

Abstract
Celia’s encephalopathy (Progressive Encephalopathy with/without Lipodystrophy, PELD) is a childhood neurodegenerative disorder with a fatal prognosis before the age of 10, due to the variant c.985C>T in the BSCL2 gene that causes a cryptic splicing site leading to skipping of exon 7. For years, different authors have reported cases of congenital generalized lipodystrophy due to the variant c.974dupG in BSCL2 associated with neurological manifestations of variable severity, although some of them clearly superimposable to PELD.
Aim:
To identify the molecular mechanisms responsible for these neurological alterations in two patients with c.974dupG.
Subjects and Methods:
Clinical characterization, biochemistry and neuroimaging studies of two girls carrying this variant. In silico analysis, PCR amplification and BSCL2 cDNA sequencing. BSCL2-201 transcript expression, which lacks exon 7, by qPCR in fibroblasts from the index case, from a healthy child as a control and from two patients with PELD, and in leukocytes from the index case and her parents.
Results:
One of the children presented with a severe encephalopathy including a picture of intellectual deficiency, severe language impairment, myoclonic epilepsy and lipodystrophy as described in PELD, dying at 9 years and 9 months of age. The other 2-year-old patient showed incipient signs of neurological involvement. In silico and cDNA sequencing studies showed that variant c.974dupG gives rise to skipping of exon 7. The expression of BSCL2-201 in fibroblasts was significantly higher in the index case than in the healthy child, although less than in the case with homozygous PELD due to c.985C>T variant. The expression of this transcript was approximately half in the healthy carrier parents of this patient.
Conclusions:
The c.974dupG variant leads to the skipping of exon 7 of the BSCL2 gene, and is responsible for a variant of Celia’s encephalopathy, with variable phenotypic expression.
Keywords: BSCL2, PELD, neurodegeneration, congenital generalized lipodystrophy, cryptic splicing
INTRODUCTION
Seipin is a resident protein of the endoplasmic reticulum (ER), encoded by the BSCL2 gene (NM_001122955.3), which is directly involved in the biogenesis of lipid droplets [1]. This gene mainly encodes three seipin isoforms, 462 (BSCL2-203, ENST00000360796.9; CCDS44627), 398 (BSCL2-205/207/210, ENST00000403550.5; ENST00000407022.7; ENST00000421906.5; CCDS8031), and 287 (BSCL2-201, ENST00000278893.11; CCDS55769) amino acids long, respectively [2]. In humans, BSCL2-203 is expressed primarily in the brain (78.9%), whereas BSCL2-205/207/210 expression is similar to BSCL2-203 in extraneural tissues and the peripheral nervous system (≈50%), and BSCL2-201 expression is low in all tissues (<1%) [3]. The 462 and 398 amino acids isoforms have 2 cytoplasmic amino and carboxy terminal domains, one intraluminal loop, and two transmembrane domains [4].
Celia’s encephalopathy or PELD (Progressive Encephalopathy with/without Lipodystrophy, MIM: # 615924) [5] is a very rare pediatric neurodegenerative disorder with a fatal prognosis caused by the variant c.985C>T (rs587777606) in the BSCL2 gene in homozygosis or compound heterozygosis. This variant results in skipping of exon 7 due to the appearance of a canonical branch site in this exon [5]. The intronization of exon 7 leads to the overexpression of the 287 amino acid seipin isoform (ENST00000278893.11) in the brain of these patients [3, 5]. This isoform, which lacks the second transmembrane domain and the carboxy-terminal cytoplasmic end, forms macroaggregates in neurons leading to ER stress and nuclear accumulation, and finally to apoptosis, which is the mechanism responsible for the neurological deterioration in these patients [6].
Patients with PELD have a psychomotor delay that appears after 2 years of age. After age three to four years they develop a process of neurological deterioration associated with refractory or drug-resistant myoclonic epilepsy, leading to death before age 9. Compound heterozygous patients, in addition to the neurodegenerative picture, have congenital generalized lipodystrophy (CGL, also named type 2 Berardinelli-Seip syndrome) associated with the metabolic and hepatic complications characteristic of this disorder [5, 7, 8]. To date, only 8 cases of PELD have been reported worldwide, 6 in Spain [5], one in Iran [9] and another in Brazil [Dr. L. Reis, personal communication], of which only 2 remain alive.
For years, more than 40 pathogenic variants have been reported in the BSCL2 gene (https://databases.lovd.nl/shared/genes/BSCL2), which may lead to congenital generalized lipodystrophy (recessive disorder, MIM: #269700) [10] or to a upper and/or lower motor neuron disease (dominant disorder) [11, 12]. Strikingly, and unlike other subtypes of Berardinelli-Seip syndrome caused by variants in other genes (AGPAT2, MIM: # 608594; CAV1, MIM: #612526; PTRF, MIM: #613327) [13–15], the lipodystrophy due to BSCL2 variants is usually associated with a certain degree of intellectual disability [16].
In 2003, the c.974dupG variant in BSCL2 (rs749890533, previously called c.1126insG (p.G271fsX283)) [17] was reported for the first time as responsible for CGL with mental retardation in a 9 years old girl. Theoretically, this variant would result in p.(Ile326HisfsTer12) in seipin. Subsequently, this same variant, referred to as c.783insG, was reported in two Chinese patients [18, 19]. One of these patients, aged 28 years, presented with dystonia in addition to CGL [18]; the other, at approximately 2 years of age, had generalized lipodystrophy with metabolic derangements without reported neurologic signs or symptoms [19].
More recently, Opri et al. (2016) [20] reported three cases of lethal epileptic encephalopathy associated with CGL caused by c.974dupG in BSCL2 (rs749890533, in the original referred to as c.782_783dupG). These patients had a neurological picture very similar to Celia’s encephalopathy, with death occurring between 8 and 12 years of age [20].
In this study, we present two female patients, one dead at 9 years and 9 months of age, harboring the c.974dupG BSCL2 biallelic variant, and other one aged 2, carrying the BSCL2 variants c.[974dupG];[1015C>T] (rs749890533; rs137852974). The first patient had a severe progressive encephalopathy with generalized lipodystrophy, superimposable with PELD, while the second one, at present, beside generalized lipodystrophy, shows a degree of speech delay and some tensing movements before sleep that appear to be self-soothing behaviors. We unveil in this work the molecular mechanisms responsible for this neurological disorder.
SUBJECTS AND METHODS
The regional IRBs approved this study, which was conducted according to the ethical guidelines of the Helsinki Declaration. The patients’ parents gave informed written consent for the subjects’ participation in the study and for the publication of clinical, imaging and genetic information.
Case reports
Case #1 (index case).
The child is a girl born to non-consanguineous Caucasian parents after an uneventful pregnancy and delivery at 38 weeks of gestation (birth weight: 2855 gr (−0.34 SD); length: 48 cm (−0.41 SD)). During the first days after birth she was hyporeactive presenting feeding difficulties. During the second week of life failure to thrive, a lipodystrophic phenotype, hepatomegaly and hypertriglyceridemia (22.6 mmol/L) were noticed. Hepatomegaly and hypertriglyceridemia were normalized with diet at the age of 10–11 months. The family pointed out that she was always hyporeactive and somnolent. Despite this, early developmental milestones were achieved appropriately until 18 months of age. She sat by herself by 7–8 months, she walked at 14 months and started developing language by 18 months. By age 2 language delay was evident, while social and communication abilities were preserved. Language development evolved positively, she learnt some children’s song and developed symbolic play by age 5. Since age 6 she showed motor and language regression. By age 7 she lost the ability to speak properly and gait became unsteady with frequent falls. Also stereotyped movements appeared, including orolingual dystonia with tongue protrusion. Motor regression continued in childhood, with progressive motor, language and cognitive deterioration. By age 8 she had lost the ability to walk and seat independently. Also, signs of dysphagia with frequent chokes were present at this time. After age 9 the disease progression was rapid over a few months presenting myoclonus and a prolonged generalized tonic clonic seizure. Electroencephalogram was abnormal showing frequent multifocal epileptiform anomalies. At this time valproate was initiated controlling the myoclonic seizures only partially.
At the age of 9 she presented severe cognitive impairment, myoclonic epilepsy, and absence of language abilities; she was wheelchair bounded and totally dependent for all the daily life activities.
The dysphagia contributed to the severe deterioration of her nutritional status at that time and a nasogastric tube for feeding was required. Finally, the patient died at the age of 9 years and 9 months.
On physical examination, she always had marked generalized lipoatrophy (Figure S1, electronic supplementary material), with minimal fat in the palms and soles, without phlebomegaly, acanthosis nigricans or muscular hypertrophy. The face was triangular without coarse features. Hepatomegaly was not present. She had severe axial hypotonia with no independent sedestation and gait. Pyramidal signs were present, showing hyperreflexia in upper and lower limbs, lower limb spasticity, lower limb distal retraction and extensor plantar reflex.
By age 2 the diagnosis of type 2 Berardinelli-Seip syndrome (c.974dupG, p.(Ile326HisfsTer12) in BSCL2 in homozygosis, rs749890533) was confirmed. The analytical parameters during the first year were normal, except for hypertriglyceridemia, and leptinemia which was always low (<0.1 ng/mL). The anthropometric and analytical data are shown in Table 1.
Table 1.
Anthropometric and analytical data of case #1.
| Age (y) | 4 | 7 | 8 | 9 |
|---|---|---|---|---|
| Weight (kg, P) | 15.7 (P.25) | N.D. | 18.9 (P.3) | 20 (P.3) |
| Height (cm) | 103 (P.75) | N.D. | 124 (P.25) | 130 (P.30) |
| Total fat (%) | N.D. | N.D. | 12.8 | N.D. |
| FFM (%) | N.D. | N.D. | 80 | N.D. |
| Tricipital S. (mm) | N.D. | N.D. | 2 | N.D. |
| Bicipital S. (mm) | N.D. | N.D. | 2 | N.D. |
| Subscapular S. (mm) | N.D. | N.D. | 3 | N.D. |
| Suprailiac S. (mm) | N.D. | N.D. | 4 | N.D. |
| Thigh S. (mm) | N.D. | N.D. | 2 | N.D. |
| Calf S. (mm) | N.D. | N.D. | 2 | N.D. |
| Glucose (mmol/L) | 4.6 | 5.2 | 4.3 | 3.9 |
| A1c (%) | 4.8 | N.D. | 4.7 | 4.1 |
| Creatinine (µmol/L) | 40.7 | 59.5 | 30 | 46 |
| Total cholesterol (mmol/L) | 4.40 | N.D. | 5.31 | 5.51 |
| LDL-cholesterol (mmol/L) | 2.93 | N.D. | 3.86 | N.D. |
| HDL-cholesterol (mmol/L) | 0.88 | N.D. | 1.19 | N.D. |
| Triglycerides (mmol/L) | 1.28 | N.D. | 0.57 | 0.6 |
| Insulin (mIU/L | N.D. | N.D. | 9.2 | N.D. |
| Leptin (ng/mL) | N.D. | N.D. | <1.0 | N.D. |
| AST (IU/L) | 25 | 38 | 16 | 20 |
| ALT (IU/L) | 21 | 114 | 28 | 27 |
| GGT (IU/L) | N.D. | 31 | 22 | 33 |
| CK (IU/L) | N.D. | N.D. | 70 | 38 |
| TSH (mUI/L) | N.D. | 2.78 | 2.84 | 1.7 |
P: Percentile; N.D.: Not determined
At age 9 electrophysiological studies were abnormal, showing in the electroencephalography a diffuse distribution of monomorphic brain activity formed by theta and delta waves and low voltage beta rhythm, associated with frequent multifocal epileptiform anomalies consisting of acute waves and irregular spike-and-wave complexes of medium and high amplitude. Electromyography and nerve conduction studies showed signs of an axonal-type sensorimotor neuropathy mainly in lower limbs.
Neuroimaging studies were obtained at the age of 7.8 and 9 years. At 7.8 years of life, the brain MRI-PET study showed bilateral caudate volume loss and a marked glucose hypometabolism in parietal and occipital lobes of both hemispheres, while the other structures included in the study were in within normal parameters (Figure S2A, electronic supplementary material). By age 9 an MRI was obtained showing a progression of the volume loss of the structures of both striated regions, specially the caudate nucleus. Moreover, globus pallidus hypointensity was observed in the susceptibility-weighted image sequence (Figure S2B, electronic supplementary material).
Case #2.
The patient is a 2 year 6 month old girl who carried the variants c.[974dupG];[1015C>T] (rs749890533; rs137852974) in BSCL2 gene. She was the product of a full-term, uncomplicated pregnancy. She presented to medical attention at age 6 weeks due to failure-to-thrive, with a decrease in weight-for-length centile from approximately the 10th centile in the first week of life, to <1st centile. During hospitalization at age 6 weeks, she was noted to have hypertriglyceridemia (>28.25 mmol/L), triangular facies, and generalized lack of adipose tissue, leading to the diagnosis of congenital generalized lipodystrophy. At age 6 months, she was doing well on high-calorie MCT oil based formula, with normal motor, social, and language development. Physical exam showed mild acanthosis nigricans and generalized absence of subcutaneous fat. Laboratory findings included high triglycerides (3.68 mmol/L), low HDL-c (0.39 mmol/L), high insulin (431 pmol/L) with normal hemoglobin A1c (4.8%), and increased liver echogenicity on ultrasound consistent with steatosis. She was initiated on metreleptin, 0.07 mg/kg per day.
At 7–8 months of age she was noted to have episodes of stiffening that would only occur while she was lying on her stomach, especially before falling asleep. She would tense her pelvis, legs and arms, but the activity was suppressible. These were thought to be self-soothing behaviors. By age 2 she was also identified to have some mild speech delay. The anthropometric and analytical data are shown in Table 2.
Table 2.
Anthropometric and analytical data of case #2.
| Age (y) | 6 months | 13 months | 24 months | 30 months |
|---|---|---|---|---|
| Weight (kg, P) | 6.83 (P.27) | 8.2 (P.5) | 11.1 (P.24) | 12.5 (P.35) |
| Height (cm, P) | 71.8 (P.>95) | 78.8 (P.92) | 89.6 (P.84) | 94.7 (P.80) |
| Glucose (mmol/L) | 4.2 | 4.7 | 4.4 | 4.3 |
| A1c (%) | 4.8 | 4.8 | 4.8 | 5 |
| Creatinine (µmol/L) | 17.7 | 13.3 | 18.6 | 20.3 |
| Total cholesterol (mmol/L) | 4.01 | 3.83 | 2.59 | 2.97 |
| LDL-cholesterol (mmol/L) | 1.93 | 2.46 | 1.6 | 1.91 |
| HDL-cholesterol (mmol/L) | 0.39 | 0.67 | 0.78 | 0.9 |
| Triglycerides (mmol/L) | 3.68 | 1.54 | 0.43 | 0.35 |
| Insulin (mIU/L) | 62.1 | 7.3 | 5.7 | 1.6 |
| AST (IU/L) | 39 | 37 | 36 | 38 |
| ALT (IU/L) | 74 | 39 | 21 | 18 |
P: Percentile
Genetic studies
For the variant analyses, genomic DNA was isolated from peripheral leukocytes using standard procedures [21]. BSCL2 exons 1–11, and the surrounding intronic sequences from the subjects, were PCR-amplified and sequenced. PCR conditions and primer sequences are available upon request.
Splicing analysis of the BSCL2 c.974dupG variant
The following in silico programs were used to predict the effect of the variants on splicing: Human Splice Finder (HSF, version 3.1, http://www.umd.be/HSF3/), NetGene2 (version 2.4, http://www.cbs.dtu.dk/services/NetGene2/), NNSplice (version 0.9, http://www.fruitfly.org/seq_tools/splice.html), Alternative Splice Site Predictor (ASSP, http://wangcomputing.com/assp/), and MutPred splice (version 1.3.2, http://www.mutdb.org/mutpredsplice/submit.htm). The latter was used to identify single base substitutions in exonic regions that disrupt pre-mRNA. They were accessed individually and the analyses were done using default settings.
BSCL2 expression studies
Total RNA was extracted from leukocytes and primary fibroblasts and reverse-transcribed as previously reported [22]. BSCL2 cDNA was amplified with primers designed with the Primer3Plus software (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) (forward: 5’-CAGATGCTGGACACACTGGT-3’, reverse: 5’-ATCACTGGCCTCAGGCTCTA-3’). PCR conditions are available upon request. Amplification fragments obtained were separated out by low-melting agarose gel electrophoresis (1%). The resulting bands were excised from agarose gel, and cDNA was extracted using the Qiaex II gel extraction kit (Qiagen, Chatsworth, CA, USA). cDNA was then amplified with the same primers and conditions as used for the first PCR; fragments were separated by agarose gel electrophoresis and directly sequenced.
Expression of BSCL2 mRNA was quantified in a LightCycler 2.0 (Roche Diagnostics, Sant Cugat del Vallès, Spain) using specific probes and oligonucleotide primers designed by the Universal ProbeLibrary (Roche Diagnostics). Specific designed primers and probe were used for BSCL2 spliced transcript. Details are available upon request. Results were normalized to the RNA polymerase II and 18S genes, using the 2-∆∆CT method [23].
Skin biopsy and cell cultures
A small sample of skin from the abdominal area was obtained from the case 1 at age 7 years. A control skin sample was obtained from a 6-year-old normal boy during cryptorchidism surgery, in accordance with the ethical regulations of current Spanish legislation. Small pieces of tissue were rinsed with PBS and placed on a 60 mm dish (cat. 353004, Falcon™, Mississauga, ON, Canada) containing Dulbecco’s modified Eagle’s medium (DMEM, cat. D5796, Sigma-Aldrich, St. Louis, Missouri, USA) supplemented with 30% fetal bovine serum (FBS, cat. 10270, Gibco Life Technologies, Carlsbad, California, USA) and gentamicin 50 µg/ml (cat. G1397, Sigma-Aldrich), and incubated at 37 °C with 5% CO2 in a Water-Jacket CO2 incubator (Galaxy 170 S, Eppendorf New Brunswick, Hamburg, Germany). Fibroblasts were cultured with fresh medium, changed every three days, until a 70% confluent monolayer was obtained. Afterwards, they were tripsinized (cat. 12604–021, TrypLE™ Express, Gibco Life Technologies) and cultured on 100 mm dishes in DMEM containing 10% FBS and penicillin-streptomycin 1% (cat. 15140–122, Gibco Life Technologies).
Statistical analysis
Real-time PCR analyses were performed in duplicate. Statistical significance was determined using a non-parametric Kruskall-Wallis and Mann–Whitney tests. Data are presented as means ± SD with the statistical significance set at p < 0.05. All statistical analyses were performed using SPSS for Mac (release 22.0; SPSS, Chicago, IL, USA).
RESULTS
The case #1 was homozygous for a guanine insertion in NM_001122955.3: c.974dupG, p.(Ile326HisfsTer12) (rs749890533) in BSCL2 gene. Her parents were asymptomatic heterozygous carriers, while his brother was not carrier. The case #2 was a compound heterozygote for BSCL2: c.974dupG (rs749890533) in one allele and c.1015C>T (rs137852974), p.(Arg339Ter) in the other allele.
Given the great similarity between case #1 neurological disorder and Celia’ encephalopathy, we hypothesized that the variant c.974dupG (rs749890533) could be given rise also to the skipping of exon 7 in BSCL2. As a first approach, we carried out different in silico analysis of this variant. Whereas we did not find any variations for splice sites (acceptor and donor values remain unchanged), HSF detected a potential alteration of the splicing in the branch point motif CCCCGAC/GCCCCGA and in some auxiliary sequences, known as the exonic splicing enhancer (ESE) and the exonic splicing silencer (ESS) sites. These sequences, either enhance or repress splicing by modulating the assembly of the early-stage spliceosome complex. The variant c.974dupG (rs749890533) appears to alter an ESE site and to create a new ESS. All these disruptions might possibly compromise the splicing. Afterwards, in order to check these findings, we performed a BSCL2 cDNA PCR including from exon 5 to exon 10, finding a 431 bp extra-band in case #1 (Figure 1A). Subsequently, this band was sequenced (Figure 1B), finding that the whole exon 7 disappeared, exactly as it happened with c.985C>T variant (rs587777606) [5].
Figure 1.
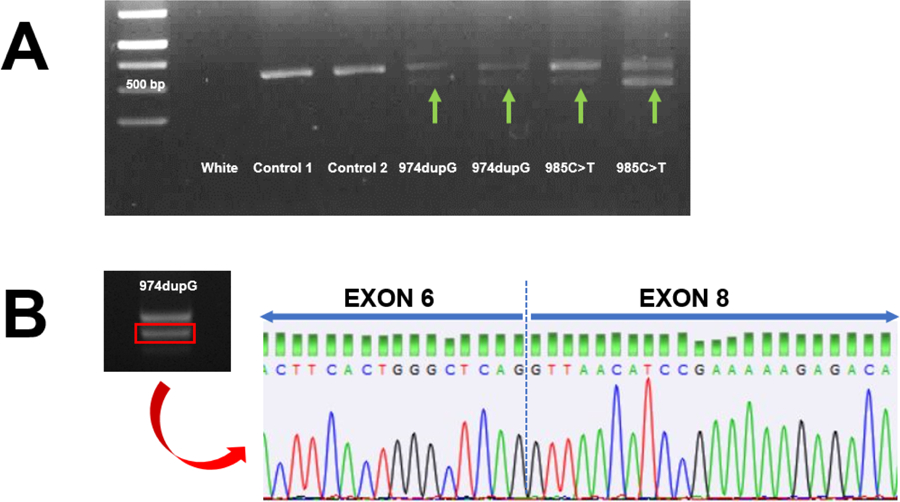
A. Results of the amplification of a 573 bp region of cDNA from leukocytes from the index case and different subjects. Samples from the 974dupG index case and 985C>T PELD index case show the presence of an additional 431 base pair-long band. B. Sequencing of the 431 base pair band showed complete skipping of exon 7.
In order to confirm these results, we measured the relative expression of BSCL2-201 transcript (which encodes the 287 amino acids seipin isoform and that lacks exon 7) in primary fibroblasts from case #1 and compared with control and Celia’s encephalopathy primary fibroblasts. The expression of this transcript was 4 times higher than control, it was similar to that of the compound heterozygote patient for c.985C>T, and significantly lower than that quantified in the samples from the homozygous patient with classical Celia’s encephalopathy (Figure 2A). When we compared BSCL2-201 expression in white blood cells from case #1 with those from her heterozygous parents and normal brother, the results were as expected (Figure 2B): approximately half of expression in parents and around 20% in the wild type brother.
Figure 2.
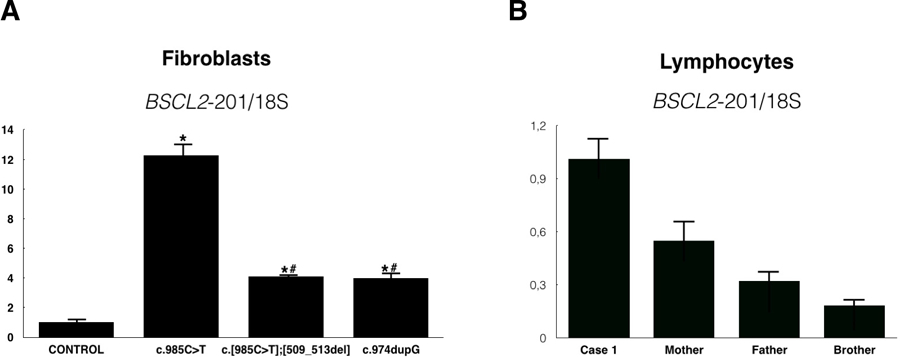
A. Relative expression of BSCL2-201 transcript in fibroblasts from a control children, a homozygous PELD patient (c.985C>T), a compound heterozygous patient (c.[985C>T];[509_5013del] and case #1 (c.974dupG). *: p<0.05 compared with control; #: p<0.05 compared with homozygous PELD patient. B. Relative expression of BSCL2-201 transcript in leukocytes from case #1, her mother (simple heterozygous), her father (simple heterozygous) and brother (wt).
DISCUSSION
In this study we reported two cases of girls carrying the c.974dupG (rs749890533) variant in BSCL2 gene, one homozygous and the other one a compound heterozygote. The first of these, who died before reaching 10 years of age, had, in addition to generalized lipodystrophy, a neurodegenerative picture starting at an early age, rapidly progressive after 8 years of age, associated with myoclonic epilepsy. The second, currently aged 2 years and 6 months, has generalized lipodystrophy and seems to be in the early stages of the neurological picture, although we cannot rule out the possible beneficial effect of metreleptin on the neurological clinic, as previously reported [24]. The first case corresponds clinically, electrophysiologically, and according to the neuroimaging tests with those reported in 2016 by Opri et al. (Table 3) [20], although the description in the original paper of the genotype of these patients corresponds to another BSCL2 transcript, (NM_032667.6, which encodes the 398 amino acids seipin isoform) which may lead to confusion. The variant found by these authors was c.782_783dupG, which corresponds with c.974dupG (rs749890533) in NM_001122955.3 transcript, which encodes the 462 amino acids seipin isoform (https://databases.lovd.nl/shared/genes/BSCL2). Interestingly, this patient developed an axonal neuropathy, as it is described in other BSCL2 related disorders [25], showing that BSCL2 variants lead to different but overlapping phenotypes affecting both upper and lower neurons.
Table 3.
Clinical features of patients carrying the c.974dupG variant in BSCL2 gene
| Patient | Genotype | Intellectual deficiency (age) | Language delay | Myoclonus(age) | Dystonia (age) | Seizures (age) | Gait ataxia (age) | Abnormal behavior (age) | Severe encephalopathy (age) | Lipodystrophy (age) | Hyper-triglyceridemia (age) | Hepatomegaly (age) | Exitus (age) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case #1 | Hmzg | Yes (3) | Yes (2) | Yes (6) | Yes (7) | Yes (7) | Yes (7.8) | Yes (7.8) | Yes (9) | Yes (birth) | Yes (birth) | Yes (birth) | Yes (9.8) |
| Case #2 | Comp Htzg | No | Yes (2) | No | No | No | No | Yes (0.7) | No | Yes (birth) | Yes (birth) | Yes (birth) | No |
| Opri et al. 2016 [20] case #2 |
Hmzg | Yes (4) | N.R. | Yes (4) | N.R. | Yes (4) | Yes (6) | N.R. | Yes (7.5) | Yes (birth) | Yes (birth) | Yes (3) | Yes (7.8) |
| Opri et al. 2016 [20] case #3 |
Hmzg | Yes (early) | N.R. | Yes (5) | N.R. | Yes (5) | Yes (10) | N.R. | Yes (10) | Yes (birth) | Yes (birth) | N.R. | Yes (11) |
| Huang et al. 2010 [19] | Hmzg | No | N.R. | N.R. | N.R. | N.R. | N.R. | N.R. | N.R. | Yes (birth) | Yes (birth) | Yes (birth) | No (1.8) |
| Opri et al. 2016 [20] case #1 |
Comp Htzg (fs stop codon) |
Yes (?) | Yes (1) | Yes (1) | Yes (1) | Yes (1) | Yes (5) | N.R. | Yes (7) | Yes (5) | Yes (birth) | Yes (5) | Yes (9.9) |
| Wu et al. 2009 [18] | Comp Htzg (stop codon) |
Yes (?) | Yes (?) | No | Yes (?) | No | Yes (?) | Yes (?) | No | Yes (?) | N.R. | Yes (?) | No (28) |
| Poisson et al. 2018 [27] | Comp Htzg (missense) |
Yes (4–5) | Yes (3,5) | N.R. | Yes (16) | N.R. | Yes (16) | Yes (3) | Yes (23) | N.R. | N.R. | N.R. | Yes (28) |
| Agarwal et al. 2003 [17] | Simple Htzg | Yes (?) | N.R. | N.R. | N.R. | N.R. | N.R. | N.R. | N.R. | Yes (?) | N.R. | N.R. | No (9) |
N.R.: Not reported
The molecular studies carried out both in leukocytes and fibroblasts of case #1 showed that the c.974dupG (rs749890533) variant in BSCL2 gave rise to the skipping of exon 7 in this gene. This mechanism is identical to the one reported years ago by us in the case of Celia’s encephalopathy, caused by the variant c.985C>T (rs587777606) in BSCL2 [5]. The clinical features of Celia encephalopathy are superimposable to those of case #1, and also to those of case #2 in its initial stages. Briefly, patients with classic Celia’s encephalopathy are normal at birth; by age 2 years these children show psychomotor delay, with both poor motor coordination and language and unsteady gait. As a rule, around 3 years old these patients showed a rapid psychomotor regression, with lost all language and showed severe cognitive impairment. By age 4 years, they have ataxic gait, tremor, dystonia, sleep disturbances and myoclonic epileptic seizures. Usually, before 6 years they have severe encephalopathy and progressive deterioration, leading to passed away before 8 [5]. In the same way, PELD patients show similar epileptic features and neuroimaging findings than case #1, particularly caudate atrophy and a marked decrease in glucose uptake in the parietal and occipital lobes [5, 24]. On the other hand, expression of BSCL2-201 transcript in the index case of Celia’s encephalopathy [5] in non-neural tissues was increased, but the abnormal transcript was much more highly expressed in brain.
In Celia’s encephalopathy, the expression of the BSCL2-201 transcript (which lacks exon 7), which is hardly expressed in normal brain (<1%) [3, 5], was markedly elevated in the different areas of the brain analyzed in the index case of our initial study [5]. Although we did not have samples from the encephalon of case #1, we analyzed the expression of this transcript in fibroblasts from this patient, and compared it with that observed in fibroblasts from a healthy control child, being the expression of such a transcript 4 times higher than that of the control. On the other hand, and as expected, the expression of BSCL2-201 was lower in the leukocytes from the simple heterozygous healthy parents of case #1.
Taken together, the clinical, electrophysiological, imaging and molecular data of these patients, together with that previously reported in the literature (Table 3), suggest that the loss of exon 7 of BSCL2 induced by the c.974dupG (rs749890533) variant is the cause of the neurodegenerative picture suffered by these patients. In previous studies of our group [5, 6], we showed that seipin encoded by this transcript tends to aggregate abnormally resulting in ER stress, intranuclear seipin inclusions and neuronal apoptosis [6].
The variant c.974dupG (rs749890533) had already been reported in 2003 [17] although with another notation (c.1126insG, p.(G271fsX283)). In this case, the patient (GC1100), aged 9 years, had mental retardation in addition to lipodystrophy and diabetes, but no reference was made to a neurodegenerative picture. It is noteworthy that in this case this variant was in simple heterozygosis and no other pathogenic variant was found in BSCL2. Later, Wu et al. (2009) [18] and Huang et al. (2010) [19] reported 2 cases of CLG associated with the c.975dupG variant (according to https://databases.lovd.nl/shared/variants/0000021148#00023833), although in the original articles it was reported to as c.783insG. Theoretically this variant would originate the same change in seipin than c.974dupG, that is, p.(Ile326HisfsTer12). However, after a careful analysis of BSCL2-203 sequence (Figure 3), we found that the correct notation is c.974dupG, as the 975 position in the ENST00000360796.9 transcript corresponds to a cytosine. Furthermore, the HGVS notation prescribes that, like in this case where there is one guanine insertion among the seven guanines from 968 to 974 positions, the notation insertion must be referred to the last 3’ guanine (3’ rule). Strikingly, the study by Wu et al. (2009) [18] describes a 26-year-old man, compound heterozygote (c.[757G>T];[974dupG] or c.[565G>T];[783insG] in the original, rs137852975; rs749890533), with generalized lipodystrophy and a picture of intellectual disability and generalized dystonia. The variant c.757G>T (rs137852975, exon 5) would give rise to a premature stop codon, p.(Glu253Ter). On the other hand, Huang et al. (2010) [19] reported the case of a 1.8-year-old child carrying the same variant c.974dupG (c.783insG in the original) than Wu et al. [18] in homozygosity in BSCL2, with a clear CGL picture, but he did not show any intellectual impairment at that age. However, we do not know how the neurological evolution of this patient was over the years.
Figure 3.
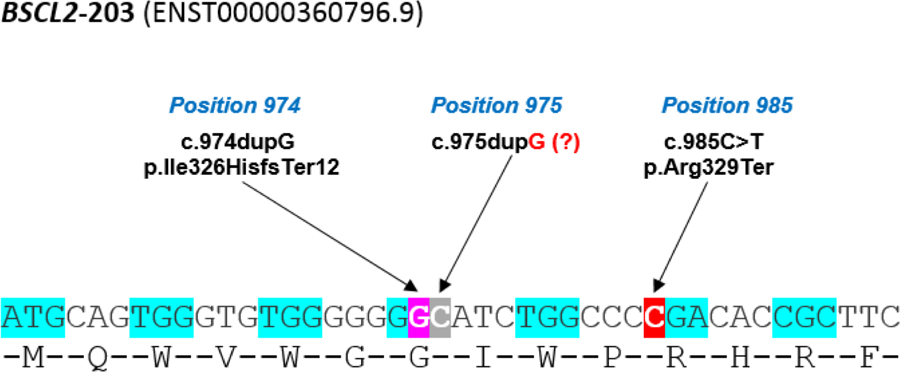
BSCL2-203 transcript sequence surrounding the c.974dupG (purple shading) and c.985C>T (red shading) variants, and the incorrectly annotated c.975dupG variant (grey shading).
A review of other reported variants which could lead to the skipping exon 7 suggests that the variant c.864–2A>G (rs766061024, NM_001122955.3), originally reported as IVS6–2A>G [17], is especially interesting because is a splice acceptor variant (intron 6–7). This might lead to a cryptic splice site, although this never has not been studied. This variant was associated to congenital generalized lipodystrophy with mental retardation, but not with neurodegeneration [17]. Very recently, the bialellic variant c.1076dupC (p.Glu360*), located in exon 9 of the BSCL2 gene, has been associated with congenital generalized lipodystrophy, myoclonic seizures, and progressive neurological degeneration [26].
Supporting our hypothesis that the skipping of exon 7 is critical for explaining the neurological symptoms of these patients, recently, Poisson et al. (2018) [27] reported the case of a patient, who died at 28 years of age, and who during the first years was diagnosed with a regressive autism spectrum disorder, although at the age of 6 years he presented motor stereotypies and lower limbs hypertonia; from 16 to 23 years of age, the neurological picture was getting worse, and a clear frontal lobe syndrome appeared [27]. The brain MRI disclosed bilateral atrophy of the caudate nucleus. Interestingly, the authors report no lipodystrophy or metabolic alterations in this patient. This patient was a carrier of the variants c.[985C>T];[1004A>C] in BSCL2. The first is the classic variant responsible for Celia’s encephalopathy. The second variant (c.1004A>C) would theoretically give rise to the missense change p.(Gln335Pro), which according to PolyPhen-2 is possibly damaging (PolyPhen-2, score: 0.522), although it has never been reported as a cause of CGL (https://databases.lovd.nl/shared/genes/BSCL2). In silico analysis with MutPred splice software for c.1004A>C variant indicates a score of 0.84 predicting to be a splice affecting variant with high confident hypothesis (p<0.000001), whereas HSF did not detect any significant splicing motif alteration. Based on in silico and RNA retrotranscription studies (although not direct sequencing), the authors concluded that the variant c.1004A>C also gives rise to the skipping of exon 7 in the BSCL2 gene. The fact that this patient did not present with lipodystrophy could suggest that the variant c.1004A>C does not impair the lipid droplets biogenesis. On the other hand, the late appearance of neurodegenerative symptoms and long survival suggest that the splicing rate induced by c.1004A>C could be low. Our in vitro studies [6] showed that if the amount of wt seipin is sufficient (as in the simple heterozygotes), the aberrant seipin would be sequestered by the homo-oligomers of wt protein. According to all these data, we can speculate that the neurodegenerative process would be more or less aggressive depending on two factors, the rate of splicing induced by a particular variant in the exon 7, and of the presence or not of variants in the other allele which allow the translation of an abnormal seipin isoform with certain capability for kidnaping the 287 amino acids seipin.
In summary, certain variants in exon 7 of BSCL2 gene originate splicing alterations that lead to the skipping of this exon and consequently to the excessive production of the 287 amino acids seipin isoform at a variable rate, either in homozygosis or in compound heterozygosis, generally leading to a lethal neurodegenerative condition, with special involvement of the striated nuclei, particularly the caudate. However, at present, we have not a clear explanation for the different neurological phenotype previously reported.
Supplementary Material
ACKNOWLEDGEMENTS
We are indebted to the patients and their parents for their collaboration in this study. This work was supported by the Instituto de Salud Carlos III and the European Regional Development Fund, FEDER (grants number PI10/02873 and PI13/00314), by the Consellería de Industria, Xunta de Galicia (grants number 10PXIB208013PR and ED341b 2017/19), and by Fundación Mutua Madrileña (Call 2015). S.S-I was awarded a Research Fellowship, granted by the Asociación Española de Familiares y Afectados de Lipodistrofias (AELIP). Evaluation of Case 2 was supported by the intramural research program of the National Institute of Diabetes and Digestive and Kidney Diseases. Control 18F-FDG PET brain of subject at 7 years of age was a courtesy of A. Niñerola-Baizán and X. Setoain from Hospital Clínic Barcelona.
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
REFERENCES
- 1.Wang H, et al. , Seipin is required for converting nascent to mature lipid droplets. Elife, 2016. 5: p. e16582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cartwright BR and Goodman JM, Seipin: from human disease to molecular mechanism. J Lipid Res, 2012. 53(6): p. 1042–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanchez-Iglesias S, et al. , Does Seipin Play a Role in Oxidative Stress Protection and Peroxisome Biogenesis? New Insights from Human Brain Autopsies. Neuroscience, 2019. 396: p. 119–137. [DOI] [PubMed] [Google Scholar]
- 4.Lundin C, et al. , Membrane topology of the human seipin protein. FEBS Lett, 2006. 580(9): p. 2281–4. [DOI] [PubMed] [Google Scholar]
- 5.Guillen-Navarro E, et al. , A new seipin-associated neurodegenerative syndrome. J Med Genet, 2013. 50(6): p. 401–9. [DOI] [PubMed] [Google Scholar]
- 6.Ruiz-Riquelme A, et al. , Larger aggregates of mutant seipin in Celia’s Encephalopathy, a new protein misfolding neurodegenerative disease. Neurobiol Dis, 2015. 83: p. 44–53. [DOI] [PubMed] [Google Scholar]
- 7.Brown RJ, et al. , The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J Clin Endocrinol Metab, 2016. 101(12): p. 4500–4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Araujo-Vilar D and Santini F, Diagnosis and treatment of lipodystrophy: a step-by-step approach. J Endocrinol Invest, 2018. [DOI] [PMC free article] [PubMed]
- 9.Alaei MR, et al. , Whole Exome Sequencing Reveals a BSCL2 Mutation Causing Progressive Encephalopathy with Lipodystrophy (PELD) in an Iranian Pediatric Patient. Iran Biomed J, 2016. 20(5): p. 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Magre J, et al. , Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet, 2001. 28(4): p. 365–70. [DOI] [PubMed] [Google Scholar]
- 11.Auer-Grumbach M, et al. , Phenotypes of the N88S Berardinelli-Seip congenital lipodystrophy 2 mutation. Ann Neurol, 2005. 57(3): p. 415–24. [DOI] [PubMed] [Google Scholar]
- 12.Irobi J, et al. , The phenotype of motor neuropathies associated with BSCL2 mutations is broader than Silver syndrome and distal HMN type V. Brain, 2004. 127(Pt 9): p. 2124–30. [DOI] [PubMed] [Google Scholar]
- 13.Agarwal AK, et al. , AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet, 2002. 31(1): p. 21–3. [DOI] [PubMed] [Google Scholar]
- 14.Kim CA, et al. , Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab, 2008. 93(4): p. 1129–34. [DOI] [PubMed] [Google Scholar]
- 15.Hayashi YK, et al. , Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest, 2009. 119(9): p. 2623–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Maldergem L, et al. , Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet, 2002. 39(10): p. 722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agarwal AK, et al. , Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J Clin Endocrinol Metab, 2003. 88(10): p. 4840–7. [DOI] [PubMed] [Google Scholar]
- 18.Wu YR, et al. , Complementary mutations in seipin gene in a patient with Berardinelli-Seip congenital lipodystrophy and dystonia: phenotype variability suggests multiple roles of seipin gene. J Neurol Neurosurg Psychiatry, 2009. 80(10): p. 1180–1. [DOI] [PubMed] [Google Scholar]
- 19.Huang HH, et al. , A Taiwanese boy with congenital generalized lipodystrophy caused by homozygous Ile262fs mutation in the BSCL2 gene. Kaohsiung J Med Sci, 2010. 26(11): p. 615–20. [DOI] [PubMed] [Google Scholar]
- 20.Opri R, et al. , Progressive Myoclonus Epilepsy in Congenital Generalized Lipodystrophy type 2: Report of 3 cases and literature review. Seizure, 2016. 42: p. 1–6. [DOI] [PubMed] [Google Scholar]
- 21.Sambrook J and Russell DW, Molecular cloning : a laboratory manual. 3rd ed. 2001, Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 22.Victoria B, et al. , Reduced adipogenic gene expression in fibroblasts from a patient with type 2 congenital generalized lipodystrophy. Diabet Med, 2010. 27(10): p. 1178–87. [DOI] [PubMed] [Google Scholar]
- 23.Livak KJ and Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 2001. 25(4): p. 402–8. [DOI] [PubMed] [Google Scholar]
- 24.Araujo-Vilar D, et al. , Association of metreleptin treatment and dietary intervention with neurological outcomes in Celia’s encephalopathy. Eur J Hum Genet, 2018. 26(3): p. 396–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi BO, et al. , Clinical and histopathological study of Charcot-Marie-Tooth neuropathy with a novel S90W mutation in BSCL2. Neurogenetics, 2013. 14(1): p. 35–42. [DOI] [PubMed] [Google Scholar]
- 26.Serino D, et al. , Berardinelli-Seip syndrome and progressive myoclonus epilepsy. Epileptic Disord, 2019. 21(1): p. 117–121. [DOI] [PubMed] [Google Scholar]
- 27.Poisson A, et al. , Regressive Autism Spectrum Disorder Expands the Phenotype of BSCL2/Seipin-Associated Neurodegeneration. Biol Psychiatry, 2018. S0006–3223(18): p. 31524–31525. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.