

Abstract
Peptide identification algorithms rely on the comparison between the experimental tandem mass spectrometry spectrum and the theoretical spectrum to identify a peptide from the tandem mass spectra. Hence, it is important to understand the fragmentation process and predict the tandem mass spectra for high-throughput proteomics research. In this study, a novel method was developed to predict the theoretical ion trap collision-induced dissociation (CID) tandem mass spectra of the singly, doubly, and triply charged tryptic peptides. The fragmentation statistics of the ion trap CID spectra were used to predict the theoretical tandem mass spectra of the peptide sequence. The study estimated the relative cleavage frequency for each pair of adjacent amino acids along the peptide length. The study showed that the cleavage frequency can be directly used to predict the tandem mass spectra. The predicted spectra show a high correlation with the experimental spectra used in this study; 99.73% of the high-quality reference spectra have correlation scores greater than 0.8. The new method predicts the theoretical spectrum and correlates significantly better with the experimental spectrum as compared to the existing spectrum prediction tools OpenMS_Simulator, MS2PIP, and MS2PBPI, where only 80, 85.76, and 85.80% of the spectral count, respectively, has a correlation score greater than 0.8.
1. Introduction
Tandem mass spectrometry is a widely used technique for proteomics data analysis, which includes sequencing and identification of peptides and proteins. In this approach, the proteins are digested by protease, and the peptides are subjected to precursor mass scans (MS1) to isolate the precursor. Tandem MS fragments the selected precursor ion from MS1 into smaller ions. Widely used methods for dissociating peptides along the peptide backbone in tandem MS include collision-induced dissociation (CID), and high-energy CID (HCD) generates predominantly b- and y-ions, the electron capture dissociation and electron transfer dissociation (ETD) generate predominantly c- and z–ions corresponding to N-terminal and C-terminal fragment ions, respectively. The mass to charge ratio and intensities of the fragment ions are recorded in the tandem mass spectrum.1,2 The mass of the fragment ions for any peptide is predictable, hence theoretical mass spectra can be constructed from the peptide sequence. The similarity-based peptide-identification method usually predicts the theoretical spectrum based on the sequence of a peptide and then compares it with the experimental spectrum.3 The predicted theoretical spectrum must be sufficiently similar to the experimental spectrum, for the accurate identification of peptides.
Peptide spectrum search algorithms such as SEQUEST3 and MASCOT4 identify the peptides by matching the experimental spectra with the predicted theoretical spectra of the candidate peptides available in the peptide sequence database. Many of such algorithms assign fixed theoretical intensities to the matched ions regardless of the sequences of the peptides, thus neglecting the intensity information contained in the experimental spectrum. The protein identification algorithm, SeQuence IDentfication (SQID), made use of intensity information obtained from the statistical analysis. It was shown that incorporating the fragment ion probabilities between amino acid pairs in the scoring algorithms would increase the peptide-identification rate.5 For many peptides, only some dominant peaks of fragment ions appear in the spectrum. The methods that are used to calculate the probabilistic score based on these assumptions may cause error in peptide spectrum match (PSM) ranking. Thus, the investigation of peptide-fragmentation patterns and prediction of accurate theoretical spectra of the peptide is an essential step in the peptide-identification algorithm.
Extensive research has been done to understand the relationship between the fragment ion intensities and the fragmentation pathway.6−8 Based on the mobile proton hypothesis,8 a kinetic model was developed by Zhang9 to predict the low-energy CID spectra from sequences. This is based on the fragmentation pathway and the rate of dissociation. Zhou et al.10 used a machine-learning technique, such as the Bayesian neural network approach, to find the features that potentially influence the peptide fragmentation and the subsequent intensity pattern of the fragmentation spectra. The PeptideART11 tool predicts the theoretical spectrum by learning the probability of the occurrence of each peak using a shallow feed-forward neural network. PepNOVO predicts the intensity ranks instead of relative intensities using learning-to-rank algorithms.12 Many such models are often specific to their training data and need to be retrained for specific laboratory conditions. These algorithms further require improvement in the perfect prediction of the spectrum for boosting the accuracy for identification of the peptides. The method based on the decision tree model and the hidden markov model was employed to predict the intensity of fragment ions.13−15 Furthermore, more advanced technologies like the deep neural network architecture were also implemented to predict the tandem mass spectrum of peptides.16 The decision tree model and deep learning requires a vast amount of data and requires a computationally demanding training process. Accurate prediction of the theoretical spectra can enhance the identification of peptides.5,17 However, the peptide-fragmentation behavior is a complex process and the fragmentation pattern also has dependency on the sequence of the peptide, charge state, and residue content18,19 causing difficulty in the accurate prediction of the tandem mass spectra.
The current study exploits the information from the vast amount of the PSM mass spectral data collected over the recent years for improved prediction of the tandem mass spectra. In our previously reported study of the CID fragmentation pattern,20,21 a large number of spectra with known sequences were analyzed, based on the cleavage position along the peptide backbone and pairwise amino acid at the cleavage site. The relative frequency of the occurrence of the fragment ion peaks was recorded based on the cleavage position and the residue pair at the fragmentation site. The results have verified the presence of the residue-specific cleavage preferences known earlier and have found new residual and positional cleavage preferences of the CID fragmentation pattern. In this work, the intensity of the fragment ion for a particular residue pair at a specific position along the length of the peptide is checked to be consistent with its statistical value of relative frequency of occurrence of the fragment ion peak. This approach has been followed in the current study to predict the tandem mass spectra of the peptide. The approach is analogous to the manual process of analyzing the tandem mass spectra by looking for known fragmentation motifs using the statistical information gathered. In order to measure the accuracy of the predicted spectra, the dot product is used to measure the similarity between the predicted theoretical spectra and the experimental spectra. The study also compared the new method with the existing tools like OpenMS_Simulator,17 MS2PIP,22 and MS2PBPI15 tool based on the similarity score.
2. Method
2.1. Datasets
The dataset 1 used to extract the frequency of occurrence of fragment ion peaks is collected from the NIST peptide spectral library. The library contains tandem mass spectra with known sequences. Also, each spectral peak is annotated with fragment ion labels b, y, and a—ions along with isotopic and neutral loss labels. The main intention of this collection is to demonstrate the utility of peptide ion fragmentation libraries, for the development of peptide-identifying applications.23 Out of 340,357 ion trap CID tandem mass spectra available in the human peptide spectral library in NIST, 131,601 ion trap CID tandem mass spectra consist of 87,661 mass spectra of doubly charged tryptic peptides, 14787 singly charged tryptic peptide, and 29153 triply charged tryptic peptides with no missed cleavage and of length 6–21 residues are used as dataset 1 for this study. Dataset 1 is used for training and extracting the frequency matrix, and datasets 2 & 3 are used for evaluating the performance of the new method.
The dataset 2 is collected from the spectral library of the ProteomeTool project (http://www.proteometools.org/).24 The spectral library contains the high-quality reference MS/MS spectra of synthetic peptides of the human proteome. From the spectral library, 11,122 ion trap CID spectra consist of 292, 5830, and 5000 distinct singly, doubly, and triply charged tryptic peptides, respectively, which are not available in dataset 1 and are randomly selected.
The dataset 3 is extracted from the Standard Protein Mix Database collected by Institute of System Biology, from a mixture of 18 purified proteins using Thermo Finnigan ESI-ITMS.25 8622 ion trap CID spectra identified using search engines, SEQUEST, and X!Tandem,26,27 with the validation score of peptide prophet probability > 0.99, are selected for this study. Dataset consists of 865, 5840, and 1917 singly, doubly, and triply charged precursor tandem mass spectra, respectively.
2.2. Methodology for Prediction and Validation of the Tandem Mass Spectra
The fragmentation pattern of the ion trap CID spectra was efficiently studied from the NIST reference spectral dataset. Using the statistical analysis method, the relative frequency of occurrence of fragment ion peaks with respect to the position of the cleavage site, and with respect to the residue pair of these sites, was calculated.20 The current study showed that the relative frequency information obtained can be directly used to predict the tandem mass spectra of the given peptide. A simple sequence-based method is used here to predict the spectrum with high accuracy. The spectrum prediction using frequency information has the following steps:
Step 1: Generate the relative frequency table: the relative frequency of the occurrence of fragment ions calculated from the dataset using eq 1(21)
| 1 |
where “t” represents the ion type, b-, a-, and y-ions. “rp” represents the residue pair at the cleavage site, and “p” represents the location of the fragmentation site along the length of the peptide or the number of residues present in the fragment ion. n ⊆ {peaks of (b-ion, bi, b + 18, b-17, b-18, b-34, b-35, b-36, b-44, b-45, b-46) when t = b-ion, peaks of (y-ion, yi, y-17, y-18, y-35, y-36, y-44, y-45, y-46) when t = y-ion-type peaks of (a-ion) when t = a-ion type }. Cn,rp,pt denotes the number of times each peak occurred in each position, corresponding to each amide bond residue pair at the fragmentation site calculated from the spectra. Nrp,p denotes the number of times each amide bond residue pair is observed in each position calculated from the peptide sequence.21 The relative frequency information provides the extent of occurrence of fragmentation and the generation of the neutral loss fragment ion on both N- and C- terminal sides of the amino acid residue in each position along the peptide length.
The frequency matrices for each of the b-ion, y-ion, and a-ion were created. The rows of the matrix contain residue pairs at the cleavage site. The columns of the matrix contain the position of the cleavage of fragment ions and their neutral loss peaks along the length of the peptide. The study focuses on tryptic peptides without any missed cleavages. Therefore, 362 possible residue pairs fill the rows of the matrix.20,21 The maximum length of the peptide selected for this study is 21. Therefore, there are 20 possible cleavage sites along the length of the peptide. For the b-ion, there are 11 possible peaks considered, such as b-ion, bi, b + 18, b-17, b-18, b-34, b-35, b-36, b-44, b-45, and b-46 which create 11 × 20 columns in the b-ion frequency table. For the y-ion, 9 possible peaks were considered such as y-ion, yi, y-17, y-18, y-35, y-36, y-44, y-45, and y-46, which create 9 × 20 columns in the y-ion frequency table. The a-ion matrix has 20 columns for each position along the peptide length. The matrix is filled with frequency information Fnt(rp, p) obtained using eq 1.
Step 2: Generate the tandem mass spectra of the given peptide: for the given peptide, the mass of the fragment ion produced by the CID fragmentation pattern is predictable. Hence, the theoretical mass spectra can be constructed from the peptide sequence. The mass of the fragment ions b-, y-, and a- and their neutral loss peaks are calculated along the length of the peptide. To those masses of the fragment ions of the peptide are assigned the corresponding frequency values obtained from the relative frequency table. The frequency values of the fragment ion saved in the frequency matrix form the intensity of the peaks in the predicted tandem mass spectrum.
Step 3: Correlating the predicted spectra with the experimental spectra: experimental spectrum is preprocessed using a two-step procedure. First, all peaks related to parent mass are removed. That is, the precursor mass peaks and their corresponding neutral loss peaks are removed. Next, the experimental spectral peak intensities are transformed into the natural logarithmic scale. The predicted spectrum and the experimental spectrum are converted into vectors, where the m/z (mass/charge) ratio forms the index of the vector. The maximum value of the intensity or frequency within that mass index tolerance is taken as the value of the vector. Intensity forms the values of the vector in the experimental spectral vector and relative frequency forms the values of the vector in the predicted spectrum vector. The dot product is used as an efficient method for spectral matching.28
The correlation of the predicted spectra with the experimental spectra is found using eq 2.
| 2 |
where IE denotes the intensity of the ions in the experimental spectra, FT denotes the corresponding frequency value of the fragment ion assigned in the theoretical predicted spectra.
3. Results and Discussion
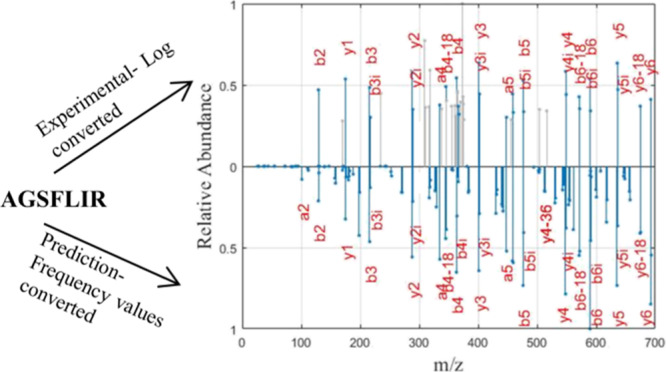
Our previous study shows the detailed study of the CID fragmentation pattern and the influence of the position and residue-specific cleavage preferences of CID fragmentation using the frequency values calculated from the large set of ion trap CID spectrum.21 In the present work, it is shown that the frequency values can be directly used to predict the theoretical spectrum. Because the mass of the fragment ion produced by the CID fragmentation pattern is predictable, the m/z value of the peaks of the theoretical mass spectra can be constructed from the peptide sequence. The frequency of occurrence of fragment ion values is used as the intensity of the theoretical predicted mass spectrum of a peptide, as mentioned in step 2 above. This is a very simplified and efficient method for predicting the tandem mass spectrum. The spectrum with these frequency values as intensity is shown to have a high correlation with the experimental spectrum. For example, the experimental and predicted spectra of the peptide “AHLWTYK” are shown in Figure 1.
Figure 1.

Experimental and predicted spectra of the peptide “AHLWTYK”: the x-axis represents m/z, and the y-axis represents the relative abundance of the fragment ions. The upper portion shows the experimental spectrum with the y-axis represented as the normalized value of the natural logarithm of the intensity of fragment ions. The lower portion represents the predicted spectrum with the y-axis representing the normalized value of the frequency values of fragment ions.
The experimental spectrum of peptide “AHLWTYK” shown in Figure 1 is the high-quality spectrum with fragment ions annotated. The spectrum is extracted from the proteome spectral library. From Figure 1, it can be seen that all the fragment ion peaks annotated in the experimental spectrum are also obtained in the predicted spectra with a similar magnitude of relative abundance. The fragment ion peaks corresponding to b-, a-, and y-, their isotopic peak, and their neutral loss fragments are also seen in the new theoretical spectrum. It is also noted that the correlation score between the two spectral vectors is 0.92. Hence, the new frequency-based method reliably predicts the spectrum of a given peptide and is highly correlated with the natural logarithmic transformed intensity of the experimental spectrum. A tandem mass spectrum usually has some dominant peaks with high intensity, and other informative peaks with much less intensity. The dominant peaks always diminish the information from less-intensity peaks and these factors add error to the intensity-based methods. By considering the frequency of occurrence of the fragment ion peaks, the intensity dominance can be reduced producing all the informative peaks in the spectrum. This provides how frequently a fragment ion occurs in the experimental spectrum related to the amino acids at the cleavage site and the position of the fragmentation site. The results shown in this study elucidate that fragment ion peaks in the predicted spectrum with the relative frequency values have a strong correlation with the log-transformed intensity of the peaks in the experimental spectrum.
Using the new frequency-based method, the 11,122 high-quality reference peptide spectra in dataset 2 and 8622 spectra in dataset 3 are evaluated, and the correlation scores are calculated. Distribution of correlation scores between the experimental and predicted spectra obtained for two datasets 2 and 3 are shown in Figure 2a,b, respectively. Figure 2a shows that for the high-quality reference spectra in dataset 2 extracted from proteomics DB, 99.73% predicted spectra have a correlation score greater than 0.8. For the dataset 3 having 8622 tandem mass spectra from the ISB protein mix database, 87.94% of the predicted spectra have a correlation score greater than 0.8.
Figure 2.

Distribution of correlation values: distribution of correlation scores of the predicted spectra of multi charged peptides, with respect to the experimental spectra in the datasets 2 & 3 are shown in Figure 2a,b, respectively. The x-axis shows the correlation score, and the y-axis shows the number of spectra. (a) Dataset 2: proteome DB spectral library, (b) dataset 3: ISB protein mix.
The predicted theoretical spectra of singly, doubly, and triply charged tryptic peptides have a strong match with the reference spectra in the proteome library and also have good correlation with the realtime experimental spectra in the ISB protein mix database. The correlation scores are confined to the higher values. Hence, the new frequency-based method can be reliably used to predict the tandem mass spectra of the peptide. Because the frequency values are directly used as the intensity of the predicted spectrum, it is proved that the frequency of occurrence of fragment ions is consistent with the log-transformed intensity of the experimental spectrum for the dataset tested in this study.
3.1. Comparison with the Existing Methods
The new frequency-based method is compared with the existing methods—OpenMS_Simulator, MS2PIP, and MS2PBPI. OpenMS_Simulator17 is based on the mobile proton peptide-fragmentation model. It supports the prediction of the CID spectrum from doubly charged peptides. The decision tree model is implemented in MS2PBPI,17 and MS2PIP14,22 is based on stochastic gradient boosting tree regression and random forest regression model, respectively. The theoretical tandem MS spectra are predicted using these methods for the peptides in dataset 2 and dataset 3. The correlation score using eq 2 is calculated between the experimental spectra and the theoretical spectra for peptides in dataset 2 and 3.
Distribution of the correlation scores obtained for the high-quality reference peptide spectra in dataset 2 with respect to the predicted spectra and the percentage of the peptide spectra having a correlation score greater than a threshold, obtained using the new frequency-based method, OpenMS_Simulator, MS2PIP, and MS2PBPI, are plotted in Figure 3a,b and values are tabulated in Table 1. Figure 3(i–iii) shows the same plots for singly, doubly, and triply charged peptides. In the case of a singly charged peptide spectrum, the novel frequency-based approach has predicted 99.66% of the spectra with a correlation score greater than 0.8, while the existing methods MS2PIP and MS2PBPI could predict only 88.35 and 93.15% of the spectra, respectively, with the same threshold. In the case of the doubly charged peptide spectrum, the frequency-based approach has predicted 100% of the spectra with a correlation threshold of 0.8, while the existing methods OpenMS_Simulator, MS2PIP, and MS2PBPI predicted only 84.8, 95.95, and 94.47% of the spectra, respectively. For the triply charged peptide spectrum, the frequency-based approach has predicted 99.42% of the spectra with a correlation threshold of 0.8, while the existing methods MS2PIP and MS2PBPI could only predict 73.72 and 75.3% of the spectra, respectively, with the same threshold.
Figure 3.

Correlation score distribution of the existing methods—OpenMS_Simulator, MS2PIP, MS2PBPI, and the new frequency-based method for dataset 2 and the percentage of spectral count with correlation score > x for charge 1, 2, and 3 peptides are shown in 3(i–iii), respectively.
Table 1. Percentage of Spectral Counts for Different Correlation Scores Obtained Using the Frequency-Based Method and Existing Methods—OpenMS_Simulator, MS2PIP, and MS2PBPI.
| percentage of spectral count having correlation score > x | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| dataset 2: spectral library | ||||||||||
| charge 1 |
charge 2 |
charge 3 |
||||||||
| correlation score | frequency-based method | MS2PIP | MS2PBPI | frequency-based method | MS2PIP | MS2PBPI | OpenMS_Simulator | frequency-based method | MS2PIP | MS2PBPI |
| >0.9 | 89.04 | 72.60 | 76.37 | 94.38 | 83.8 | 75.23 | 68.63 | 30.52 | 45.76 | 52.98 |
| >0.85 | 98.63 | 82.19 | 88.36 | 99.58 | 92.14 | 89.53 | 78.75 | 85.28 | 62.44 | 66.96 |
| >0.8 | 99.66 | 88.36 | 93.15 | 100 | 95.95 | 94.47 | 84.80 | 99.42 | 73.72 | 75.3 |
| >0.75 | 100.00 | 93.84 | 94.86 | 97.53 | 96.91 | 88.44 | 100 | 82.38 | 81.72 | |
| >0.7 | 95.89 | 96.58 | 98.38 | 98.16 | 91.37 | 88.5 | 85.92 | |||
| >0.65 | 97.60 | 97.95 | 99.05 | 99.1 | 93.72 | 92.06 | 89.04 | |||
| >0.6 | 98.29 | 98.29 | 99.39 | 99.33 | 94.97 | 94.86 | 91.64 | |||
| >0.55 | 98.97 | 98.97 | 99.57 | 99.55 | 96.00 | 96.62 | 93.56 | |||
Distribution graph of correlation scores obtained while correlating the predicted theoretical spectra, obtained using the new frequency-based method and existing methods, with the experimental spectra in the dataset 3, the ISB protein mix database is shown Figure 4a. The percentage of the spectral count obtained with a correlation score greater than a threshold is also shown in Figure 4b, and it is tabulated in Table 2. Figure 4(i–iii) shows the same plots for singly, doubly, and triply charged peptides. For the singly charged peptide spectrum, the frequency-based approach has predicted 95.72% of the spectra with a correlation score greater than 0.8, while the existing methods MS2PIP and MS2PBPI predicted 82.66 and 92.14% of the spectra, respectively, with a correlation score greater than 0.8. In the case of the doubly charged peptide spectrum, the frequency-based approach has predicted 95.39% of the spectra with a correlation score greater than 0.8, while the existing methods OpenMS_Simulator, MS2PIP, and MS2PBPI predicted only 72.59, 80.34, and 86.06% of the spectra, respectively, with the same threshold. In the case of the triply charged peptide spectrum, the frequency-based approach has predicted 61.76% of the spectra with a correlation score greater than 0.8. While the existing methods MS2PIP and MS2PBPI predicted 56.85 and 52.63% of the spectra, respectively, with a correlation score greater than 0.8. Reducing the correlation threshold to 0.7, triply charged peptide spectrum has predicted 97.96, 79.65, and 67.34% of the spectra, using the new frequency-based method, MS2PIP, and MS2PBPI, respectively.
Figure 4.

Correlation score distribution of existing methods OpenMS_Simulator, MS2PIP, MS2PBPI, and the new frequency-based methods—for datset 3 and the percentage of spectral counts having correlation score > x for charge 1, 2, 3 peptides is shown in 4(i–iii), respectively.
Table 2. Percentage of the Spectral Count for Different Correlation Scores Obtained Using the Frequency-Based Method and Existing Methods—OpenMS_Simulator, MS2PIP, and MS2PBPI.
| percentage of spectral count having correlation score > x | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| dataset 3: ISB protein mix | ||||||||||
| charge 1 |
charge 2 |
charge 3 |
||||||||
| correlation Score | frequency-based method | MS2PIP | MS2PBPI | frequency-based method | MS2PIP | MS2PBPI | OpenMS_Simulator | frequency-based method | MS2PIP | MS2PBPI |
| >0.9 | 27.86 | 59.31 | 77.92 | 27.03 | 67.07 | 62.69 | 58.13 | 1.04 | 28.79 | 22.69 |
| >0.85 | 84.97 | 75.38 | 87.51 | 78.85 | 74.57 | 78.96 | 67.55 | 20.24 | 44.39 | 39.33 |
| > 0.8 | 95.72 | 82.66 | 92.14 | 95.39 | 80.34 | 86.06 | 72.59 | 61.76 | 56.86 | 52.63 |
| >0.75 | 98.84 | 86.47 | 96.42 | 99.12 | 83.51 | 90.75 | 76.46 | 91.29 | 65.94 | 60.93 |
| >0.7 | 99.88 | 88.90 | 98.15 | 99.76 | 85.89 | 92.69 | 80.34 | 97.97 | 79.66 | 67.34 |
| >0.65 | 100.00 | 89.36 | 98.84 | 99.94 | 87.91 | 94.35 | 83.48 | 99.27 | 83.83 | 73.24 |
| >0.6 | 90.64 | 99.54 | 100.00 | 89.61 | 96.15 | 84.91 | 99.79 | 87.06 | 77.99 | |
| >0.55 | 92.72 | 99.54 | 91.32 | 97.36 | 86.44 | 100.00 | 89.15 | 82.26 | ||
The analysis shows that the novel frequency-based method predicted the spectrum of all peptides in the dataset with a strong correlation with the reference spectrum in the library, compared to other existing methods OpenMS_Simulator, MS2PIP, and MS2PBPI. Even though the existing methods have efficiently predicted a number of peptide spectra with a correlation threshold > 0.85; there are many predicted peptide spectra with much less similarity score compared with those of the spectra in the database. In the case of triply charged peptides, even though there is a slight shift in the correlation score to a lower value (0.75) for the new proposed method, the percentage of spectral count is much higher than those of the existing methods. The correlation scores of peptides obtained for new frequency-based methods are confined within the higher end of the correlation distribution graph, while the other methods have some scores spread to the lower correlation score. Thus, the new method has predicted the spectra with reliable match, than the existing methods OpenMS_Simulator, MS2PIP, and MS2PBPI. The correlation score distribution substantiates that the new frequency-based method has a higher accuracy in predicting the CID MS/MS spectrum of the tryptic peptides considered in this study.
4. Conclusion
A simple sequence-based method was developed and implemented to predict the theoretical tandem mass spectra of tryptic peptides. The theoretical spectrum was derived from the estimated relative cleavage frequency for each pair of adjacent amino acids along the peptide length. These rates were derived from a collection of reliably identified peptide spectra from the NIST library. The new method predicted the theoretical spectrum with a higher accuracy. The study showed that the relative cleavage frequency can be directly used to predict the intensity of the theoretical spectrum of the peptide. The predicted spectra show high correlation with the experimental spectra used in this study. 99.73% of the high-quality reference spectra have correlation scores greater than 0.8 whereas the existing methods like MS2PIP and MS2PBPI have only 85.76 and 85.80% of the spectral count, respectively. The existing method OpenMS_Simulator supports only doubly charged peptides and predicted 84.80% of the spectral count with a correlation score greater than 0.8. The correlation scores obtained using the new frequency-based method are confined to higher values. The study shows that the new method predicts the theoretical spectrum and correlates significantly better with the experimental spectrum as compared to the existing spectrum prediction tool methods OpenMS_Simulator, MS2PIP, and MS2PBPI. Because the predicted spectra have a high correlation with the experimental spectrum, more reliable confirmation of the peptide sequence can be obtained. The current study focuses on the prediction of the ion trap CID spectra of singly, doubly, and triply charged tryptic peptides. The method can be further extended to suit HCD, ETD spectra, and support multicharged peptides in future studies. For implementing this, a frequency matrix has to be created for each dissociation pattern and requires enough training data having possible fragment ions and residue pairs along the length of the peptide.
Acknowledgments
The authors would like to acknowledge the support from the Department of Electronics, Cochin University of Science and Technology.
The authors declare no competing financial interest.
References
- Aebersold R.; Mann M. Mass Spectrometry-Based Proteomics. Nature 2003, 422, 198–207. 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Aebersold R. A Mass Spectrometric Journey into Protein and Proteome Research. J. Am. Soc. Mass Spectrom. 2003, 14, 685–695. 10.1016/s1044-0305(03)00289-7. [DOI] [PubMed] [Google Scholar]
- Eng J. K.; McCormack A. L.; Yates J. R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Perkins D. N.; Pappin D. J. C.; Creasy D. M.; Cottrell J. S. Probability-Based Protein Identification by Searching Sequence Databases Using Mass Spectrometry Data. Electrophoresis 1999, 20, 3551–3567. . [DOI] [PubMed] [Google Scholar]
- Li W.; Ji L.; Goya J.; Tan G.; Wysocki V. H. SQID: An Intensity-Incorporated Protein Identification Algorithm for Tandem Mass Spectrometry. J. Proteome Res. 2011, 10, 1593–1602. 10.1021/pr100959y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paizs B. l.; Suhai S. n. Towards Understanding Some Ion Intensity Relationships for the Tandem Mass Spectra of Protonated Peptides. Rapid Commun. Mass Spectrom. 2002, 16, 1699–1702. 10.1002/rcm.747. [DOI] [PubMed] [Google Scholar]
- Paizs B.; Suhai S. Towards Understanding the Tandem Mass Spectra of Protonated Oligopeptides. 1: Mechanism of Amide Bond Cleavage. J. Am. Soc. Mass Spectrom. 2004, 15, 103–113. 10.1016/j.jasms.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Wysocki V. H.; Tsaprailis G.; Smith L. L.; Breci L. A. Mobile and Localized Protons: A Framework for Understanding Peptide Dissociation. J. Mass Spectrom. 2000, 35, 1399–1406. . [DOI] [PubMed] [Google Scholar]
- Zhang Z. Prediction of Low-Energy Collision-Induced Dissociation Spectra of Peptides. Anal. Chem. 2004, 76, 3908–3922. 10.1021/ac049951b. [DOI] [PubMed] [Google Scholar]
- Zhou C.; Bowler L. D.; Feng J. A Machine Learning Approach to Explore the Spectra Intensity Pattern of Peptides Using Tandem Mass Spectrometry Data. BMC Bioinf. 2008, 9, 325. 10.1186/1471-2105-9-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Arnold R. J.; Tang H.; Radivojac P. On the Accuracy and Limits of Peptide Fragmentation Spectrum Prediction. Anal. Chem. 2011, 83, 790–796. 10.1021/ac102272r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank A. M. Predicting Intensity Ranks of Peptide Fragment Ions. J. Proteome Res. 2009, 8, 2226–2240. 10.1021/pr800677f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik U.; Refsgaard J. C.; Jensen L. J. Improving Peptide-Spectrum Matching by Fragmentation Prediction Using Hidden Markov Models. J. Proteome Res. 2019, 18, 2385–2396. 10.1021/acs.jproteome.8b00499. [DOI] [PubMed] [Google Scholar]
- Degroeve S.; Martens L.; Jurisica I. MS2PIP: A Tool for MS/MS Peak Intensity Prediction. Bioinformatics 2013, 29, 3199–3203. 10.1093/bioinformatics/btt544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong N.-p.; Liang Y.-Z.; Xu Q.-s.; Mok D. K. W.; Yi L.-z.; Lu H.-m.; He M.; Fan W. Prediction of Peptide Fragment Ion Mass Spectra by Data Mining Techniques. Anal. Chem. 2014, 86, 7446–7454. 10.1021/ac501094m. [DOI] [PubMed] [Google Scholar]
- Zhou X.-X.; Zeng W.-F.; Chi H.; Luo C.; Liu C.; Zhan J.; He S.-M.; Zhang Z. PDeep: Predicting MS/MS Spectra of Peptides with Deep Learning. Anal. Chem. 2017, 89, 12690–12697. 10.1021/acs.analchem.7b02566. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Yang F.; Wu P.; Bu D.; Sun S. OpenMS-Simulator: An Open-Source Software for Theoretical Tandem Mass Spectrum Prediction. BMC Bioinf. 2015, 16, 110. 10.1186/s12859-015-0540-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Triscari J. M.; Tseng G. C.; Pasa-Tolic L.; Lipton M. S.; Smith R. D.; Wysocki V. H. Statistical Characterization of the Charge State and Residue Dependence of Low-Energy CID Peptide Dissociation Patterns. Anal. Chem. 2005, 77, 5800–5813. 10.1021/ac0480949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Tseng G. C.; Yuan S.; Pasa-Tolic L.; Lipton M. S.; Smith R. D.; Wysocki V. H. A Data-Mining Scheme for Identifying Peptide Structural Motifs Responsible for Different MS/MS Fragmentation Intensity Patterns. J. Proteome Res. 2008, 7, 70–79. 10.1021/pr070106u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S.; Thomas T. Characterization of the Fragmentation Pattern of Peptide from Tandem Mass Spectra. Mass Spectrom. Lett. 2019, 10, 50–55. 10.5478/MSL.2019.10.2.50. [DOI] [Google Scholar]
- Ramachandran S.; Thomas T. Characterization of Neutral Loss Peaks in the Fragmentation Pattern of Peptide in Collision Induced Dissociation Spectra. Int. J. Mass Spectrom. 2020, 448, 116270. 10.1016/j.ijms.2019.116270. [DOI] [Google Scholar]
- Gabriels R.; Martens L.; Degroeve S. Updated MS2PIP Web Server Delivers Fast and Accurate MS2 Peak Intensity Prediction for Multiple Fragmentation Methods, Instruments and Labeling Techniques. Nucleic Acids Res. 2019, 47, W295–W299. 10.1093/nar/gkz299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein S. E.; Pudnick P. A.. NIST Peptide Tandem Mass Spectral Libraries. Human Peptide Mass Spectral Reference Data; National Institute of Standards and Technology: Gaithersburg, MD, 2008. NIST/EPA/NIH Mass Spectral Library: http://chemdata.nist.gov/.
- Zolg D. P.; Wilhelm M.; Schnatbaum K.; Zerweck J.; Knaute T.; Delanghe B.; Bailey D. J.; Gessulat S.; Ehrlich H.-C.; Weininger M.; et al. Building ProteomeTools Based on a Complete Synthetic Human Proteome. Nat. Methods 2017, 14, 259–262. 10.1038/nmeth.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimek J.; Eddes J. S.; Hohmann L.; Jackson J.; Peterson A.; Letarte S.; Gafken P. R.; Katz J. E.; Mallick P.; Lee H.; et al. The Standard Protein Mix Database: A Diverse Data Set to Assist in the Production of Improved Peptide and Protein Identification Software Tools. J. Proteome Res. 2008, 7, 96–103. 10.1021/pr070244j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig R.; Beavis R. C. TANDEM: Matching Proteins with Tandem Mass Spectra. Bioinformatics 2004, 20, 1466–1467. 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- Deutsch E. W.; Mendoza L.; Shteynberg D.; Farrah T.; Lam H.; Tasman N.; Sun Z.; Nilsson E.; Pratt B.; Prazen B.; et al. A Guided Tour of the Trans-Proteomic Pipeline. Proteomics 2010, 10, 1150–1159. 10.1002/pmic.200900375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W.; Lam H. Tandem Mass Spectral Libraries of Peptides and Their Roles in Proteomics Research. Mass Spectrom. Rev. 2017, 36, 634–648. 10.1002/mas.21512. [DOI] [PubMed] [Google Scholar]