

Abstract
Delayed afterdepolarizations (DADs) and spontaneous depolarizations (SDs) are typically triggered by spontaneous diastolic Ca2+ release from the sarcoplasmic reticulum (SR) which is caused by an elevated SR Ca2+-ATPase (SERCA) uptake and dysfunctional ryanodine receptors. However, recent studies on the T-box transcription factor gene (TBX5) demonstrated that abnormal depolarizations could occur despite a reduced SERCA uptake. Similar findings have also been reported in experimental or clinical studies of diabetes and heart failure. To investigate the sensitivity of SERCA in the genesis of DADs/SDs as well as its dependence on other Ca2+ handling channels, we performed systematic analyses using the Maleckar et al. model. Results showed that the modulation of SERCA alone cannot trigger abnormal depolarizations, but can instead affect the interdependency of other Ca2+ handling channels in triggering DADs/SDs. Furthermore, we discovered the existence of a threshold value for the intracellular concentration of Ca2+ ([Ca2+]i) for abnormal depolarizations, which is modulated by the maximum SERCA uptake and the concentration of Ca2+ in the uptake and release compartments in the SR ([Ca2+]up and [Ca2+]rel). For the first time, our modelling study reconciles different mechanisms of abnormal depolarizations in the setting of ‘lone’ AF, reduced TBX5, diabetes and heart failure, and may lead to more targeted treatment for these patients.
This article is part of the theme issue ‘Uncertainty quantification in cardiac and cardiovascular modelling and simulation’.
Keywords: SERCA uptake, delayed afterdepolarizations, spontaneous depolarizations, altered calcium handling, bifurcation analysis, atrial fibrillation
1. Introduction
Atrial fibrillation (AF) leads to the rapid and irregular contraction of the upper chambers of the heart, characterized by the disorganized electrical activity within the atria [1]. It is the most encountered arrhythmia in clinical practice, and if left untreated, can lead to an increased risk of heart failure, stroke, and mortality [2]. AF is associated with an increased risk from conditions such as diabetes, obesity, and hypertension [3,4] or from old age [5]. Altered Ca2+ homeostasis is strongly implicated in the initiation of AF [6,7]. In paroxysmal AF, self-terminating episodes of arrhythmia occur in structurally normal atria. This has been linked with increased diastolic Ca2+ leak from the sarcoplasmic reticulum (SR) and triggered activity from upregulated SR Ca2+ ATPase2a (SERCA) and type 2 ryanodine receptor (RyR) dysregulation [7–10]. Similarly, an elevated SERCA uptake was observed in patients with persistent AF [10].
Recent studies from referral populations and in the community suggest that the familial clustering of AF may be genetically caused [11–15]. Furthermore, epidemiological studies have found that individuals who have a first-degree relative with lone AF carry an eightfold increased risk of AF [16]. Linkage and candidate gene sequencing methods have identified several rare genetic variants in genes encoding proteins which alter the kinetics of ion channels, leading to AF [17,18]. With the advent of the genome-wide association studies (GWAS) approach, studies were instead predominantly focused on identifying the common genetic variants associated with AF in the general population [19].
Among recent large-scale GWAS studies, multiple transcription factor genes including the T-box transcription factor gene TBX5 were found to be strongly associated with the arrhythmia [20–24]. TBX5 is known primarily for its role in cardiac development and rhythm control during embryogenesis [25]. Mutations in TBX5 have been reported to underlie Holt-Oram syndrome, features of which include conduction abnormalities and AF [26]. Through a crucial knockout mice study by Nadadur et al. [27], they demonstrated that impairment in TBX5 expression resulted in delayed afterdepolarizations (DADs) and spontaneous depolarizations (SDs) which can lead to AF (figure 1a) [28]. Dai et al. hypothesized that these abnormalities were caused by the remodelling of Ca2+ handling channels and they conducted a subsequent study to investigate their activity in TBX5 knockout mice [29]. Using an experimental protocol with caffeine-containing, Na+-free, Tyrode solution to block Ca2+ extrusion through INaCa, and then a protocol with Na+ containing caffeine solution to prevent uptake through SERCA in the subsequent experiment, they found that the activity of SERCA and INaCa had decreased and increased respectively. The conductivity of the L-type Ca2+ current (ICaL) was also shown to have increased through a voltage-clamp experiment. In addition, they found that if the SERCA inhibitor phospholamban (PLN) was reduced in these TBX5 knockout atrial myocytes, then this instead restored SERCA activity and abolished DADs/SDs. This is intriguing, as these DADs and SDs were caused by a reduction in SERCA instead.
Figure 1.

(a) Example of experimental traces of the membrane potential from the experiment by Nadadur et al. [27]. Delayed afterdepolarizations (DADs) and spontaneous depolarizations (SDs) occur for a majority of (homogeneous TBX5 knockout) mice atrial myocytes, but none for control mice atrial myocytes. (b) Simplified schematic highlighting that reduced expression of TBX5 reduces sarcoplasmic reticulum Ca2+ ATPase 2a (SERCA) and increases INaCa and ICaL, respectively. (Online version in colour.)
In relation to this, experimental studies on heart failure and diabetes have also reported a reduction in SERCA and an increase in INaCa [6,30–33], as well as the elevated occurrence of DADs/SDs [34,35]. Furthermore, restoration of SERCA function through viral transfection of SERCA into cardiac tissue in vivo or knockout of PLN has also been shown to improve cardiomyocyte contractility and suppress triggered activities in both animal and clinical studies of heart failure and diabetes [36–39]. Together, these studies highlight our incomplete understanding of the sensitivity of SERCA in DAD/SD genesis as well as its dependence on other key Ca2+ handling channels such as the RyR, INaCa, and ICaL.
There has also been much debate into the actual mechanisms by which spontaneous diastolic Ca2+ release from the SR can occur to trigger DADs/SDs [40]. Several reports have hypothesized that this is due to the overload of Ca2+ in the SR, termed store-overload induced Ca2+ release [41–43]. Some experimental studies have instead indicated that a more loaded SR is not the only prerequisite for spontaneous diastolic Ca2+ release [44]. Using heart failure as an example, there have been reports that DADs/SDs can appear during this condition despite low Ca2+ levels in the SR [33,45–47].
Altered Ca2+ handling and its implications on the mechanisms of triggered activity can only be explored through a computer model as it is a powerful tool for quantitative dissection of the fundamental mechanisms and in investigating their individual role in DADs/SDs [48]. In this study, we use the Maleckar et al. model [49] to explore the complex relationship between SERCA and other Ca2+ handling channels, and how changes in this relationship affect the Ca2+ dynamics and the susceptibility of the atrial myocyte to DADs/SDs. We define an SD as a self-depolarization that has reached -50 mV or higher, and a DAD as a self-depolarization that is below −50 mV. Throughout this study, we consider only the occurrence of SDs in our results as it is easier to identify in simulation than DADs. However, all our analysis and results for SDs are applicable to DADs as well.
2. Methods
(a). The Maleckar et al. model
The Maleckar et al. model was based on the human atrial myocyte model of Nygren et al. [50,51], and was designed to accurately capture the repolarization phase of an action potential (AP) at different cycle lengths [49,52]. In the Maleckar et al. model (figure 2), the time and voltage-dependent currents INa (Na+ current), ICaL, It, IKur, IK1, IKr, and IKs (Ca2+ independent transient outward, ultra-rapid, inward rectifier and delayed rectifier K+ currents, respectively) are the main attributors to the generation of the human atrial AP.
Figure 2.

Schematic of the full Maleckar et al. model [49]. The sarcoplasmic reticulum Ca2+ ATPase2a (SERCA) pump is bidirectional, and the activation of the ryanodine receptor is modulated by [Ca2+]d and [Ca2+]i. (Online version in colour.)
Ca2+ cycling occurs through SERCA and RyR, located on the SR. The uptake of Ca2+ from the intracellular domain follows a bidirectional formulation [53], where if the concentration of Ca2+ in the uptake compartment ([Ca2+]up) becomes high relative to the concentration of Ca2+ in the intracellular ([Ca2+]i), then the pump will operate more prominently in the reverse direction to reduce the net flux from SERCA. In addition, Ca2+ is transferred from the uptake compartment to the release compartment through Itr (transfer current). Activation of the RyR is modulated by the concentration of Ca2+ in the dyadic space and intracellular ([Ca2+]d and [Ca2+]i, respectively). Release of Ca2+ from the SR is triggered when [Ca2+]d and/or [Ca2+]i is sufficiently elevated, known as Ca2+-induced Ca2+ release. Once activated, the concentration difference between [Ca2+]rel and [Ca2+]i determines the magnitude of Irel.
Currently, the only reported functional changes due to a reduction in TBX5 are from experimental studies by Dai et al. [29] and Laforest et al. [54]. Voltage-clamp experiments show an increase in ICaL in atrial myocytes with reduced expression of TBX5, and separate caffeine-based protocols demonstrated a reduction in SERCA and an elevation in INaCa activity through measurements in the decay rate of [Ca2+]i. Similar findings have also been reported in the setting of diabetes and heart failure [32,35,38,55]. Hence, we model functional changes in SERCA as simply a multiplicative change in maximum SERCA uptake, and changes in ICaL and INaCa as a change in their conductivity. In addition, because the underlying commonality behind these conditions is disrupted Ca2+ handling, we have also included RyR in this study, and changes in its activity will also be modelled as a change in its conductivity. The study will be carried out across a wide parameter space to assess how changes in these parameters impact Ca2+ handling.
Throughout our experiments, we define the control as the original full Maleckar et al. model with its original parameters and equations, i.e. normal human atrial myocytes under physiological conditions. In each of our simulations with the full cellular model, a train of stimuli with a duration of 6 ms and magnitude of −15 pA/pF was applied at a frequency of 1 Hz up to 100 s into the simulation.
(b). Bifurcation analysis and sensitivity of SERCA
We first investigated how the individual sensitivity of SERCA compared with that of ICaL, INaCa, and RyR in the genesis of SDs by running simulations where we individually varied the conductivities of these channels from a range of 0 to 2 times its control value. We then probed for the minimum increase or decrease required for each channel for an SD to occur in increments of 0.01 until it was identified.
Afterwards, we conducted a type of analysis known as a bifurcation analysis to investigate how changes in the parameter of a Ca2+ handling channel affected that of another channel in triggering SDs. The first bifurcation analysis we performed was between ICaL and INaCa with SERCA at its control value. We varied the conductivity of ICaL from a range of 0.2 to 2 times its control value in increments of 0.2, and for each of these values of ICaL, we identified the minimum multiplier that must be applied to INaCa to trigger at least one SD. Following this, we then conducted three other bifurcation analyses and identified minimum multipliers for RyR, ICaL, and INaCa for varying levels of SERCA using the same approach.
Minimum multipliers were determined through a trial and error approach. For example, if we were interested in finding the minimum multiplier that must be applied to RyR to trigger at least one SD, then we ran simulations of the full Maleckar et al. model with SERCA set at a certain condition, e.g. 120% of its original value, for varying levels of RyR until we ascertain a rough estimate of the parameter region where the SDs occur. After this, we honed in onto the minimum multiplier value for RyR that could cause at least one SD to occur for 120% SERCA. Through this approach, we derived the SD boundary lines for each of the corresponding bifurcation diagrams which illustrated the relationship between Ca2+ handling parameter pairs in the genesis of at least one SD.
(c). Calcium subsystem analysis
In the full Maleckar et al. model, it can be quite complex to identify the exact mechanisms to trigger an SD since many factors can influence this. However, by reducing the model down to only the essentials in Ca2+ handling, i.e. Ca2+ subsystem (electronic supplementary material, figure S1), we can instead probe for the conditions for Ca2+ oscillations instead which is far simpler to identify as they only take place when the myocyte is overloaded with Ca2+. These conditions can then be applied to the full Maleckar et al. model to investigate the mechanisms of SDs as such abnormal depolarizations are triggered by the occurrence of Ca2+ oscillations in the Ca2+ subsystem [56–60]. To carry out this Ca2+ subsystem analysis, we used the approach developed by Fink et al. [40].
To reduce the Maleckar et al. model down to its Ca2+ subsystem, we set all transmembrane currents apart from INaCa to zero, and set the membrane potential and intracellular and cleft space Na+ concentrations to a constant value as determined by their end-diastolic values after 3000 s of 1 Hz pacing in the full Maleckar et al. model. Note that ICaL was also removed despite playing an important role in Ca2+ handling in the full model because since the membrane potential was clamped, ICaL would not activate, and hence would provide a negligible amount of Ca2+ into the reduced model. Since the bulk space and K+ concentrations were not required in the Ca2+ subsystem, we removed them from the reduced model (electronic supplementary material, figure S1).
To induce an overload of Ca2+, a multiplier was applied to clamped [Na+]i. This multiplier caused a rise in Ca2+ in the myocyte, which triggered INaCa to extrude Na+ out of the cell and Ca2+ into the cell. A minimum multiplier was required to sufficiently elevate the total Ca2+ in the myocyte (which we defined as [Ca2+]tot = [Ca2+]up + [Ca2+]rel + [Ca2+]d + [Ca2+]i) so that Ca2+ oscillations occurred. During this phenomenon, [Ca2+]up, [Ca2+]rel, [Ca2+]d, and [Ca2+]i oscillated between a maximum and minimum value as time progressed. These oscillations would persist since [Na+]i remained fixed at that elevated value.
We considered the system as having successfully generated Ca2+ oscillations if the average range during the simulation time-course of 5000 s was at least 0.2 mM for [Ca2+]up, [Ca2+]rel, and 0.00002 mM for [Ca2+]d, and [Ca2+]i. Note that if the multiplier for [Na+]i could not sufficiently elevate [Ca2+]tot to trigger SDs, then [Ca2+]up, [Ca2+]rel, [Ca2+]d, and [Ca2+]i would not oscillate and would immediately reach steady state instead.
Using the reduced model, we implemented the changes in SERCA, RyR, and INaCa from a range of 0.2 to 2 times their respective control values in increments of 0.2 individually. For each increase for a particular channel, we then found the minimum multiplier required for clamped [Na+]i to subsequently elevate the total Ca2+ concentration in the atrial myocyte sufficiently to trigger Ca2+ oscillations. The corresponding minimum [Ca2+]tot that triggered Ca2+ oscillations was derived by finding the steady state for [Ca2+]up, [Ca2+]rel, [Ca2+]d, and [Ca2+]i during these oscillations. Steady state for each compartment was obtained by computing their mean value over the simulation time-course of 5000 s. This approach was then used to investigate how changes in the Ca2+ handling channels along the points on the SD boundary line for SERCA-RyR and SERCA-INaCa bifurcations impacted the minimum [Ca2+]tot for Ca2+ oscillations and hence SDs.
(d). The dependency test of ryanodine receptors and SERCA on the genesis of spontaneous depolarizations
As previously mentioned, there is much debate in experimental studies regarding the precise mechanisms by which diastolic spontaneous Ca2+ release occurs to trigger SDs. However, in the study by Fink et al., the importance of the modulation of [Ca2+]i and [Ca2+]up on SERCA, and [Ca2+]i and [Ca2+]d on RyR in the genesis of SDs was also investigated.
We conducted this dependency test on the full Maleckar et al. model to determine the necessary properties for the initiation of SDs in this model. To carry this out, we first applied a train of stimuli with a duration of 6 ms and magnitude of −15 pA/pF at a frequency of 1 Hz up to 3000 s into the simulation. At 3000 s, we recorded the values of [Ca2+]up, [Ca2+]i, and [Ca2+]d, and labelled them as [Ca2+]up, 3000s, [Ca2+]i, 3000s, and [Ca2+]d, 3000 s, respectively.
To illustrate the subsequent steps, we then used the RyR as an example, the modulation terms of the RyR in the full Maleckar et al. model were given by
| 2.1 |
| 2.2 |
| 2.3 |
| 2.4 |
To remove the modulation of [Ca2+]i on RyR to test its dependence on the genesis of SDs, we substituted [Ca2+]i in equation (2.2) with [Ca2+]i, 3000s. This gave
| 2.5 |
To test the dependence of [Ca2+]d on RyR, we substituted [Ca2+]d in equation (2.1) with [Ca2+]d, 3000 s. This gave
| 2.6 |
By a similar approach, the dependence of [Ca2+]i and [Ca2+]up on SERCA in the full Maleckar et al. model was also tested.
(e). Intracellular calcium injection protocols
The first protocol was designed to investigate the threshold value for [Ca2+]i in triggering SDs, and whether this threshold followed a biphasic trend as SERCA decreased from 200% to 20% of its control value. The second protocol was designed to assess whether [Ca2+]up and [Ca2+]rel were also determining factors in [Ca2+]i SD threshold for various levels of SERCA, and to ascertain how changes in SERCA impacted Ca2+ handling and hence gave rise to the biphasic trend in the [Ca2+]i SD threshold.
(i). Intracellular calcium injection protocol 1
We applied a train of stimuli with a duration of 6 ms, magnitude of −15 pA/pF and frequency of 1 Hz up to 100 s into the simulation. At exactly 100 s into the simulation, we recorded the value of [Ca2+]i, then applied a multiplier to this value. This new value was then used to replace the original value of [Ca2+]i, and the simulation was then resumed except no external stimulus was applied. This protocol was repeated until we found the minimum multiplier required to increase [Ca2+]i to the threshold value needed to trigger an SD at 100 s. The protocol was performed for values of SERCA from 20% to 200% control SERCA in increments of 20%.
(ii). Intracellular calcium injection protocol 2
For this protocol, we followed the same steps as outlined above, except exactly 100 s into the simulation, we adjusted [Ca2+]up and [Ca2+]rel to their value at 20% SERCA at 100 s in protocol 1 before we applied the multiplier to [Ca2+]i. The same protocol in protocol 1 was repeated but with [Ca2+]up and [Ca2+]rel adjusted to their value at 200% at 100 s instead.
3. Results
(a). The impact of individual calcium handling channels on spontaneous depolarizations
From our series of simulations where we individually implemented the remodelling of each Ca2+ handling channel from a range of 0% to 200% of their control value, we identified a minimum value that INaCa, ICaL, RyR, and SERCA must be increased or decreased to trigger SDs. For INaCa, SDs occurred when it was decreased by 11%, while for ICaL and RyR, they instead occurred when they were increased by 13% and 38%, respectively (figure 3a). We were unable to identify a minimum value by which we needed to increase or decrease SERCA alone to generate SDs. In figure 3b–f, we show the time-course of the membrane potential, Irel, [Ca2+]i, [Ca2+]up, and [Ca2+]rel after the last stimulated AP (black triangle) and the SDs occurred at their respective minimum percentage for INaCa, ICaL, and RyR.
Figure 3.

(a) The minimum percentage change to control individual INaCa, ICaL and RyR to trigger SDs. (b–f) Time-course of the membrane potential, Irel, [Ca2+]i, [Ca2+]up, and [Ca2+]rel, for control and for 89% INaCa, 113% ICaL, and 138% RyR after the last stimulated AP (black triangle). SERCA, sarcoplasmic reticulum Ca2+-ATPase; SD, spontaneous depolarization; RyR, ryanodine receptors. (Online version in colour.)
(b). The sensitivity of spontaneous depolarizations to SERCA
We examined the relationship between ICaL and INaCa in SDs through a bifurcation analysis (figure 4a). As shown by the blue SD boundary quadratic line in figure 4a, as we increased the multiplier to ICaL, the minimum multiplier for INaCa required to generate SDs also increased. The region below the blue SD boundary line illustrates the parameter range between ICaL and INaCa where SDs can occur.
Figure 4.

(a) Bifurcation diagram of ICaL and INaCa at 100% SERCA. Shaded region illustrates the parameter range where SDs occur. (b–f) Time-course of the membrane potential, Irel, [Ca2+]i, [Ca2+]up, and [Ca2+]rel for control and at 140% ICaL and 125% INaCa for 100%, 54%, and 107% SERCA after the last stimulated AP (black triangle). SERCA, sarcoplasmic reticulum Ca2+-ATPase; SD, spontaneous depolarization; AP, action potential. (Online version in colour.)
We then chose a data point on the blue line of the ICaL-INaCa bifurcation (figure 4a), e.g. 1.4 × ICaL, 1.25 × INaCa, and ran a series of simulations where we changed SERCA from 0% to 200% of its control value to ascertain how changes in SERCA affected the capacity of ICaL, and INaCa in generating SDs. As expected, SDs were observed when we kept SERCA at its control value with 140% ICaL and 125% INaCa since we used a pair of values on the SD threshold of ICaL and INaCa. However, SDs were abolished when SERCA was reduced below 55% or increased above 106% of its control value (figure 4b). This illustrates an interdependency between SERCA with ICaL and INaCa, as SDs only occurred within a certain parameter range for SERCA. In figure 4c–f, we show the time-course of Irel, [Ca2+]i, [Ca2+]up, and [Ca2+]rel after the last stimulated AP with SERCA at 100%, 54%, 107% of its control value.
(c). Bifurcation analysis between SERCA and other calcium handling channels
As observed in the previous subsection, changes in SERCA can have an impact on ICaL, RyR, and INaCa in generating SDs. To investigate this issue systematically, we conducted a SERCA-ICaL, SERCA-RyR and SERCA-INaCa bifurcation. For both SERCA-RyR and SERCA-ICaL bifurcations (figure 5a,b), at less than approximately 80% SERCA, the minimum multiplier for ICaL and RyR required to generate at least an SD decreased linearly with increasing SERCA. However, at greater than approximately 80%, the minimum multiplier for ICaL and RyR instead increased linearly with increasing SERCA. For the SERCA-INaCa bifurcation, at less than approximately 80% SERCA, the minimum multiplier for INaCa instead increased linearly with increasing SERCA, but decreased linearly at greater than approximately 80% (figure 5c). Together, this implied that at approximately 80% SERCA, there was a shift in the Ca2+ handling of the atrial myocyte since that was roughly the point where the trend changed direction for all three bifurcation diagrams.
Figure 5.

Bifurcation analysis for SERCA-RyR (a) and SERCA-ICal (b), respectively. (c) Bifurcation analysis for SERCA-INaCa. Shaded region illustrates the parameter range where SDs occur. The cross marks roughly where the shift in the Ca2+ handling took place. SERCA, sarcoplasmic reticulum Ca2+-ATPase; SD, spontaneous depolarization; RyR, ryanodine receptors. (Online version in colour.)
(d). The calcium subsystem sensitivity analysis of SERCA
Under this subsection, we conducted a Ca2+ subsystem sensitivity analysis to investigate how changes in SERCA, RyR, and INaCa alone affected the minimum multiplier to clamped [Na+]i (and hence the total Ca2+ concentration threshold) to generate Ca2+ oscillations. For either SERCA or INaCa, as it decreased from 200% of its control value to 40% of its control value, the minimum multiplier to clamped [Na+]i also decreased (electronic supplementary material, figure S2). By contrast, the minimum multiplier to clamped [Na+]i instead increased as RyR decreased. Since the existence of Ca2+ oscillations in the Ca2+ subsystem corresponded to the occurrence of SDs in the full model, this showed that reducing SERCA or INaCa alone decreased the minimum total Ca2+ concentration required for Ca2+ oscillations, while reducing RyR instead has the opposite effect.
We then conducted a subsequent analysis to find how changes along the SD boundary in the direction of increasing SERCA for both SERCA-RyR and SERCA-INaCa bifurcations affected the minimum multiplier for clamped [Na+]i for Ca2+ oscillations to occur. The obtained data points for the minimum multiplier for clamped [Na+]i from the changes in SERCA and INaCa, and changes in SERCA and RyR along the SD boundary line in their respective bifurcation diagrams were then compared with the points obtained from changes in SERCA alone (electronic supplementary material, figure S2).
As shown in electronic supplementary material, figure S3A, as we went in the direction of decreasing SERCA, the minimum multiplier for clamped [Na+]i for Ca2+ oscillations decreased for all three cases. Correspondingly, decreasing SERCA uptake function reduced the minimum [Ca2+]tot needed for SDs to occur, and thus increased the myocyte's susceptibility to SDs in the full Maleckar et al. model (electronic supplementary material, figure S3B). Interestingly, electronic supplementary material, figure S3B also showed that only the minimum [Ca2+]tot for SDs was significantly different for changes in both SERCA and RyR based upon the points in the SD boundary of the SERCA-RyR bifurcation. This, along with the linear decreasing tend among the three cases suggested that reducing the minimum [Ca2+]tot was not the only requirement for SDs to occur, and that a deeper mechanism existed alongside this.
(e). The necessary components of the genesis of spontaneous depolarizations
One plausible explanation for the biphasic trend in the SERCA-RyR, SERCA-ICaL, and SERCA-INaCa bifurcation diagrams was due to the Ca2+ threshold required to trigger SDs changing in a biphasic manner as SERCA changes, and thus different minimum increases in RyR and ICaL, or decreases in INaCa were required to meet this Ca2+ threshold for various levels of SERCA. To be able to determine if this was the case, we first needed to identify the necessary components in the Maleckar et al. model to give rise to an SD. Hence we performed the dependency test proposed by Fink et al. where we tested the modulation of [Ca2+]i and [Ca2+]up on SERCA, and [Ca2+]i and [Ca2+]d on RyR to see which ones were essential for the genesis of SDs. Through this test, we found that only the modulation of RyR by [Ca2+]i was crucial for the initiation of SDs (figure 6b).
Figure 6.

The dependency test of SERCA and RyR in the genesis of SDs. (a) Time-course of the membrane potential, [Ca2+]up, [Ca2+]rel, and Irel for modulated SERCA and RyR, removed modulation of [Ca2+]i on SERCA, removed modulation of [Ca2+]up on SERCA, and removed modulation of [Ca2+]d on RyR. (b) Time-course of the membrane potential, [Ca2+]up, [Ca2+]rel, and Irel for removed modulation of [Ca2+]i on RyR. The black triangle indicates the time when a stimulus was applied. SERCA, sarcoplasmic reticulum Ca2+-ATPase; SD, spontaneous depolarization; RyR, ryanodine receptors. (Online version in colour.)
(f). The impact of SERCA on the intracellular calcium threshold for spontaneous depolarizations
Having determined that it was the modulation of [Ca2+]i on RyR that was essential in the genesis of SDs in the full Maleckar et al. model, we were then interested in ascertaining whether there existed a threshold value for [Ca2+]i in triggering SDs, and whether this threshold followed a biphasic trend as SERCA decreased from 200% to 20% of its control value. Hence, we performed the intracellular Ca2+ injection protocol 1 for various levels of SERCA. As shown in figure 7, the [Ca2+]i threshold to trigger SDs followed a biphasic trend as SERCA decreased from 200% to 20% of its control value, demonstrating that this threshold was different for varying levels of SERCA. This also confirmed that when SERCA was sufficiently inhibited or elevated, to trigger SDS, an increase in RyR, ICaL, or decrease in INaCa was required to meet the elevated [Ca2+]i threshold to trigger SDs.
Figure 7.

The [Ca2+]i SD threshold for various levels of SERCA. The cross represents the point where the trend changed. SERCA, sarcoplasmic reticulum Ca2+-ATPase. (Online version in colour.)
(g). The relationship between calcium concentrations in the uptake and release compartment and the intracellular calcium threshold
Having confirmed that there existed a threshold value for [Ca2+]i in triggering SDs and that the threshold followed a biphasic trend as SERCA decreased, we were then interested in knowing the Ca2+ handling mechanisms which gave rise to the biphasic relationship between the [Ca2+]i SD threshold and SERCA. We hypothesized that this mechanism not only involved SERCA but also [Ca2+]up and [Ca2+]rel, because as we were conducting the experiment under protocol 1, we noted that they kept decreasing as SERCA decreased (electronic supplementary material, figure S4A and B). Hence, there was the possibility that different levels in [Ca2+]up and [Ca2+]rel could therefore also have an impact on the [Ca2+]i SD threshold.
To investigate this, we conducted two more series of simulations under intracellular calcium injection protocol 2. These series of simulations respectively assessed how the [Ca2+]i SD threshold was impacted when [Ca2+]up and [Ca2+]rel was reduced or increased beyond their intrinsic value for a particular level of SERCA. As shown in figure 8, when [Ca2+]rel and [Ca2+]up were elevated to their values for 200% SERCA, the [Ca2+]i SD threshold decreased relative to the threshold obtained in the default case for all values of SERCA. By contrast, when [Ca2+]rel and [Ca2+]up were reduced to their values for 20% SERCA, the [Ca2+]i SD threshold instead increased relative to the threshold obtained in the default case for all values of SERCA.
Figure 8.

The [Ca2+]i SD threshold for various levels of SERCA for three different cases. The default case is when no change was made to [Ca2+]up and [Ca2+]rel at 100 s into the simulation (blue). The other two cases are when at 100 s into the simulation, prior to intracellular Ca2+ injection, [Ca2+]up and [Ca2+]rel were elevated and reduced to their values at 200% SERCA (bottom) and 20% SERCA (top) at 100 s respectively in the default case (see electronic supplementary material, figure S4A and B). The coloured crosses indicate where a change in the trend occurs in the corresponding case. SERCA, sarcoplasmic reticulum Ca2+-ATPase; SD, spontaneous depolarization. (Online version in colour.)
4. Discussion
It is generally accepted that SDs/DADs are caused by spontaneous release of Ca2+ from the SR due to an upregulation in SERCA uptake and dysfunctional RyR in atrial cells [8,9]. However, recent mice knockout experiments of TBX5 have demonstrated that SDs could also occur through a reduced SERCA uptake, and increased INaCa and RyR [27,29,54]. Similar findings have also been reported in experimental and clinical studies of diabetes [6,31] and heart failure [30,32,33]. Surprisingly, in all these cases, the rescue of impaired SERCA uptake through the knockout of PLN or SERCA2a gene therapy was able to suppress these SDs [29,36–39]. To our knowledge, this is the first computational study that systematically investigates the sensitivity of SERCA in relation to other Ca2+ handling channels in the genesis of SDs/DADs and the mechanisms which are involved.
Though the results shown in this study are specific for the occurrence of SDs as they are easier to identify in simulations than DADs, the conclusions on the dynamic relationship between the key Ca2+ proteins, i.e. SERCA, ICaL, INaCa, and RyR can be drawn similarly for DADs. Generally, in our simulations, DADs occur only in the form of a slight depolarization if changes to channels are close to the changes required to trigger an SD. For example, if we increased ICaL to 112% of its control value instead (figure 3), then after the last stimulated AP, a self-depolarization up to −72 mV was observed. By contrast, at 113% of the control value of ICaL and greater, SDs were observed.
Our four bifurcation analyses together illustrate that variations in SERCA can have a pronounced impact on Ca2+ handling. When SERCA is significantly inhibited (i.e. less than approx. 80% SERCA) or elevated (i.e. greater than approx. 100%), the minimum increase in RyR or ICaL, or decrease in INaCa that is required to trigger SDs is greater than when SERCA is only slightly inhibited (i.e. greater than approx. 80% SERCA and less than approx. 100% SERCA). This highlights that the myocyte's susceptibility to abnormal depolarizations is greatest when SERCA is slightly inhibited, because changes in RyR, ICaL, and INaCa needed to trigger SDs/DADs in this parameter range for SERCA are at a minimum. These bifurcation diagrams also illustrate that increased RyR or ICaL triggers SDs/DADs, but increased INaCa suppresses them instead. This is interesting, because in the setting of reduced TBX5, diabetes, and heart failure, SDs/DADs was observed alongside an elevation in INaCa. Therefore, for SDs/DADs to occur in conditions with an increased INaCa, there must also be an increase in ICaL to overcome the suppression effects of INaCa.
In the dependency test by Fink et al., we found that in the full Maleckar et al. model, only the modulation of the RyR by [Ca2+]i was crucial. This differs from the 37 previously published mathematical models of atrial and ventricular myocytes that they tested, where they found that in most models, it was the modulation of RyR through Ca2+ in the dyadic space ([Ca2+]d) that triggered SDs [40]. This could be because in most models they tested, the RyR is connected to the dyadic space rather than the intracellular space as is the case in the Maleckar et al. model.
During our experiment with intracellular Ca2+ injection protocol 1, we observed that diastolic [Ca2+]up and [Ca2+]rel at the time point where intracellular Ca2+ is injected to trigger spontaneous Ca2+ release decreased as SERCA decreased. This is consistent with experimental findings by Fernandez-Tenorio and Niggli, where they applied a range of pharmacological SERCA stimulators and a PLB antibody to mouse cardiomyocytes, and found that mice with non-stimulated SERCA had a lower diastolic [Ca2+]SR prior to spontaneous Ca2+ release. Interestingly, they considered [Ca2+]SR as being the threshold for spontaneous Ca2+ release, and this threshold increased as SERCA was elevated [61].
With the second intracellular Ca2+ injection protocol, we probed whether [Ca2+]up and [Ca2+]rel were indeed also factors in determining the [Ca2+]i SD threshold. Results from this experiment revealed that in general, if [Ca2+]up and [Ca2+]rel are increased above their intrinsic value for a particular value of SERCA, then the [Ca2+]i SD threshold will be lowered. However, if [Ca2+]up and [Ca2+]rel are decreased below their intrinsic value instead, then the [Ca2+]i SD threshold will be elevated instead. From the point of view of this study, it is hence better to view [Ca2+]up and [Ca2+]rel alongside SERCA to be regulators of the [Ca2+]i SD threshold rather than act as a threshold itself for spontaneous Ca2+ release and hence SDs/DADs as proposed by Fernandez-Tenorio and Niggli.
On closer inspection, this experiment also provides some insights into why the [Ca2+]i SD threshold increases when SERCA is significantly inhibited (i.e. less than approx. 80% SERCA) or elevated (i.e. greater than approx. 100% SERCA). To illustrate the former situation, we consider the default case starting at 80% of its control value of SERCA in figure 8. If we reduce SERCA from 80% to 60% of its control value, the [Ca2+]i SD threshold will increase slightly (80% SERCA to 60% SERCA on the blue line). If at 60% SERCA, [Ca2+]up and [Ca2+]rel are artificially reduced below its intrinsic value, the increase in the [Ca2+]i SD threshold from 80% SERCA to 60% will be much greater (80% SERCA on the blue line to 60% SERCA on the red line). However, if at 60% SERCA, [Ca2+]up and [Ca2+]rel are artificially elevated above its intrinsic value, the [Ca2+]i SD threshold from 80% SERCA to 60% will instead decrease (80% SERCA on the blue line to 60% SERCA on the green line). This seems to illustrate that in the default case, the [Ca2+]i SD threshold starts to increase at less than approximately 80% SERCA because the maximum SERCA uptake is no longer high enough to maintain [Ca2+]up and [Ca2+]rel at sufficient levels. As a result, greater [Ca2+]i is required to compensate this for spontaneous Ca2+ release and subsequently SDs to occur. In a similar fashion, we can illustrate the latter situation. However, the key difference here is that raising [Ca2+]up and [Ca2+]rel has a negligible impact on the [Ca2+]i SD threshold (160% SERCA on the blue line to 160% SERCA on the green line).
This latter situation likely occurs because of the properties of the bidirectional SERCA pump. If maximum SERCA uptake is elevated, then this would result in an increase of [Ca2+]up and [Ca2+]rel. Because [Ca2+]up and [Ca2+]rel are now elevated, the reverse mode of the bidirectional pump activates, reducing the net Ca2+ uptake into the SR by SERCA. Further increasing [Ca2+]up and [Ca2+]rel would therefore have a negligible effect on the net Ca2+ uptake into the SR by SERCA. In this situation, an elevation in [Ca2+]i is required to reduce the reverse direction of SERCA and restore SERCA uptake function such that spontaneous Ca2+ release and hence SDs/DADs can occur. In electronic supplementary material, figure S7, we have demonstrated that by replacing the bidirectional SERCA pump in the Maleckar et al. model with the unidirectional SERCA pump in the CRN model and a generic leak in the uptake compartment (electronic supplemental materials, figure S6), the minimum multiplier for RyR and ICaL to trigger at least one SD is significantly less than in the bidirectional case, and significantly greater for INaCa for elevated SERCA (greater than approx. 100% SERCA), illustrating that the [Ca2+]i SD threshold is much lower with a unidirectional SERCA pump than a bidirectional one in this parameter range.
Based on these findings together, we therefore hypothesize that there are three main phases in the Ca2+ handling involving SERCA, [Ca2+]up, and [Ca2+]rel which attribute to the [Ca2+]i SD threshold in the full Maleckar et al. model at varying levels of maximum SERCA uptake. If SERCA is significantly inhibited (i.e. less than approx. 80% SERCA), the bidirectional SERCA pump becomes too inhibited to uptake enough calcium into the SR necessary to aid in the initiation of spontaneous Ca2+ release and hence SDs, and so an elevation in [Ca2+]i is required to compensate (figure 9a).
Figure 9.
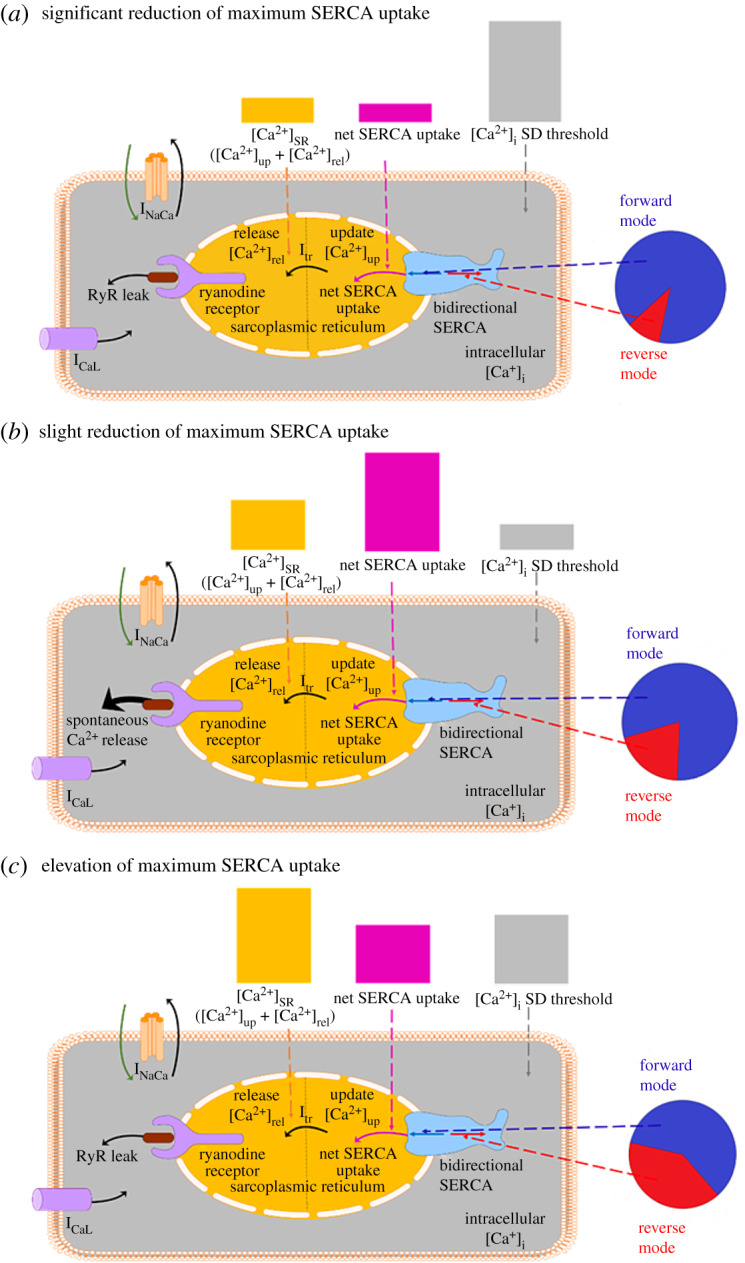
Schematic illustrating the general differences in Ca2+ handling for maximum SERCA uptake ranges between (a) approximately 20–80% (b) approximately 80–100% and (c) greater than approximately 100% of its control value in the Maleckar et al. model. Pie graph inserts show the relative magnitude of the forward and reverse mode of the bidirectional SERCA pump in each range. Bar graph inserts qualitatively highlight the relative magnitude of the net SERCA uptake and [Ca2+]SR in each range, and how this impacts the magnitude of the [Ca2+]i threshold for SDs. SERCA, sarcoplasmic reticulum Ca2+-ATPase; SD, spontaneous depolarization. (Online version in colour.)
On the other hand, when SERCA is elevated (e.g. PLN knockout or SERCA2a gene therapy) (i.e. greater than approx. 100% SERCA), the properties of the bidirectional SERCA pump becomes pronounced. The resultant [Ca2+]up and [Ca2+]rel become relatively high, which trigger the reverse direction of the pump. This in turn reduces the net uptake of Ca2+ into the SR and disrupts the ability of SERCA to contribute to spontaneous Ca2+ release and hence SDs. To promote the forward direction of the bidirectional SERCA pump in this situation, there needs to be an increase in [Ca2+]i such that [Ca2+]up and [Ca2+]rel do not appear relatively high (figure 9c).
Figure 9b illustrates when SERCA is in the ideal range to trigger SDs (i.e. greater than approx. 80% SERCA and less than approx. 100% SERCA). In this range, SERCA has sufficient uptake into the SR such that [Ca2+]up and [Ca2+]rel reach appropriate levels without triggering the reverse mode of the SERCA pump and without a compensatory increase in [Ca2+]i. As a result, the [Ca2+]i SD threshold is at a minimal in this range, and only small increases in RyR and ICaL, or decreases in INaCa are required.
5. Limitations
There are several limitations to this modelling study. The first issue concerns the insufficient details of spatial Ca2+ handling in the Maleckar et al. model. The internal structure of Ca2+ handling in cardiac myocytes is vastly more complicated than is represented in common pool models. The SR is a complex structural network within the myocyte and consists of clusters of RyRs which can open and close collectively. To be able to expand the scope of this study to include the mechanisms by which a modulation of SERCA can increase the incidence and spatial dynamics of Ca2+ waves, and how normalization or elevation of SERCA can reduce this, the descriptions of Ca2+ handling in the Maleckar et al. model need to be further developed to include these structural and spatial aspects of Ca2+ handling, such as the Voigt et al. [7] and Thul et al. models [62,63]. Additionally, the peak systolic [Ca2+]i level and decay of [Ca2+]i in our simulated results of the Maleckar et al. model are different from those in the experimental studies by Neef et al. and Voigt et al. [7,64,65]. This is because the Maleckar et al. model was based on the Nygren et al. model, which was validated through a different experimental study on human atrial myocytes. Further investigation is required to ascertain the modifications which are required in the formulations of the ionic and Ca2+ handling channels such that the model can accurately capture the experimental results by Neef et al. and Voigt et al. as well as the phenomena in SERCA as detailed in this study.
Limited availability of data for SERCA and other Ca2+ handling channels in the setting of reduced TBX5, diabetes, and heart failure is also an obstacle of this study. Across these settings, studies only provide the voltage-clamp recordings of ICaL and caffeine-based protocol results of SERCA and INaCa for a select few myocytes [6,29,54,55,66], and so it would be difficult to verify if any parameters that we assigned would fall under physiological ranges. In our study, we instead conducted a modelling analysis of how changes in the conductivity of ICaL, INaCa, and RyR, and the uptake of SERCA impacts Ca2+ handling in the atrial myocyte across a wide parameter space to qualitatively examine the mechanisms by which a reduction in SERCA can trigger SDs, and how normalization or elevation of SERCA through knockout of PLN or SERCA2a gene therapy can suppress SDs.
While this approach provides insights as to how Ca2+ handling is altered as we vary maximum SERCA uptake and the relationships between SERCA and other Ca2+ handling channels in triggering SDs, it can only make predictions into the physiological ranges where these phenomena can occur. This may be where it would be more advantageous to use a population-based approach combined with a complementary animal study [67] to investigate if deviations from the limited data that we have on Ca2+ handling channels under reduced TBX5, diabetes, and heart failure can still give rise to SDs, and for a better estimate of the parameter ranges where SDs can be triggered or suppressed. Experimental studies in reduced TBX5, diabetes, and heart failure used varying experimental and pacing protocols [6,27,29,54,55,66]. In our simulations with the Maleckar et al. model, a pacing frequency of 1 Hz was used instead as we are interested in how reduced SERCA impacts Ca2+ handling to trigger SDs, and how normalization or elevation of SERCA can suppress SDs in human atrial myocytes. While we currently lack the experimental data to verify our results, it would, however, be of benefit to this study if simulations were conducted at multiple different pacing frequencies. This could be because changing the pacing frequency would likely also affect [Ca2+]up and [Ca2+]rel, causing an increase in the [Ca2+]i SD threshold.
In terms of the suppression of SDs due to the knockout of PLN, this was regarded as an increase in maximum SERCA uptake back to normal or elevated levels. This is analogous to changing the maximum conductance through agents such as a pharmacological inhibitor or through SERCA2a gene therapy. However, disruption of PLN-mediated SERCA inhibition can also cause an increased affinity to [Ca2+]i and therefore likely shift the balance between the forward and reverse mode of SERCA. Hence for our hypothesis to hold, it is assumed that changes in SERCA activity are exclusive to changes in its maximum SERCA uptake. Disruption of PLN-mediated SERCA inhibition could be more rigorously modelled as changes in the phosphorylation rates of SERCA and could potentially shed more insight into the Ca2+ handling mechanisms attributing to the suppression of SDs/DADs if these parameters were also investigated through a bifurcation analysis and intracellular Ca2+ injection protocol to gauge how changes in phosphorylation rates impacted the [Ca2+]i threshold. Similarly, the impact of Ca2+ buffering on Ca2+ handling warrants investigation through these analytical methods.
The second caveat in our hypothesis is that for spontaneous Ca2+ release to occur, there must also be sufficient diffusion of Ca2+ in the SR. Diffusion of Ca2+ through the SR has been shown to be a critical factor of spontaneous Ca2+ release in atrial myocytes [68]. Rapid Ca2+ diffusion within the SR is necessary to stabilize and balance [Ca2+]SR within the myocyte. In the context of this study, reducing Ca2+ diffusion could potentially cause an overall reduction in [Ca2+]up and [Ca2+]rel, which subsequently increases the [Ca2+]i SD threshold.
The other intermediate pathway that was not considered in the Maleckar et al. model or in our mathematical analyses was the Ca2+/calmodulin-dependent protein kinase II (CaMKII). A reduction in SERCA uptake would increase diastolic [Ca2+]i, and thus lead to greater phosphorylation of PLN on SERCA or RyR. This in turn could mitigate the reduced SERCA uptake and increase the open probability of RyR and could potentially also explain the onset of SDs from a reduced SERCA, and the suppression of SDs from PLN knockout or SERCA2a gene therapy [69]. More recently, Alsina et al. discovered an intermediate pathway involving a novel protein phosphatase 1 (PP1)- regulatory subunit PPP1R3A (PP1 regulatory subunit type 3A) within the RyR2 channel complex. Through the knockout of PPP1R3A, they demonstrated that reduced expression of this novel PP1 regulatory subunit reduces the binding of PP1 to both RyR2 and PLN, which subsequently increases SR Ca2+ release and hence increases the susceptibility of the myocyte to SDs [70]. Changes in Ca2+ handling channels can therefore have an impact on the dynamics of this intermediate pathway and thus the [Ca2+]i SD threshold, and would need to be incorporated along with the intermediate pathway involving CaMKII in a future study.
6. Conclusion
In summary, a reduction in SERCA alone cannot cause SDs/DADs, but instead affects the [Ca2+]i threshold required to trigger SDs/DADs. Compensatory changes in other Ca2+ handling channels, such as increasing RyR/ ICaL or decreasing INaCa alongside SERCA are therefore required to raise [Ca2+]i to its threshold level to trigger spontaneous Ca2+ release and hence SDs/DADs. Furthermore, this study also demonstrates that a bidirectional SERCA pump is necessary to capture the effects of SD/DAD initiation particularly due to a reduction in SERCA and [Ca2+]SR load. For the first time, our modelling study reconciles different mechanisms of abnormal depolarizations in the setting of ‘lone’ AF, TBX5, diabetes, and heart failure, and it may lead to more targeted treatment for these patients, such as the suppression of SDs/DADs from the normalization or elevation of SERCA from PLN knockout or SERCA2a gene therapy.
Supplementary Material
Data accessibility
All supporting data and computer simulation source code can be accessed at the CELLML depository (https://models.physiomeproject.org/exposure/bbd802c6a6d6e69b746244f83b4fb89b/maleckar_greenstein_trayanova_giles_2009.cellml) and the GitHub repository (https://github.com/SirAndii/AndyHub/blob/master/MaleckarCaSubsystemControl.m).
Authors' contributions
A.L. and J.Z. conceived and designed the study. A.L. conducted the experiments and drafted the manuscript. A.L., J.B., P.A.G., V.V.F. and J.Z. interpreted the data, and reviewed, revised and approved the final version of this manuscript.
Competing interests
The authors have no competing interests.
Funding
This work was supported by the Health Research Council of New Zealand (16/385), the National Institutes of Health grant nos (HL115580 and HL135109), the National Natural Science Foundation of China (grant no. 61901192) and the National Key R&D Program of China (grant nos 2019YFC0120100 and 2019YFC0121900).
References
- 1.Feghaly J, Zakka P, London B, MacRae CA, Refaat MM. 2018. Genetics of atrial fibrillation. J. Am. Heart Assoc. 7, e009884 ( 10.1161/JAHA.118.009884) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benjamin EJ, Chen P-S, Bild DE, Mascette AM, Albert CM, Alonso A et al. . 2009. Prevention of atrial fibrillation: report from a national heart, lung, and blood institute workshop. Circulation 119, 606–618. ( 10.1161/CIRCULATIONAHA.108.825380) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Magnani JW, Hylek EM, Apovian CM. 2013. Obesity begets atrial fibrillation: a contemporary summary. Circulation 128, 401–405. ( 10.1161/CIRCULATIONAHA.113.001840) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asghar O, Alam U, Hayat SA, Aghamohammadzadeh R, Heagerty AM, Malik RA. 2012. Obesity, diabetes and atrial fibrillation; epidemiology, mechanisms and interventions. Curr. Cardiol. Rev. 8, 253–264. ( 10.2174/157340312803760749) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wasmer K, Eckardt L, Breithardt G. 2017. Predisposing factors for atrial fibrillation in the elderly. J. Geriatr. Cardiol. JGC. 14, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamilton S, Terentyev D. 2018. Proarrhythmic remodeling of calcium homeostasis in cardiac disease; implications for diabetes and obesity. Front. Physiol. 9, 1517 ( 10.3389/fphys.2018.01517) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, Wehrens XH, Nattel S, Dobrev D. 2014. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation 129, 145–156. ( 10.1161/CIRCULATIONAHA.113.006641) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwasaki Y-k, Nishida K, Kato T, Nattel S. 2011. Atrial fibrillation pathophysiology: implications for management. Circulation 124, 2264–2274. ( 10.1161/CIRCULATIONAHA.111.019893) [DOI] [PubMed] [Google Scholar]
- 9.Nattel S, Burstein B, Dobrev D. 2008. Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ. Arrhythmia Electrophysiol. 1, 62–73. ( 10.1161/CIRCEP.107.754564) [DOI] [PubMed] [Google Scholar]
- 10.Shanmugam M, Molina CE, Gao S, Severac-Bastide R, Fischmeister R, Babu GJ. 2011. Decreased sarcolipin protein expression and enhanced sarco (endo) plasmic reticulum Ca2+ uptake in human atrial fibrillation. Biochem. Biophys. Res. Commun. 410, 97–101. ( 10.1016/j.bbrc.2011.05.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Potpara TS, Lip GYH. 2015. A brief history of ‘lone’ atrial fibrillation: from ‘a peculiar pulse irregularity’ to a modern public health concern. Curr. Pharm. Des. 21, 679–696. ( 10.2174/1381612820666140929100209) [DOI] [PubMed] [Google Scholar]
- 12.Fox CS, Parise H, D'Agostino RB Sr, Lloyd-Jones DM, Vasan RS, Wang TJ, Levy D, Wolf PA, Benjamin EJ. 2004. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA 291, 2851–2855. ( 10.1001/jama.291.23.2851) [DOI] [PubMed] [Google Scholar]
- 13.Ellinor PT, Yoerger DM, Ruskin JN, MacRae CA. 2005. Familial aggregation in lone atrial fibrillation. Hum. Genet. 118, 179–184. ( 10.1007/s00439-005-0034-8) [DOI] [PubMed] [Google Scholar]
- 14.Arnar DO, Thorvaldsson S, Manolio TA, Thorgeirsson G, Kristjansson K, Hakonarson H, Stefansson K. 2006. Familial aggregation of atrial fibrillation in Iceland. Eur. Heart J. 27, 708–712. ( 10.1093/eurheartj/ehi727) [DOI] [PubMed] [Google Scholar]
- 15.Øyen N, Ranthe MF, Carstensen L, Boyd HA, Olesen MS, Olesen S-P, Wohlfahrt J, Melbye M. 2012. Familial aggregation of lone atrial fibrillation in young persons. J. Am. Coll. Cardiol. 60, 917–921. ( 10.1016/j.jacc.2012.03.046) [DOI] [PubMed] [Google Scholar]
- 16.Marcus GM, Smith LM, Vittinghoff E, Tseng ZH, Badhwar N, Lee BK, Lee RJ, Scheinman MM, Olgin JE. 2008. A first-degree family history in lone atrial fibrillation patients. Heart Rhythm 5, 826–830. ( 10.1016/j.hrthm.2008.02.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahida S. 2014. Genetic discoveries in atrial fibrillation and implications for clinical practice. ArrhythmiaElectrophysiol. Rev. 3, 69 ( 10.15420/aer.2014.3.2.69) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hucker WJ, Saini H, Lubitz SA, Ellinor PT. 2016. Atrial fibrillation genetics: is there a practical clinical value now or in the future? Can. J. Cardiol. 32, 1300–1305. ( 10.1016/j.cjca.2016.02.032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalstø SM, Siland JE, Rienstra M, Christophersen IE. 2019. Atrial fibrillation genetics update: towards clinical implementation. Front. Cardiovasc. Med. 6, 127 ( 10.3389/fcvm.2019.00127) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zang X, et al. 2013. SNP rs3825214 in TBX5 is associated with lone atrial fibrillation in Chinese Han population. PLoS ONE 8, e64966 ( 10.1371/journal.pone.0064966) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christophersen IE, et al. 2017. Fifteen genetic loci associated with the electrocardiographic P wave. Circ. Cardiovas. Genet. 10, e001667 ( 10.1161/CIRCGENETICS.116.001667) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tucker NR, Ellinor PT. 2014. Emerging directions in the genetics of atrial fibrillation. Circ. Res. 114, 1469–1482. ( 10.1161/CIRCRESAHA.114.302225) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahida S, Ellinor PT. 2012. New advances in the genetic basis of atrial fibrillation. J. Cardiovasc. Electrophysiol. 23, 1400–1406. ( 10.1111/j.1540-8167.2012.02445.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holm H, et al. 2010. Several common variants modulate heart rate, PR interval and QRS duration. Nat. Genet. 42, 117 ( 10.1038/ng.511) [DOI] [PubMed] [Google Scholar]
- 25.Garg V, et al. 2003. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424, 443 ( 10.1038/nature01827) [DOI] [PubMed] [Google Scholar]
- 26.Guo DF, et al. 2016. TBX5 loss-of-function mutation contributes to atrial fibrillation and atypical Holt-Oram syndrome. Mol. Med. Rep. 13, 4349–4356. ( 10.3892/mmr.2016.5043) [DOI] [PubMed] [Google Scholar]
- 27.Nadadur RD, et al. 2016. Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm. Sci. Trans. Med. 8, 354ra115 ( 10.1126/scitranslmed.aaf4891) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antzelevitch C, Burashnikov A. 2011. Overview of basic mechanisms of cardiac arrhythmia. Cardiac. Electrophysiol. Clin. 3, 23–45. ( 10.1016/j.ccep.2010.10.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai W, et al. 2019. A calcium transport mechanism for atrial fibrillation in Tbx5-mutant mice. eLife 8, e41814 ( 10.7554/eLife.41814) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marks AR. 2013. Calcium cycling proteins and heart failure: mechanisms and therapeutics. J. Clin. Invest. 123, 46–52. ( 10.1172/JCI62834) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lacombe VA, Viatchenko-Karpinski S, Terentyev D, Sridhar A, Emani S, Bonagura JD, Feldman DS, Gyorke S, Carnes CA. 2007. Mechanisms of impaired calcium handling underlying subclinical diastolic dysfunction in diabetes. Am. J. Physiol. Regul., Integr. Comp. Physiol. 293, R1787–R1797. ( 10.1152/ajpregu.00059.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Priebe L, Beuckelmann DJ. 1998. Simulation study of cellular electric properties in heart failure. Circ. Res. 82, 1206–1223. ( 10.1161/01.RES.82.11.1206) [DOI] [PubMed] [Google Scholar]
- 33.Bers DM, Eisner DA, Valdivia HH. 2003. Sarcoplasmic reticulum Ca2+ and heart failure: roles of diastolic leak and Ca2+ transport. Am. Heart Assoc. [DOI] [PubMed] [Google Scholar]
- 34.Popescu I, Yin G, Velmurugan S, Erickson JR, Despa F, Despa S. 2019. Lower sarcoplasmic reticulum Ca2+ threshold for triggering afterdepolarizations in diabetic rat hearts. Heart Rhythm 16, 765–772. ( 10.1016/j.hrthm.2018.11.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verkerk AO, Veldkamp MW, Baartscheer A, Schumacher CA, Klöpping C, van Ginneken AC, Ravesloot JH. 2001. Ionic mechanism of delayed afterdepolarizations in ventricular cells isolated from human end-stage failing hearts. Circulation 104, 2728–2733. ( 10.1161/hc4701.099577) [DOI] [PubMed] [Google Scholar]
- 36.Shanmugam M, Gao S, Hong C, Fefelova N, Nowycky MC, Xie L-H, Periasamy M, Babu GJ. 2010. Ablation of phospholamban and sarcolipin results in cardiac hypertrophy and decreased cardiac contractility. Cardiovasc. Res. 89, 353–361. ( 10.1093/cvr/cvq294) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karakikes I, Kim M, Hadri L, Sakata S, Sun Y, Zhang W, Chemaly ER, Hajjar RJ, Lebeche D. 2009. Gene remodeling in type 2 diabetic cardiomyopathy and its phenotypic rescue with SERCA2a. PLoS ONE 4, e6474 ( 10.1371/journal.pone.0006474) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lyon AR, et al. 2011. SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ. Arrhythmia Electrophysiol. 4, 362–372. ( 10.1161/CIRCEP.110.961615) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roe AT, Frisk M, Louch WE. 2015. Targeting cardiomyocyte Ca2+ homeostasis in heart failure. Curr. Pharm. Des 21, 431–448. ( 10.2174/138161282104141204124129) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fink M, Noble PJ, Noble D. 2011. Ca2+-induced delayed afterdepolarizations are triggered by dyadic subspace Ca2+ affirming that increasing SERCA reduces aftercontractions. Am. J. Physiol. Heart Circ. Physiol. 301, H921–HH35. ( 10.1152/ajpheart.01055.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.January CT, Fozzard H. 1988. Delayed afterdepolarizations in heart muscle: mechanisms and relevance. Pharmacol. Rev. 40, 219–227. [PubMed] [Google Scholar]
- 42.Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, Chen SRW. 2004. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc. Natl Acad. Sci. USA. 101, 13 062–13 067. ( 10.1073/pnas.0402388101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, Zhang L, Chen SRW. 2005. Enhanced store overload–induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ. Res. 97, 1173–1181. ( 10.1161/01.RES.0000192146.85173.4b) [DOI] [PubMed] [Google Scholar]
- 44.Tweedie D, Harding SE, MacLeod KT. 2000. Sarcoplasmic reticulum Ca2+ content, sarcolemmal Ca2+ influx and the genesis of arrhythmias in isolated guinea-pig cardiomyocytes. J. Mol. Cell. Cardiol. 32, 261–272. ( 10.1006/jmcc.1999.1070) [DOI] [PubMed] [Google Scholar]
- 45.Louch WE, et al. 2010. Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J. Physiol. 588, 465–478. ( 10.1113/jphysiol.2009.183517) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andersson KB, et al. 2009. Moderate heart dysfunction in mice with inducible cardiomyocyte-specific excision of the Serca2 gene. J. Mol. Cell. Cardiol. 47, 180–187. ( 10.1016/j.yjmcc.2009.03.013) [DOI] [PubMed] [Google Scholar]
- 47.Li L, Louch WE, Niederer SA, Aronsen JM, Christensen G, Sejersted OM, Smith NÂP. 2012. Sodium accumulation in SERCA knockout-induced heart failure. Biophys. J. 102, 2039–2048. ( 10.1016/j.bpj.2012.03.045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bai J, Gladding PA, Stiles MK, Fedorov VV, Zhao J. 2018. Ionic and cellular mechanisms underlying TBX5/PITX2 insufficiency-induced atrial fibrillation: insights from mathematical models of human atrial cells. Sci. Rep. 8, 15642 ( 10.1038/s41598-018-33958-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maleckar MM, Greenstein JL, Trayanova NA, Giles WR. 2008. Mathematical simulations of ligand-gated and cell-type specific effects on the action potential of human atrium. Prog. Biophys. Mol. Biol. 98, 161–170. ( 10.1016/j.pbiomolbio.2009.01.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nygren A, Fiset C, Firek L, Clark JW, Lindblad DS, Clark RB, Giles WR. 1998. Mathematical model of an adult human atrial cell: the role of K+ currents in repolarization. Circ. Res. 82, 63–81. ( 10.1161/01.RES.82.1.63) [DOI] [PubMed] [Google Scholar]
- 51.Nygren A, Leon L, Giles W. 2001. Simulations of the human atrial action potential. Phil. Trans. R. Soc. A 359, 1111–1125. ( 10.1098/rsta.2001.0819) [DOI] [Google Scholar]
- 52.Wilhelms M, Hettmann H, Maleckar MMC, Koivumäki JT, Dössel O, Seemann G. 2013. Benchmarking electrophysiological models of human atrial myocytes. Front. Physiol. 3, 487 ( 10.3389/fphys.2012.00487) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koivumäki JT, Takalo J, Korhonen T, Tavi P, Weckström M. 2009. Modelling sarcoplasmic reticulum calcium ATPase and its regulation in cardiac myocytes. Phil. Trans. R. Soc. A 367, 2181–2202. ( 10.1098/rsta.2008.0304) [DOI] [PubMed] [Google Scholar]
- 54.Laforest B, Dai W, Tyan L, Lazarevic S, Shen KM, Gadek M, Broman MT, Weber CR, Moskowitz IP. 2019. Atrial fibrillation risk loci interact to modulate Ca2+-dependent atrial rhythm homeostasis. J. Clin. Invest. 129, 4937–4950. ( 10.1172/JCI124231) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. 2001. Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, and residual β-adrenergic responsiveness. Circ. Res. 88, 1159–1167. ( 10.1161/hh1101.091193) [DOI] [PubMed] [Google Scholar]
- 56.Stambler BS, Fenelon G, Shepard RK, Clemo HF, Guiraudon CM. 2003. Characterization of sustained atrial tachycardia in dogs with rapid ventricular pacing-induced heart failure. J. Cardiovasc. Electrophysiol. 14, 499–507. ( 10.1046/j.1540-8167.2003.02519.x) [DOI] [PubMed] [Google Scholar]
- 57.Rizzi N, et al. 2008. Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: a complex arrhythmogenic cascade in a knock in mouse model. Circ. Res. 103, 298–306. ( 10.1161/CIRCRESAHA.108.171660) [DOI] [PubMed] [Google Scholar]
- 58.Song Y, Shryock JC, Belardinelli L. 2008. An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am. J. Physiol. Heart Circ. Physiol. 294, H2031–H20H9. ( 10.1152/ajpheart.01357.2007) [DOI] [PubMed] [Google Scholar]
- 59.Marban E, Robinson SW, Wier WG. 1986. Mechanisms of arrhythmogenic delayed and early afterdepolarizations in ferret ventricular muscle. J. Clin. Invest. 78, 1185–1192. ( 10.1172/JCI112701) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kass RS, Tsien RW. 1982. Fluctuations in membrane current driven by intracellular calcium in cardiac Purkinje fibers. Biophys. J. 38, 259–269. ( 10.1016/S0006-3495(82)84557-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fernandez-Tenorio M, Niggli E. 2018. Stabilization of Ca2+ signaling in cardiac muscle by stimulation of SERCA. J. Mol. Cell. Cardiol. 119, 87–95. ( 10.1016/j.yjmcc.2018.04.015) [DOI] [PubMed] [Google Scholar]
- 62.Thul R, Coombe S, Roderick HL, Bootman MD. 2012. Subcellular calcium dynamics in a whole-cell model of an atrial myocyte. Proc. Natl Acad. Sci. USA 109, 2150–2155. ( 10.1073/pnas.1115855109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marchena M, Echebarria B. 2018. Computational model of calcium signaling in cardiac atrial cells at the submicron scale. Front. Physiol. 9, 1760 ( 10.3389/fphys.2018.01760) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Neef S, et al. 2010. Novelty and significance. Circ. Res. 106, 1134–1144. ( 10.1161/CIRCRESAHA.109.203836) [DOI] [PubMed] [Google Scholar]
- 65.Voigt N, et al. 2012. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 125, 2059–2070. ( 10.1161/CIRCULATIONAHA.111.067306) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Puglisi JL, Bers DM. 2001. LabHEART: an interactive computer model of rabbit ventricular myocyte ion channels and Ca transport. Am. J. Physiol. Cell Physiol. 281, C2049–C2C60. ( 10.1152/ajpcell.2001.281.6.C2049) [DOI] [PubMed] [Google Scholar]
- 67.Devenyi RA, Sobie EA. 2016. There and back again: iterating between population-based modeling and experiments reveals surprising regulation of calcium transients in rat cardiac myocytes. J. Mol. Cell. Cardiol. 96, 38–48. ( 10.1016/j.yjmcc.2015.07.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bers DM, Shannon TR. 2013. Calcium movements inside the sarcoplasmic reticulum of cardiac myocytes. J. Mol. Cell. Cardiol. 58, 59–66. ( 10.1016/j.yjmcc.2013.01.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Louch WE, Stokke MK, Sjaastad I, Christensen G, Sejersted OM. 2012. No rest for the weary: diastolic calcium homeostasis in the normal and failing myocardium. Physiology 27, 308–323. ( 10.1152/physiol.00021.2012) [DOI] [PubMed] [Google Scholar]
- 70.Alsina KM, et al. 2019. Loss of protein phosphatase 1 regulatory subunit PPP1R3A promotes atrial fibrillation. Circulation 140, 681–693. ( 10.1161/CIRCULATIONAHA.119.039642) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All supporting data and computer simulation source code can be accessed at the CELLML depository (https://models.physiomeproject.org/exposure/bbd802c6a6d6e69b746244f83b4fb89b/maleckar_greenstein_trayanova_giles_2009.cellml) and the GitHub repository (https://github.com/SirAndii/AndyHub/blob/master/MaleckarCaSubsystemControl.m).