

Abstract
Lessons Learned
Concurrent ETBX‐011, ETBX‐051, and ETBX‐061 can be safely administered to patients with advanced cancer.
All patients developed CD4+ and/or CD8+ T‐cell responses after vaccination to at least one tumor‐associated antigen (TAA) encoded by the vaccine; 5/6 patients (83%) developed MUC1‐specific T cells, 4/6 (67%) developed CEA‐specific T cells, and 3/6 (50%) developed brachyury‐specific T cells.
The presence of adenovirus 5‐neutralizing antibodies did not prevent the generation of TAA‐specific T cells.
Background
A novel adenovirus‐based vaccine targeting three human tumor‐associated antigens—CEA, MUC1, and brachyury—has demonstrated antitumor cytolytic T‐cell responses in preclinical animal models of cancer.
Methods
This open‐label, phase I trial evaluated concurrent administration of three therapeutic vaccines (ETBX‐011 = CEA, ETBX‐061 = MUC1 and ETBX‐051 = brachyury). All three vaccines used the same modified adenovirus 5 (Ad5) vector backbone and were administered at a single dose level (DL) of 5 × 1011 viral particles (VP) per vector. The vaccine regimen consisting of all three vaccines was given every 3 weeks for three doses then every 8 weeks for up to 1 year. Clinical and immune responses were evaluated.
Results
Ten patients enrolled on trial (DL1 = 6 with 4 in the DL1 expansion cohort). All treatment‐related adverse events were temporary, self‐limiting, grade 1/2 and included injection site reactions and flu‐like symptoms. Antigen‐specific T cells to MUC1, CEA, and/or brachyury were generated in all patients. There was no evidence of antigenic competition. The administration of the vaccine regimen produced stable disease as the best clinical response.
Conclusion
Concurrent ETBX‐011, ETBX‐051, and ETBX‐061 can be safely administered to patients with advanced cancer. Further studies of the vaccine regimen in combination with other agents, including immune checkpoint blockade, are planned.
Discussion
The TriAdeno vaccine regimen (TAV) uses Ad5 vaccines containing tumor‐associated antigens (TAAs) CEA, MUC1, and brachyury. In preclinical studies, TAV induced immune responses directed against TAAs with minimal to no “antigenic competition” 1. A prior clinical trial in metastatic colorectal cancer showed that the CEA ETBX‐011 vaccine was safe and had clinical benefit 2, 3. The primary objectives of this trial were to assess the safety of TAV in advanced solid malignancies and to identify the recommended dose for future trials.
Ten patients enrolled on this open label, phase I trial from January 31, 2018, to April 24, 2018 (DL1, n = 6; expansion, n = 4). The data cutoff date for final analysis was October 23, 2018. All patients were monitored for dose‐limiting toxicities (DLTs) for 3 weeks after the first dose. Reported adverse events (AEs) were graded according to the Common Terminology Criteria for Adverse Events v5.0. Computed tomography of the thorax, abdomen, and pelvis was performed at baseline, week 6, and then every 8 weeks.
Five patients were female. Median age was 51.7 years. Nine patients had colorectal cancer and one had cholangiocarcinoma. All patients were evaluable for clinical, safety, and immune responses. TAV was well tolerated with no DLTs. When given concurrently, the recommended phase II dose of TAV (ETBX‐011, ETBX‐051 and ETBX‐061) is 5 × 1011 VP per vaccine. There were no grade ≥3 AEs. All AEs attributed to TAV were temporary and self‐limiting. Grade 1 or 2 injection site reactions occurred in all patients, with most reporting injection site pain (n = 9; 90%), erythema (n = 8; 80%), and induration (n = 7; 70%). These reactions generally occurred within 24 hours of administration and resolved within 7 days without intervention. Pyrexia (n = 5; 50%) and chills (n = 8; 80%) were common. Myalgias, nausea, and fatigue were also reported. The average time on treatment was 13.6 weeks (range 3–34 weeks). The best radiographic response was stable disease per RECIST v1.1.
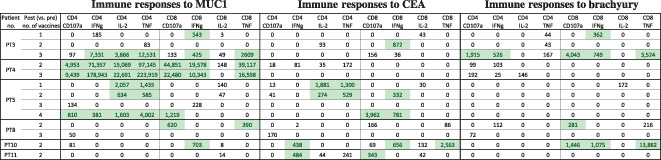
After vaccination, all patients developed CD4+ and/or CD8+ T‐cell responses 4 to at least one TAA encoded by the vaccine; 5/6 (83%) developed MUC1‐specific T cells, 4/6 (67%) developed CEA‐specific T cells, and 3/6 (50%) developed brachyury‐specific T cells (Table 1). Two patients developed responses to all TAAs in the vaccines. Induction of antigen‐specific T cells was rapid, with most occurring by week 6. Polyfunctional T cells (i.e., T cells positive for two or more of the following: interferon gamma, tumor necrosis factor, interleukin‐2, or CD107a) specific for MUC1, CEA, or brachyury were generated in 50%, 33%, and 17% of patients, respectively. The presence of Ad5‐neutralizing antibodies did not prevent the generation of TAA‐specific T cells.
Table 1.
Tumor‐associated antigen T‐cell responses developed after treatment with the TriAdeno vaccine regimen
|
Immune responses reported in this table are calculated by comparing the absolute number of CD4+ or CD8+ T cells producing cytokine (IFN, IL‐2, TNFa) or positive for CD107a per 1 × 106 PBMCs plated at the start of the in vitro stimulation at the specified time points after vaccine. Background (obtained with the negative control peptide pool, human leukocyte antigen [HLA]) and any response prior to vaccine are subtracted: [TAA after vaccine – HLA after vaccine] – [TAA before vaccine – HLA before vaccine]. Positive immune responses are defined as >250 (highlighted).
Abbreviations: IFNg, interferon gamma; IL‐2, interleukin‐2; PT, patient; TNF, tumor necrosis factor.
Although TAV does not appear to have single‐agent activity, it has a manageable safety profile and generates TAA‐specific T‐cell responses in patients with cancer (Table 1). Future immuno‐oncology trials aimed at enhancing synergistic antitumor mechanisms with TAV are planned.
Trial Information
| Disease | Advanced cancer/solid tumor only |
| Stage of Disease/Treatment | Metastatic/advanced |
| Prior Therapy | 1 prior regimen |
| Type of Study – 1 | Phase I |
| Type of Study – 2 | Dose evaluation and cohort expansion |
| Primary Endpoint | Safety |
| Primary Endpoint | Tolerability |
| Secondary Endpoint | Efficacy |
| Investigator's Analysis | Drug tolerable, efficacy indeterminant |
Drug Information
| Drug 1 | |
| Generic/Working Name | ETBX‐011 |
| Trade Name | None |
| Company Name | Etubics (a wholly owned subsidiary of ImmunityBio) |
| Drug Type | Vaccine |
| Drug Class | Immune therapy |
| Dose | 5 × 1011 viral particles per flat dose |
| Route | Other; subcutaneous |
| Schedule of Administration |
ETBX‐011 = CEA Every 3 weeks for three doses, then every 8 weeks for 1 year |
| Drug 2 | |
| Generic/Working Name | ETBX‐051 |
| Trade Name | None |
| Company Name | Etubics (a wholly owned subsidiary of ImmunityBio) |
| Drug Type | Vaccine |
| Drug Class | Immune therapy |
| Dose | 5 × 1011 viral particles per flat dose |
| Route | Other; subcutaneous |
| Schedule of Administration |
ETBX‐061 = MUC1 Every 3 weeks for three doses, then every 8 weeks for 1 year |
| Drug 3 | |
| Generic/Working Name | ETBX‐061 |
| Trade Name | None |
| Company Name | Etubics (a wholly owned subsidiary of ImmunityBio) |
| Drug Type | Vaccine |
| Drug Class | Immune therapy |
| Dose | 5 × 1011 viral particles per flat dose |
| Route | Other; subcutaneous |
| Schedule of Administration |
ETBX‐051 = brachyury Every 3 weeks for three doses, then every 8 weeks for 1 year |
Dose Escalation Table
| Dose level | Dose of drug: ETBX‐011 | Dose of drug: ETBX‐051 | Dose of drug: ETBX‐061 | No. enrolled | No. evaluable for toxicity |
|---|---|---|---|---|---|
| 1 | 5 × 1011 VP | 5 × 1011 VP | 5 × 1011 VP | 10 | 10 |
| −1 | 1 × 1011 VP | 1 × 1011 VP | 1 × 1011 VP | 0 |
Abbreviation: VP, viral particles.
Patient Characteristics
| Number of Patients, Male | 5 |
| Number of Patients, Female | 5 |
| Stage | Advanced or metastatic solid tumor |
| Age | Median (range): 51.7 (36.1–65.6) |
| Number of Prior Systemic Therapies | Median (range): 2 (0–12) |
| Performance Status: ECOG |
0 — 5 1 — 5 2 — 0 3 — 0 Unknown — 0 |
| Other | Race: white, 7; Asian, 3 |
| Cancer Types or Histologic Subtypes | Microsatellite stable colorectal cancer, 9; cholangiocarcinoma, 1 |
Primary Assessment Method
| Title | Secondary objective: efficacy |
| Number of Patients Screened | 11 |
| Number of Patients Enrolled | 10 |
| Number of Patients Evaluable for Toxicity | 10 |
| Number of Patients Evaluated for Efficacy | 10 |
| Evaluation Method | RECIST 1.1 |
| Response Assessment CR | n = 0 (0%) |
| Response Assessment PR | n = 0 (0%) |
| Response Assessment SD | n = 6 (60%) |
| Response Assessment PD | n = 4 (40%) |
| Response Assessment OTHER | n = 0 (0%) |
| (Median) Duration Assessments TTP | 13.6 weeks |
| Outcome Notes | Secondary objective |
Adverse Events
| All Cycles | |||||||
|---|---|---|---|---|---|---|---|
| Name | NC/NA | 1 | 2 | 3 | 4 | 5 | All grades |
| Injection site reaction | 0% | 30% | 70% | 0% | 0% | 0% | 100% |
| Chills | 20% | 80% | 0% | 0% | 0% | 0% | 80% |
| Fever | 50% | 50% | 0% | 0% | 0% | 0% | 50% |
| Fatigue | 60% | 40% | 0% | 0% | 0% | 0% | 40% |
| Nausea | 90% | 10% | 0% | 0% | 0% | 0% | 10% |
| Myalgia | 90% | 10% | 0% | 0% | 0% | 0% | 10% |
Common Terminology Criteria for Adverse Events v5 used.
Grade 1 or 2 injection site reactions occurred in all patients, with most reporting injection site pain (n = 9; 90%), erythema (n = 8; 80%), and induration (n = 7; 70%). Some adverse events (AEs) were reported more than once by a single patient.
There were two grade 3 AEs reported by two separate patients on the trial (anal pain in a previously radiated area and gram‐negative rod bacteremia). Neither AE was attributed to the vaccines.
Abbreviation: NC/NA, no change from baseline/no adverse event.
Dose‐Limiting Toxicities
| Dose level | No. enrolled | No. evaluable for toxicity | No. with a dose‐limiting toxicity |
|---|---|---|---|
| 1 | 10 | 10 | 0 |
Assessment, Analysis, and Discussion
| Completion | Study completed |
| Investigator's Assessment | Drug tolerable, efficacy indeterminant |
This open‐label, phase I trial demonstrated that the TriAdeno vaccine regimen (TAV) is safe and well tolerated. The recommended dose of TAV for use in future trials is 5 × 1011 viral particles of each vaccine (ETBX‐011, ETBX‐051, and ETBX‐061). The dosing schedule used in this study was every 3 weeks for three doses and then every 8 weeks; however, other studies using TAV are employing other dosing schedules. TAV induced antigen‐specific immune responses directed against all three tumor‐associated antigens (TAAs) with minimal to no “antigenic competition” 1. Neutralizing antibodies to adenovirus 5 (Ad5) were measured as previously described with slight modification 2, 5, 6, 7. At baseline, two of eight patients had neutralizing Ad5 antibodies. After one or two vaccinations, all eight patients analyzed developed neutralizing Ad5 antibodies. The presence of neutralizing antibodies to Ad5 did not prevent the generation of TAA‐specific T cells.
These safety and immunologic data are consistent with findings from a prior clinical trial in metastatic colorectal cancer that showed that the ETBX‐011 vaccine (CEA) was safe and induced CEA‐specific cytotoxic T‐cell activity 2, 3. There was also some evidence of clinical benefit, with half of patients who received ETBX‐011 alive at 1 year after treatment and a little less than one third of patients alive at 18 months after treatment 8.
Currently, immune checkpoint blockade (ICB) benefits only a small percentage of patients, with response rates for Food and Drug Administration‐approved agents around 20%–30% depending on the type of cancer. However, the addition of agents such as vaccines that generate tumor‐specific immune responses and induce immunogenic cell death may be an important component to expand the benefit of ICB to more patients 9, 10, 11, 12, 13. For example, antitumor activity exceeding what would be expected historically has been observed with vaccines plus ICB in small data sets 9, 10. For several reasons, vaccines like TAV are promising candidates for generating immune responses when used in combination with other immuno‐oncology (IO) agents.
The three TAAs (CEA, MUC1, and brachyury) encoded in TAV are associated with several common malignancies. CEA is an attractive target for immunotherapy because it is overexpressed in multiple adenocarcinomas. In addition, CEA is a good target for T‐cell–mediated immunity because it contains known epitopes that are recognized via a major histocompatibility complex (MHC)—restricted fashion by human cytolytic T lymphocytes that bind to MHC loci human leukocyte antigens (HLA) A2, A3, and A24 1, 14. MUC1 is expressed on the majority of human adenocarcinomas, with high expression seen in colorectal cancer, breast cancer, non‐small cell lung cancer, bladder cancer, and pancreatic cancer. Multiple enhanced agonist epitopes of MUC1 including the C‐terminus of MUC1 act as oncogenes and can induce plasticity 15, 16. Human T‐cell lines generated using MUC1 agonist epitopes generated antigen‐specific interferon gamma (IFNγ) and lysis of tumor cells that express the native MUC1 16, 17. Brachyury is an embryonic transcription factor of the T‐box family that regulates cellular plasticity 18. High brachyury expression is found in lung cancer, colorectal cancer, breast cancer, prostate cancer, and gastrointestinal stromal tumor 19. Carcinoma cells that undergo a phenotypic transition exhibit enhanced motility and invasiveness in vitro and the propensity to metastasize in vivo 20. Using a 9‐mer peptide of the brachyury protein, brachyury‐specific CD8+ T cells were expanded in vitro from the blood of patients with cancer and then used in cytotoxic assays for effective lysis of human tumor cells that endogenously express brachyury 21, 22. Additionally, multiple studies have demonstrated that MUC1 and/or brachyury expression are markers of poor prognosis 23, 24, 25, 26, 27, treatment resistance 28, 29, 30, 31, and tumor aggressiveness 32, 33. In vitro and preclinical animal models with the TAAs demonstrated antitumor cytolytic T‐cell responses. Multiple therapeutic cancer vaccine trials have been conducted using one or two of these TAAs, but this is the first trial to our knowledge that uses this triad of TAAs.
TAV vaccination generated CD4+ and/or CD8+ T‐cell responses to at least one TAA encoded by the vaccine in all patients. Two patients developed responses after vaccination to all three TAAs in the vaccine. Furthermore, polyfunctional TAA‐specific responses, defined as CD4+ or CD8+ T cells that express ≥2 of the following markers: IFNγ, tumor necrosis factor, interleukin‐2, or CD107a, were measured before and after vaccination. Using the criteria of a >10‐fold increase after versus before vaccination, or the presence of >1,000 polyfunctional cells after vaccination per 1 × 106 peripheral blood mononuclear cells (if negative at prevaccination), polyfunctional T‐cells specific for MUC1, CEA, or brachyury were generated in 50%, 33%, and 17% of patients, respectively. Although there is no overt clinical benefit seen in this small phase I trial, the generation of long‐lasting polyfunctional T cells has been previously associated with improved overall survival 34. In patients with melanoma, polyfunctional T cells can be detected as early as after one vaccination, and these T cells can persist for years after initial vaccination in responders 34.
In conclusion, this work adds to the existing literature 11, 35, 36, 37, 38 that antitumor vaccines directed against CEA, MUC1, and brachyury are well tolerated and can generate antitumor immune responses. TAV generated antigen‐specific T cells to one or more target antigens in all patients evaluated. Planned studies are aimed at interrogating the TAV regimen's potential antitumor activity when used in combination with other IO agents such as ICB and immunocytokines.
Disclosures
The authors indicated no financial relationships.
Acknowledgments
We thank Angie Schwab for technical assistance with immune assays and Debra Weingarten for editorial assistance in the preparation of this manuscript. The National Institutes of Health and National Cancer Institute have a CRADA (Collaborative Research and Development Agreement) with the Etubics Corporation, as well as patents involving agonist epitopes, brachyury, MUC1, and CEA.
This article has been contributed to by US Government employees and their work is in the public domain in the USA.
Footnotes
- ClinicalTrials.gov Identifier: NCT03384316
- Sponsor(s): Etubics (a wholly owned subsidiary of ImmunityBio) and the NCI
- Principal Investigator: Julius Strauss
- IRB Approved: Yes
Contributor Information
Margaret E. Gatti‐Mays, Email: margaret.gatti-mays@nih.gov.
Julius Strauss, Email: julius.strauss@nih.gov.
References
- 1. Gabitzsch ES, Tsang KY, Palena C et al. The generation and analyses of a novel combination of recombinant adenovirus vaccines targeting three tumor antigens as an immunotherapeutic. Oncotarget 2015;6:31344–31359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morse MA, Chaudhry A, Gabitzsch ES et al. Novel adenoviral vector induces T‐cell responses despite anti‐adenoviral neutralizing antibodies in colorectal cancer patients. Cancer Immunol Immunother 2013;62:1293–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Balint JP, Gabitzsch ES, Rice A et al. Extended evaluation of a phase 1/2 trial on dosing, safety, immunogenicity, and overall survival after immunizations with an advanced‐generation Ad5 [E1‐, E2b‐]‐CEA(6D) vaccine in late‐stage colorectal cancer. Cancer Immunol Immunother 2015;64:977–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heery CR, Singh BH, Rauckhorst M et al. Phase I trial of a yeast‐based therapeutic cancer vaccine (GI‐6301) targeting the transcription factor brachyury. Cancer Immunol Res 2015;3:1248–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gabitzsch ES, Xu Y, Balint JP et al. Anti‐tumor immunotherapy despite immunity to adenovirus using a novel adenoviral vector Ad5 [E1‐, E2b‐]‐CEA. Cancer Immunol Immunother 2010;59:1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gabitzsch ES, Xu Y, Balcaitis S et al. An Ad5[E1‐, E2b‐]‐HER2/neu vector induces immune responses and inhibits HER2/neu expressing tumor progression in Ad5 immune mice. Cancer Gene Ther 2011;18:326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gabitzsch ES, Xu Y, Balint JP et al. Induction and comparison of SIV immunity in Ad5 naïve and Ad5 immune non‐human primates using an Ad5 [E1‐, E2b‐] based vaccine. Vaccine 2011;29:8101–8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gabitzsch ES, Morse MA, Lyerly HK et al. Immunotherapeutic treatment of metastatic colorectal cancer using ETBX‐011. J Clin Oncol 2014;32(suppl 15):3093a.25154826 [Google Scholar]
- 9. Massarelli E, William W, Johnson F et al. Combining immune checkpoint blockade and tumor‐specific vaccine for patients with incurable human papillomavirus 16‐related cancer: A phase 2 clinical trial. JAMA Oncol 2019;5:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jochems C, Tucker JA, Tsang KY et al. A combination trial of vaccine plus ipilimumab in metastatic castration‐resistant prostate cancer patients: Immune correlates. Cancer Immunol Immunother 2014;63:407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gatti‐Mays ME, Strauss J, Donahue RN et al. A phase 1 dose escalation trial of BN‐CV301, a recombinant poxviral vaccine targeting MUC1 and CEA with costimulatory molecules. Clin Cancer Res 2019;25:4933–4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gatti‐Mays ME, Redman JM, Collins JM et al. Cancer vaccines: Enhanced immunogenic modulation through therapeutic combinations. Hum Vaccin Immunother 2017;13:2561–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schlom J, Gulley JL. Vaccines as an integral component of cancer immunotherapy. JAMA 2018;320:2195–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zaremba S, Barzaga E, Zhu M et al. Identification of an enhancer agonist cytotoxic T lymphocyte peptide from human carcinoembryonic antigen. Cancer Res 1997;57:4570–4577. [PubMed] [Google Scholar]
- 15. Kufe DW. MUC1‐C oncoprotein as a target in breast cancer: Activation of signaling pathways and therapeutic approaches. Oncogene 2013;32:1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsang KY, Palena C, Gulley J et al. A human cytotoxic T‐lymphocyte epitope and its agonist epitope from the nonvariable number of tandem repeat sequence of MUC‐1. Clin Cancer Res 2004;10:2139–2149. [DOI] [PubMed] [Google Scholar]
- 17. Jochems C, Tucker JA, Vergati M et al. Identification and characterization of agonist epitopes of the MUC1‐C oncoprotein. Cancer Immunol Immunother 2014;63:161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fernando RI, Litzinger M, Trono P et al. The T‐box transcription factor brachyury promotes epithelial‐mesenchymal transition in human tumor cells. J Clin Invest 2010;120:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hamilton DH, Fernando RI, Schlom J et al. Aberrant expression of the embryonic transcription factor brachyury in human tumors detected with a novel rabbit monoclonal antibody. Oncotarget 2015;6:4853–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nieto MA, Cano A. The epithelial‐mesenchymal transition under control: Global programs to regulate epithelial plasticity. Semin Cancer Biol 2012;22:361–368. [DOI] [PubMed] [Google Scholar]
- 21. Tucker JA, Jochems C, Boyerinas B et al. Identification and characterization of a cytotoxic T‐lymphocyte agonist epitope of brachyury, a transcription factor involved in epithelial to mesenchymal transition and metastasis. Cancer Immunol Immunother 2014;63:1307–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Palena C, Polev DE, Tsang KY et al. The human T‐box mesodermal transcription factor brachyury is a candidate target for T‐cell‐mediated cancer immunotherapy. Clin Cancer Res 2007;13:2471–2478. [DOI] [PubMed] [Google Scholar]
- 23. Zeng Y, Zhang Q, Zhang Y et al. MUC1 predicts colorectal cancer metastasis: A systematic review and meta‐analysis of case controlled studies. PLoS One 2015;10:e0138049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kharbanda A, Rajabi H, Jin C et al. Oncogenic MUC1‐C promotes tamoxifen resistance in human breast cancer. Mol Cancer Res 2013;11:714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang X, Sun Q, Chen C et al. MUC1 overexpression predicts worse survival in patients with non‐small cell lung cancer: Evidence from an updated meta‐analysis. Oncotarget 2017;8:90315–90326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khodarev NN, Pitroda SP, Beckett MA et al. MUC1‐induced transcriptional programs associated with tumorigenesis predict outcome in breast and lung cancer. Cancer Res 2009;69:2833–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Palena C, Roselli M, Litzinger MT et al. Overexpression of the EMT driver brachyury in breast carcinomas: Association with poor prognosis. J Natl Cancer Inst 2014;106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maeda T, Hiraki M, Jin C et al. MUC1‐C induces PD‐L1 and immune evasion in triple‐negative breast cancer. Cancer Res 2018;78:205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bouillez A, Adeegbe D, Jin C et al. MUC1‐C promotes the suppressive immune microenvironment in non‐small cell lung cancer. Oncoimmunology 2017;6:e1338998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. David JM, Hamilton DH, Palena C. MUC1 upregulation promotes immune resistance in tumor cells undergoing brachyury‐mediated epithelial‐mesenchymal transition. Oncoimmunology 2016;5:e1117738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bouillez A, Rajabi H, Pitroda S et al. Inhibition of MUC1‐C suppresses MYC expression and attenuates malignant growth in KRAS mutant lung adenocarcinomas. Cancer Res 2016;76:1538–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rajabi H, Kufe D. MUC1‐C oncoprotein integrates a program of EMT, epigenetic reprogramming and immune evasion in human carcinomas. Biochim Biophys Acta 2017;1868:117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kharbanda A, Rajabi H, Jin C et al. MUC1‐C confers EMT and KRAS independence in mutant KRAS lung cancer cells. Oncotarget 2014;5:8893–8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wimmers F, Aarntzen EH, Duiveman‐deBoer T et al. Long‐lasting multifunctional CD8. Oncoimmunology 2016;5:e1067745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gulley JL, Arlen PM, Tsang KY et al. Pilot study of vaccination with recombinant CEA‐MUC‐1‐TRICOM poxviral‐based vaccines in patients with metastatic carcinoma. Clin Cancer Res 2008;14:3060–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mohebtash M, Tsang KY, Madan RA et al. A pilot study of MUC‐1/CEA/TRICOM poxviral‐based vaccine in patients with metastatic breast and ovarian cancer. Clin Cancer Res 2011;17:7164–7173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hamilton DH, Litzinger MT, Jales A et al. Immunological targeting of tumor cells undergoing an epithelial‐mesenchymal transition via a recombinant brachyury‐yeast vaccine. Oncotarget 2013;4:1777–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heery CR, Singh BH, Rauckhorst M et al. Phase I trial of a yeast‐based therapeutic cancer vaccine (GI‐6301) targeting the transcription factor brachyury. Cancer Immunol Res 2015;3:1248–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]