

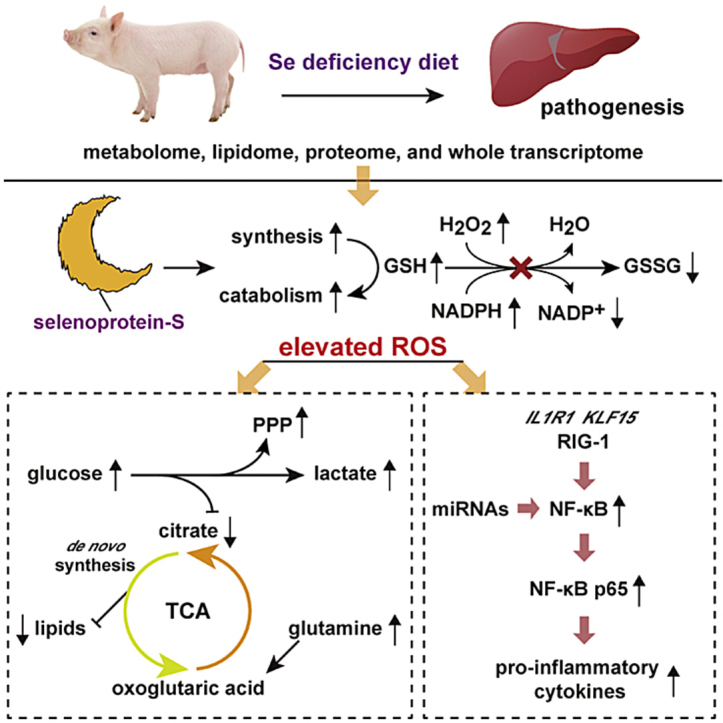
Abstract
Selenium (Se) intake disequilibrium is associated with many human diseases (e.g., Keshan disease and type 2 diabetes). To understand the mechanism of Se deficiency-induced hepatic pathogenesis, a pure line pig model was established by feeding a diet with either 0.07 mg/kg Se or 0.3 mg/kg Se for 16 weeks. The hepatic metabolome, lipidome, global proteome, and whole transcriptome were analyzed. Se deficiency causes a redox imbalance via regulation of selenoproteins at both the mRNA and protein level, and blocks the glutathione anti-oxidant system along with enhanced glutathione synthesis and catabolism. The Warburg effect was observed by enhanced activation of the glycolysis and phosphate pentose pathways. The tricarboxylic acid cycle was dysfunctional since the preliminary metabolites decreased and shifted from using glycolysis origin substrates to a glutamine catabolism-preferred metabolic mode. The reprogrammed central carbon metabolism induced widely restrained lipid synthesis. In addition, a Se deficiency initiated inflammation by activating the NF-κB pathway through multiple mechanisms. These results identified the potential metabolic vulnerability of the liver in response to a Se deficiency-induced redox imbalance and possible therapeutic or intervention targets.
Keywords: Selenium deficiency, Redox imbalance, Metabolic reprogramming, Inflammation, Pig liver
Abbreviations: Se, selenium; SELENO, selenoprotein; GSH, glutathione; GSSG, oxidized glutathione; TRX, thioredoxin; ROS, reactive oxygen species; NADPH, nicotinamide adenine dinucleotide phosphate; NADH, nicotinamide adenine dinucleotide; ATP, adenosine triphosphate; ADP, adenosine diphosphate; AMP, adenosine monophosphate; TCA, tricarboxylic acid cycle; AMPK, AMP-activated protein kinase; NF-κB, nuclear factor κ-light-chain-enhancer’ of activated B-cells; GPX, glutathione peroxidase; Se-D, Se deficient; Se-A, Se adequate; ICP-MS, inductively coupled plasma mass spectrometry; TXNRD, thioredoxin reductase; CAT, catalase; SOD, super oxide dismutase; MDA, malondialdehyde; IL, interleukin; TNF-α, tumor necrosis factor α; TGF-β, transfer growth factor β; PPP, pentose phosphate pathway; ncRNA, non-coding RNA; ceRNA, competing endogenous RNA; PCA, principal component analysis; SEPHS2, selenophosphate synthetase 2; MSRB, methionine-R-sulfoxide; GCLC, glutamate-cysteine ligase catalytic subunit; GST, glutathione S-transferase; ANPEP, aminopeptidase N; SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; Cysthi, l-Cystathionine; PAPSS2, bifunctional 3′-phosphoadenosine5′-phosphosulfate synthase 2; CTH, cystathionase; HIF-1α, hypoxia-inducible factor 1α; Ribu-5P, ribose-5-phosphate; Sed-7p, sedoheptulose 7-phosphate; 2-OG, Oxoglutaric acid; PKLR, pyruvate kinase; TKFC, dihydroxyacetone kinase; PGD, 6-phosphogluconate dehydrogenase; PDK4, pyruvate dehydrogenase kinase isozyme 4; GLUD1, glutamate dehydrogenase 1; FASN, fatty acid synthase; GPAM, glycerol-3-phosphate acyltransferase 1; ACAA2, 3-ketoacyl-CoA thiolase mitochondrial; GK, glycerol kinase; PLPP3, phosphatidate phosphatase 3; DGKZ, diacylglycerol kinase zeta; ACSL5, long-chain-fatty-acid--CoA ligase 5-like; CPT1A, carnitine O-palmitoyltransferase 1; CPT2, carnitine palmitoyltransferase 2; ACOX1, acyl-CoA oxidase 1; FA, fatty acids; TG, triglycerides; PS, phosphatidylserine; PI, phosphatidylinositol; PG, phosphatidylglycerol; DG, diglycerides; PE, phosphatidylethanolamine; PC, phosphatidylcholine; Cer, ceramides; CL, cardiolipin; IL1R1, IL-1β receptor; PLCG2, 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase gamma-2; KLF15, krueppel-like factor 15; RIG-1, antiviral innate immune response receptor RIG-I; IFIT, interferon-induced protein with tetratricopeptide repeats; Mx1, interferon-induced GTP-binding protein Mx1; ISG15, ubiquitin-like protein ISG15
Graphical abstract
Highlights
-
•
Se deficiency elevates liver ROS via selenoprotein down-regulation and blocks GSH system.
-
•
The Warburg effect was observed and the TCA cycle shifted to glutamine catabolism.
-
•
Reprogrammed central carbon metabolism widely restrains hepatic lipid synthesis.
-
•
Se deficiency activates the NF–B signaling pathway through multiple mechanisms.
1. Introduction
Selenium (Se) is considered to represent a living history of how the accumulation of knowledge has extended our understanding of nutrients and human health. Discovered by Berzelius in 1818 [1], Se was initially considered to be a toxic substance. Se deficiency was later identified to be the main cause of Keshan disease in the 19th century and recommended as an essential trace element by the WHO in 1973 [2]. Moreover, Se has recently been shown to exhibit beneficial anti-cancer and pro-longevity effects, as well as the ability to inhibit ferroptosis [[3], [4], [5], [6]]. However, excessive Se was reported to be related to type 2 diabetes, prostate cancer, and mortality [[7], [8], [9]]. Thus, the intake amount is associated with either the protective or toxic effects of Se. Moreover, Se intake is extremely variable across the world, due to a number of different factors, including the Se content in the soil, food habit, and other factors, which enable the effects of both Se deficiency and excess to be observed as a naturally-occurring phenomena [10]. In addition, a Se deficiency is a common public nutrition issue in Europe, Middle East, as well as some Asian countries [[11], [12], [13], [14]]. Thus, the underlying mechanisms of Se on human health is fundamental for tackling Se intake disequilibrium-induced health problems.
Inserting protein synthesis in the form of selenocysteine, Se exerts its biological function primarily through selenoproteins, of which humans and pigs have 25 and mice have 24. There is a range of 1–59 selenoproteins in all three domains of life (most of which are redox enzymes) [15,16]. In response to either a Se deficiency or excess, Se metabolism in an organism exhibits a selenoprotein hierarchy at both the organ and cellular level [17]. As the central organ for Se regulation, the liver produces excretory forms of Se to regulate whole body Se, and responds to dietary Se by decreasing Se excretion and secreting selenoprotein P (SELENOP) into the plasma via regulation of intracellular selenoproteins. SELENOP is distributed to the tissues in proportion to SELENOP receptor expression and is utilized by these organs [18], creating a Se tissue hierarchy. When Se is limited, the cells utilize Se to synthesize the most important selenoproteins, creating a selenoprotein hierarchy within the cell. Collectively, these factors indicate that the hepatic response to dietary Se is critical for the entire body.
Although multiple functions have been reported [19,20], the redox maintain represents the major effects of selenoproteins. These enzymes primarily function through the glutathione (GSH) and thioredoxin (TRX) systems by converting the reactive oxygen species (ROS) produced by energy metabolism to H2O using reduced nicotinamide adenine dinucleotide phosphate (NADPH) [16,21], referred to as intrinsically linked redox and energy metabolism. Energy metabolism, the process of generating adenosine triphosphate (ATP), has long been recognized to be a major source of ROS production during the transfer of electrons from reduced organic molecules to acceptor molecules (e.g., NAD+ and oxygen). ROS and other substrates (e.g., adenosine diphosphate [ADP] and adenosine monophosphate [AMP]), can also affect enzymes associated with energy metabolism. Moreover, ROS can regulate glyceraldehyde-3-phosphate dehydrogenase and tricarboxylic acid (TCA) cycle enzyme activity through various post-transcriptional mechanisms [22]. AMP and ADP can regulate energy metabolism through AMP-activated protein kinase (AMPK) signaling pathways [23]. Despite glycolysis and the TCA cycle, lipid metabolism is also tightly connected to ROS production and can also be regulated by feedback mechanisms [24,25]. The catabolism of triglycerides into fatty acids, which are then oxidized in mitochondria is a substantial contributor of ROS production (i.e., cardiomyocytes). Moreover, lipid metabolism can be feedback regulated by ROS since de novo synthesis lipid substrates (e.g., citrate and acetyl-CoA) involved in central carbon metabolism can be regulated by ROS as described above [24].
Additionally, inflammation is energy-intensive and contributes to the ROS-energy metabolism interaction network. Moreover, ROS can act as stimuli and interact with inflammatory cells at various stages within the signaling pathway. Using nuclear factor κ-light-chain-enhancer of activated B-cells (NF-κB) as an example, the transcription of NF-κB-dependent genes influences the level of ROS. In turn, the level of NF-κB activity is also regulated by the level of ROS [26]. Depending on the context, ROS can both activate and inhibit NF-κB signaling. A high degree of complexity characterizes ROS interactions with the NF-κB signaling pathway since ROS can simultaneously function via multiple mechanisms and at numerous locations [26]. Inflammation and energy metabolism are indirectly and directly linked in multiple ways. Such indirect ways are ROS-mediated as described above, whereas the direct ways are mediated via the metabolic requirement of innate immune cells. While neutrophils are more dependent on glycolysis, monocytes can use both mitochondrial oxidative phosphorylation and glycolysis, whereas lymphocytes are largely reliant on oxidative phosphorylation [27]. Taken together, these factors demonstrate the interplay of ROS-energy metabolism-inflammation. Therefore, this interaction network is crucial to our understanding of the molecular basis of disease pathogenesis [28,29].
Based on the above characteristics, hepatic metabolic disorders and pathology (e.g., elevated oxidative stress and diabetes-like changes, altered ultrastructure, and cirrhosis) have been observed in response to a Se deficiency or excess [[30], [31], [32], [33]]. For Se excess, our preliminary findings have shown that high dietary Se-induced hyperglycemia and hyperinsulinemia are associated with the suppression of sugar metabolism and elevated lipid synthesis in pig livers [34]. These findings are in line with that of other studies which have shown a relationship between high Se intake and type 2 diabetes [8,9,32,35], thereby extending the knowledge of Se excess toxicity and chronic metabolic diseases. For a Se deficiency, it has been shown that dietary Se deficiency decreased hepatic selenoprotein enzyme activity and resulted in oxidative stress [36,37], enhanced glycolysis and modulated one-carbon metabolism [36,38], and activated signaling pathways linked to nutrient sensing [5]. Of note, using a KO mice model and HT-29 cells, it has been shown that glutathione peroxidase 2 (GPx2) and GPx1 have distinct and overlapping functions in controlling inflammatory lipid mediator synthesis [39]. However, how dietary Se deficiency-induced ROS modulates hepatic energy metabolism (i.e., central carbon and lipid metabolism) and inflammation still warrant further investigation and urgently needs to be clarified to understand the molecular basis of Se with chronic metabolic disease. In addition, mice do not appear to be sensitive to a dietary Se deficiency [33]. Moreover, other mammals (e.g., pigs) that have similar selenoprotein profiles to humans and are closely related to humans in terms of their anatomy, genetics, and physiology, represent a more suitable animal model.
In this manner, we created a model using a pure line of pigs and found that a Se deficiency induced pathological changes in the liver. The hepatic metabolome, global proteomics, and the whole transcriptome were all altered. Multi-omics data revealed that a Se deficiency causes a redox imbalance via regulation of selenoproteins and blocks the GSH system, leading to central carbon and lipid metabolism reprograming. Moreover, inflammation is initiated via multiple mechanisms.
2. Materials and methods
2.1. Animal experiment
All animal procedures used in this study were approved by the Animal Care and Use Committee of the Institute of Animal Sciences of Chinese Academy of Agricultural Sciences. The pigs were pure line castrated male Yorkshire pigs (45 days old) and housed in a climate-controlled facility (20°C-25 °C) in ventilated cages (one pig per cage) and given ad libitum access to feed and water.
A total of 24 pigs (two each from the same sow, aged 45-days-old) were divided into two equal groups. Two pigs obtained from the same sow were divided into two different groups, with 12 replicates per group and one pig per replicate. One group was fed a Se deficient (Se-D) diet, composed of corn and soybeans (Supplemental Table 1) produced in the Se-deficient area of Heilongjiang, China, and the final Se content in the diet was 0.007 mg/kg as determined by inductively coupled plasma mass spectrometry (ICP-MS). The other group was fed a Se adequate (Se-A) diet, which was supplemented with SeMet (J&K Chemical, Shanghai, China). The final Se content was 0.3 mg Se/kg (recommended value by NY/Y 65–2004). The trial lasted for 16 weeks, and hair and whole blood samples were collected at 8, 12, and 16 weeks for an analysis of the Se content. At the end of the experiment, after the pigs were fasted for 12 h, blood samples were collected from the precaval vein and centrifuged at 3000 × g for 10 min at 4 °C to achieve plasma separation, and stored at −80 °C. All 24 pigs were anesthetized and exsanguinated, the liver was rapidly removed, washed in ice-cold isotonic saline, minced with surgical scissors, snap-frozen in liquid nitrogen, and stored at −80 °C before further use. The tissues used for pathological examination were fixed in 4% paraformaldehyde for approximately 24 h, dehydrated, embedded in paraffin, and sectioned into slices. The tissue sections were stained with H&E for further microscopic observation.
2.2. Se content, redox parameters, cytokines, and selenoprotein mRNA expression analysis
The hair, whole-blood, and liver Se content were determined by ICP-MS using our previously reported methods [40]. The enzyme activity, including GPXs, thioredoxin reductase (TXNRD), catalase (CAT), and super oxide dismutase (SOD) were measured using commercial kits (Nanjing Jiancheng Bioengineering Institute, Jiangsu, China). The amount of H2O2, NO, and malondialdehyde (MDA) were also measured using commercial kits (Nanjing Jiancheng Bioengineering Institute, Jiangsu, China). Inflammatory molecules, including interleukin-1β (IL-1β), IL-6, IL-8, IL-12, tumor necrosis factor α (TNF-α), transfer growth factor β (TGF- β), NF-κB, and NF-κB p65 were determined using commercial kits (Shanghai Enzyme-linked Biotechnology Co. Ltd. Shanghai, China). Inflammatory parameters in both plasma and liver and redox parameters in liver were normalized by protein concentrations which were measured with a bicinchoninic acid assay.
The total RNA was isolated from liver samples with an RNAprep pure tissue kit (Tiangen Biotech Co. Ltd. Beijing, China) and the RNA purity and integrity were evaluated with agarose gel (1.2%) electrophoresis and the OD260/OD280 value. The cDNA was synthesized from 2 μg of the total RNA with a PrimeScript RT reagent Kit (TaKaRa, Japan), according to the manufacturer's protocol. The expression of selected mRNAs was detected with an Applied Biosystems QuantStudioTM Real-Time PCR system (Thermos Fisher Scientific, USA), with SYBR® Premix Ex TaqTM reagents (TaKaRa, Japan). The primers for selenoprotein and a housekeeping gene (GAPDH) were as previously described [41] and are presented in the Supplemental Table 2. The relative mRNA abundance of the selected genes was normalized to GAPDH expression and was then calculated using the 2−ΔΔCt method.
2.3. Energy metabolism-targeted metabolomic analysis
The hydrophilic energy metabolism metabolites were extracted as follows. The liver samples (about 50 mg) were placed in a 2 mL Eppendorf tube and 500 μL of 80% (v/v) HPLC‐grade methanol (pre-chilled at −80 °C) was added. The mixture was ground for 1–2 min with a tissue grinder on dry ice in the tube, vortexed for 1 min at 4°C-8°C and incubated at −80 °C for 2 h. After centrifugation at 14,000×g for 20 min in a refrigerated centrifuge at 4 °C, and the supernatant was transferred to a new Eppendorf tube and was ready for analysis. The pellet was dissolved and the protein concentration was measured. All of the subsequent responses were normalized to protein levels.
Targeted metabolomic experiments were analyzed with TSQ Quantiva (Thermo, CA). C18-based reverse phase chromatography was used with 10 mM tributylamine and 15 mM acetate in water, and 100% methanol as mobile phases A and B, respectively. This analysis was focused on the TCA cycle, glycolysis, pentose phosphate pathway (PPP), as well as amino acid and purine metabolism. In this experiment, we used a 25-min gradient from 5% to 90% mobile B. The positive-negative ion switching mode was used for data acquisition. The resolution for both Q1 and Q3 was 0.7 FWHM. The source voltage was 3500 V for positive and 2500 V for the negative ion mode. The source parameters were as follows: spray voltage: 3000 V; capillary temperature: 320 °C; heater temperature: 300 °C; sheath gas flow rate: 35; and auxiliary gas flow rate: 10. Metabolite identification was based on a Tracefinder (Thermo Scientific, Version 3.2) search using a home-built database.
2.4. Lipidome analysis
For lipid extraction, approximately 70 mg of sample was homogenized in 700 μL chloroform/methanol (v: v = 2:1) using a superfine homogenizer. An aqueous solution (175 μL) was added for the liquid-liquid extraction. The samples were vortexed for 2 min and incubated at 4 °C for 10 min three times and centrifuged at 3000 rpm for 15 min at 4 °C. The lower chloroform layer was transferred into a new centrifuge tube (1.5 mL, Eppendorf, Wesseling-Berzdorf, Germany) and dried under nitrogen. Lipid samples were stored at −80 °C as dry pellets and redissolved in 200 μL in chloroform/methanol (v: v = 2:1) for further analysis. The pellet was dissolved and measured for protein concentration and all the later responses were normalized to the protein level. Untargeted lipidomics analysis was conducted using a Q-Exactive Orbitrap mass spectrometer coupled to a UHPLC system Ultimate 3000 (Thermo Fisher Scientific, Waltham, MA, USA) according to previously reported methods [42]. Lipid extracts were analyzed using reverse phase chromatography equipped with a Cortecs C18 column (2.1 × 100 mm, Waters, Milford, MA, USA) in the positive and negative mode. HPLC-grade water (400 mL) containing 0.77 g of ammonium acetate was mixed with 600 mL HPLC-grade acetonitrile (pH ~ 7) to generate the mobile phase A. Acetonitrile (100 mL) and isopropanol (900 mL) were mixed to prepare the mobile phase B. The gradient elution procedure was programmed as follows, 0 min, 30% B; 3 min, 30% B; 4.5 min, 33% B; 7 min, 45% B; 8 min, 52% B; 11 min, 58% B; 14 min, 66% B; 17 min, 70% B; 21 min, 75% B; 23 min, 98% B; 30 min, 98% B; 30.5 min, 30% B; and 35 min, 30% B.
The data were acquired by MS/MS through data dependent acquisition. The mass spectrometer parameters consisted of a spray voltage of 3200 V (positive) and 2800 V (negative), a capillary temperature of 320 °C, an auxiliary gas flow rate of 10 Arb, a mass range (m/z) of 240–2000 (positive) and 200–2000 (negative), an NCE of 15/30/45, a topN of 10, a full scan resolution of 70,000, a fragment spectra resolution of 17,500, and a duty cycle of 1.2 s. Lipids were identified and quantified using Lipidsearch (Thermo Fisher Scientific, Version 3.2).
2.5. Global proteomics analysis
Approximately 20 mg of the liver tissue was homogenized in 400 μL buffer (100 mM Tris-HCl, 200 mM NaCl, 4% SDS, pH 8.05) for 1.5 min using a tissue homogenizer and incubated at 95 °C for 5 min. The homogenate was centrifuged at 20,000×g for 20 min and the supernatant was transferred into a new tube and the protein concentration was measured using a bicinchoninic acid assay. Then 100 μg of the samples were reduced by 5 mM tris(2-carboxyethyl) phosphine hydrochloride in the 100 μg system and alkylation by 30 mM chloroacetamide at room temperature for 1 h in the dark. Next, 700 μL pre-cooled acetone was added and placed at −20 °C overnight to precipitate the protein. Then, 50 μL triethylamine borane was added to resuspend the protein and 2 μg trypsin (Progema, Mannheim, Germany) was added to each sample for an overnight digestion at 37 °C. Next, 20 μg peptide from each sample was used for subsequent isotope labelling. TMT 10 plex amine reactive reagents (Thermo Fisher Scientific) were resuspended in 46 μL of anhydrous acetonitrile and added to each sample and mixed briefly using a vortex. Reactions were allowed to proceed at room temperature for 1 h, and then quenched by the addition of 8 μL of 5% hydroxylamine for 15 min. The labeled samples were pooled, freeze-dried, and resuspended in 20 μL of 0.1% formic acid and 2% acetonitrile in water for a fraction. The pooled and labeled peptides were loaded onto a PAK C18 ES column (5 μm, 4.6 × 250 mm, 120 Å) with 10 mM ammonium formate in water as buffer A (pH 10) and 10 mM ammonium formate in acetonitrile as buffer B (pH 10). The peptides were separated using the following gradients: 0–3 min, 5% B; 3–40 min, 60% B; 40–48 min, 80% B; 48–52 min, 80% B; 52–53 min, 5% B; and 53–55 min, 5% B. A total of 36 fractions were collected, dried using a SpeedVac, combined into nine fractions, and resuspended in 0.1% formic acid and 2% acetonitrile for a subsequent nano LC-MS/MS analysis.
A nano LC-MS/MS analysis was performed using an Orbitrap Fusion Tribrid MS (Thermo Scientific, San Jose, CA) equipped with a nanospray flex ion source, and coupled with a Dionex UltiMate 3000 RSLC nano system (Thermo, Sunnyvale, CA). Peptide samples (2 μL) were injected into the PepMap C18 columns (27 μm × 200 mm) at 8 μL/min for on-line enrichment and then separated on a PepMap C18 column (3 μm, 75 μm × 250 mm) with 0.1% formic acid as buffer A and 0.1% formic acid in 80% acetonitrile as buffer B at 300 nL/min. The peptides were eluted with the followed gradients: 0–5 min, 5–12% B; 5–65 min, 12%–38% B; 65–72 min, 38–95% B; 72–80 min, 95% B; 80–81 min, 95–5% B; and 81–95 min, 5% B. The raw data files were searched using Proteome Discoverer (Thermo Fisher Scientific, Version 2.3) against the SuS scrofa protein database from UniProt Proteomes. Mass tolerances for precursor and fragment ions were 0.02 Da and 10 ppm, respectively. The proteins and peptides were filtered with a false discovery rate (FDR) < 1%. The enzyme parameter was limited to semi-tryptic peptides with a maximum miscleavage of 2. Carbamidomethyl (C) of the peptides was set as fixed modifications; oxidation (M) and deamidated (NQ) on the N-terminus of proteins were set as variable modifications. TMT quantification was obtained for the reporter ion intensities per peptide following the manufacturer's instructions.
2.6. Whole-transcriptome analysis
2.6.1. The mRNA and non-coding RNA sequencing and analysis
After the total RNA was extracted, rRNAs were removed to retain mRNAs and non-coding RNAs (ncRNAs). The enriched mRNAs and ncRNAs were fragmented into short fragments using fragmentation buffer and reverse transcribed into cDNA using random primers. Second-strand cDNA were synthesized using DNA polymerase I, RNase H, dNTP (dUTP instead of dTTP), and buffer. Next, the cDNA fragments were purified with a QiaQuick PCR extraction kit, end repaired. The poly(A) was added and ligated to Illumina sequencing adapters. Then UNG (Uracil-N-Glycosylase) was used to digest the second-strand cDNA. The digested products were size selected by agarose gel electrophoresis, PCR amplified, and sequenced using Illumina NovaSeq 6000 platform. After sequencing, the data were subjected to the following preliminary analyses and procedures: filtering of clean reads, alignment with rRNA, alignment with reference genome, transcripts reconstruction, as well as the identification and annotation of novel transcripts. The CNCI (http://www.bioinfo.org/software/cnci) and CPC (http://cpc.cbi.pku.edu.cn/) software was used to assess the protein-coding potential of novel transcripts using default parameters. The level of transcript expression was normalized using the Fragments Per Kilobase of transcript per Million mapped reads (FPKM) method. For circRNA, after filtering the clean reads, rRNA alignment, and alignment with the reference genome. For identification, 20mers from both ends of the unmapped reads were extracted and aligned to the reference genome to identify unique anchor positions within the splice site. Anchor reads aligned in the reversed orientation (head-to-tail) indicated circRNA splicing and were then subjected to find_circ to identify the circRNAs. The anchor alignments were then extended such that the complete aligned reads and the breakpoints were flanked by GU/AG splice sites. Candidate circRNA was termed if it was supported by at least two unique back spliced reads in at least one sample. To quantify the circRNAs, back-spliced junction reads were scaled to reads per million mapped reads (RPM).
2.6.2. Small RNA sequencing and analysis
After the total RNA was extracted using TRIzol, the RNA molecules in a size range of 18 nt–30 nt was enriched by polyacrylamide gel electrophoresis. Then the 3′ adapters were added and the 36 nt–44 nt RNAs were enriched. The 5′ adapters were then also ligated to the RNAs. The ligation products were reverse transcribed by PCR amplification and the 140 bp–160 bp-sized PCR products were enriched to generate a cDNA library and sequenced using an Illumina HiSeq 2500 platform. After sequencing, the data were subjected to the following preliminary analyses and procedures: filtering of clean tags, alignment and identification of small RNA (including existing, known, and novel). The existing small RNA were identified against miRBase (http://www.mirbase.org/, Release 21). All of the unannotated tags were aligned with the reference genome. The novel miRNA candidates were identified according to their genomic positions and hairpin structures predicted by software Mireap (http://sourceforge.net/projects/mireap/ Version 0.2). The level of miRNA expression was calculated and normalized to transcripts per million. For targeted gene prediction, the software, RNAhybrid (v2.1.2) + svm_light (v6.01), Miranda (http://www.microrna.org/microrna/home.do, v3.3a), and TargetScan (http://www.targetscan.org/vert_70/, Version 7.0) were used to predict the various targets. The intersection of the results were selected as the predicted miRNA target genes.
2.7. Data analysis
The data are presented as the mean and standard error of the mean and bar graph were drawn using GraphPad Prism 7.0 (GraphPad Software, Inc. San Diego, USA). The significance of the Se content, redox and inflammatory factors, and the relative selenoprotein mRNA expression obtained from Real-Time PCR was examined with a Student's t-test and p < 0.05 were used to indicate significance. The principal component analysis (PCA) of the metabolome, lipidome, and proteome was performed using Metaboanalyst 4.0 [43]. Hydrophilic and lipidomics metabolite lipids were normalized to the proteins level and analyzed with a Student's t-test, and the significant hydrophilic metabolites (p < 0.05) were imported into Metaboanalyst 4.0 [43] for an enrichment analysis. For the proteomic data, the protein abundance was compared using a Student's t-test, carried out on a log2-transferred ratio, p < 0.05 and fold-change (>1.13 or < 0.87, inter-error was defined as the value of the fold-change at which 90% of all proteins did not deviate from each other) were used as criteria, and the genes of significant proteins were imported into Metascape [44] for an enrichment analysis. The RNA sequencing data were analyzed by R, a fold-change of 1.5 and FDR 0.05 threshold were set for the mRNA sequencing data, and a fold-change of 2.0 and FDR 0.05 threshold were set for ncRNA. The differential gene expression was imported into Metascape [44] for an enrichment analysis. The miRNA-mRNA targeted network was visualized in Cytoscape (http://apps.cytoscape.org/apps/keggscape, Version 3.7.1), and the upregulated and downregulated mRNA were enrichened by KEGG analysis. The competing endogenous RNA (ceRNA) network was constructed based on: 1) expression of the correlation coefficient between mRNA-miRNA or lncRNA-miRNA < −0.7 were selected as negatively co-expressed lncRNA–miRNA or mRNA-miRNA pairs, both mRNA and lncRNA were miRNA target genes, and all RNAs were differentially expressed; 2) the expression correlation coefficient between lncRNA-mRNA > 0.9 was selected as the co-expressed lncRNA-mRNA pairs, and both the mRNA and lncRNA in this pair were targeted and negatively co-expressed with a common miRNA; and 3) a hypergeometric cumulative distribution function test was used to test whether the common miRNA sponges between the two genes were significant, and only the gene pairs with a p-value less than 0.05 were selected.
3. Results
3.1. Se deficiency induces a redox imbalance and inflammation in the blood and causes pathological changes in the liver
The Se hair content was increased and decreased in the Se-A and Se-D group, respectively, both with a time-dependent manner throughout the whole trial. Se in both hair and blood of the Se-D group was significantly lower at all three time points compared with that in the Se-A group (Figure 1A and B). Dietary Se deficiency also decreased the hepatic Se content in the liver (Fig. 1C). This implied decreased Se availability in the animals in the Se-D group. Decreased Se availability weakens the plasma anti-oxidant capacity by decreasing GPXs, SOD, and CAT, and increased the level of NO, H2O2, and the level of the lipid peroxidation product, MDA (Fig. 1D–I). Pro-inflammatory molecules (IL-1β, IL-6, IL-8, NF-κB, NF-κB p65) along with a TGF-β levels in the Se-D group were all significantly higher than those in the Se-A group (Fig. 1J–O). The histological stains showed that a dietary Se deficiency induced hepatic sinus expansion (black arrow), lymphocyte infiltration (blue arrow), and stripe-like hyperplasia (yellow arrow), as shown in Figure 1P. All of these results show that a Se deficiency induces a systematic redox imbalance and inflammation in the blood and causes pathological changes in the liver.
Fig. 1.

Se deficiency induces a redox imbalance and inflammation in the blood and causes pathological changes in the liver. (A–C) A Se deficiency decreases the Se content in the hair, blood, and liver (n = 6). (D–O) Se deficiency induced a blood redox imbalance by decreasing the anti-oxidant capacity, as well as increasing ROS levels and blood pro-inflammatory factors (n = 12). (P) Se deficiency causes hepatic stripe-like hyperplasia (yellow arrow), sinus expansion (black arrow), and lymphocyte infiltration (blue arrow). Data are represented as mean ± SEM. The white and black bar represents the Se adequate (Se-A) and deficient (Se-D) group, respectively. *P < 0.05; **P < 0.01; ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.2. Se deficiency alters the hepatic metabolome, proteome, and whole-transcriptome
To explore the mechanisms related to the phenotype changes, we analyzed the hepatic metabolome, global proteome, and whole-transcriptome. Using an in-house mass spectrometry-based targeted metabolomics approach, 161 hydrophilic metabolites were detected (Supplemental Data 1) and the profiles between the two groups were found to differ (Fig. 2A). A total of 70 hydrophilic metabolites were found to be significantly altered by a Se deficiency (Fig. 2B). These metabolites were involved in the anti-oxidant system (GSH metabolism), central carbon metabolism (e.g., Warburg effect, PPP, transfer of acetyl groups to the mitochondria, mitochondrial electron transport chain, glycolysis, and TCA cycle), one carbon metabolism (e.g., betaine metabolism and methionine metabolism), amino acid metabolism (e.g., glutamate, arginine, and proline metabolism), and glycerolipid metabolism, according to an enrichment analysis using MetaboAnalyst 4.0 (Fig. 2C). Lipidomics detected 1601 and 967 lipid molecules in the positive and negative mode, of which 531 and 434 lipid molecules were confirmed, respectively (Supplemental Data 2 and Supplemental Data 3). The lipid profiles between the two groups all differed (Fig. 2D) and the Se deficiency extensively induced lipid reduction (Fig. 2 E). Together these findings showed that both hydrophilic and lipophilic metabolites are greatly altered by a Se deficiency.
Fig. 2.

Both the hepatic hydrophilic and lipophilic metabolome are affected by Sedeficiency (n = 10). (A and B) The PCA and heatmap show that the hepatic hydrophilic metabolome were affected by a Se deficiency. The red dots and green dots represent the Se-A and Se-D group, respectively. (C) An enrichment analysis of significant metabolites using Metaboanalyst 4.0, and top 50 clusters with their representative enriched terms are presented here, using a discrete color scale to represent statistical significance. (D) PCA shows that the hepatic lipidome was affected by a Se deficiency, the red and green dots represent the Se-A and Se-D group, respectively. (E) The heatmap shows that the hepatic lipids were extensively decreased by a Se deficiency, since the red cells and green cells represent increases and decreases, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Global proteomics identified 2900 proteins (Supplemental Data 4), and the protein profiles were separated according to the treatment (Fig. 3A), and 100 proteins were significantly expressed between the two groups (Fig. 3B and C). The genes of significantly expressed proteins were imported into Metascape [44] for an enrichment analysis. The top nonredundant enrichment clusters were related to cofactor metabolism, lipid metabolism, cellular amino acid metabolism, fatty acid derivative metabolism, biological oxidation, cellular response to interferon-γ, mitotic DNA integrity checkpoint, and others (Fig. 3D). Enrichment networks are created by representing each enriched term as a node and connecting pairs of nodes with Kappa similarities above 0.3, to capture the inter-cluster similarities and intra-clusters redundancies (Fig. 3E). To analyze the protein interactions, a mature complex identification algorithm was applied, and two complexes were automatically identified (Fig. 3E). To link the changes in the metabolome and proteome with those in the transcriptome, the liver was subjected to both mRNA and ncRNA sequencing. PCA showed that the hepatic mRNA profiles were moderately affected (Fig. 4A) and 608 transcripts were significantly altered (Fig. 4B and C). The top non-redundant enrichment clusters showed that the genes involved in central carbon metabolism (e.g., carbohydrate metabolic process, carbon metabolism, pyruvate metabolism, and citric acid cycle), lipid metabolism (fatty acid metabolism, regulation of lipid metabolism, fatty acid metabolism, glycerolipid metabolism, and PPARα-activated gene expression), cofactor metabolism, cellular amino acid metabolism, and others were altered by a Se deficiency (Fig. 4D). For ncRNA, the miRNA profiles were only moderately affected (Figs. 4E) and 82 miRNAs were significantly altered (Fig. 4F and G). The targeted mRNAs of miRNAs were predicted and the miRNA-mRNA target network was constructed based on a Pearson's correlation coefficient < −0.7 (Fig. 4H). The KEGG enrichment analysis showed that for the downregulated mRNA and up-regulated miRNA network was characterized by selenocompound metabolism, cysteine and methionine metabolism, lipid metabolism (e.g., peroxisome proliferator activated receptor signaling pathway, glycerolipid metabolism, glycerophospholipid metabolism, fatty acid degradation, and fatty acid elongation), and others. The upregulated mRNA and downregulated miRNA network is primarily characterized by energy production (carbon metabolism, glycolysis, and the AMPK signaling pathway) and the inflammatory pathway (NF-κB signaling pathway). In addition, a total of 247 lncRNAs and 90 circRNAs were significantly expressed between the two groups. The ceRNAs were constructed using all significant mRNA, lncRNA, circRNA, and miRNA (Supplemental Table 3). Together these data provided comprehensive datasets for understanding how the liver responds to a dietary Se deficiency at the metabolome, global proteome, and whole-transcriptome level.
Fig. 3.

The hepatic global protein expression profiles were altered by a dietary Se deficiency (n = 5). (A) The PCA shows that the global proteome expression profile of the liver from Se-A and Se-D groups differ. The red dots and green dots represent the Se-A and Se-D group, respectively. (B and C) The volcano plot and heatmap represent significant proteins between the Se-A and Se-D groups, the red dots and cells are increased. The blue dots and green cells were decreased. (D) An enrichment analysis of the genes for significant proteins was performed using Metascape. The top 20 clusters with their representative enriched terms were presented here using a discrete color scale to represent a statistical significance. (E) The Metascape enrichment network visualization showed the intra-cluster and inter-cluster similarities of the enriched terms. Cluster annotations are shown in the color code. Metascape visualization of the interactome network was formed by all the genes of significant proteins, in which the Molecular Complex Detection (MCODE) complexes were colored according to their identities. Two MCODE complexes were automatically identified. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Fig. 4.

Transcriptome analysis of the liver from the Se-A and Se-D groups. (A) The PCA shows the global mRNA profiles of the liver from the Se-A and Se-D groups differ. The red dots and green triangles represent the Se-A and Se-D groups, respectively (n = 3). (B and C) The volcano plot and heatmap present the significant mRNAs between the Se-A and Se-D groups. The red dots and cells were significantly increased and the blue dots and green cells are significantly decreased. (D) An enrichment analysis of the significantly expressed genes between the Se-A and Se-D groups using Metascape. The top 20 clusters with their representative enriched terms are presented here using a discrete color scale to represent statistical significance. (E) The PCA of the miRNA profiles in the liver from the Se-A and Se-D groups. The red dots and green triangles represent the Se-A and Se-D group, respectively (n = 5). (F–G) A volcano plot and heatmap presented significant miRNAs between the Se-A and Se-D groups. The red dots and cells were significantly increased and the blue dots and green cells were significantly decreased. (H) Networks describing post-transcriptional regulation in the liver. Dots, mRNA; triangles, miRNA; colors reflect their expression levels. The green and red represent decreased and increased mRNA, yellow and blue represent increased and decreased levels of miRNA. A KEGG pathway analysis was performed on the genes in the two sub-networks, and some of the significant pathways of interest are listed. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.3. Se deficiency causes a redox imbalance by selenoprotein regulation and blocks the GSH system in the liver
To assess the redox state at the hepatic level and understand how a dietary Se deficiency causes a redox imbalance, we performed a targeted analysis and using the data obtained from omics. A dietary Se deficiency decreased the hepatic anti-oxidant capacity by decreasing the GPXs, TXNRD, and CAT, and increased the level of H2O2, NO and MDA (Fig. 5A–F), showed an elevated oxidative stress, a typical metabolic phenotype of Se deficiency which was widely reported [58]. The mRNA expression of three selenoprotein genes (SELENOP, GPX1, and GPX3) was significantly decreased in the RNA sequencing data, and the mRNA expression of 14 selenoprotein genes was significantly decreased, except for one (selenophosphate synthetase 2, SEPHS2), was significantly increased as determined by quantitative real-time PCR (Fig. 5G–I). At the protein level, all four selenoproteins (GPX1, GPX4, TXNRD1, and SELENOF) identified by global proteomics were found to be significantly decreased (Fig. 5J). These data demonstrate the robust data achieved using the different techniques and shows that Se decreases selenoprotein expression at both the mRNA and protein level.
Fig. 5.

Se deficiency induced a hepatic redox imbalance via the regulation of selenoproteins at both the mRNA and protein level and blocks the GSH system. (A–F) A Se deficiency induced a hepatic redox imbalance by decreasing the anti-oxidant capacity and increasing ROS levels (n = 12). (G–I) The mRNA expression of 14 selenoprotein genes were significantly decreased in response to a Se deficiency, while one was significantly increased (SEPHS2) (n = 9). (J) Global proteomics identified five selenoproteins, which were all significantly decreased and in line with the transcription (n = 5). (K, L, M, and N) Targeted metabolomics and RNA sequencing showed that the metabolites and mRNA of the genes involved in the GSH system and related pathways were substantially changed, indicating a blocked GSH system (n = 10 and n = 3 for metabolites and mRNA, respectively). (O) The integrated redox parameters, targeted metabolome, global proteome, and transcriptome revealed that a Se deficiency induced a redox imbalance via regulation of selenoproteins at both the mRNA and protein level and their post-transcriptional regulations, which then blocked the GSH system and increased ROS. The green and red color represents the decreased and increased metabolites or transcripts in the Se-D group, compared with that in the Se-A group. Data are represented as mean ± SEM. The white and black bar represents the Se adequate (Se-A) and deficient (Se-D) group, respectively. *P < 0.05; **P < 0.01; ***P < 0.001). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Since selenoproteins maintain redox homeostasis primarily through the GSH and TRX systems by transferring the reducing power from NADPH to ROS, we picked up metabolites and mRNAs that were significantly changed as obtained from targeted metabolomics and RNA sequencing to illustrate how decreased selenoproteins caused a redox imbalance. The downregulated selenoprotein mRNAs and proteins by Se deficiency decreased GPXs and TXNRD activity, which hindered the GSH system, and resulted in increased substrates, GSH, NAPDH, and ROS, as well as decreased products, oxidized glutathione (GSSG), and NADP+ (Fig. 5K). The blocked GSH system also fed back an enhanced GSH synthesis and catabolism, because the gene expression of the key enzymes, such as glutamate-cysteine ligase catalytic subunit (GCLC), glutathione S-transferase (GST), and aminopeptidase N (ANPEP), were all increased (Figure 5N) and the substrate of GSH synthesis, Gly, choline, Met, Thr, and cystathionine (Cysthi) were decreased, along with increased S-adenosylmethionine (SAM) and decreased S-adenosylhomocysteine (SAH) in response to a Se deficiency (Figure L and M). In addition, two miRNAs (novel-m0064–5p and miR-146-x) targeting SEPHS2 and GCLC were identified. Together these data demonstrate that a Se deficiency down-regulates selenoproteins at both the mRNA and protein level and blocks the GSH system, which results in a redox imbalance, enhanced GSH synthesis and catabolism (Figure 5O).
3.4. Se deficiency-induced redox imbalance leads to hepatic central carbon and lipid metabolic reprogramming
The interplay between ROS and metabolism is crucial to understanding the molecular basis of metabolic diseases and designing therapeutic strategies [28]. Products like ROS and AMP can feedback regulate energy metabolism in multiple pathways. AMP can activate AMPK signaling pathways, which results in increased glycolysis [45]. Moreover, ROS is a physiological activator of AMPK that triggers AMPK-dependent metabolic adaptations specific to mitochondrial homeostasis and control of the Warburg effect through a hypoxia-inducible factor 1α (HIF-1α) dependent pathway [46]. We found that the AMPK signaling pathway was upregulated at the transcriptome level (Fig. 4H), which may be activated by the increased AMP/ATP:ATP ratios (Fig. 6A). We observed enhanced glycolysis, as the initial substrate, glucose, and intermediates, glucose-1-phosphate (glucose-1P), glucose-6-phosphate (glucose-6P), fructose-6-phosphate (fructose-6P), phosphor-dihydroxyacetone (DHAP), and the end products, lactate, were all significantly increased, along with pyruvate kinase (PKLR) and dihydroxyacetone kinase (TKFC) mRNA expression (Fig. 6B, C, and 6F). Activation of PPP was also noted, as the protein expression of the rate-limiting enzyme, 6-phosphogluconate dehydrogenase (PGD), and concentrations of the metabolites, sedoheptulose 7-phosphate (Sed-7P) and ribulose-5-phospahte (Ribu-5P), were all increased (Fig. 6C and F). All results revealed greater NADPH production (a blockade in the GSH system also contributed to the NADPH accumulation). The TCA cycle was reprogrammed as the initial metabolites, citrate, cis-aconitate, and isocitrate, were deceased (Fig. 6D). Oxoglutaric acid (2-OG) was not significantly changed, while glutamine, malic acid, and fumarate were increased, along with up-regulated levels of glutamate dehydrogenase 1 (GLUD1) mRNA (Fig. 6D–F). These results showed that a Se deficiency induced a Warburg effect in the liver, in which glycolysis and PPP were enhanced. The TCA cycle shifted to a preferred metabolic mode of glutamine catabolism.
Fig. 6.

Se deficiency-induced redox imbalance leads to hepatic central carbon and lipid metabolic reprogramming (A) A Se deficiency decreased the level of hepatic ATP, ADP, AMP, and NADH. (B and C) The glycolysis and pentose phosphate pathways were enhanced as the intermediate metabolites and end products were increased. (D and E) The TCA cycle was disturbed as the preliminary metabolites were decreased, and the level of glutamine, fumarate, and malic acid were increased. (F) The mRNA expression of enzymes involved in the above pathways were in line with changes in metabolites. (G, H, K, and L) A Se deficiency decreased the levels of hepatic lipids both at the species (G and H) and lipid molecule levels (K and L), as identified in the mass spectrometry positive (G and K) and negative modes (H and L), and the positive and negative numbers represent the amount of increased and decreased lipid molecules, respectively. (I and J) The relative mRNA expression of genes involved in lipid biosynthesis and degradation. (M) Integration of the targeted metabolome, lipidome, global proteome, and transcriptome revealed that a Se deficiency-induced redox imbalance leads to enhanced glycolysis and PPP, disturbed TCA cycle, and hindered lipid synthesis. The green and red color represents decreased and increased metabolites, proteins, or transcripts in the Se-D group compared with that in the Se-A group. Data are represented as mean ± SEM. The white and black bar represents the Se adequate (Se-A) and deficient (Se-D) group, respectively. *P < 0.05; **P < 0.01; ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Since the central carbon and lipid metabolism are linked, the disruption in central carbon metabolism also affects lipid metabolism. We found that the lipids were extensively decreased in response to a Se deficiency. From the lipid species level, fatty acids (FA), triglycerides (TG), phosphatidylserine (PS), phosphatidylinositol (PI), and phosphatidylglycerol (PG) were all significantly decreased. Other species also had a tendency to decrease (Fig. 6G and H). At the lipid molecule levels, in the positive mode, each of the 89 significantly altered lipids were decreased. The dominate lipids were TG, diglycerides (DG), phosphatidylethanolamine (PE), phosphatidylcholine (PC), PI, and others (Fig. 6K). In the negative mode, 131 of the 135 significantly altered lipids were decreased, which primarily consisted of FA, ceramides (Cer), PC, PE, PI, cardiolipin (CL), PG, and others (Figure 6L). Regarding the expression of lipid metabolism genes, the mRNA expression of lipid degradation genes were decreased (Fig. 6J), and those involved in lipid synthesis were mainly down-regulated, except for fatty acid synthase (FASN) and glycerol-3-phosphate acyltransferase 1 (GPAM) (Fig. 6I). Taken together, these results demonstrated that a Se deficiency-induced redox imbalance led to hepatic energy consumption that was more reliant on glycolysis (Warburg effect). Moreover, PPP was activated and the TCA cycle shifted to a preferred metabolic mode of glutamine catabolism. Lipid synthesis was widely suppressed (Figure 6M).
3.5. Se deficiency-induced redox imbalance initiates liver inflammation via multiple mechanisms
Despite a disruption in energy metabolism, ROS is also an important signal for the initiation of inflammation, which was histopathologically observed as lymphocyte infiltration in the Se-D group. Hepatic pro-inflammatory factors and molecules involved in the inflammatory pathway (IL-1β, IL-6, IL-12, NF-κB, and NF-κB p65) were all significantly increased in the Se-D group (Fig. 7A–F), which indicated that a Se deficiency-induced redox imbalance initiates liver inflammation at the molecular level. For the pathways attributed to the initiation of inflammation, the mRNA expression of IL-1β receptor (IL1R1), and its downstream molecules, 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase gamma-2 (PLCG2), were upregulated (Fig. 7G). Three micRNAs (miR-11631-x, miR-6238-y, and miR-9277-y) which targeted NF-κB were identified. In addition, the transcriptional regulator, krueppel-like factor 15 (KLF15), which could inhibit NF-κB activation, was down-regulated at the mRNA level (Fig. 7G). The target miRNAs were also identified (miR-8970-y, miR-11971-y, miR-2904-x, and ssc-miR-29b). Additionally, six proteins, including antiviral innate immune response receptor RIG-I (RIG-1), interferon-induced protein with tetratricopeptide repeats 1 (IFIT1), FIFIT3, IFIT5, interferon-induced GTP-binding protein Mx1 (Mx1), and ubiquitin-like protein ISG15 (ISG15), were involved in the cytosolic RNA-mediated activation of the NF-κB signaling pathway were all upregulated as identified by global proteomics (Fig. 7H and I). Thus, all four signaling pathways may contribute to the activation of the NF-κB pathway under a Se deficiency-induced redox imbalance, and result in the initiation of inflammation (Fig. 7J).
Fig. 7.

Se deficiency-induced redox imbalance initiates hepatic inflammation in multiple potential pathways. (A–F) A Se deficiency-induced redox imbalance led to increased levels of hepatic pro-inflammatory factors (n = 12). (G, H and I) The level of expression of mRNA and proteins involved in the NF–B pathway and related upstream and downstream pathways were regulated by a Se deficiency-induced redox imbalance, as identified by the global proteome and transcriptome analysis. (J) Illustration of the initiation of Se deficiency-induced hepatic inflammation by multiple potential signaling pathways, including IL1R1, KLF15, RIG-1, and micro RNAs. Green and red represents decreased and increased cytokines, proteins, mRNA, and miRNA, respectively in the Se-D group, compared with that in the Se-A group. Data are represented as mean ± SEM. The white and black bar represents the Se adequate (Se-A) and deficient (Se-D) group, respectively. *P < 0.05; ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
4. Discussion
The report of Se deficiency-induced hepatic pathological changes (necrotic degeneration) in 1950s was a milestone that initiated research into the benefits of Se on health. However, despite the redox maintenance effect, only recently studies have preliminary demonstrated that Se deficiency can modulate liver one-carbon metabolism and glycolysis [36,38]. In this study, our global metabolome, proteome, and transcriptome findings extended the essential physiological perspective on Se biology, and suggested that suboptimal dietary Se induced a systematic redox imbalance. Such suboptimal dietary Se has the unexpected ability to drive both central carbon and lipid metabolic reprogramming, and initiating inflammation via multiple pathways in the liver. Knowledge of how to regulate metabolic disorders and inflammation induced by a redox imbalance using Se has wide intervention implications for multiple human diseases [21,47].
The biological function of Se is primarily processed by insertion into selenoprotein synthesis in the form of selenocysteine. Selenoproteins process anti-oxidant effects primarily via two of the most important systems in an organism: the GSH and TRX system. The decreased activities and expression of selenoproteins, as well as the elevated oxidative stress in response to dietary Se has been widely reported [5,37] and confirmed by our findings. Of note, the selenoprotein mRNA was also regulated by a dietary Se deficiency, as the mRNA of 14 selenoproteins was decreased and one (SEPHS2) was up-regulated, which indicated that dietary Se could modulate selenoproteins at both the mRNA and protein level. Although the impact of dietary Se on selenoproteins synthesis has already been established, the mechanism of selenoprotein mRNA regulated by dietary Se remains unclear. It was recently found that Se induces a protective selenome via activating the transcription factors, TFAP2c and Sp, which prevents ferroptosis and ferroptosis-independent models of cell death [3]. We also predicted some ncRNA that target selenoprotein mRNA, which could be used for further investigation on how Se modulates selenoproteins at the mRNA level. Due to the “bridge”function of GPXs in the anti-oxidant system, a decrease in GPXs resulted in enhanced GSH synthesis and catabolism, which appeared to be a compensatory effect. However, this effect was insufficient to combat the elevated levels of ROS induced by a Se deficiency. Additionally, the dysregulation of the GSH system also altered one-carbon metabolism, since methionine, choline, and SAH were decreased, SAM was increased. These findings may impact the hepatic methylation process, which has been proven elsewhere [38].
ROS are primarily produced from oxidative phosphorylation and can act as a signaling feedback to regulate energy metabolism, and the interaction between ROS and metabolism is related to human disease pathogenesis [48,49]. In this study, we found that a dietary Se deficiency induced oxidative stress to enhance glycolysis, diverted significant amounts of glycolytic intermediates to the PPP for NADPH production. Furthermore, the TCA cycle was found to be dysfunctional since the preliminary metabolites were decreased, whereas the glutamine and backend metabolites (malic acid and fumarate) were increased. All of these findings imply that the hepatic central carbon metabolism shifted to a Warburg mode and glutamine catabolism preferred the TCA cycle metabolism mode, rather than the glycolysis origin substrates during a Se deficiency. The Warburg mode enables the cells to resist metabolic stress [50], and was mainly observed in cancer cells [51]. Regarding whether the Warburg effect was the cause or consequence of carcinogenesis remains controversial, and whether the switch to aerobic glycolysis may represent the very point in time when a normal cell becomes cancerous warrants further investigation [52,53]. Moreover, it has also been shown that efficient mitochondrial oxidative phosphorylation supplies a chronic energy demand primarily for macromolecule synthesis and glycolysis necessary to supply the rapid energy demands of normal cells [54]. Thus, the above results may provide therapeutic or nutritional restoration targets for Se deficiency-induced metabolic diseases. In addition, the level of lipids were substantially decreased in several of the detected species, which may impair membrane function and signaling transduction. The reduction of lipids may attribute to the decreased availability of the de novo synthesis substrate (e.g., acetyl enzyme CoA and citrate) due to a shifted carbon metabolism mode, as they are intrinsically linked [24].
Despite metabolic reprogramming, the initiation of inflammation was also observed in our study, as the NF-κB pathway was activated and pro-inflammatory factors were increased at both the plasma and liver level, which were in line with a recent study, which showed that GPX2 and GPX1 have distinct and overlapping functions in controlling inflammatory lipid mediator synthesis [39]. NF-κB pathways may be activated by multiple mechanisms, as the IL-1R1 and downstream genes were upregulated, with some miRNA that targeted NF-κB. In addition, KLF 15 expression was found to be down-regulated and possibly involved in the activation of the NF-κB pathway, because KLF15 was proven to alter the acetylation status and activity of the proinflammatory factor NF-κB through direct interaction with the histone acetyltransferase, p300, and is critical for vascular inflammation [55]. Of note, auto inflammation or autoimmunity protected the organism against inappropriate immune recognition of self-nucleic acids by cytosolic sensors through metabolizing or processing intracellular DNA or RNA [56]. Such pathways were triggered, as evidenced by the upregulation of several molecules (RIG-1, ISG15, IFITs, and Mx1) that may give rise to the initiation of NF-κB activation and inflammation. Together, these findings suggest that multiple mechanisms may be responsible for activation of the NF-κB pathway and result in Se deficiency-induced hepatic inflammation. Furthermore, investigations to clarify the main upstream activation of the NF-κB pathway on a cellular level would be beneficial, and studies on different ages and elderly organism are also required, since Se metabolism functions in a sex- and age-specific manner [57].
5. Conclusion
Using a pig model, we generated a global picture of how the global metabolome, proteome, and transcriptome were altered in dietary Se deficiency-induced hepatic disease. Multi-omics data revealed that a Se deficiency induced a redox imbalance by regulating selenoproteins at both the mRNA and protein level blocked the GSH system in conjunction with enhanced GSH synthesis and catabolism. Glycolysis was enhanced, revealing a Warburg effect, and significant amount of glycolytic intermediates were diverted into PPP. The TCA cycle shifted to a glutamine catabolism preferred metabolism mode, rather than the glycolysis origin substrates during a Se deficiency. In addition, lipid synthesis was greatly suppressed by the reprogrammed central carbon metabolism. We also propose that a Se deficiency-induced redox imbalance initiated inflammation by activating the NF-κB pathway through multiple mechanisms at an organ level, and further studies on a cell-specific level (e.g., hepatocytes versus endothelial or Kupfer cells) are warranted. Taken together, these results extend the comprehensive knowledge of the essential physiological perspective on Se biology related to liver function, and may provide intervention targets for Se deficiency-induced disease in the future.
Funding
This study was supported by the National Natural Science Foundation of China 31802073, National Key Research and Development Program of China 2018YFD050040001-02/03, and the Agricultural Science and Technology Innovation Program ASTIP-IAS12.
Declaration of competing interest
The authors declare no competing interests.
Acknowledgement
The authors highly acknowledge the excellent technical support by Dr. Xiaohui Liu from Tsinghua University.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101519.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Berzelius J.J. Undersokning af en ny Mineral-kropp, funnen i de orenare sorterna af det i Falun tillverkade svaflet. Afhandlingar i fysik. kemi och mineralogi. 1818;6:42. [Google Scholar]
- 2.Longo DL and Loscalzo J. Keshan disease, selenium deficiency, and the selenoproteome. N. Engl. J. Med.; 370(18): 1756-1760. [DOI] [PubMed]
- 3.Alim I., Caulfield J.T., Chen Y., Swarup V., Geschwind D.H., Ivanova E., Seravalli J., Ai Y., Sansing L.H., Ste, Marie E.J., Hondal R.J., Mukherjee S., Cave J.W., Sagdullaev B.T., Karuppagounder S.S., Ratan R.R. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell. 2019;177(5):1262–1279. doi: 10.1016/j.cell.2019.03.032. e25. [DOI] [PubMed] [Google Scholar]
- 4.Ingold I., Berndt C., Schmitt S., Doll S., Poschmann G., Buday K., Roveri A., Peng X., Porto Freitas F., Seibt T., Mehr L., Aichler M., Walch A., Lamp D., Jastroch M., Miyamoto S., Wurst W., Ursini F., Arnér E.S.J., Fradejas-Villar N., Schweizer U., Zischka H., Friedmann Angeli J.P., Conrad M. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. 2018;172(3):409–422. doi: 10.1016/j.cell.2017.11.048. e21. [DOI] [PubMed] [Google Scholar]
- 5.Yim S.H., Clish C.B., Gladyshev V.N. Selenium deficiency is associated with pro-longevity mechanisms. Cell Rep. 2019;27(9):2785–2797. doi: 10.1016/j.celrep.2019.05.001. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang L., Zeng H.W., Cheng W.H. Beneficial and paradoxical roles of selenium at nutritional levels of intake in healthspan and longevity. Free Radic. Biol. Med. 2018;127:3–13. doi: 10.1016/j.freeradbiomed.2018.05.067. [DOI] [PubMed] [Google Scholar]
- 7.Rayman M.P., Winther K.H., Pastor-Barriuso R., Cold F., Thvilum M., Stranges S., Guallar E., Cold S. Effect of long-term selenium supplementation on mortality: results from a multiple-dose, randomised controlled trial. Free Radic. Biol. Med. 2018;127:46–54. doi: 10.1016/j.freeradbiomed.2018.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Stranges S., Marshall J.R., Natarajan R., Donahue R.P., Trevisan M., Combs G.F., Cappuccio F.P., Ceriello A., Reid M.E. Effects of long-term selenium supplementation on the incidence of type 2 diabetes: a randomized trial. Ann. Intern. Med. 2007;147(4):217–223. doi: 10.7326/0003-4819-147-4-200708210-00175. [DOI] [PubMed] [Google Scholar]
- 9.Zhou J., Huang K., Lei X.G. Selenium and diabetes—evidence from animal studies. Free Radic. Biol. Med. 2013;65:1548–1556. doi: 10.1016/j.freeradbiomed.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rayman M.P. Selenium and human health. Lancet. 2012;379(9822):1256–1268. doi: 10.1016/S0140-6736(11)61452-9. [DOI] [PubMed] [Google Scholar]
- 11.Sun J.W., Shu X.O., Li H.L., Zhang W., Gao J., Zhao L.G., Zheng W., Xiang Y.B. Dietary selenium intake and mortality in two population-based cohort studies of 133 957 Chinese men and women. Publ. Health Nutr. 2016;19(16):2991–2998. doi: 10.1017/S1368980016001130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stoffaneller R., Morse N.L. A review of dietary selenium intake and selenium status in Europe and the Middle East. Nutrients. 2015;7(3):1494–1537. doi: 10.3390/nu7031494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alehagen U., Johansson P., Björnstedt M., Rosén A., Post C., Aaseth J. Relatively high mortality risk in elderly Swedish subjects with low selenium status. Eur. J. Clin. Nutr. 2016;70(1):91–96. doi: 10.1038/ejcn.2015.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang J., Wang N. Peking University Medical Publishing House; 2016. Comprehensive Report on the Monitoring of Nutrition and Health Status of Chinese Residents 2010-2013. [Google Scholar]
- 15.Chen X.D., Zhao Z.P., Zhou J.C., Lei X.G. Evolution, regulation, and function of porcine selenogenome. Free Radic. Biol. Med. 2018;127:116–123. doi: 10.1016/j.freeradbiomed.2018.04.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandes J., Hu X., Smith M.R., Go Y.M., Jones D.P. Selenium at the redox interface of the genome, metabolome and exposome. Free Radic. Biol. Med. 2018;127:215–227. doi: 10.1016/j.freeradbiomed.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burk R.F., Hill K.E. Regulation of selenium metabolism and transport. Annu. Rev. Nutr. 2015;35(35):109. doi: 10.1146/annurev-nutr-071714-034250. [DOI] [PubMed] [Google Scholar]
- 18.Hill K.E., Wu S., Motley A.K., Stevenson T.D., Winfrey V.P., Capecchi M.R., Atkins J.F., Burk R.F. Production of selenoprotein P (Sepp1) by hepatocytes is central to selenium homeostasis. J. Biol. Chem. 2012;287(48):40414–40424. doi: 10.1074/jbc.M112.421404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Speckmann B., Grune T. Epigenetic effects of selenium and their implications for health. Epigenetics. 2015;10(3):179–190. doi: 10.1080/15592294.2015.1013792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hatfield D., Berry M., Gladyshev V. 2012. Selenium: its Molecular Biology and Role in Human Health. [Google Scholar]
- 21.Ren X., Zou L., Zhang X., Branco V., Wang J., Carvalho C., Holmgren A., Lu J. Redox signaling mediated by thioredoxin and glutathione systems in the central nervous system. Antioxidants Redox Signal. 2017;27(13):989–1010. doi: 10.1089/ars.2016.6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Reest J., Lilla S., Zheng L., Zanivan S., Gottlieb E. Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat. Commun. 2018;9(1):1581. doi: 10.1038/s41467-018-04003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shao D., Oka S., Liu T., Zhai P., Ago T., Sciarretta S., Li H., Sadoshima J. A redox-dependent mechanism for regulation of AMPK activation by thioredoxin1 during energy starvation. Cell Metabol. 2014;19(2):232–245. doi: 10.1016/j.cmet.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Currie E., Schulze A., Zechner R., Walther T.C., Farese R.V. Cellular fatty acid metabolism and cancer. Cell Metabol. 2013;18(2):153–161. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zechner R., Zimmermann R., Eichmann Thomas O., Kohlwein Sepp D., Haemmerle G., Lass A., Madeo F. Fat signals - lipases and lipolysis in lipid metabolism and signaling. Cell Metabol. 2012;15(3):279–291. doi: 10.1016/j.cmet.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morgan M.J., Liu Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21(1):103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griffiths H.R., Gao D., Pararasa C. Redox regulation in metabolic programming and inflammation. Redox Biol. 2017;12:50–57. doi: 10.1016/j.redox.2017.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quijano C., Trujillo M., Castro L., Trostchansky A. Interplay between oxidant species and energy metabolism. Redox Biol. 2016;8:28–42. doi: 10.1016/j.redox.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L., Wang X., Cueto R., Effi C., Zhang Y., Tan H., Qin X., Ji Y., Yang X., Wang H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019;26:101284. doi: 10.1016/j.redox.2019.101284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Bader A., Abul H., Hussain T., Al-Moosawi M., Mathew T.C., Dashti H. Selenium and liver cirrhosis. Mol. Cell. Biochem. 1998;185(1–2):1–5. doi: 10.1023/a:1006850514295. [DOI] [PubMed] [Google Scholar]
- 31.Han J., Liang H., Yi J., Tan W., He S., Wang S., Li F., Wu X., Ma J., Shi X. Long-term selenium-deficient diet induces liver damage by altering hepatocyte ultrastructure and MMP1/3 and TIMP1/3 expression in growing rats. Biol. Trace Elem. Res. 2017;175(2):396–404. doi: 10.1007/s12011-016-0781-5. [DOI] [PubMed] [Google Scholar]
- 32.Misu H., Takamura T., Takayama H., Hayashi H., Matsuzawa-Nagata N., Kurita S., Ishikura K., Ando H., Takeshita Y., Ota T., Sakurai M., Yamashita T., Mizukoshi E., Yamashita T., Honda M., Miyamoto K-i, Kubota T., Kubota N., Kadowaki T., Kim H.-J., Lee I-k, Minokoshi Y., Saito Y., Takahashi K., Yamada Y., Takakura N., Kaneko S. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metabol. 2010;12(5):483–495. doi: 10.1016/j.cmet.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 33.Hu X., Chandler J.D., Orr M.L., Hao L., Liu K., Uppal K., Go Y.M., Jones D.P. Selenium supplementation alters hepatic energy and fatty acid metabolism in mice. J. Nutr. 2018;148(5):675–684. doi: 10.1093/jn/nxy036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang K., Han Y., Zhao Q., Zhan T., Li Y., Sun W., Li S., Sun D., Si X., Yu X., Qin Y., Tang C., Zhang J. Targeted metabolomics analysis reveals that dietary supranutritional selenium regulates sugar and acylcarnitine metabolism homeostasis in pig liver. J. Nutr. 2019 doi: 10.1093/jn/nxz317. [DOI] [PubMed] [Google Scholar]
- 35.Galan-Chilet I., Grau-Perez M., De Marco G., Guallar E., Martin-Escudero J.C., Dominguez-Lucas A., Gonzalez-Manzano I., Lopez-Izquierdo R., Briongos-Figuero L.S., Redon J., Chaves F.J., Tellez-Plaza M. A gene-environment interaction analysis of plasma selenium with prevalent and incident diabetes: the Hortega study. Redox Biol. 2017;12:798–805. doi: 10.1016/j.redox.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lennicke C., Rahn J., Kipp A.P., Dojčinović B.P., Müller A.S., Wessjohann L.A., Lichtenfels R., Seliger B. Individual effects of different selenocompounds on the hepatic proteome and energy metabolism of mice. Biochim. Biophys. Acta Gen. Subj. 2017;1861(1):3323–3334. doi: 10.1016/j.bbagen.2016.08.015. Part A. [DOI] [PubMed] [Google Scholar]
- 37.Burk R.F., Hill K.E., Nakayama A., Mostert V., Levander X.A., Motley A.K., Johnson D.A., Johnson J.A., Freeman M.L., Austin L.M. Selenium deficiency activates mouse liver Nrf2–ARE but vitamin E deficiency does not. Free Radic. Biol. Med. 2008;44(8):1617–1623. doi: 10.1016/j.freeradbiomed.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Speckmann B., Schulz S., Hiller F., Hesse D., Schumacher F., Kleuser B., Geisel J., Obeid R., Grune T., Kipp A.P. Selenium increases hepatic DNA methylation and modulates one-carbon metabolism in the liver of mice. J. Nutr. Biochem. 2017;48:112–119. doi: 10.1016/j.jnutbio.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 39.Koeberle S.C., Gollowitzer A., Laoukili J., Kranenburg O., Werz O., Koeberle A., Kipp A.P. Distinct and overlapping functions of glutathione peroxidases 1 and 2 in limiting NF-κB-driven inflammation through redox-active mechanisms. Redox Biol. 2020;28:101388. doi: 10.1016/j.redox.2019.101388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang K., Guo X., Zhao Q., Han Y., Zhan T., Li Y., Tang C., Zhang J. Development and application of a HPLC-ICP-MS method to determine selenium speciation in muscle of pigs treated with different selenium supplements. Food Chem. 2020;302:125371. doi: 10.1016/j.foodchem.2019.125371. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y., Zhao H., Zhang Q.S., Tang J.Y., Li K., Xia X.J., Wang K.N., Li K., Lei X.G. Prolonged dietary selenium deficiency or excess does not globally affect selenoprotein gene expression and/or protein production in various tissues of pigs. J. Nutr. 2012;142(8):1410–1416. doi: 10.3945/jn.112.159020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang H., Wang X., Xu L., Ran X., Li X., Chen L., Zhao X., Deng H., Liu X. Establishment of local searching methods for orbitrap-based high throughput metabolomics analysis. Talanta. 2016;156–157:163–171. doi: 10.1016/j.talanta.2016.04.051. [DOI] [PubMed] [Google Scholar]
- 43.Chong J., Soufan O., Li C., Caraus I., Li S., Bourque G., Wishart D.S., Xia J. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018;46(W1):W486–W494. doi: 10.1093/nar/gky310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10(1):1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia D., Shaw R.J. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell. 2017;66(6):789–800. doi: 10.1016/j.molcel.2017.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rabinovitch R.C., Samborska B., Faubert B., Ma E.H., Gravel S.P., Andrzejewski S., Raissi T.C., Pause A., St-Pierre J, Jones R.G. AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. 2017;21(1):1–9. doi: 10.1016/j.celrep.2017.09.026. [DOI] [PubMed] [Google Scholar]
- 47.Crosas-Molist E., Fabregat I. Role of NADPH oxidases in the redox biology of liver fibrosis. Redox Biol. 2015;6:106–111. doi: 10.1016/j.redox.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamanaka R.B., Chandel N.S. Warburg effect and redox balance. Science. 2011;334(6060):1219–1220. doi: 10.1126/science.1215637. [DOI] [PubMed] [Google Scholar]
- 49.DeBerardinis R.J., Thompson C.B. Cellular metabolism and disease: what do metabolic outliers teach us? Cell. 2012;148(6):1132–1144. doi: 10.1016/j.cell.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang C., Ko B., Hensley C.T., Jiang L., Wasti A.T., Kim J., Sudderth J., Calvaruso M.A., Lumata L., Mitsche M., Rutter J., Merritt M.E., DeBerardinis R.J. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell. 2014;56(3):414–424. doi: 10.1016/j.molcel.2014.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith B., Schafer X.L., Ambeskovic A., Spencer C.M., Land H., Munger J. Addiction to coupling of the Warburg effect with glutamine catabolism in cancer cells. Cell Rep. 2016;17(3):821–836. doi: 10.1016/j.celrep.2016.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Devic S. Warburg effect - a consequence or the cause of carcinogenesis? J. Canc. 2016;7(7):817–822. doi: 10.7150/jca.14274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen X., Qian Y., Wu S. The Warburg effect: evolving interpretations of an established concept. Free Radic. Biol. Med. 2015;79:253–263. doi: 10.1016/j.freeradbiomed.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Epstein T., Xu L., Gillies R.J., Gatenby R.A. Separation of metabolic supply and demand: aerobic glycolysis as a normal physiological response to fluctuating energetic demands in the membrane. Canc. Metabol. 2014;2(1):7. doi: 10.1186/2049-3002-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu Y., Zhang L., Liao X., Sangwung P., Prosdocimo D.A., Zhou G., Votruba A.R., Brian L., Han Y.J., Gao H., Wang Y., Shimizu K., Weinert-Stein K., Khrestian M., Simon D.I., Freedman N.J., Jain M.K. Kruppel-like factor 15 is critical for vascular inflammation. J. Clin. Invest. 2013;123(10):4232–4241. doi: 10.1172/JCI68552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kretschmer S., Lee-Kirsch M.A. Type I interferon-mediated autoinflammation and autoimmunity. Curr. Opin. Immunol. 2017;49:96–102. doi: 10.1016/j.coi.2017.09.003. [DOI] [PubMed] [Google Scholar]
- 57.Hybsier S., Schulz T., Wu Z., Demuth I., Minich W.B., Renko K., Rijntjes E., Köhrle J., Strasburger C.J., Steinhagen-Thiessen E., Schomburg L. Sex-specific and inter-individual differences in biomarkers of selenium status identified by a calibrated ELISA for selenoprotein P. Redox Biol. 2017;11:403–414. doi: 10.1016/j.redox.2016.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y., Yui D., Zhang J., Bao J., Tang C., Zhang Z. The role of necroptosis and apoptosis through the oxidative stress pathway in the liver of selenium-deficient swine. Metallomics. 2020 doi: 10.1039/c9mt00295b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.