

Abstract
A variety of structurally and functionally distinct progestins is used in contraception and menopausal hormone therapy (MHT). Some progestins elicit off-target effects by binding to steroid receptors other than the progesterone receptor, which may impact their therapeutic and side-effect profiles. We directly compared the binding affinities, efficacies and potencies of selected progestins via the mineralocorticoid receptor (MR). We did not detect a significant difference in the affinities of medroxyprogesterone acetate (MPA), norethisterone acetate (NET-A), levonorgestrel (LNG), gestodene (GES), etonogestrel (ETG), nestorone (NES) and nomegestrel acetate (NoMAC) for the MR, while these were significantly lower compared to drospirenone (DRSP). While GES and NoMAC display affinities indistinguishable from progesterone (P4), the binding affinity of DRSP is significantly greater and all other progestins significantly lower than that of P4. Dose-response analyses showed that P4, GES and ETG display indistinguishable MR antagonist potencies for transactivation to the well-known MR antagonist spironolactone, while LNG, NoMAC and DRSP are significantly more potent than spironolactone and MPA, NET-A and NES are significantly less potent. Similar to our previous findings for NET-A, we show that LNG, GES, ETG and NES dissociate between transactivation and transrepression via the MR. Together our results provide strong evidence for progestin- and promoter-specific transcriptional effects via the MR, which are poorly predicted by relative binding affinities. A comparison of the binding affinities and potencies with reported free serum concentrations of progestins relative to the endogenous mineralocorticoid aldosterone, suggest that all progestins except MPA, NET-A and NES will likely compete with aldosterone for binding to the MR in vivo at doses used in hormonal therapy to elicit physiologically significant off-target effects.
Keywords: Mineralocorticoid receptor, progestins, contraception, menopausal hormone therapy
1. Introduction
For more than five decades, women have relied on hormonal therapies containing progestins as means of preventing unwanted pregnancies and to prevent endometrial hyperplasia induced by the estrogen component of menopausal hormone therapy (MHT) (reviewed in [1,2]). The beneficial effects of progestins have however been challenged by evidence of increased risk of developing invasive breast cancer, cardiovascular complications, venous thromboembolism and susceptibility to genital tract infections (reviewed in [1–3]). A more detailed understanding of progestin mechanisms are thus needed to understand these and other possible risks.
Progestins are synthetic compounds designed to mimic the actions of the endogenous female sex steroid progesterone (P4) by binding to the progesterone receptor (PR) [1,2]. However, some progestins also bind to other members of the steroid receptor family such as the glucocorticoid- (GR), androgen- (AR), mineralocorticoid receptor (MR) and estrogen- (ER)-α [4–7] (reviewed in [1–3]). It is thought that these off-target biological effects of progestins are responsible for some of the side-effects associated with the clinical use of progestins (reviewed in [1,2]). Not all progestins have been pharmacologically characterized in terms of their binding affinities, and relative efficacies and potencies for transactivation and transrepression via these steroid receptors. Our earlier studies characterized medroxyprogesterone acetate (MPA) and norethisterone acetate (NET-A), two of the oldest progestins available and extensively used in contraception by women in sub-Saharan Africa and South Africa, via the GR [4], MR [5] and AR [6]. Given that a number of other progestins with distinct structures are clinically available, we recently also characterized levonorgestrel (LNG), gestodene (GES), nestorone (NES), nomegestrel acetate (NoMAC) and drospirenone (DRSP), in parallel, in terms of binding affinities, relative efficacies and potencies via the AR and ER subtypes [7]. Collectively, results from these studies highlight the fact that progestins can have similar or differential effects via different steroid receptors, and that these effects do not always mimic those of P4. Given that progestins have been associated with negative effects on cardiovascular health (reviewed in [1,2,8]), and that activation of the MR by aldosterone (Ald) may be associated with harmful effects on the cardiovascular system [9,10] an understanding of the actions of individual progestins via the MR is crucial.
The MR is expressed in a number of tissues including the heart, kidney and vasculature, and its abnormal activation by Ald has been reported to result in increased blood pressure, a known risk factor for cardiovascular disease (CVD). Indeed, MR antagonists are suggested to be beneficial in the treatment of patients with CVD [11]. The ligand-activated MR can regulate the transcription of target genes by either increasing (transactivation) or decreasing (transrepression) transcription. In general, transactivation occurs when the ligand-activated MR binds to mineralocorticoid response elements (MREs) in the promoters of target genes, while transrepression occurs when the ligand-activated MR binds to DNA-bound transcription factors such as activator protein (AP)-1 and nuclear factor kappa B (NFκB) (reviewed in [12]). Findings from our previous study showed that while both MPA and NET-A are weak MR antagonists for transactivation, NET-A but not MPA is an agonist for transrepression via the MR [5]. Importantly, the study concluded that it is unlikely that MPA and NET-A would elicit effects via the MR in vivo. Whether these finding are also true for LNG, GES, etonogestrel (ETG), NES, NoMAC and DRSP is not known. The present study thus aimed to directly compare the binding affinities and transcriptional activities of these selected progestins relative to each other and P4 via the human MR exogenously expressed in COS-1 cells. MPA and NET-A were also included to perform a parallel comparison to the other progestins.
2. Materials and Methods
2.1. Cell culture and inducing compounds
The COS-1 monkey kidney cell line was obtained from the ATCC and was previously described [6]. Only mycoplasma-negative cells were used in experiments. P4, MPA, NET-A, LNG, GES, ETG, NES, NoMAC, DRSP, Ald, spironolactone and phorbol 12-myristate13-acetate (PMA) were obtained from Sigma-Aldrich, RSA. [3H]-Ald (83.4 Ci/mmol) was purchased from PerkinElmer Life and Analytical Science, RSA.
2.2. Plasmids
The human MR expression vector (pRS-hMR) [13] and pTAT-2xPRE-E1b-luciferase promoter-reporter construct [14] were kind gifts from Prof. R. Evans (Howard Hughes Medical Institute, La Jolla, USA) and Prof G. Jenster (Erasmus University of Rotterdam, Netherlands), respectively. The 7xAP-1-luciferase and 5xNFκB-luciferase plasmids were purchased from Stratagene (Houston, USA).
2.3. Whole cell binding assay
Competitive whole cell binding assays were performed in the COS-1 cell line as previously described [5,7]. Total binding ([3H]-Ald only) was determined by scintillation counting as counts per minute (cpm) and set as 100%. Specific bound [3H]-Ald was calculated as the difference between total and non-specific binding ([3H]-Ald plus 10 μM unlabelled Ald) and expressed as a relative % of total binding. Cpm values were normalized to the protein concentration (mg/ml) determined using the Bradford protein assay [15]. Ki ± SEM values for the competing ligands were determined from heterologous displacement curves using the EC50 value, the published Kd value for Ald [5] and the concentration of radiolabelled Ald, according to the equation by Cheng and Prusoff [16].
2.4. Luciferase reporter assays
Promoter-reporter assays were performed essentially as previously described for the MR [5], with a few modifications. Briefly, COS-1 cells expressing a human MR expression vector and pTAT-2xPRE-E1b-luciferase (transactivation) or 7xAP-1-luciferase or 5xNFκB-luciferase (transrepression) were treated for 24 hours as follows:
For transactivation: serum-free DMEM containing increasing concentrations of the test compounds (agonist activity) or 1 nM Ald in the absence and presence of increasing concentrations of the test compounds (antagonist activity); For transrepression: serum-free DMEM containing 10 ng/ml PMA and increasing concentrations of the test compounds (agonist activity). Cells were lysed and analyzed as previously described [7].
2.5. Data analysis and statistical analysis
Graph Pad Prism® software was used for data analysis. Non-linear regression and one site competition were used for binding assays, while non-linear regression and sigmoidal dose-response were used for luciferase reporter assays. Fixed Hill slopes of 1 (transactivation) or −1 (competitive binding and transrepression) were chosen. One-way ANOVA analysis of variance and Newman-Keuls (compares all pairs of columns) post-test were used for statistical analysis. Statistically significant differences are either indicated by different letters (a, b, c, etc.) where all those values that differ significantly from each other are assigned a different letter, while ns denotes no statistical significance (p>0.05). Figures show pooled results from at least three independent experiments performed in triplicate and error bars represent the standard error of the mean (SEM).
3. Results
3.1. All progestins, except DRSP, display binding affinities indistinguishable from each other for the human MR
Competitive whole cell binding assays were performed in COS-1 cells expressing exogenous human MR to determine and directly compare the equilibrium dissociation constants (Ki values) of the selected progestins relative to each other and P4 in the same model system. We show that MPA, NET-A, LNG, GES, ETG, NES and NoMAC have binding affinities statistically indistinguishable from each other, but have significantly lower affinities compared to DRSP (Fig. 1, Supplementary Table 1). Interestingly, results show that NoMAC displays an atypical binding curve in displacing only ~57% of the 3H-Ald. The affinities of GES and NoMAC are also indistinguishable from those of P4 and Ald, while that of DRSP is significantly greater, and that of all other progestins is significantly lower, than for P4 and Ald.
Fig. 1.
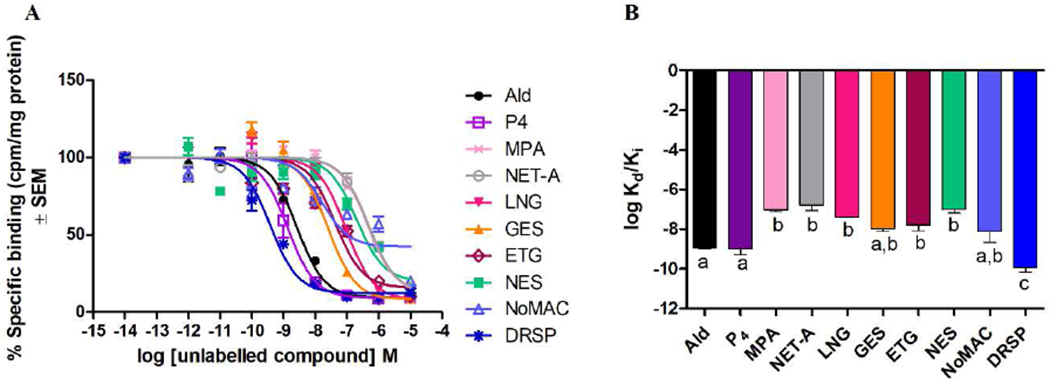
(A) Like P4, all the selected progestins compete with [3H]-Ald for binding to the human MR. COS-1 cells were transiently transfected with 11.25 μg of the human MR expression vector and incubated for 16 hours with 0.2 nM [3H]-Ald in the absence or presence of increasing concentrations of unlabelled ligands. Counts per minute (cpm) were normalized to the protein concentration (mg/ml). Total specific binding of [3H]-Ald only was expressed as 100% and the binding of unlabelled competitors expressed as a percentage relative to this. (B) Log Kd/Ki values of the ligands for the MR are plotted.
3.2. Most progestins display differential anti-mineralocorticoid potencies compared to each other and P4
Having shown that the selected progestins all bind the human MR, we next directly compared their relative agonist and antagonist efficacies (maximal responses) and potencies (EC50 values) for transactivation in COS-1 cells transiently transfected with the human MR and a MRE-driven reporter (Fig. 2, Supplementary Table 2). Unlike DRSP and P4 that display very weak agonist activity at high concentrations, no significant agonist activity is observed for any of the other progestins at concentrations of 0.1 nM to 10 μM (Fig. 2A and 2B). In contrast, all the progestins, like P4, can antagonize the effects of Ald (Fig. 2C). When comparing the different progestins to each other and P4, no significant difference in efficacy (Fig. 2D) is observed between P4, MPA, NET-A, LNG, GES, ETG, NES and DRSP. While NoMAC displays a significantly lower efficacy compared to GES and DRSP, it is as efficacious as P4 and the other progestins investigated in this study. As expected, the spironolactone-derived progestin, DRSP, displayed the greatest MR antagonist potency (Fig. 2E). This was followed by LNG and NoMAC, which are significantly more potent than P4, ETG and GES. Like MPA and NET-A, NES is a weak MR antagonist. Interestingly, NoMAC is significantly less efficacious than the well-known MR antagonist spironolactone, while P4 and all other progestins have efficacies indistinguishable from that of spironolactone. In terms of potency, however, LNG, NoMAC and DRSP are significantly more potent than spironolactone, MPA, NET-A and NES significantly less potent, and P4, GES and ETG indistinguishable from spironolactone.
Fig. 2.
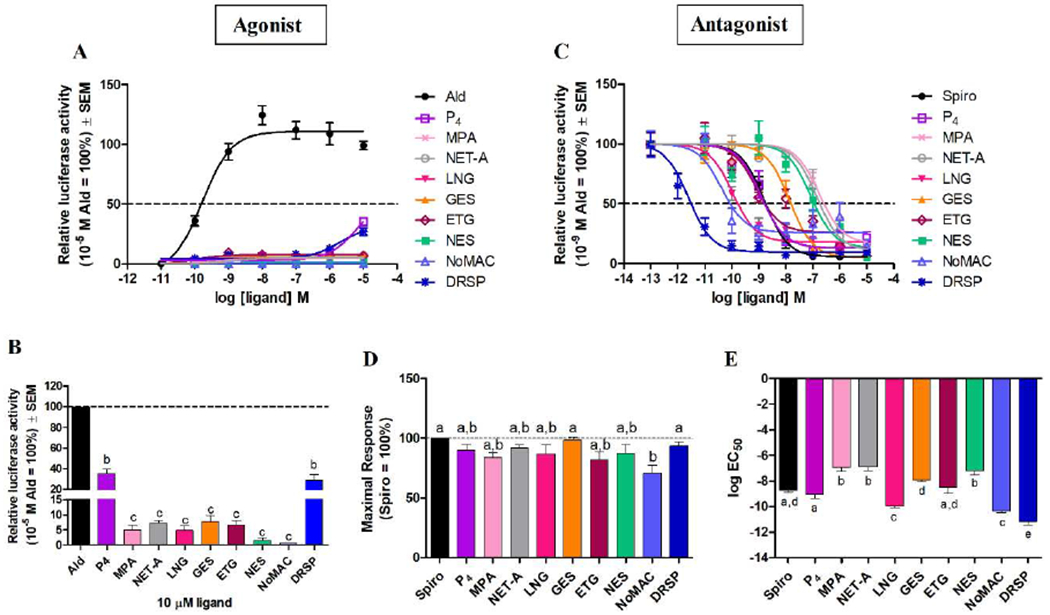
Most progestins display differential antagonist potencies for transactivation via the MR, while DRSP and P4 are weak partial MR agonists for transactivation. COS-1 cells, transiently transfected with 1 μg of the human MR expression vector and 10 μg of the pTAT-2xPRE-E1b-luciferase reporter plasmid, were treated with increasing concentrations of Ald, P4 or progestins in the absence (A) or (C) presence of 1 nM Ald (set as 100%) for 24 hours. (B) Induction by P4 and the progestins at 10 μM (from A) is shown as a percentage relative to 10 μM Ald expressed as 100%. Luciferase activity was measured in relative light units and normalized to the protein concentration. (D) Maximal responses and (E) log EC50 values of the ligands for the MR (from C) were plotted.
3.3. Progestins display indistinguishable MR agonist activity for transrepression via the synthetic NFκB promoter, but differential activity via the AP-1 promoter
We next investigated the comparative agonist efficacies (Supplementary Table 3) and potencies (Supplementary Table 4) of the selected progestins and P4 for transrepression in COS-1 cells with exogenously expressed human MR via AP-1 (Fig. 3A–3B) and NFκB (Fig. 3C–3D) containing promoter-reporter constructs. Like P4, all progestins except MPA and NoMAC are MR agonists via an AP-1-containing promoter-reporter construct (Fig. 3A and Supplementary Fig. 1A). Statistical analysis indicates that LNG, GES, ETG, NES and DRSP, but not NET-A, display agonist efficacies (Fig. 3A and Supplementary Fig. 1A) and potencies (Fig. 3B) that are indistinguishable from that of Ald via an AP-1 promoter. NET-A is as efficacious, but significantly less potent than P4, LNG, GES, ETG, NES and DRSP. All progestins and P4 display indistinguishable agonist efficacies (Fig. 3C and Supplementary Fig. 1B) compared to each other and to Ald, but significantly different potencies (Fig. 3D), via the NFκB-containing promoter. ETG is significantly more potent than MPA, NET-A and DRSP, but displays an indistinguishable potency compared to LNG, GES, NES and NoMAC.
Fig. 3.
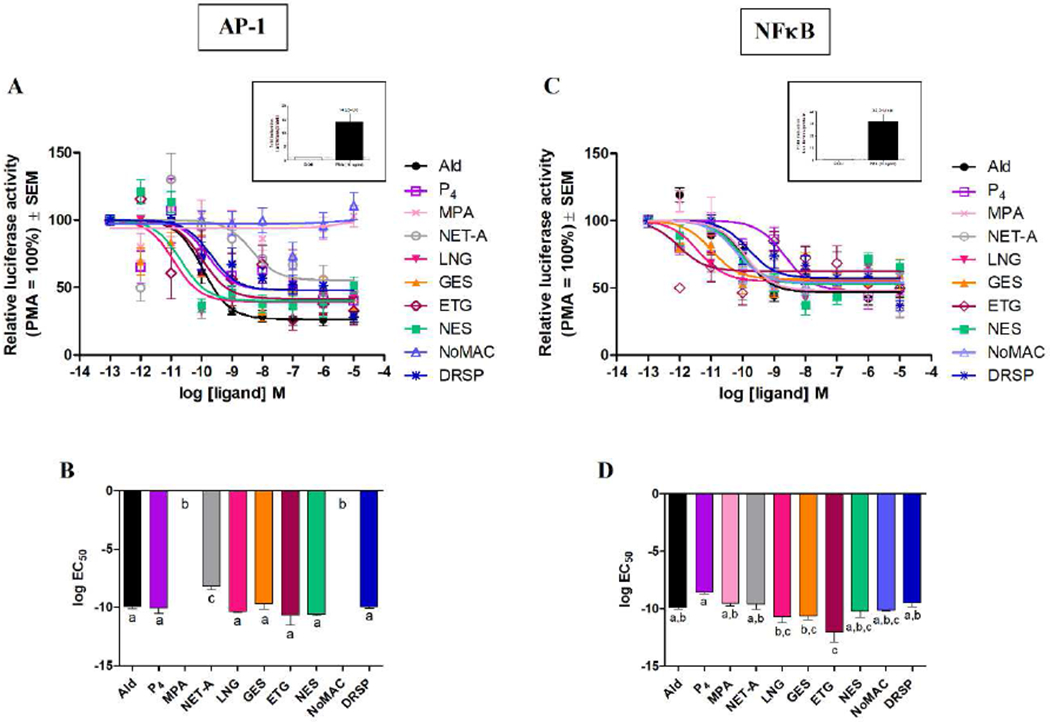
Most progestins display indistinguishable agonist activity for transrepression via the MR on the synthetic NFκB, but not the AP-1, promoter. COS-1 cells transiently transfected with 1.35 μg of the human MR expression vector and 2.7 μg of the (A) 7xAP-1-luciferase or (C) 5xNFκB-luciferase reporter plasmid, were treated with EtOH and 10 ng/ml PMA in the absence (set as 100%) or presence of increasing concentrations of ligands for 24 hours. Luciferase activity was measured and normalized as in Fig. 2. Treatment with 10 ng/ml PMA resulted in a ~14-fold and ~32-fold induction, respectively (Fig. 3A and 3C inserts). (B and D) Log EC50 values of the ligands for the MR were plotted.
4. Discussion
Few studies have investigated the binding and transcriptional activities of progestins via the human MR [17–22]; however, these available studies have several shortcomings. For example, relative binding affinities rather than precise equilibrium dissociation constants (Ki values) were determined [17,19,21]. In terms of transcriptional activity, most studies [17,18,20,22] except one previously published by our group [5], investigated the transactivation, but not transrepression, potential of some progestins via the MR. Although some of the available studies have pharmacologically characterized some progestins via the MR [17,18,20], studies directly comparing different progestins in parallel and in the same model system are lacking. Our study is the first to directly compare in parallel the Ki values, relative efficacies and potencies for transactivation and transrepression of a number of different progestins in a cell line model overexpressing the human MR and deficient of competing receptors. Although our study has limitations including the use of expressed MR and synthetic promoters, these are offset by the ability to directly compare effects via the MR in the absence of significant levels of competing steroid receptors [2,23], as well as allowing direct investigation of transcriptional effects via a specific cis-element, which is not possible by measuring mRNA levels of endogenous genes.
It has previously been shown that LNG, GES, ETG and DRSP can bind the human MR [17,19,21]; however, our study is the first to show binding of NES and NoMAC to the human MR and report accurate Ki values for LNG, GES, ETG, NES, NoMAC and DRSP. Although it has previously been shown that the structurally unique progestin DRSP has a higher affinity for the MR than Ald [19], we show that the affinity is also significantly greater than the rest of the progestins investigated. MPA, NET-A, LNG, GES, ETG, NES and NoMAC have binding affinities indistinguishable from each other. Since activation of the MR by Ald has been suggested to be linked to detrimental effects on the cardiovascular system [9,10], and MR antagonists are thought to be beneficial in terms of CVD [11], it is likely that a progestin with potent anti-mineralocorticoid activity may have a reduced CVD risk profile than progestins lacking anti-mineralocorticoid activity. Given that the binding of a ligand to a receptor does not always correlate with its biological activity [24], this study aimed to evaluate the biological activity of a number of different progestins in parallel. In agreement with previous studies [5,20] ([23] and references therein), our detailed dose-response analysis show that P4 elicits weak partial MR agonist activity at high concentrations, but MR antagonist activity at concentrations as low as that found in the serum of premenopausal women during the follicular phase (0.4 – 1.6 nM) [3]. Here we show, consistent with previous studies, that DRSP displays weak partial agonist activity indistinguishable from P4 [18], but is a significantly more potent anti-mineralocorticoid than P4 [22] for transactivation. The latter is not surprising as it is known that DRSP is a derivative of the well-known MR antagonist spironolactone [18,22]. Similar to previous studies, our results show that MPA, NET-A, LNG, GES, ETG and NoMAC have negligible MR agonist activity, while MPA, NET-A, LNG and GES are weak MR antagonists for transactivation [5,17,20,23,25,26]. Our study is however, the first to show that NoMAC and ETG display anti-mineralocorticoid properties and that NES displays negligible MR agonist activity, and like MPA and NET-A, is a weak MR antagonist. We show that DRSP displays the greatest MR antagonist potency, followed by LNG and NoMAC, which are more potent than ETG and GES, while MPA, NET-A and NES are the weakest MR antagonists. These findings suggest that although DRSP may be the ideal progestin to use in terms of CVD risk, the use of LNG, NoMAC, ETG and GES, but not MPA, NET-A and NES, may also be favorable. Like us, Sasagawa and co-workers also showed , that P4 has a similar potency to spironolactone, while MPA and NET are ~ 100-fold less potent [20]. However, our study is the first to show no significant difference between the potencies of GES, ETG and spironolactone, while NES is significantly less potent and LNG, NoMAC and DRSP significantly more potent, than spironolactone. While only statistically significant differences are discussed, it is important to note that there may be other significant differences that are not within the statistical power of the current experiments.
This study is the first to directly compare efficacy and potency values for a number of different progestins for transrepression via the MR. Although we have previously shown that MPA cannot transrepress via a synthetic AP-1 promoter [5], results from the current study show that it can however repress via a synthetic NFκB promoter. Similarly, NoMAC displays agonist activity via the synthetic NFκB, but not the AP-1, promoter. Moreover, we show that the agonist efficacies of all the progestins are indistinguishable from P4 and Ald for transpression via a synthetic NFκB promoter, while significant differential efficacies were observed for some via the synthetic AP-1 promoter. Although NET-A was significantly less efficacies than Ald, no difference was detected in its efficacy compared to P4, LNG, GES, ETG, NES and DRSP for transrepression via AP-1. In terms of potency, NET-A was the least potent, while the potencies of P4, LNG, GES, ETG, NES and DRSP were indistinguishable via the AP-1 promoter. Via the NFκB promoter, ETG was as potent as LNG, GES, NES and NoMAC, but significantly more potent than MPA, NET-A and DRSP. Our results showing differential effects of progestins highlights the importance of investigating multiple progestins in parallel in the same model system and that progestins should not be classified as a single treatment group. Collectively our results show progestin- and promoter-specific transcriptional effects via the MR and provide evidence that relative binding affinity is a poor predictor of biological activity.
Taken together, our results show that similar to our previous findings for NET-A, the progestins LNG, GES, ETG and NES can dissociate between transactivation and transrepression via the MR (Supplementary Table 5). Considering that the MR plays a role in inflammation [9,27] and that inflammation is a driver of CVD [28], these results could indicate, as suggested for NET-A in our previous study [5], that LNG, GES, ETG and NES may be promising anti-inflammatory agents where the MR is involved. Whether this is in fact physiologically possible for these progestins will depend on their serum concentrations, whether they bind to serum binding proteins, and their likelihood to compete with Ald in vivo for binding to the MR. The availabilities and serum concentrations reported for Ald and the selected progestins in both contraception and MHT users are summarized in Supplementary Table 6. Thus, considering that the serum concentration (0.24 – 0.46 nM) of Ald is ~10-500 fold lower than that of the progestins evaluated in the current study, together with the fact that the Ki and EC50 values we determined for LNG, GES, ETG, NoMAC and DRSP are similar or even lower than the serum concentrations reported (Supplementary Table 6), it is likely that these progestins will compete with Ald for binding to the MR in vivo. Our results suggest that NES, like MPA and NET-A [5], is however unlikely to elicit significant biological effects via the MR in vivo. While it remains to be determined whether the transcriptional effects observed in our study are mimicked on endogenous genes via the endogenous MR, our study provides proof of concept that these effects are likely to be progestin- and promoter-specific.
Supplementary Material
Highlights.
Progestin binding affinities for the MR do not predict their biological activities
Like P4, all progestins except MPA, NET-A and NES are potent MR antagonists
All progestins and P4 transrepress via a NFκB, but not AP-1, promoter reporter gene
LNG, GES, ETG and NES dissociate between transactivation and transrepression via MR
Acknowledgments
Funding: This work was supported by a grant to JH from the National Institute of Health (Grant No: R01 HD083026-02S1), where DA is a subawardee.
Abbreviations:
- AP-1
activator protein 1
- Ald
aldosterone
- DRSP
drospirenone
- ETG
etonogestrel
- GES
gestodene
- LNG
levonorgestrel
- MPA
medroxyprogesterone acetate
- MHT
menopausal hormone therapy
- MR
mineralocorticoid receptor
- MRE
mineralocorticoid response element
- NES
nestorone
- NoMAC
nomegestrol acetate
- NET-A
norethisterone acetate
- NFκB
nuclear factor kappa B
- PMA
phorbol 12-myristate 13-acetate
- P4
progesterone
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Africander D, Verhoog N, Hapgood JP, Molecular mechanisms of steroid receptor-mediated actions by synthetic progestins used in HRT and contraception, Steroids. 76 (2011) 636–652. 10.1016/j.steroids.2011.03.001. [DOI] [PubMed] [Google Scholar]
- [2].Stanczyk FZ, Hapgood JP, Winer S, Mishell DR, Progestogens Used in Postmenopausal Hormone Therapy: Differences in Their Pharmacological Properties, Intracellular Actions, and Clinical Effects, Endocr. Rev 34 (2013) 171–208. 10.1210/er.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hapgood JP, Kaushic C, Hel Z, Hormonal contraception and HIV-1 acquisition: Biological mechanisms, Endocr. Rev 39 (2018) 36–78. 10.1210/er.2017-00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Koubovec D, Ronacher K, Stubsrud E, Louw A, Hapgood JP, Synthetic progestins used in HRT have different glucocorticoid agonist properties, Mol. Cell. Endocrinol 242 (2005) 23–32. 10.1016/j.mce.2005.07.001. [DOI] [PubMed] [Google Scholar]
- [5].Africander D, Louw R, Hapgood JP, Investigating the anti-mineralocorticoid properties of synthetic progestins used in hormone therapy, Biochem. Biophys. Res. Commun 433 (2013) 305–310. 10.1016/j.bbrc.2013.02.086. [DOI] [PubMed] [Google Scholar]
- [6].Africander DJ, Storbeck K-H, Hapgood JP, A comparative study of the androgenic properties of progesterone and the progestins, medroxyprogesterone acetate (MPA) and norethisterone acetate (NET-A), J. Steroid Biochem. Mol. Biol 143 (2014) 404–415. 10.1016/j.jsbmb.2014.05.007. [DOI] [PubMed] [Google Scholar]
- [7].Louw-du Toit R, Perkins MS, Hapgood JP, Africander D, Comparing the androgenic and estrogenic properties of progestins used in contraception and hormone therapy, Biochem. Biophys. Res. Commun 491 (2017) 140–146. 10.1016/j.bbrc.2017.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lopez-Pier MA, Lipovka Y, Koppinger MP, Harris PR, Konhilas JP, The clinical impact of estrogen loss on cardiovascular disease in menopausal females, Med. Res. Arch 6 (2018) 1–18. 10.18103/mra.v6i2.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gorini S, Kim SK, Infante M, Mammi C, La Vignera S, Fabbri A, Jaffe IZ, Caprio M, Role of Aldosterone and Mineralocorticoid Receptor in Cardiovascular Aging, Front. Endocrinol. (Lausanne). 10 (2019) 1–11. 10.3389/fendo.2019.00584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cannavo A, Bencivenga L, Liccardo D, Elia A, Marzano F, Gambino G, D’Amico ML, Perna C, Ferrara N, Rengo G, Paolocci N, Aldosterone and mineralocorticoid receptor system in cardiovascular physiology and pathophysiology, Oxid. Med. Cell. Longev 2018 (2018). 10.1155/2018/1204598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pitt B, Ferreira JP, Zannad F, Mineralocorticoid receptor antagonists in patients with heart failure: Current experience and future perspectives, Eur. Hear. J. - Cardiovasc. Pharmacother 3 (2017) 48–57. 10.1093/ehjcvp/pvw016. [DOI] [PubMed] [Google Scholar]
- [12].Deng L, Hein L, Lother A, Transcriptional Regulation and Epigenetics in Cardiovascular Cells: Role of the Mineralocorticoid Receptor, in: Cell Biol. to Transl. Med. Brian Harvey Frederic Jaisser, IntechOpen, 2019: pp. 1–17. [Google Scholar]
- [13].Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM, Cloning of Human Mineralocorticoid Receptor Complementary DNA : Structural and Functional Kinship with the Glucocorticoid Receptor, Science (80-. ). 237 (1987) 268–275. [DOI] [PubMed] [Google Scholar]
- [14].Jenster G, Spencer TE, Burcin MM, Tsai SY, Tsai M-J, O’Malley BW, Steroid receptor induction of gene transcription: A two-step model, Proc. Natl. Acad. Sci 94 (1997) 7879–7884. 10.1073/pnas.94.15.7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bradford MM, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem 72 (1976) 248–254. 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- [16].Yung-Chi C, Prusoff WH, Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction, Biochem. Pharmacol 22 (1973) 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- [17].Fuhrmann U, Slater EP, Fritzemeier KH, Characterization of the novel progestin gestodene by receptor binding studies and transactivation assays, Contraception. 51 (1995) 45–52. 10.1016/0010-7824(94)00003-F. [DOI] [PubMed] [Google Scholar]
- [18].Fuhrmann U, Krattenmacher R, Slater EP, Fritzemeier K-H, The novel progestin drospirenone and its natural counterpart progesterone: Biochemical profile and antiandrogenic potential, Contraception. 54 (1996) 243–251. 10.1016/S0010-7824(96)00195-3. [DOI] [PubMed] [Google Scholar]
- [19].Krattenmacher R, Drospirenone: Pharmacology and pharmacokinetics of a unique progestogen, Contraception. 62 (2000) 29–38. 10.1016/S0010-7824(00)00133-5. [DOI] [PubMed] [Google Scholar]
- [20].Sasagawa S, Shimizu Y, Kami H, Takeuchi T, Mita S, Imada K, Kato S, Mizuguchi K, Dienogest is a selective progesterone receptor agonist in transactivation analysis with potent oral endometrial activity due to its efficient pharmacokinetic profile, Steroids. 73 (2008) 222–231. 10.1016/j.steroids.2007.10.003. [DOI] [PubMed] [Google Scholar]
- [21].Philibert D, Bouchoux F, Degryse M, Lecaque D, Petit F, Gaillard M, The pharmacological profile of a novel norpregnane progestin (trimegestone), Gynecol. Endocrinol 13 (1999) 316–326. 10.3109/09513599909167574. [DOI] [PubMed] [Google Scholar]
- [22].Winneker RC, Bitran D, Zhang Z, The preclinical biology of a new potent and selective progestin: Trimegestone, Steroids. 68 (2003) 915–920. 10.1016/S0039-128X(03)00142-9. [DOI] [PubMed] [Google Scholar]
- [23].Hapgood JP, Africander D, Louw R, Ray RM, Rohwer JM, Potency of progestogens used in hormonal therapy: Toward understanding differential actions, J. Steroid Biochem. Mol. Biol 142 (2014) 39–47. 10.1016/j.jsbmb.2013.08.001. [DOI] [PubMed] [Google Scholar]
- [24].Ronacher K, Hadley K, Avenant C, Stubsrud E, Simons SS, Louw A, Hapgood JP, Ligand-selective transactivation and transrepression via the glucocorticoid receptor: Role of cofactor interaction, Mol. Cell. Endocrinol 299 (2009) 219–231. 10.1016/j.mce.2008.10.008. [DOI] [PubMed] [Google Scholar]
- [25].van Diepen HA, Preclinical pharmacological profile of nomegestrol acetate, a synthetic 19-nor-progesterone derivative, Reprod. Biol. Endocrinol 10 (2012) 1–12. 10.1186/1477-7827-10-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Van Diepen HA, Lam TW, Kuil CW, Nomegestrol acetate: Steroid receptor transactivation profile in Chinese hamster ovary cells and ovulation inhibition in rat and monkey, Contraception. 84 (2011) 199–204. 10.1016/j.contraception.2010.11.017. [DOI] [PubMed] [Google Scholar]
- [27].Yagi S, Akaike M, Aihara KI, Fukuda D, Ishida M, Ise T, Niki T, Sumitomo-Ueda Y, Yamaguchi K, Iwase T, Taketani Y, Yamada H, Soeki T, Wakatsuki T, Shimabukuro M, Sata M, Pharmacology of aldosterone and the effects of mineralocorticoid receptor blockade on cardiovascular systems, Acta Cardiol. Sin 29 (2013) 201–207. [PMC free article] [PubMed] [Google Scholar]
- [28].Blake GJ, Ridker PM, Inflammatory bio-markers and cardiovascular risk prediction, J. Intern. Med 252 (2002) 283–294. 10.1046/j.1365-2796.2002.01019.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.