.Blasberg RG, Fenstermacher JD, Patlak CS. Transport of alpha-aminoisobutyric acid across brain capillary and cellular membranes. J. Cereb. blood flow. metabolism: Off. J. Int. Soc. Cereb. Blood Flow. Metab. 1983;3:8–32. doi: 10.1038/jcbfm.1983.2. [DOI] [PubMed] [Google Scholar]
- 49.Patlak CS, Blasberg RG, Fenstermacher JD. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J. Cereb. blood flow. metabolism: Off. J. Int. Soc. Cereb. Blood Flow. Metab. 1983;3:1–7. doi: 10.1038/jcbfm.1983.1. [DOI] [PubMed] [Google Scholar]
- 50.Banks WA, et al. Lipopolysaccharide-induced blood-brain barrier disruption: roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J. neuroinflammation. 2015;12:223. doi: 10.1186/s12974-015-0434-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banks WA, et al. Nitric oxide isoenzymes regulate lipopolysaccharide-enhanced insulin transport across the blood-brain barrier. Endocrinology. 2008;149:1514–1523. doi: 10.1210/en.2007-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xaio H, Banks WA, Niehoff ML, Morley JE. Effect of LPS on the permeability of the blood-brain barrier to insulin. Brain Res. 2001;896:36–42. doi: 10.1016/S0006-8993(00)03247-9. [DOI] [PubMed] [Google Scholar]
- 53.Xu S, Zhou X, Yuan D, Xu Y, He P. Caveolin-1 scaffolding domain promotes leukocyte adhesion by reduced basal endothelial nitric oxide-mediated ICAM-1 phosphorylation in rat mesenteric venules. Am. J. Physiol. Heart Circ. Physiol. 2013;305:H1484–1493. doi: 10.1152/ajpheart.00382.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao F, et al. Reduction of Endothelial Nitric Oxide Increases the Adhesiveness of Constitutive Endothelial Membrane ICAM-1 through Src-Mediated Phosphorylation. Front. Physiol. 2017;8:1124. doi: 10.3389/fphys.2017.01124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu M, Tsirka SE. Endothelial NOS-deficient mice reveal dual roles for nitric oxide during experimental autoimmune encephalomyelitis. Glia. 2009;57:1204–1215. doi: 10.1002/glia.20842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McKee CA, Lukens JR. Emerging Roles for the Immune System in Traumatic. BraInjury. Front. immunology. 2016;7:556. doi: 10.3389/fimmu.2016.00556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Engelhardt B, Conley FK, Butcher EC. Cell adhesion molecules on vessels during inflammation in the mouse central nervous system. J. Neuroimmunol. 1994;51:199–208. doi: 10.1016/0165-5728(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 58.Wong D, Prameya R, Wu V, Dorovini-Zis K, Vincent SR. Nitric oxide reduces T lymphocyte adhesion to human brain microvessel endothelial cells via a cGMP-dependent pathway. Eur. J. pharmacology. 2005;514:91–98. doi: 10.1016/j.ejphar.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 59.Steiner O, et al. Differential roles for endothelial ICAM-1, ICAM-2, and VCAM-1 in shear-resistant T cell arrest, polarization, and directed crawling on blood-brain barrier endothelium. J. Immunol. 2010;185:4846–4855. doi: 10.4049/jimmunol.0903732. [DOI] [PubMed] [Google Scholar]
- 60.Rahner-Welsch S, Vogel J, Kuschinsky W. Regional congruence and divergence of glucose transporters (GLUT1) and capillaries in rat brains. J. Cereb. blood flow. metabolism: Off. J. Int. Soc. Cereb. Blood Flow. Metab. 1995;15:681–686. doi: 10.1038/jcbfm.1995.84. [DOI] [PubMed] [Google Scholar]
- 61.Dehnes Y, et al. The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: a glutamate-gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J. Neurosci. 1998;18:3606–3619. doi: 10.1523/JNEUROSCI.18-10-03606.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature. 1995;375:599–603. doi: 10.1038/375599a0. [DOI] [PubMed] [Google Scholar]
- 63.Liu W, et al. Perfusion deficits in patients with mild traumatic brain injury characterized by dynamic susceptibility contrast MRI. NMR biomedicine. 2013;26:651–663. doi: 10.1002/nbm.2910. [DOI] [PubMed] [Google Scholar]
- 64.Fischer BL, et al. Neural activation during response inhibition differentiates blast from mechanical causes of mild to moderate traumatic brain injury. J. neurotrauma. 2014;31:169–179. doi: 10.1089/neu.2013.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yeh PH, et al. Postconcussional disorder and PTSD symptoms of military-related traumatic brain injury associated with compromised neurocircuitry. Hum. brain Mapp. 2014;35:2652–2673. doi: 10.1002/hbm.22358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Petrie EC, et al. Neuroimaging, Behavioral, and Psychological Sequelae of Repetitive Combined Blast/Impact Mild Traumatic Brain Injury in Iraq and Afghanistan War Veterans. J. neurotrauma. 2014;31:425–436. doi: 10.1089/neu.2013.2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mac Donald C, et al. Cerebellar white matter abnormalities following primary blast injury in US military personnel. PLoS one. 2013;8:e55823. doi: 10.1371/journal.pone.0055823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robinson ME, et al. Positron emission tomography of tau in Iraq and Afghanistan Veterans with blast neurotrauma. Neuroimage Clin. 2019;21:101651. doi: 10.1016/j.nicl.2019.101651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu J, et al. Effect of blast exposure on the brain structure and cognition in Macaca fascicularis. J. neurotrauma. 2012;29:1434–1454. doi: 10.1089/neu.2010.1591. [DOI] [PubMed] [Google Scholar]
- 70.Koliatsos VE, et al. A mouse model of blast injury to brain: initial pathological, neuropathological, and behavioral characterization. J. neuropathology Exp. Neurol. 2011;70:399–416. doi: 10.1097/NEN.0b013e3182189f06. [DOI] [PubMed] [Google Scholar]
- 71.Baldwin SA, Fugaccia I, Brown DR, Brown LV, Scheff SW. Blood-brain barrier breach following cortical contusion in the rat. J. Neurosurg. 1996;85:476–481. doi: 10.3171/jns.1996.85.3.0476. [DOI] [PubMed] [Google Scholar]
- 72.Baskaya MK, Rao AM, Dogan A, Donaldson D, Dempsey RJ. The biphasic opening of the blood-brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci. Lett. 1997;226:33–36. doi: 10.1016/S0304-3940(97)00239-5. [DOI] [PubMed] [Google Scholar]
- 73.Louveau A, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–341. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Braun M, et al. Activation of Myeloid TLR4 Mediates T Lymphocyte Polarization after Traumatic Brain Injury. J. Immunol. 2017;198:3615–3626. doi: 10.4049/jimmunol.1601948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tompkins P, et al. Brain injury: neuro-inflammation, cognitive deficit, and magnetic resonance imaging in a model of blast induced traumatic brain injury. J. neurotrauma. 2013;30:1888–1897. doi: 10.1089/neu.2012.2674. [DOI] [PubMed] [Google Scholar]
- 76.Braun M, et al. White matter damage after traumatic brain injury: A role for damage associated molecular patterns. Biochimica et biophysica acta. Mol. basis Dis. 2017;1863:2614–2626. doi: 10.1016/j.bbadis.2017.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wertheimer SJ, Myers CL, Wallace RW, Parks TP. Intercellular adhesion molecule-1 gene expression in human endothelial cells. Differential regulation by tumor necrosis factor-alpha and phorbol myristate acetate. J. Biol. Chem. 1992;267:12030–12035. [PubMed] [Google Scholar]
- 78.Hatakeyama T, et al. Endothelial nitric oxide synthase regulates microvascular hyperpermeability in vivo. J. Physiol. 2006;574:275–281. doi: 10.1113/jphysiol.2006.108175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc. Natl Acad. Sci. USA. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abdul-Muneer PM, et al. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free. Radic. Biol. Med. 2013;60:282–291. doi: 10.1016/j.freeradbiomed.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Avontuur JA, Buijk SL, Bruining HA. Distribution and metabolism of N(G)-nitro-L-arginine methyl ester in patients with septic shock. Eur. J. Clin. Pharmacol. 1998;54:627–631. doi: 10.1007/s002280050525. [DOI] [PubMed] [Google Scholar]
- 82.Khaldi A, Chiueh CC, Bullock MR, Woodward JJ. The significance of nitric oxide production in the brain after injury. Ann. N. Y. Acad. Sci. 2002;962:53–59. doi: 10.1111/j.1749-6632.2002.tb04055.x. [DOI] [PubMed] [Google Scholar]
- 83.Mishra V, et al. Primary blast causes mild, moderate, severe and lethal TBI with increasing blast overpressures: Experimental rat injury model. Sci. Rep. 2016;6:26992. doi: 10.1038/srep26992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rao VL, Dogan A, Bowen KK, Dempsey RJ. Traumatic injury to rat brain upregulates neuronal nitric oxide synthase expression and L-[3H]nitroarginine binding. J. neurotrauma. 1999;16:865–877. doi: 10.1089/neu.1999.16.865. [DOI] [PubMed] [Google Scholar]
- 85.Tweedie D, et al. Changes in mouse cognition and hippocampal gene expression observed in a mild physical- and blast-traumatic brain injury. Neurobiol. Dis. 2013;54:1–11. doi: 10.1016/j.nbd.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gahm, C. et al. Reduced neuronal injury after treatment with NG-nitro-L-arginine methyl ester (L-NAME) or 2-sulfo-phenyl-N-tert-butyl nitrone (S-PBN) following experimental brain contusion. Neurosurgery 57, 1272–1281; discussion 1272–1281 (2005). [DOI] [PubMed]
- 87.Townsend, M. T., Alay, E., Skotak, M. & Chandra, N. Effect of Tissue Material Properties in Blast Loading: Coupled Experimentation and Finite Element Simulation. Annals of biomedical engineering, 10.1007/s10439-018-02178-w (2018). [DOI] [PubMed]
- 88.Nonaka H, et al. The microvasculature of the human cerebellar meninges. Acta Neuropathol. 2002;104:608–614. doi: 10.1007/s00401-002-0592-y. [DOI] [PubMed] [Google Scholar]
- 89.Ruiz de Almodovar C, Lambrechts D, Mazzone M, Carmeliet P. Role and therapeutic potential of VEGF in the nervous system. Physiol. Rev. 2009;89:607–648. doi: 10.1152/physrev.00031.2008. [DOI] [PubMed] [Google Scholar]
- 90.Macdonald A, Monson CM, Doron-Lamarca S, Resick PA, Palfai TP. Identifying patterns of symptom change during a randomized controlled trial of cognitive processing therapy for military-related posttraumatic stress disorder. J. Trauma. Stress. 2011;24:268–276. doi: 10.1002/jts.20642. [DOI] [PubMed] [Google Scholar]
- 91.MacDonald CL, et al. Functional status after blast-plus-impact complex concussive traumatic brain injury in evacuated United States military personnel. J. neurotrauma. 2014;31:889–898. doi: 10.1089/neu.2013.3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Winer, B. J., Brown, D. R. & Michels, K. M. Statistical Principles in Experimental Design. 3rd edn, (McGraw-Hill Inc. (1991).
- 93.Field, A. Discovering Statistics Using SPSS. 2nd edn, (Sage Publications (2005).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Authors will provide published data and relevant protocols upon request to the Principal Investigator by phone or email.