

Abstract
The incidence of cystic fibrosis (CF) and the frequency of the variants reported for CFTR depend on the population; furthermore, CF symptomatology is characterized by obstructive lung disease and pancreatic insufficiency among other symptoms, which are reliant on the individual's genotype. The Ecuadorian population is a mixture of Native Americans, Europeans, and Africans. That population admixture could be the reason for the new mutations reported in a previous study by Ruiz et al. (2019). A panel of 46 Ancestry Informative Markers was used to estimate the ancestral proportions of each available sample (12 samples in total). As a result, the Native American ancestry proportion was the most prevalent in almost all individuals, except for three patients from Guayaquil with the mutation [c.757G>A:p.Gly253Arg; c.1352G>T:p.Gly451Val] who had the highest European composition.
1. Introduction
Cystic fibrosis (CF) is an autosomal recessive disorder that has been extensively studied among populations [1]. It is characterized by obstructive lung disease, pancreatic insufficiency, diabetes, and liver disease, among others [2]. The most frequent worldwide mutation in Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) protein gene is c.1521_1523delCTT (p.Phe508del) [3] which originated between 11,000 and 34,000 years ago in Europeans, then it spread across all Europe [4]. CF occurs in 1 out of 2,500 live births with high prevalence in the European ancestry, and the frequency of the heterozygotes has been reported as 1 in 25 in Europeans [5] [6]. There are plenty of studies in CF, yet the majority in Europeans, underrepresenting the Latin Americans [4] [6] [7] [8]. In the United States, a study reported the CF incidence to be 1 in 9,200 Hispanics and 1 in 10,900 Native Americans, yet the USA has a different population structure to South America [1] [6] [9]. In general, in Latin America, the incidence is 1 per 6,000 live newborns; specifically, Ecuador exhibits an incidence of 1 in 11,252 newborns [10–12].
The Ecuadorian population, located in the northwest of South America, is a mixed population conformed by Native Americans, Europeans who arrived in the 16th century during the conquest, and Africans who came with them as slaves. According to the last census, the population projection for 2020 was estimated as 17,510,643 Ecuadorians. Moreover, Ecuadorian self-identified as “mestizos” 71.9%, “montubios” 7.4%, Afro-Ecuadorian 7.2%, “indígenas” 7%, “blancos” 6.1%, and others (0.4%) [13]. There are also reports of the Ecuadorian ancestry using AIMs in the mestizo population where Native American was the most prevalent ancestry (59.6%), followed by European (28.8%) and lastly African (11.6%) [14] [15].
Like other South American studies, Ecuador is underrepresented in cystic fibrosis research, and none of them involve the comparison of the mutations with the population's origin. Paz-y-Miño et.al (1999) reported 10 cases of Ecuadorian CF patients; at least 60% of the mutations differ from c.1521_1523delCTT (p.Phe508del) [16]. Valle et al. (2007) analyzed 62 Ecuadorian CF patients; the most prevalent mutation was F508del (37.1%) [12]. The last report by Ortiz et al. (2017), which included 48 Ecuadorian individuals with CF, reported F508del with the highest frequency (20.27%) [17]. These studies, however, are mainly focused on the particular F508 mutation, revealing that the percentage is not relatively high as in Europeans. The incidence and the frequency of the CF mutation depend on the population under study; Ecuadorians are a mestizo population, and the population's composition is not clear yet.
Here, we provide the ancestry origin data of 46 Ancestry Informative Markers of the individuals with the new mutations reported in a previous study of CF patients from Ecuador [18]. We aimed to elucidate if the mutations reported are mainly from European ancestry, due to the previous data of the main incidence.
2. Main Text
2.1. Methods
2.1.1. Samples and DNA Extraction
Twelve CF patients from Guayaquil (coast) and Cuenca (highland) who were available and had new CFTR disease-causing variants reported in a previous study were selected: one patient from Guayaquil with c.1473T>A:p.Cys491∗, one patient from Guayaquil and two from Cuenca with c.2672del:p.Asp891Alafs∗15, one patient from Cuenca with c.1486T>C:p.Trp496Arg, and six patients from Guayaquil and one from Cuenca with [c.757G>A:p.Gly253Arg; c.1352G>T:p.Gly451Val] were selected [18]. DNA was extracted using Chelex 100 (Bio-Rad) (10%) from peripheral blood samples collected on FTA cards (GE Healthcare Life Sciences) and quantified using NanoDrop (Thermo Scientific). To protect the identity of the individuals, the samples were anonymized.
2.1.2. DNA Amplification
PCR amplification of the twelve CF samples and controls (positive: 2800 and negative) was performed using 46 AIMs-INDELs: MID-1470, MID-777, MID-196, MID-881, MID-3122, MID-548, MID-659, MID-2011, MID-2929, MID-593, MID-798, MID-1193, MID-1871, MID-17, MID-2538, MID-1644, MID-3854, MID-2275, MID-94, MID-3072, MID-772, MID-2313, MID-397, MID-1636, MID-51, MID-2431, MID-2264, MID-2256, MID-128, MID-15, MID-2241, MID-419, MID-943, MID-159, MID-2005, MID-250, MID-1802, MID-1607, MID-1734, MID-406, MID-1386, MID-1726, MID-3626, MID-360, MID-1603, and MID-2719 [19], in one multiplex reaction and following the standardized protocol of the laboratory. The fragment separation was carried out in 3500 Genetic Analyzers (Applied Biosystems). Data were collected with Data Collection v3 and visualized with GeneMapper v5.
2.2. Statistical Analyses
Data were analyzed with Structure v2.3.4 in order to estimate the ancestral proportions in the population; the runs consisted of a burn-in length of 10,000 followed by 10,000 Markov Chain Monte Carlo (MCMC) interactions. The option used was the admixture model (“use population information to test for migrants”). The cluster considered for the analysis was one to three (k = 1, k = 2, and k = 3) due to the historical background of the Ecuadorian population and according to the cluster identification by Zambrano et al. and Evanno et al. [20] [14].
Principal component analysis (PCA) was built with RStudio v1.1.453 to visualize the CF individuals' structure: the correlation between individuals under analysis and the reference population from HGDP-CEPH (Native Americans, Europeans, and Africans) subset H952 [19].
3. Results
The DNA quantification was optimal to perform the PCR (5-20 ng/μl). After the amplification, complete profiles were obtained. A total of 339 individuals (reference population and samples) were analyzed, assuming a clustering of three using the information to test for migrants, permuting 10,000 burn-in periods and 10,000 interactions, and a bar plot was obtained showing the main ancestral population analyzed (Figure 1).
Figure 1.
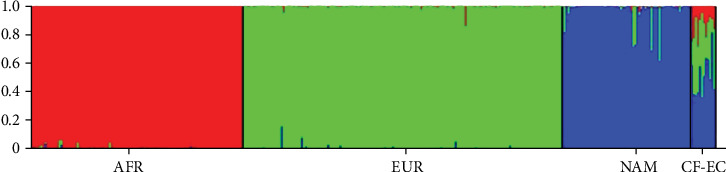
. Bar plot grouped by population identification. AFR: African ancestry; EUR: European ancestry; NAM: Native American ancestry; CF-EC: Ecuadorian cystic fibrosis patients. Three inferred clusters (K = 3).
Principal component analysis (PCA) results showed the three reference populations clearly differentiate between them. The CF Ecuadorian population is in the middle of them but mainly between the European and Native American reference populations. The two main principal components represented 38.86% of the total (Figure 2).
Figure 2.
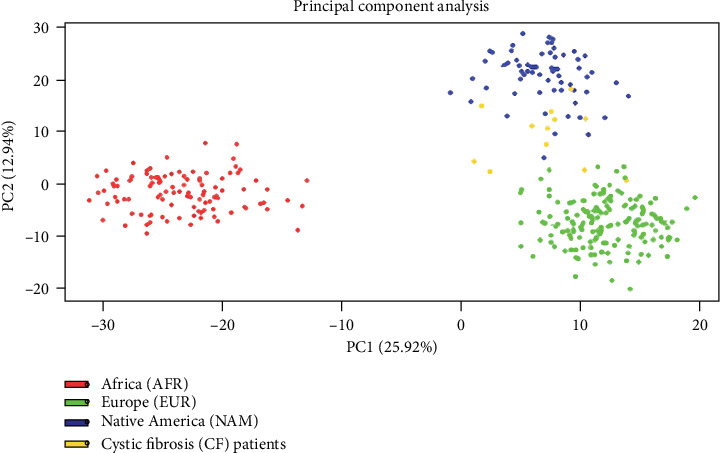
. Principal component analysis of Ecuadorian cystic fibrosis patients.
A percentage of the ancestral composition of each individual was obtained; as a result, a heterogeneous percentage was found depending on the individual and the region under study, thus clearly showing the admixture of the Ecuadorian population according to history (Table 1). The global ancestry composition of CF patients was the Native American 50% (standard deviation of 14.03), the European 35% (standard deviation of 15.5), and the African 11.5% (standard deviation of 7.82). The Native American ancestry was the first origin of almost all individuals, except for three patients from Guayaquil with the mutation [c.757G>A:p.Gly253Arg; c.1352G>T:p.Gly451Val] with the highest European composition.
Table 1.
Percentage of the ancestral composition of each individual under study.
| Patient | AFR | EUR | NAM | Mutation | Mutations' reference |
|---|---|---|---|---|---|
| 01 | 24.6 | 16.7 | 58.7 | c.1473T>A:p.Cys491∗ | [18] |
| 02 | 7.6 | 43.5 | 48.9 | c.2672del:p.Asp891Alafs∗15 | [18] |
| 03 | 21.6 | 14.8 | 63.6 | c.2672del:p.Asp891Alafs∗15 | [18] |
| 04 | 16 | 42.2 | 41.8 | c.2672del:p.Asp891Alafs∗15 | [18] |
| 05 | 9.1 | 8.9 | 82 | c.1486T>C:p.Trp496Arg | [18] |
| 06 | 22.5 | 39.6 | 37.9 | [c.757G>A:p.Gly253Arg; c.1352G>T:p.Gly451Val] | [18] |
| 07 | 7.2 | 55.3 | 37.5 | [c.757G>A:p.Gly253Arg; c.1352G>T:p.Gly451Val] | [18] |
| 08 | 28.2 | 31.9 | 39.9 | [c.757G>A:p.Gly253Arg; c.1352G>T:p.Gly451Val] | [18] |
| 09 | 5 | 58.7 | 36.3 | [c.757G>A:p.Gly253Arg; c.1352G>T:p.Gly451Val] | [18] |
| 10 | 11.9 | 37.2 | 50.9 | [c.757G>A:p.Gly253Arg; c.1352G>T:p.Gly451Val] | [18] |
| 11 | 9.3 | 32.1 | 58.6 | [c.757G>A:p.Gly253Arg; c.1352G>T:p.Gly451Val] | [18] |
| 12 | 11 | 25.7 | 63.3 | [c.757G>A:p.Gly253Arg; c.1352G>T:p.Gly451Val] | [18] |
AFR: African ancestry; EUR: European ancestry; NAM: Native American ancestry.
4. Discussion
The present study is the first report of the ancestral composition of CF Ecuadorian patients with new CFTR mutations. There are plenty of CF studies among different populations that revealed the differences between gender, age, and symptoms in CF patients. Some studies compared CF patients of different ages and gender describing that the incidence of CF in Europe is higher in children than in adults, approximately 4 CF children per 3 adults and around 1.1 males per each female [21] [22].
There are reports that evidenced CF prevalence differs depending on ethnicity. For instance, the incidence in Native Americans, whites and black individuals is 37.2, 38.8, and 17.1 per 100,000, respectively [23]. Moreover, other studies revealed the incidence among diverse ethnicities: as an example, the prevalence of CF reported by Rohlfs et al. (2011) is 1 in 242 Asian, 1 in 28 Caucasian, 1 in 59 Hispanic, and 1 in 70 Native American [2]. That study clearly revealed the ethnic differences in the incidence and the distribution of CF worldwide.
In Ecuador, there are studies about the ancestral origin; for instance, the Ecuadorian was reported to be composed of 59.6% Native American, 28.8% European, and lastly 11.6% African origin [14] [15].
In addition to the variable predisposition of CF among populations, reports exhibit a total of 2,063 mutations listed on the CFTR mutation database [24], while in the CFTR2 database, the most recent file updated on 8 December 2017 shows a total of 374 variants [25]. Those variants were identified in different populations in diverse frequencies. For instance, the frequencies of the most common variant c.1521_1523delCTT (p.Phe508del) depend on ethnicity; it was reported as 72% in US Caucasians, ~41% in African Americans, and 18% in Iranians, yet it also differs among Caucasians [26] [2] [27] [1] [28].
There are some mutations that have been commonly reported in ethnic groups; as examples, c.1624G>T (p.Gly542X) was reported in 43% of Turkish origin [29], while in a study in Peruvian patients, the frequency was 6.9% [30], c.3846G>A (p.Trp1282Ter) was reported in 43% of Ashkenazi patients [31], c.2988+1G>A (3120+1G>A) was reported in 12.3% of native African CF patients [32] [33], and c.3909C>G (p.Asn1303Lys) was described in 1.7% of the total number of CF analyzed from Europeans and the United States population [34], while in the Algerian population, the frequency was 20% [35]; c.1652G>A (p.Gly551Asp) presented a frequency of 3% in a north Brazilian population [36]. Furthermore, some mutations have been found in a specific ethnic group, like c.16C>G (p.Leu6Val) was found in one Argentinian and c.3294G>C (p.Trp1098Cys) was found in one Mexican, among other variations described [4]; c.3276C>G (p.Tyr1092Ter) was found in Jews from Iraq [31] [2].
In conclusion, the identification of ethnicity-dependent mutations would be an important aspect of CF testing in Ecuador. The present study exhibited a greater ancestral composition of Native American, followed by European and lastly African; the mixed population origin could possibly explain the new CF mutations reported.
4.1. Limitations
Although we have found the ancestral proportions of the majority of CF patients with new mutations previously reported, we could not access all the samples due to the available conditions of the patients. Moreover, a larger CF patient study with the commonly reported mutation should be conducted to better approximate the ancestral proportions of the patients.
Acknowledgments
The authors are grateful to the patients for their selfless participation in the present study and to all the people and institutions involved in the development of the present study.
Abbreviations
- CF:
Cystic fibrosis
- CFTR:
Cystic Fibrosis Transmembrane Conductance Regulator
- AIMs:
Ancestry Informative Markers
- INDELs:
Insertions and deletions
- PCA:
Principal component analysis
- AFR:
African ancestry
- EUR:
European ancestry
- NAM:
Native American ancestry.
Data Availability
The authors confirm that the data supporting the findings of this study are available within the article. The complete raw data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethical Approval
The patients were registered with the Ecuadorian Cystic Fibrosis Foundation at Guayaquil and Cuenca. The investigation was approved with the number 2018-127E by the “Comité de Ética de Investigación en Seres Humanos Universidad San Francisco de Quito.”
Consent
Patients provided informed consent, including the paternal signed authorization to participate in the study.
Conflicts of Interest
The authors declare that they have no competing interests.
Authors' Contributions
César Paz-y-Miño was responsible for coordination and followed up with the development of the article. Ana Karina Zambrano was responsible for the design, experimental procedure, data analysis, and writing. Juan Carlos Ruiz-Cabezas participated in the samples collection and written edition. Isaac Armendáriz-Castillo participated in the written edition. Jennyfer García-Cárdenas participated in the written edition. Santiago Guerrero participated in the written edition. Andrés López-Cortés participated in the written edition. Andy Pérez-Villa participated in the writing and formatting of the article. Patricia Guevara-Ramírez participated in the written edition. Verónica Yumiceba participated in the written edition. Paola E. Leone participated in the written edition. César Paz-y-Miño and Ana Karina Zambrano contributed equally to this work.
References
- 1.Heim R. A., Sugarman E. A., Allitto B. A. Improved detection of cystic fibrosis mutations in the heterogeneous U.S. population using an expanded, pan-ethnic mutation panel. Genetics in Medicine. 2001;3(3):168–176. doi: 10.1097/00125817-200105000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Rohlfs E. M., Zhou Z., Heim R. A., et al. Cystic fibrosis carrier testing in an ethnically diverse US population. Clinical Chemistry. 2011;57(6):841–848. doi: 10.1373/clinchem.2010.159285. [DOI] [PubMed] [Google Scholar]
- 3.Raskin S., Pereira-Ferrari L., Reis F. C., et al. Incidence of cystic fibrosis in five different states of Brazil as determined by screening of p.F508del, mutation at the CFTR gene in newborns and patients. Journal of Cystic Fibrosis. 2008;7(1):15–22. doi: 10.1016/j.jcf.2007.03.006. https://www.cysticfibrosisjournal.com/article/S1569-1993(07)00036-7/pdf. [DOI] [PubMed] [Google Scholar]
- 4.Pérez M. M., Luna M. C., Pivetta O. H., Keyeux G. CFTR gene analysis in Latin American CF patients: heterogeneous origin and distribution of mutations across the continent. Journal of Cystic Fibrosis. 2007;6(3):194–208. doi: 10.1016/j.jcf.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Pereira L., Raskin S., Freund A. A., et al. Cystic fibrosis mutations R1162X and 2183AA<FONT FACE=Symbol>®</FONT>G in two southern Brasilian states. Genetics and Molecular Biology. 1999;22(3):291–294. doi: 10.1590/S1415-47571999000300002. [DOI] [Google Scholar]
- 6.World Health Organization. The molecular genetic epidemiology of cystic fibrosis. 1983. pp. 1–24. https://www.who.int/genomics/publications/en/HGN_WB_04.02_report.pdf.
- 7.Ioannou L., McClaren B. J., Massie J., et al. Population-based carrier screening for cystic fibrosis: a systematic review of 23 years of research. Genetics in Medicine. 2014;16(3):207–216. doi: 10.1038/gim.2013.125. [DOI] [PubMed] [Google Scholar]
- 8.Ogino S., Flodman P., Wilson R. B., Gold B., Grody W. W. Risk calculations for cystic fibrosis in neonatal screening by immunoreactive trypsinogen and CFTR mutation tests. Genetics in Medicine. 2005;7(5):317–327. doi: 10.1097/01.GIM.0000162871.68167.8A. [DOI] [PubMed] [Google Scholar]
- 9.Hamosh A., FitzSimmons S. C., Macek M., Knowles M. R., Rosenstein B. J., Cutting G. R. Comparison of the clinical manifestations of cystic fibrosis in black and white patients. The Journal of Pediatrics. 1998;132(2):255–259. doi: 10.1016/S0022-3476(98)70441-X. [DOI] [PubMed] [Google Scholar]
- 10.Hernández P., González-Andrade F. Guía de práctica clínica (GPC) y manual de procedimientos fibrosis quística. 2013. p. p. 94. Jun 2018 http://instituciones.msp.gob.ec/documentos/Guias/fibrosis.pdf.
- 11.Silva Filho L. V. R. F., Castaños C., Ruíz H. H. Cystic fibrosis in Latin America—improving the awareness. Journal of Cystic Fibrosis. 2016;15(6):791–793. doi: 10.1016/j.jcf.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 12.Valle E. P., Burgos R. I., Valle J. R., Egas Béjar D., Ruiz-Cabezas J. C. Analysis of CFTR gene mutations and cystic fibrosis incidence in the Ecuadorian population. Investigación Clínica. 2007;48(1):91–98. [PubMed] [Google Scholar]
- 13.INEC Poster INEC. Censo de población y vivienda. 2010. http://www.ecuadorencifras.gob.ec/censo-de-poblacion-y-vivienda.
- 14.Zambrano A. K., Gaviria A., Cobos-Navarrete S., et al. The three-hybrid genetic composition of an Ecuadorian population using AIMs- InDels compared with autosomes, mitochondrial DNA and Y chromosome data. Scientific Reports. 2019;9(1):p. 9247. doi: 10.1038/s41598-019-45723-w. http://www.nature.com/scientificreports. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zambrano A. K., Gaviria A., Vela M., et al. Ancestry characterization of Ecuador’s highland mestizo population using autosomal AIM-INDELs. Forensic Science International: Genetics Supplement Series. 2017;6:e477–e478. doi: 10.1016/j.fsigss.2017.09.191. [DOI] [Google Scholar]
- 16.Paz-y-Mino C., Perez J. C., Burgos R., Davalos M. V., Leone P. E. The ΔF508 mutation in Ecuador, South America. Human Mutation. 1999;14(4):348–350. doi: 10.1002/(SICI)1098-1004(199910)14:4<348::AID-HUMU11>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 17.Ortiz S. C., Aguirre S. J., Flores S., Maldonado C., Mejía J., Salinas L. Spectrum of CFTR gene mutations in Ecuadorian cystic fibrosis patients: the second report of the p.H609R mutation. Molecular Genetics & Genomic Medicine. 2017;5(6):751–757. doi: 10.1002/mgg3.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruiz-Cabezas J. C., Barros F., Sobrino B., et al. Mutational analysis of CFTR in the Ecuadorian population using next-generation sequencing. Gene. 2019;696:28–32. doi: 10.1016/j.gene.2019.02.015. [DOI] [PubMed] [Google Scholar]
- 19.Pereira R., Phillips C., Pinto N., et al. Straightforward inference of ancestry and admixture proportions through ancestry-informative insertion deletion multiplexing. PLoS One. 2012;7(1):p. e29684. doi: 10.1371/journal.pone.0029684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evanno G., Regnaut S., Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology. 2005;14(8):2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 21.di Sant’Agnese P. A., Davis P. B. Cystic fibrosis in adults: 75 cases and a review of 232 cases in the literature. The American Journal of Medicine. 1979;66(1):121–132. doi: 10.1016/0002-9343(79)90491-1. [DOI] [PubMed] [Google Scholar]
- 22.McCormick J., Green M. W., Mehta G., Culross F., Mehta A. Demographics of the UK cystic fibrosis population: implications for neonatal screening. European Journal of Human Genetics. 2002;10(10):583–590. doi: 10.1038/sj.ejhg.5200850. May 2018, https://www.nature.com/articles/5200850.pdf. [DOI] [PubMed] [Google Scholar]
- 23.Spoonhower K. A., Davis P. B. Epidemiology of cystic fibrosis. Clinics in Chest Medicine. 2016;37(1):1–8. doi: 10.1016/j.ccm.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 24.Dorfman R. Cystic Fibrosis Mutation Database: Statistics. Cystic Fibrosis Mutation Database. 2015 Apr 2019, http://www.genet.sickkids.on.ca/StatisticsPage.html. [Google Scholar]
- 25.The Hospital for Sick Children, Johns Hopkins University, The Hospital for Sick Children. CFTR2 clinical and functional translation if CFTR. 2019. Apr 2019, https://www.cftr2.org/
- 26.Palomaki G. E., Fitzsimmons S. C., Haddow J. E. Clinical sensitivity of prenatal screening for cystic fibrosis via _CFTR_ carrier testing in a United States panethnic population. Genetics in Medicine. 2004;6(5):405–414. doi: 10.1097/01.gim.0000139505.06194.39. [DOI] [PubMed] [Google Scholar]
- 27.Sugarman E. A., Rohlfs E. M., Silverman L. M., Allitto B. A. CFTR mutation distribution among U.S. Hispanic and African American individuals: Evaluation in cystic fibrosis patient and carrier screening populations. Genetics in Medicine. 2004;6(5):392–399. doi: 10.1097/01.gim.0000139503.22088.66. [DOI] [PubMed] [Google Scholar]
- 28.Alibakhshi R., Kianishirazi R., Cassiman J.-J., Zamani M., Cuppens H. Analysis of the CFTR gene in Iranian cystic fibrosis patients: identification of eight novel mutations. Journal of Cystic Fibrosis. 2008;7(2):102–109. doi: 10.1016/j.jcf.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 29.Lerer I., Sagi M., Cutting G. R., Abeliovich D. Cystic fibrosis mutations delta F508 and G542X in Jewish patients. Journal of Medical Genetics. 1992;29(2):131–133. doi: 10.1136/jmg.29.2.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aquino R., Protzel A., Rivera J., et al. Frequency of the most common mutations of the CFTR gene in Peruvian patients with cystic fibrosis using the ARMS-PCR technique. Revista Peruana de Medicina Experimental y Salud Pública. 2017;34(1):62–69. doi: 10.17843/rpmesp.2017.341.2767. [DOI] [PubMed] [Google Scholar]
- 31.Quint A., Lerer I., Sagi M., Abeliovich D. Mutation spectrum in Jewish cystic fibrosis patients in israel: implication to carrier screening. American Journal of Medical Genetics Part A. 2005;136A(3):246–248. doi: 10.1002/ajmg.a.30823. [DOI] [PubMed] [Google Scholar]
- 32.Padoa C., Goldman A., Jenkins T., Ramsay M. Cystic fibrosis carrier frequencies in populations of African origin. Journal of Medical Genetics. 1999;36(1):41–44. May 2018, http://www.ncbi.nlm.nih.gov/pubmed/9950364. [PMC free article] [PubMed] [Google Scholar]
- 33.Macek M., Jr., Mackova A., Hamosh A., et al. Identification of common cystic fibrosis mutations in African-Americans with cystic fibrosis increases the detection rate to 75% American Journal of Human Genetics. 1997;60(5):1122–1127. [PMC free article] [PubMed] [Google Scholar]
- 34.Osborne L., Santis G., Schwarz M., et al. Incidence and expression of the N1303K mutation of the cystic fibrosis (CFTR) gene. Human Genetics. 2004;89(6):653–658. doi: 10.1007/BF00221957. [DOI] [PubMed] [Google Scholar]
- 35.Loumi O., Baghriche M., Delpech M., Kaplan J. C., Bienvenu T. Analysis of the complete coding region of the CFTR gene in ten Algerian cystic fibrosis families. Human Heredity. 1999;49(2):81–84. doi: 10.1159/000022849. [DOI] [PubMed] [Google Scholar]
- 36.de Araújo F. G., Novaes F. C., dos Santos N. P. C., et al. Prevalence of deltaF508, G551D, G542X, and R553X mutations among cystic fibrosis patients in the north of Brazil. Brazilian Journal of Medical and Biological Research. 2005;38(1):11–15. doi: 10.1590/S0100-879X2005000100003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article. The complete raw data that support the findings of this study are available from the corresponding author upon reasonable request.