

Abstract
Introduction
Drug induced steatohepatitis (DISH), a form of drug induced liver injury (DILI) is characterized by intracellular accumulation of lipids in hepatocytes and subsequent inflammatory events, in some ways similar to the pathology seen with other metabolic, viral and genetic causes of non alcoholic fatty liver disease and steatohepatitis (NAFLD and NASH).
Areas covered
This paper provides a comprehensive review of the main underlying mechanisms by which various drugs cause DISH, and outlines existing preclinical tools to predict it and study underlying pathways involved. The translational hurdles of these models are discussed, with the example of an organotypic liver system designed to address them. Finally, we describe the clinical assessment and management of DISH.
Expert Opinion
The complexity of the interconnected mechanistic pathways underlying DISH makes it important that preclinical evaluation of drugs is done in a physiologically and metabolically relevant context. Advanced organotypic tissue models, coupled with translational functional biomarkers and next-generational pan-omic measurements, may offer the best shot at gathering mechanistic knowledge and potential of a drug causing steatohepatitis. Ultimately this information could also help predict, detect or guide the development of specific treatments for DISH, which is an unmet need as of today.
Keywords: Drug-induced steatohepatitis, liver, organotypic, DILI, NASH
1. Introduction
Drug induced steatohepatitis (DISH), is a form of drug induced liver injury (DILI) that is characterized by characteristic pathological patterns of intracellular accumulation of lipids in hepatocytes, often accompanied by oxidative stress and inflammatory events that involving secondary cell types[1]. This is in many ways similar to the pathology seen with other metabolic, viral and genetic causes of non alcoholic fatty liver disease and steatohepatitis (NAFLD and NASH). Though many include DISH under the spectrum of NAFLD and NASH (Figure 1), recognized as the leading cause of liver disease in the western world, a majority of NASH cases are attributed to metabolic causes, due to the burgeoning obesity epidemic[2]. While DISH accounts for fewer than 2% of all cases of NASH[3], there are a significant number of drugs that can acutely cause a spectrum of steatohepatitic progressive changes. This pattern of DISH has pathogenic mechanisms and phenotypes that are distinct from other forms of DILI such as hepatocellular, cholestatic and immunologic hepatotoxicity induced by drugs. In this article, we review the drugs and mechanisms implicated in causing DISH, existing models and endpoints to preclinically predict the potential of drugs causing it and study underlying pathways, as well as the clinical assessment and management of the condition. We also discuss an organotypic model for studying underlying mechanisms in a more physiologically relevant milieu to provide the context for an expert opinion that frames assessment approaches for the future.
Figure 1.
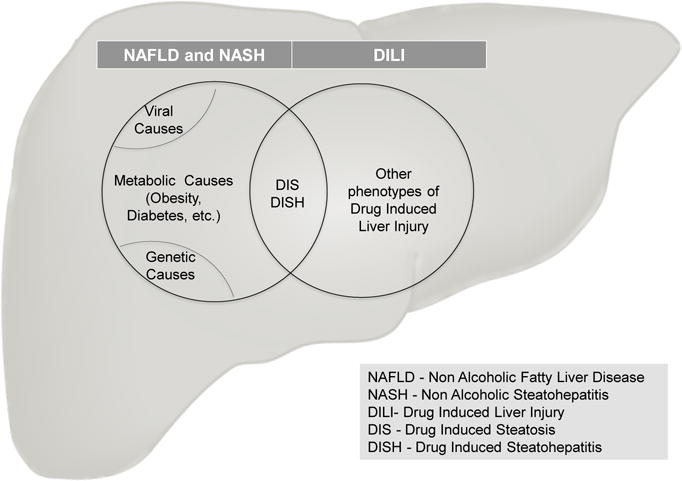
Drug Induced Steatohepatitis (DISH) is a form of drug induced liver injury (DILI) that shares pathological features characterized by lipid accumulation and inflammatory changes, similar to non alcoholic steatohepatitis arising due to other causes.
2. DISH: Drugs and Underlying Mechanisms
A number of excellent recent reviews have taken disparate approaches towards the classification of drugs that cause DISH. While none of these classifications is arbitrary, they often reflect the particular interest of the authors as well as the complex nature of a phenomenon where few commonalities may appear to exist amongst drugs responsible for the induction of steatohepatitis. The basis for classification has included systematic groupings based on the phenotype of the resulting hepatic injury[4, 5], the underlying mechanisms of injury[5, 6], dependence, or lack thereof on an underlying metabolic phenotype or pre-existing NAFLD[3], or the chemical properties of the drugs themselves (Schumacher, 2015). Attempts to refine the classification of DISH have also led to the sub-classification of drugs based on their class affiliation and the genesis of such terms as “CASH” (chemotherapy-associated steatohepatitis;[7]) and “TASH” (toxicant-associated steatohepatitis;[8]).
2.1. Cationic Amphiphilic Drugs
Many of the drugs that induce DISH possess characteristic chemical features and are known as cationic amphiphilic drugs (CADs). CADs include such drugs as amiodarone, perhexiline, propranolol and tamoxifen[9]. These drugs tend to be sequestered in the liver, probably due to trapping in mitochondria and lysosomes[10, 11]. CADs typically have a lipophilic ring structure (LogP > 1) and possess one or more substituent groups containing secondary or tertiary amines (pKa > 6.5). In the acidic intracellular environment of the lysosome these drugs become increasingly ionized and unable to readily diffuse across membranes. This phenomenon can result in the intracellular accumulation of very high levels of drug and sets the stage for hepatotoxic interactions. From a drug development perspective, this can make the interpretation of pharmacokinetic and pharmacodynamic data difficult since sequestration of these compounds can be mistaken for active transport[11]. Lysosomal sequestration of CADs has been implicated in connection with phospholipidosis, the excess accumulation of phospholipids, after forming tight complexes with phospholipids and interfering with their catabolism[12, 13]. Amiodarone and perhexiline are both known to induce phospholipidosis[6, 9]. Similarly, after crossing the outer membrane of the mitochondria, CADs are protonated in the acidic intermembrane space and then driven into the matrix by the mitochondrial membrane potential causing mitochondrial dysfunction through a number of mechanisms including disruption of fatty acid β-oxidation and resulting in steatosis[6].
2.2. Mechanisms underlying intracellular lipid accumulation
A consensus view among many authors is that the mechanisms of drugs that cause steatosis or intracellular lipid accumulation in DISH can be aligned with the four broad, basic mechanisms associated with the induction of steatosis: 1) increased fatty acid synthesis; 2) decreased fatty acid β-oxidation; 3) decreased lipoprotein export; and 4) increased mobilization and uptake of fatty acids[6, 9, 14]. The progression of steatosis into steatohepatitis is thought to further involve pathophysiologic mechanisms that result in the production of reactive oxygen species that drive oxidative stress[4, 5].
2.3. Increased fatty acid synthesis
The mechanisms by which xenobiotics induce de novo lipogenesis are not well understood. De novo synthesis of fatty acids is regulated by glucose and insulin. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein (SREBP) are transcription factors that respond to glucose and insulin, respectively[14]. Tamoxifen is thought to increase fatty acid synthesis through upregulation of SREBP and activation of its downstream target genes fatty acid synthase, acetyl-CoA carboxylase and stearoyl-CoA desaturase [15, 16]. Many hepatic nuclear receptors also play a prominent role in energy homeostasis and lipid metabolism and the steatogenic potential of some nuclear receptors such as PXR, LXR and PPARγ has been clearly demonstrated [17]. Some drugs are known to activate these lipogenic transcription factors leading to the subsequent induction of the enzymes directly involved in fatty acid synthesis. Drugs that may utilize this mechanism include the glucocorticoids through activation of glucocorticoid receptor, the PXR activator nifedipine, and the PPARγ activator troglitazone[4, 6].
2.4. Decreased fatty acid β-oxidation
Drugs that severely impair mitochondrial β-oxidation can induce the accumulation of free fatty acid and triglycerides resulting in microvesicular steatosis. Unlike the more common and reversible, large droplet macrovesicular steatosis associated with alcohol and metabolic disease, microvesicular steatosis is more serious and manifests as diffuse accumulation of small fat droplets without peripheral displacement of the nucleus or evidence of significant inflammation[4]. A number of distinct mechanisms have been identified for the inhibition of β-oxidation by xenobiotics. Direct inhibition of mitochondrial enzymes involved in β-oxidation can be induced by amiodarone, tamoxifen and valproic acid[6]. In addition, indirect inhibition of β-oxidation can occur through the inhibition of carnitine palmitoyltransferase I (CPT-1) (tamoxifen, amiodarone, perhexiline, valproic acid), sequestration of essential cofactors for the esterification of fatty acids such as coenzyme A and carnitine (valproic acid, salicylic acid), inhibition of the mitochondrial respiratory chain (amiodarone, methotrexate, perhexiline, tamoxifen, tetracycline) and disruption of mitochondrial DNA (tamoxifen, troglitazone) [4–6, 9, 18, 19].
2.5. Decreased lipoprotein export
Once synthesized, fatty acids are esterified to glycerol to form triglycerides, incorporated into very low density lipoproteins (VLDL) and secreted into plasma. Another potential mechanism driving DISH is a reduction in the rate of lipoprotein export. This can be a result of decreased fatty acid incorporation into VLDL or a reduction in VLDL secretion [4]. Synthesis of VLDL particles is dependent on apolipoprotein B (apoB), the primary lipoprotein of VLDL, as well as upon microsomal triglyceride transfer protein (MTP) which plays a critical role in lipoprotein assembly (Hussain, 2008). Several steatogenic drugs, including amiodarone and tetracycline, have been demonstrated to inhibit MTP activity, the lipidation of apoB into VLDL and hepatic lipoprotein secretion [14]. This mechanism has also been demonstrated for tamoxifen [16].
2.6. Increased mobilization and uptake of fatty acids
Hepatocellular uptake of non-esterified fatty acids is a major contributor to the excess accumulation of triglycerides associated with steatosis [20]. One of the best characterized regulators of this process is fatty acid translocase, CD36 [21]. Adenosine monophosphate-activated protein kinase (AMPK) is universally recognized as a major regulator of fat metabolism and promotes fatty acid uptake into cells via activation of CD36 [6, 22]. This mechanism has been implicated for the non-nucleoside reverse transcriptase inhibitor, efavirenz, where the activation of AMPK is likely a compensatory response to the inhibition of complex I of the mitochondrial respiratory chain [23]. CD36 has also been defined as a shared target of the hepatic nuclear receptors LXR and PXR, as well as PPARα and PPARγ [14, 24]. This may provide a link between activators of these transcription factors and increased steatotic potential.
It is imperative that strategies be developed for use in the early preclinical stages of drug development in order to effectively evaluate the potential for steatohepatitis of drugs with recognizable risk factors such as cationic amphiphilic structures or mechanisms of action which involve interaction with nuclear receptors or other key regulators of hepatic lipid metabolism. A number of groups have attempted to define a transcriptomic signature that is predictive of DISH for the purpose of providing a specific and sensitive assay for pre-clinical drug development. Sahini et al [25] defined nine genes regulated in common by drugs which induced DISH related to lipid transport and excess triglyceride accumulation in the form of lipid droplets. Benet et al [26] have described a common signature for steatotic drugs likely mediated by repression of a complex network of transcription factors including three key regulators of liver metabolism: FOXA1, HEX and SREBP1C. Most recently, Liu et al [27] have suggested that serum miRNAs can be used to distinguish between DISH and NAFLD. Despite these efforts at defining molecular signatures for the identification of DISH, there remains an unmet need for human-relevant in vitro systems for the preclinical assessment of the steatohepatitic potential of drugs in development.
3. Preclinical and in vitro DISH Models
Preclinical assessment of DISH can be a routine part of animal toxicity studies as well as include various in vitro and cellular assays aimed at screening the potential of drugs to induce the accumulation of fat and subsequent inflammatory effects in hepatocytes.
3.1. Animal models
A variety of animal models have been specifically developed to recreate NASH arising from metabolic causes using dietary and genetic approaches and have been previously covered extensively in multiple reviews[28–31]. However animal models of DISH are generally described in literature secondary to, or as part of in vivo toxicity testing of compounds. Most of these reported studies include the drugs commonly associated with DISH listed in the previous section. For instance, Choi et al investigated the role of fatty acid transport and esterification in tetracycline-induced steatosis male ICR mice[32]. Intracellular lipid accumulation and the protein expression of fatty acid translocase (FAT or CD36) and diacylglycerol acyltransferase (DGAT) 2 were increased in the mouse liver. Tamoxifen is one of the most widely assessed drugs in animal models for its ability to cause DISH. Larosche et al. treated male Crl:CD-1(ICR)BR Swiss mice with Tamoxifen for 28 days and demonstrated its ability to induce steatosis by inhibiting hepatic triglyceride secretion as well as mitochondrial β-oxidation and respiration[33]. Other studies have similarly demonstrated the ability of Tamoxifen to induce microvesicular steatosis in male Wistar rats[34] or increased triacylglycerol biosynthesis with resulting accumulation in female rats[35]. Le et al 2014 co-administered Uridine, a pyrimidine nucleoside was tested along with tamoxifen in C57BL/6J mice to evaluate its reported protective effects against drug-induced fatty liver. Liver lipid levels were evaluated with lipid visualization using coherent anti-Stokes Raman scatting (CARS) microscopy, biochemical assay measurement of triacylglyceride (TAG), and liquid chromatography coupled with mass spectrometry (LC-MS) measurement of membrane phospholipid. Blood TAG and cholesterol levels were measured. Uridine co-administration prevented tamoxifen-induced liver lipid droplet accumulation in mice. Others have used publically available microarray databases from drug treated animals as in the the Japanese Toxicogenomics Project (TGP) to explore mechanisms underlying drug-induced steatotic signatures by studying comprehensively whole genome gene expression changes in the liver of treated rats[25]. In this study Sahini et al examined a total of 36 drugs diverse in molecular structure and mode of action that included 17 and 12 drugs with the ability to cause either steatosis or phospholipidosis respectively, and 7 drugs negative controls based on histopathological findings. They focused on 200 genes that they had previously deemed to be mechanistically relevant in the process of lipid droplet biogenesis in hepatocytes. In the study, they identified 19 genes that exhibited dose dependent responses and defined a unique signature made up of 9 genes (ANGPTL4, FABP7, FADS1, FGF21, GOT1, LDLR, GK, STAT3, and PKLR) to predict drug induced steatosis. Other strongly regulated genes included genes linked to glucose metabolism, lipid transport and lipogenesis signalling. They were able to identify 26 genes to be regulated in common between drugs causing phospholipidosis and/or steatosis.
3.2. Cellular Models of DISH
Cellular models to identify efficacious drugs and compounds that lack the potential to cause hepatotoxicity in man have been around for decades. These offer a way to predict effects that may be missed in animal models due to inherent species differences and can be a medium throughput screen to identify cytotoxic potential at different doses. To be optimally successful in reproducing key processes that underlie specific phenotypic hepatotoxic outcome such as DISH, the culture models need to be metabolically competent while possessing a lifespan long enough to build up the pathologic change, e.g steatosis. Cellular models also need to be coupled with endpoint assays that measure the phenotytpic changes or underlying perturbations with high sensitivity, specificity and reproducibility. Some of these approaches have been described in a previous review[18]. Cell types typically used include primary hepatocytes from multiple species, immortalized cell lines as well as iPSC derived hepatocytes (Table).
Table 1.
Comparison of Some Steatohepatitic Drugs.
| Drug | Chemical class | Indication | Primary Mechanism of Action | Histological Type of Injury | DISH Mechanisms involved |
|---|---|---|---|---|---|
| Amiodarone | Benzofuran | Antianginal, Antiarrhythmic (Class III) | Prolongs myoardial action potential, Antagonism of α- and β-adrenergic receptors | Microvesicular, macrovesicular, steatohepatitis | 1, 2, 3, 4 |
| Cocaine | Alkaloid ester | Local anesthetic | Blockade of the dopamine transporter protein | Microvesicular | 2 |
| 5-Fluorouracil | Fluoropyrimidine | Antineoplastic, | Inhibits thymidylate synthase | Macrovesicular | 2 |
| Glucocorticoids | Steroid | Antiinflammatory, Immunosupression | Glucocorticoid receptor agonist | Microvesicular, macrovesicular | 1,2 |
| Methotrexate | Pteridine | Antirheumatic, Antineoplastic, Antimetabolite | Inhibits dihydrofolate reductase, Immune modulation | Macrovesicular, steatohepatitis | 2 |
| Nucleoside Reverse Transcriptase Inhibitors (NRTIs) | Multiple | HIV | Inhibits Reverse transcriptase | Microvesicular | 1,2,3 |
| Perhexiline | Piperidine | Antianginal | Inhibits carnitine palmitoyltransferase I | 2, 4 | |
| Tamoxifen | Stilbene | Antineoplastic | Selective estrogen receptor modulator | Macrovesicular, steatohepatitis | 1, 2, 3 |
| Tetracycline | Tetracylcine | Broad spectrum antibiotic | Inhibition of bacterial protein synthesis | Microvesicular | 2,3 |
| Valproic acid | Fatty acyl | Antiepileptic, Mood stabilizer | Increased brain concentrations of GABA | Microvesicular | 2 |
DISH Mechanisms:
Increased fatty acid synthesis.
Decreased fatty acid β-oxidation:
Decreased lipoprotein export and
Increased mobilization and uptake of fatty acids.
While the use of human primary hepatocytes is ideal, cell limes and rodent hepatocytes are often a cheaper alternative making their use more common. Summeren et al [36] used primary mouse hepatocytes to overcome issues of availability and inter-individual variation faced with human hepatocytes and coupled them with differential in gel electrophoresis (DIGE) to study large-scale protein expression following exposure to amiodarone. Le and colleagues [37] interestingly used mouse hepatocytes to demonstrate the effect of uridine in preventing tamoxifen-induced liver lipid droplet accumulation. Primary rat hepatocytes have also been used extensively and as early as 1994 when Deschamps et al [38] demonstrated the ability of perhexiline to induce microvesicular steatosis via inhibition of oxidative phosphorylation and the beta-oxidation of fatty acids, by the measurement of ATP and carbon 14 labeled fatty acids. Other drugs like amiodarone, diethylaminoethoxyhexestrol, valproate, tetracycline and tamoxifen have also been assessed for steatotic potential using a variety of endpoints such as triglycerides, lipid peroxidation, mitochondrial respiration dysfunction and oxidative stress [32, 34, 39, 40]. Recently Germano and colleagues exposed rat hepatocytes in Collagen I-Matrigel™ sandwich cultures to chronic non-cytotoxic concentrations of various drugs over 14 days. While they were able to induce phospholipidosis similar to that seen in vivo, they had limited success in reproducing steatosis. Tetracycline-induced steatosis has been demonstrated in primary canine beagle hepatocyte cultures by increases in the Oil Red O-stained lipid inclusions and an increase in intracellular triglyceride following a 24-h exposure[41].
To overcome the lack of species relevance of animal cells, various human hepatocyte cell lines are commonly employed to assess steatosis. Hep G2 cells, a cell line derived from a well-differentiated hepatocellular carcinoma are used for toxicity as they possess many of the genotypic features of primary hepatocytes[42]. HepG2 cells have been demonstrated to pick up the steatotic effects of oleic acid, amiodarone, doxycycline, tetracycline, valproate and tamoxifen in combination of a variety of endpoints such as oil red O, Nile red, BODIPY493/503 dyes, cellular ATP content and lipid peroxidation assays [16, 43–46]. A challenge with HepG2 cells however is that their metabolic activity is significantly lower than primary hepatocytes, translating into decreased sensitivity that is less than a fifth of primary hepatocytes while testing the toxicity of drugs [47]. HepaRG cells, also derived from a human hepatocellular carcinoma cell line [48] retain a drug metabolism capacity that is more comparable to primary hepatocytes [49] and are shown to maintain hepatic functions and expression of liver-specific genes at levels comparable to human hepatocytes[50]. However they are more expensive than HepG2 cells. HepaRG cells have been used to study the steatotic potential of amiodarone and tetracycline using Oil Red O and triglyceride as endpoints in combination to a lipid metabolism gene panel [50]. Others have used HepaRG cells to demonstrate the ability of agents such as PPAR agonists or Farnesol to reduce steatosis following oleic acid-overloading [51, 52].
Human induced pluripotent stem cell (iPSC)–derived hepatocytes hold the promise of possessing a primary hepatocyte-like phenotype, and can offer an unlimited source of genotype-specific cells from different individuals [53, 54]. Sirenko and colleagues recently used iPSC derived hepatocytes to demonstrate the potential of amiodarone, chloroquine and propranolol to induce phospholipidosis and steatosis using the using LipidTOX reagent[55]. However, a major drawback of iPSC derived hepatocytes is that they exhibit aspects of fetal phenotype [56] including significantly lower albumin secretion than primary hepatocytes with simultaneous persistence of alpha-fetoprotein (AFP) expression. This is also associated with lower expression and activities of specific CYP450 enzymes (e.g. the CYP3A family), responsible for metabolizing almost 50% of drugs including amiodarone, reflective of a limited ability to generate toxic intermediates [57].
3.3. Translational Challenges
Though animal models such as those described in section 3.1 can often pick up the ability of drugs that induce fatty liver and steatohepatitis, they could be compromised in instances where the drug effects on metabolic pathways are dependent on enzymes whose isoforms may be species dependent (e.g. CYP4A family cytochrome). Alternately the drug-metabolizing CYP enzymes responsible for generating a specific metabolite that could induce the hepatotoxic effect may not be present in the animal species or the equivalent isoform may have a vastly different spectrum of activity challenging the translatability of the effect. With cellular models, the biggest hurdles in translatability relate to four important aspects: 1) the lack of metabolic equivalence of the cell type when non-primary cells are used. For instance, HepG2 cells and Huh7 cells are compromised in the levels of various CYP enzyme activities relative to primary hepatocytes[58]. 2) dedifferentiation and loss of metabolic and liver specific functions over time in cell culture[59–61]. 3) non-physiological drug and metabolite concentration profiles in conventional cultures due to the static design and lack of in vivo-like flow and transport and 4) the non-physiological levels of concentrations of hormones, enzymes, growth factors and drugs used in cell culture maintenance media and for treatments, relative to the in vivo context. The last aspect could be a consequence of the earlier three limitations and results in routine use extremely high baseline levels of glucose and insulin for cell survival and maintenance in many of these culture systems (Table 2). While the glucose is at diabetic levels, the concentration of insulin is often over 10,000 fold greater than the fasting to post-prandial physiological range of 50–300 pmol of insulin[62]. This particularly poses a major translational challenge while assessing drugs that either cause steatohepatitis or even those may be used to treat NASH. The non-physiological levels of insulin and diabetic levels of glucose used to counter the lack of insulin responsiveness not only create an altered metabolic baseline which is not reflective of the healthy state, but also influence other processes such as inflammatory signaling that are instrumental to the mechanisms driving steatohepatitis and complexly regulated by insulin. For instance, there is evidence that insulin activates the inflammatory NF-κB in mammalian cells through a post-translational mechanism via insulin receptor tyrosine kinase and Raf-1 kinase activities [63]. Conversely, there are reports that NF-κB can induce insulin resistance [64] and inactivation of NF-κB increases insulin sensitivity [65]. This makes it imperative to develop culture systems that retain the in vivo hepatic phenotype and responsiveness adequately to allow survival in more physiological milieus and a more relevant baseline to study drugs impacting metabolic pathways.
Table 2.
In vitro models Used to Assess Steatohepatitis
| Species | Cell Type | Drugs Tested | Endpoints Used | Insulin concentration | Glucose concentration | References |
|---|---|---|---|---|---|---|
| Mouse | Primary Hepatocytes | Amiodarone, Tamoxifen | Microarrays, Differential in gel electrophoresis, Bioenergetics | 3.5 μmol/L | 4500 mg/L | Summeren 2013, Le et al 2014 |
| Precision Cut Liver Slices | Amiodarone, Valproic Acid, and Tetracycline | – | 2000 mg/L | Szalowska et al 2014 | ||
| Rat | Primary Hepatocytes | Diethylaminoethoxyhexestrol, amiodarone, perhexiline, valproate, tetracycline, tamoxifen | ATP, β-oxidation, triglycerides, lipid peroxidation, mitochondrial respiration dysfunction, oxidative stress | 0.7 μmol/L | 2000 mg/L | Deschamps et al 1994, Berson et al 1998, Tong et al 2005 and Choi 2015, Lelliott at al 2005. |
| NT H35 hepatoma cell line | Oleic acid, linoleic acid | Oil red O, triglycerides, Reactive Oxygen Species, Mitochondrial Membrane Potential | 3 μmol/L | – | Janorkar 2009 | |
| Dog | Primary Hepatocytes | Tetracycline | Oil red O, triglyceride content Nile red, β-oxidation | 0.05 μmol/L | 10000mg/L | David E. Amacher and Barbara-Anne Martin 1997 |
| Human | Primary Hepatocytes | 76 nuclear receptor ligands | Lipid accumulation, mRNA | – | – | Moya et al 2010 |
| HepG2 cancer cell line | Oleic acid, Amiodarone, Doxycycline, Tetracycline, Valproate, Tamoxifen | Mitochondrial respiration dysfunction, oxidative stress, Oil red O, mRNA | – | – | Donato 2009, Choi 2015, Kamalian 2015, Zhao et al 2014. | |
| Huh7 cell line | u-PA, valproate | Triglyceride, lipid droplets | – | – | Paland 2014, Elphick 2012 | |
| HepaRG Cells | Amiodarone and Tetracycline | Oil red O | 0.86 μmol/L | 2000 mg/L | Antherieu 2011 | |
| iPSC derived hepatocytes | Amiodarone, Propranolol | Mitochondrial potential, LipidTOX (phospholipids and neutral lipids) | – | – | Sirenko et al 2014 | |
| hSKP-HPCs postnatal skin precursor derived Hepatocytes | Sodium Valproate | Microarrays, LipidTox stains, transmission electron microscopy | 1.0 μmol/L | 4500 mg/L | Rodrigues 2015 |
Another challenge is the lack of adequate clinical biomarkers that are specific to the steatohepatitic process and translate from the pre-clinical models to the clinic. Most of the endpoints used in pre-clinical and in vitro models (Table 2) are either cytotoxicity or cell injury measures similar to those routinely used for DILI, measurement of intracellular lipids and phospholipids, mitochondrial β-oxidation activity or ATP and respiratory activity. Many of the clinical serum biomarkers are still in the exploratory stage, and are mainly being evaluated for metabolic causes of NASH (detailed in another review [18]). These include serum adipokines and inflammatory cytokine panels (including TNF-α IL-6 and IL-8), cytokeratin 18 (CK 18), tissue inhibitor of metalloproteinase 1 (TIMP-1), YKL-40, hyaluronic acid, serum prolidase enzyme activity, plasmapentraxin3, visfatin, chemirin, vaspin, fibrinogen chain, retinol binding protein 4, serum amyloid P component, lumican, and transgelin 2 among others. While there is no clear consensus of any single one of these being adequate to distinguish the state of steatosis from steatohepatitis, most of them are not being used during pre-clinical evaluation leaving a gap for clinical translatability.
3.4. Assessment of DISH in a human organotypic system
Flow based organotypic liver systems are designed to avoid the static nature of conventional cultures, and offer control over oxygen and nutrient transport [66–68] and prevent the build up of cytokines and toxic metabolites produced by the cells to non-physiological levels [69]. Other flow based systems used as vascular tissue model designs have used cone and plate viscometry principles to incorporate shear and waveform patterns missing in static culture conditions[70]. We previously described the adaptation of this technology, on human primary hepatocytes plated under a transwell culture modeled on the microarchitecture of the liver sinusoids (Figure 3A), to apply liver-derived blood flow parameters[71]. The restoration of transmural perfusion, circulatory hemodynamics and transport gradients missing in conventional culture systems was shown to make primary hepatocytes more in vivo-like in terms of morphology, function and responsiveness[71, 72]. This allowed primary hepatocytes to be cultured under close to physiological insulin/glucose media conditions with retention of metabolic pathways such as gluconeogenesis and glycogenolysis and exhibit de novo lipogenesis under high glucose and insulin milieus[73, 74]. The system has also been used to assess drug responses at clinically-relevant concentrations[75]. We evaluated the effects of Amiodarone in this system alongside obeticholic acid, an FXR agonist and a promising new drug in clinical trials for non-alcoholic steatohepatitis (NASH), as an inverse control. Their transcriptomic signatures of both drugs were compared, with the goal of gaining insights into overlapping mechanistic pathways involved in the development and treatment of steatohepatitis. Following restoration of biology in the system over 7 days, primary human hepatocytes from 5 separate donors were exposed to the two drugs for 48 hours at concentrations approximating both therapeutic and toxic levels, alongside DMSO controls. Whole genome transcriptomics by RNAseq, along with measurement of intracellular lipid accumulation was performed. The heatmaps of key pathways depicting differentially expressed genes relative to DMSO controls revealed that amiodarone impacts multiple pathways relevant to lipid accumulation in addition to its well appreciated effect down-regulating (blue) fatty acid beta oxidation genes (Figure 3B). This was reflected by marked down-regulation the ABCA family transporters that regulate the transfer of lipid into organelles and out of cells (Figure 3C) and simultaneous up-regulation (red) of fatty acid and triglyceride synthesis pathway genes (Figure 3D), offering additional possible explanations for its steatohepatitic potential. Interestingly, obeticholic acid strongly down-regulated most triglyceride synthesis genes (Figure 3D) while up-regulating some fatty acid metabolism genes (Figure 3B) supporting its observed beneficial effect on lowering steatosis in patients of NASH. Obeticholic acid modestly suppressed the lipid transporters but strongly up-regulated ABCG5 and ABCG8 (Figure 3C), which may reflect a potential for increased cholesterol efflux in addition to bile acids. These steatogenic potential of amiodarone was also reflected in the marked accumulation of lipid droplets evidenced by Nile red staining in the amiodarone treated hepatocytes (Figure 3F) relative to the DMSO (Figure 3E) and obeticholic acid (Figure 3G) treated conditions. Taken together, these data reflect the opposing effects of amiodarone and obeticholic acid on regulating key pathways involved in steatohepatitis.
Figure 3.
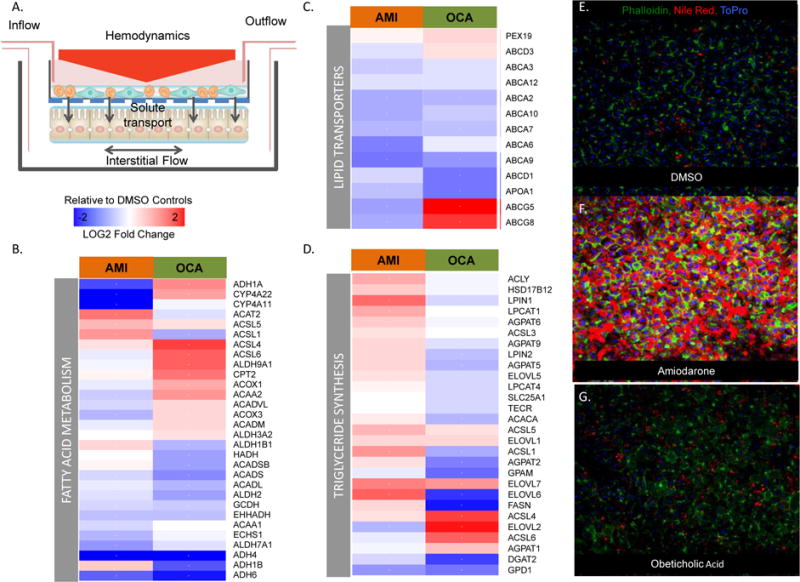
Assessment of Steatohepatitis in an Organotypic Model. (A) The previously described model uses liver-derived blood flow parameters to restore transmural perfusion, circulatory hemodynamics and transport gradients in the system. Primary hepatocytes from 5 donors were exposed to amiodarone (AMI) or obeticholic acid (OCA) in the system for 48 hours prior to performing transcriptomics and measuring lipid accumulation by Nile Red staining. (B) Heatmap of fatty acid beta oxidation genes reveals down-regulation by AMI while OCA selectively up-regulates multiple genes. (C) Both drugs down-regulated the lipid/cholesterol transporters of the ABCA family but OCA strongly up-regulated ABCG5 and ABCG8 (D) Fatty acid and triglyceride synthesis pathway genes were upregulated by AMI but mostly down-regulated by OCA. (E -G) These steatogenic potential of amiodarone was also reflected in the marked accumulation of lipid droplets evidenced by Nile red staining in the amiodarone treated hepatocytes (F) relative to the DMSO (E) and obeticholic acid (G) treated conditions.
4. Clinical Assessment and Management of Drug Induced Steatohepatitis
4.1: Identification of the subject at risk
The key consideration is assessment of the likelihood of a drug to cause hepatic steatosis or steatohepatitis is its mechanism of action. Specifically agents that increase delivery of lipids to the liver, decrease lipid oxidation, alter the incorporation of fatty acids in to other lipid classes or affect their transport out of the liver all have the potential to cause fat to accumulate in the liver. These insights should be available before moving such agents in to clinical trials. There is also a potential for off-target effects for agents designed to impact other pathways. Furthermore, distinction of DISH from background NASH or NAFLD is an important consideration especially in those with risk factors who are prescribed agents with the potential for DISH.
When considering the clinical assessment of DISH, one has to make distinctions between clinical trials. In clinical trials of agents with the potential for DISH or where steatosis of the liver has been noted in preclinical models, it is advisable to obtain baseline measures of steatosis and steatohepatitis. MR-based proton density fat fraction methods are currently considered the most accurate way to quantify hepatic fat[76]. However, such methods are expensive and not widely available and substantially increase the study burden on individual subjects. Alternatively, the fibroscan continuous attenuation parameter (CAP) score can be used at a “point-of-care” to assess the presence of hepatic steatosis[77, 78]. However, this measure is less quantitative and has not yet been validated to be sensitive to change. Regardless of the technology used, it is also advisable to obtain a liver stiffness measurement at the time of assessment of hepatic steatosis[79–81]. The presence of increased stiffness (> 7 Kp for fibroscan and > 2.9 kp for MRE) suggests the presence of underlying chronic liver disease with hepatic fibrosis. The presence of elevated AST and ALT values further increases the likelihood of underlying NASH with fibrosis, the phenotype at risk of progression to cirrhosis[82, 83]. Considering the widespread prevalence of NAFLD and NASH in the general population and particularly in those who are obese with other features of the metabolic syndrome[84, 85], it is likely that a substantial subset of subjects will have these features. This is particularly relevant for drugs being developed for cardiovascular disease and type 2 diabetes. The decision to include or exclude such individuals from clinical trials require a consideration of the need to generate generalizable data in multiple at risk populations, the potential risks to the individual subject and the likelihood of DILI or DISH with a given drug.
In clinical practice, there is growing acceptance of non-invasive tools to assess the presence of fatty liver disease and the risk of liver related outcomes. If these have not been used already in those with risk factors, they should be used prior to starting drugs with the potential for causing DISH. This allows assessment of the pre-therapeutic status and makes assessment of DISH much easier once therapy is started. Those with advanced disease, as assessed by a FIB-4, AST:platelet ratio, fibroscan or MRI should be triaged for more detailed assessment regardless of their symptom status. For clinical trials, it is probably prudent to exclude such individuals unless the study specifically targets this population.
4.2: Clinical Evaluation of DISH
In those without baseline evidence of NAFLD who are started on drugs with the potential for DISH, it seems rational to evaluate for the presence of DISH following a long-enough exposure time to the agent. Unfortunately, this is not clearly clarified for all potential agents. In such cases a 3–6 month interval from initiation of therapy could be considered since the natural course of DISH is generally slow and the probability of harm is low within this time frame for most drugs. Subsequent studies should be performed at 6–12 month intervals initially and annually after the first year of drug exposure. A measure of steatosis, changes in liver enzymes and changes in liver stiffness represent a minimal data-set for assessment of DISH. In those with new onset of steatosis, especially with elevation of liver enzymes and or increased liver stiffness should be offered a liver biopsy for verification of the presence and severity of DISH. The development of findings suggestive of steatohepatitis should lead to a consideration for drug discontinuation. The final decision for drug discontinuation must be individualized in clinical practice. In clinical trials, this decision is best made based on an a priori set of criteria established in the protocol to avoid imbalance in how subjects with new onset DISH are handled in the trial.
5.3: Management of DISH
The traditional paradigm in DILI is to stop the offending drug. However, in those who are being treated for severe life-threatening illnesses e.g. tamoxifen in those with breast cancer, this paradigm could be challenged and the treating physician must consider the competing risks of drug discontinuation versus drug toxicity. Further, with increasing literature to support the ability of various therapies to reverse NASH, one may also consider the use of such agents in managing DISH. It is however important to remember that none of current agents that have been shown to improve NASH such as vitamin E, pioglitazone etc have been systematically evaluated for DISH. Furthermore, none of these agents are approved for the treatment of NASH and their use would represent off-label use of these compounds. There is clearly a major unmet need to evaluate the ability to reverse DISH with such agents in relevant animal models and in-vitro systems and even in small scale human studies with intense monitoring of safety.
In those with evidence of NAFLD at baseline, the worsening of steatosis, elevation of liver enzymes from baseline values and increasing stiffness should be considered indicative of progressive disease. The decision to confirm this with a liver biopsy is up to the treating physician with input from the patient. In the absence of other obvious factors that the worsening disease state can be attributed to, one must consider the presence of superimposed worsening due to the drug and consider alternate agents or drug discontinuation. Certainly, progression to bridging fibrosis or cirrhosis should definitely lead to drug discontinuation.
Finally, in those where there is evidence of development of steatosis or worsening of steatosis without changes in liver enzymes or worsening of liver stiffness, the risk of adverse liver related outcomes is likely to be low. However, it may have an impact on the individual patients cardiometabolic risk. There are no long-term outcomes studies of DISH to provide data on the actual impact of DISH on cardio-metabolic outcomes and these risks have to be inferred from changes in cardio-metabolic risk factors.
If modifiable risk factors worsen, they could be managed accordingly. On the other hand, a substantial worsening of risk factors especially non-modifiable risk factors should warrant a discussion of drug discontinuation in clinical trials and at the very least a discussion of competing risks and alternate approaches in routine practice. These potential cardio-metabolic risks of drug induced fatty liver are also germane for those with DISH.
In summary, there is growing concern about the potential for many drugs to cause DISH or worsen underlying NASH. More work is needed especially in preclinical models to develop paradigms to minimize such risks, identify the problem early and develop ways to manage DISH to avoid DISH-related adverse outcomes and also allow individuals who need to be on the offending agents for other reasons to continue on their life-saving drugs while the DISH is effectively managed. Until then, when in doubt, it is advisable to stop the offending drug.
6. Conclusion
The increasing importance of DISH due to the high background of metabolic disease in the general population, and the limited management approaches for its detection and treatment make it a relevant problem. Due to complex mechanisms underlying DISH involving multiple cell types with sequential metabolic and inflammatory steps followed by cellular responses, there is a need for developing more physiologically relevant models with translational biomarkers and assays. These may eventually lead to newer management approaches for detection and treatment of DISH.
7. Expert Opinion
The phenomenon of DISH is truly complex on account of the multiple interconnected mechanistic pathways driving the process. Although the number of drugs causing DISH is a relatively small fraction of all fatty liver cases, the problem assumes a far greater significance against the background of the fast-growing metabolic NAFLD pandemic that tracks with obesity in the western world. Pre-existing NAFLD alters the baseline intracellular lipid content of hepatocytes, making the population more susceptible to drugs that affect the lipid homeostasis in the liver. It is known that steatosis can also significantly alter hepatic cytochrome p450 enzymes and drug transporters that control pharmacokinetics and drug metabolism, thereby enhancing the toxic potential of drugs. For instance, it is appreciated that the enzymatic activities of CYP1A2 and CYP2C19 decrease while CYP2A6 and CYP2C9 go up with increasing NAFLD severity in patients[86, 87]. Another significant effect of background NAFLD and NASH is the marked increase of expression and activity of the cytochrome P450 2E1 (CYP2E1) in patients[88]. This has direct consequences of increasing levels of reactive oxygen species (ROS) triggering oxidative stress and JNK overactivation[89], and could potentially impact the effects of concomitant steatohepatitic drugs. CYP2E1 can impair insulin signaling, further dysregulating the metabolic process and potentiating drug effects. Similarly, changes in expression and function of ATP-binding cassette (ABC) transporters in NASH are believed to make hepatocytes more susceptible to hepatocellular damage after administration of methotrexate[90].
Following the sequencing of the human genome, tremendous advances in the field of molecular biology have led to the -omics revolution. The shift away from a reductionist approach driven by tools to measure global cellular responses, coupled with refinements in big data analytics, have provided us unbiased ways to understand disease biology and drug pharmaco-toxicology. However, to be truly effective, the biological output from in vitro systems that feed the hypothesis generation and predictive algorithms needs to be reliable and reflect an in vivo-like response. The relationship between the insulin responsiveness and NASH particularly make it particularly important that preclinical evaluation of drugs causing DISH in vitro are done in metabolically competent cells in a patho-physiologically relevant milieu. As pointed out in this review, most in vitro systems continue to use cell culture media that are far removed from the physiological milieu experienced by cells in vivo. This may not impact a single endpoint, but assumes paramount importance while using –omics global approaches as the perturbation by the milieu can have a domino effect on multiple interconnected pathways. Another factor to consider while using in vitro systems is the stable retention of specific metabolic enzymes that can impact the underlying pathway being studied. For instance, in a recent study, the effect of obeticholic acid on NASH using human precision cut liver slices[91] noted the lack of on-target FXR response on key metabolic enzymes such as CYP7A1 and CYP8B1, and attributed it to the rapid loss of expression of these genes in the model. Often drugs causing DISH take a prolonged period to manifest pathologic changes[92, 93]. Loss of relevant lipid metabolism enzymes could severely compromise the ability of these systems to study such drugs. The use of advanced organotypic tissue models that retain in vivo-like functional responses, coupled with translational functional biomarkers and next-generational pan-omic measurements, may offer the best approach at gathering prior mechanistic knowledge and potential of a drug that can cause DISH.
Looking forward, the greatest opportunities lie in the diagnosis, monitoring and management of DISH. As of today, there is a need for better non-invasive biomarkers that not only help diagnose DISH, but also allow for monitoring DISH over a period of time. This is particularly important, as the only current management option is to withdraw the offending drug. In instances where there is significant risk with drug discontinuation and there are no suitable alternative medications, the ability to monitor DISH may offer a way to titrate treatment. Finally, the lack of definitive treatments highlights the need to develop therapies based on countering the underlying pathological mechanisms. Since there is a significant overlap with NASH, which is currently one of the most active areas of drug development, one can imagine utilizing similar targets and chemistries to be able to reverse the effects of DISH. Controlled clinical studies, with proper stratification of patient populations from other contributing causes of NAFLD would need to be conducted to confirm the benefits of these novel therapies.
Figure 2.
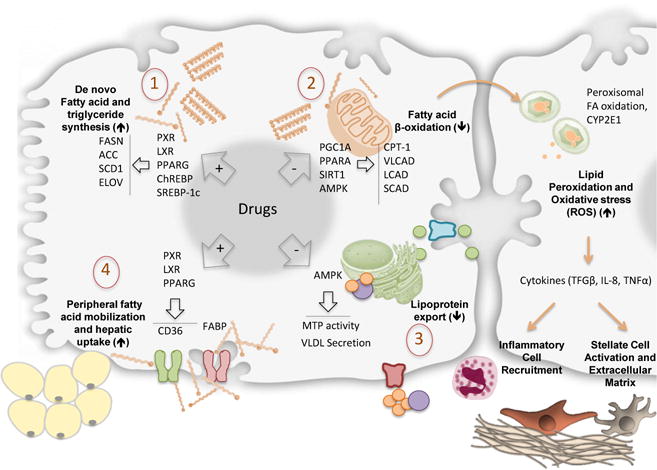
Mechanisms involved in Drug Induced Steatohepatitis (DISH). Accumulation of lipids and triglycerides within hepatocytes is driven by one or more of four different mechanisms: 1. Increased fatty acid synthesis. 2. Decreased fatty acid β-oxidation: 3. Decreased lipoprotein export and 4. Increased mobilization and uptake of fatty acids. Subsequent mitochondrial dysfunction, peroxisomal fatty acid oxidation and induction of CYP2E1 are responsible for increased lipid peroxidation and oxidative stress. The consequent induction of inflammatory cytokines like TGF-β, TNF-α and IL-8 results with inflammatory cell recruitment and stellate cell activation with extracellular matrix production.
Highlights.
Drug induced steatohepatitis (DISH) is increasingly important due to the high background of metabolic disease in the population.
Mechanisms causing DISH are complex due to involvement of multiple cell types and sequential metabolic and inflammatory steps followed by cellular responses.
Predicting and avoiding DISH pre-clinically is challenging due to limitations of existing models and endpoints.
More physiological models and translational biomarkers and assays may support discovery of newer management approaches for detection and treatment of DISH.
Acknowledgments
Funding
Part of the data presented in the paper was funded by National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases grant R44 DK091104
Footnotes
Declaration of Interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- 1.Farrell GC. Drugs and steatohepatitis. Semin Liver Dis. 2002;22:185–194. doi: 10.1055/s-2002-30106. [DOI] [PubMed] [Google Scholar]
- 2.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 3.Grieco A, Forgione A, Miele L, Vero V, Greco AV, Gasbarrini A, Gasbarrini G. Fatty liver and drugs. Eur Rev Med Pharmacol Sci. 2005;9:261–263. [PubMed] [Google Scholar]
- 4**.Rabinowich L, Shibolet O. Drug Induced Steatohepatitis: An Uncommon Culprit of a Common Disease. Biomed Res Int. 2015;2015:168905. doi: 10.1155/2015/168905. This review looks at the mechanistic underpinnings of the drugs more commonly associated with DISH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5**.Patel V, Sanyal AJ, Sterling R. Clinical Presentation and Patient Evaluation in Nonalcoholic Fatty Liver Disease. Clin Liver Dis. 2016;20:277–292. doi: 10.1016/j.cld.2015.10.006. This review covers the clinical approach to diagnosing and treating fatty liver arising from different causes including drugs. [DOI] [PubMed] [Google Scholar]
- 6.Begriche K, Massart J, Robin MA, Borgne-Sanchez A, Fromenty B. Drug-induced toxicity on mitochondria and lipid metabolism: mechanistic diversity and deleterious consequences for the liver. J Hepatol. 2011;54:773–794. doi: 10.1016/j.jhep.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Fong Y, Bentrem DJ. CASH (Chemotherapy-Associated Steatohepatitis) costs. Ann Surg. 2006;243:8–9. doi: 10.1097/01.sla.0000193599.57858.9b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wahlang B, Beier JI, Clair HB, Bellis-Jones HJ, Falkner KC, McClain CJ, Cave MC. Toxicant-associated steatohepatitis. Toxicol Pathol. 2013;41:343–360. doi: 10.1177/0192623312468517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9**.Schumacher JD, Guo GL. Mechanistic review of drug-induced steatohepatitis. Toxicol Appl Pharmacol. 2015;289:40–47. doi: 10.1016/j.taap.2015.08.022. This review is interesting as it describes specific chemical structural attributes of drugs associated with DISH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanisms of hepatotoxicity. Toxicol Sci. 2002;65:166–176. doi: 10.1093/toxsci/65.2.166. [DOI] [PubMed] [Google Scholar]
- 11.Kazmi F, Hensley T, Pope C, Funk RS, Loewen GJ, Buckley DB, Parkinson A. Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells) Drug Metab Dispos. 2013;41:897–905. doi: 10.1124/dmd.112.050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lowe R, Mussa HY, Nigsch F, Glen RC, Mitchell JB. Predicting the mechanism of phospholipidosis. J Cheminform. 2012;4:2. doi: 10.1186/1758-2946-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shayman JA, Abe A. Drug induced phospholipidosis: an acquired lysosomal storage disorder. Biochim Biophys Acta. 2013;1831:602–611. doi: 10.1016/j.bbalip.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14**.Amacher DE. The mechanistic basis for the induction of hepatic steatosis by xenobiotics. Expert Opin Drug Metab Toxicol. 2011;7:949–965. doi: 10.1517/17425255.2011.577740. This is another excellent review comparing different diverse mechanisms that are associated with DISH caused by different drugs. [DOI] [PubMed] [Google Scholar]
- 15.Cole LK, Jacobs RL, Vance DE. Tamoxifen induces triacylglycerol accumulation in the mouse liver by activation of fatty acid synthesis. Hepatology. 2010;52:1258–1265. doi: 10.1002/hep.23813. [DOI] [PubMed] [Google Scholar]
- 16.Zhao F, Xie P, Jiang J, Zhang L, An W, Zhan Y. The effect and mechanism of tamoxifen-induced hepatocyte steatosis in vitro. Int J Mol Sci. 2014;15:4019–4030. doi: 10.3390/ijms15034019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moya M, Gomez-Lechon MJ, Castell JV, Jover R. Enhanced steatosis by nuclear receptor ligands: a study in cultured human hepatocytes and hepatoma cells with a characterized nuclear receptor expression profile. Chem Biol Interact. 2010;184:376–387. doi: 10.1016/j.cbi.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 18**.Amacher DE. Strategies for the early detection of drug-induced hepatic steatosis in preclinical drug safety evaluation studies. Toxicology. 2011;279:10–18. doi: 10.1016/j.tox.2010.10.006. This review details the preclinical models and assays used to detect DISH. [DOI] [PubMed] [Google Scholar]
- 19.Labbe G, Pessayre D, Fromenty B. Drug-induced liver injury through mitochondrial dysfunction: mechanisms and detection during preclinical safety studies. Fundam Clin Pharmacol. 2008;22:335–353. doi: 10.1111/j.1472-8206.2008.00608.x. [DOI] [PubMed] [Google Scholar]
- 20.Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol. 2013;48:434–441. doi: 10.1007/s00535-013-0758-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J Lipid Res. 2009;50(Suppl):S86–90. doi: 10.1194/jlr.R800085-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomson DM, Winder WW. AMP-activated protein kinase control of fat metabolism in skeletal muscle. Acta Physiol (Oxf) 2009;196:147–154. doi: 10.1111/j.1748-1716.2009.01973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blas-Garcia A, Apostolova N, Ballesteros D, Monleon D, Morales JM, Rocha M, Victor VM, Esplugues JV. Inhibition of mitochondrial function by efavirenz increases lipid content in hepatic cells. Hepatology. 2010;52:115–125. doi: 10.1002/hep.23647. [DOI] [PubMed] [Google Scholar]
- 24.Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba R, He J, Lee JH, Khadem S, Ren S, Li S, Silverstein RL, Xie W. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology. 2008;134:556–567. doi: 10.1053/j.gastro.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 25**.Sahini N, Selvaraj S, Borlak J. Whole genome transcript profiling of drug induced steatosis in rats reveals a gene signature predictive of outcome. PLoS One. 2014;9:e114085. doi: 10.1371/journal.pone.0114085. This paper describes a transcriptomic approach using animal models to help predict DISH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benet M, Moya M, Donato MT, Lahoz A, Hervas D, Guzman C, Gomez-Lechon MJ, Castell JV, Jover R. A simple transcriptomic signature able to predict drug-induced hepatic steatosis. Arch Toxicol. 2014;88:967–982. doi: 10.1007/s00204-014-1197-7. [DOI] [PubMed] [Google Scholar]
- 27.Liu Z, Wang Y, Borlak J, Tong W. Mechanistically linked serum miRNAs distinguish between drug induced and fatty liver disease of different grades. Sci Rep. 2016;6:23709. doi: 10.1038/srep23709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diehl AM. Lessons from animal models of NASH. Hepatol Res. 2005;33:138–144. doi: 10.1016/j.hepres.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 29**.Kohli R, Feldstein AE. NASH animal models: are we there yet? J Hepatol. 2011;55:941–943. doi: 10.1016/j.jhep.2011.04.010. This paper highlights the challenges of animal models of fatty liver. [DOI] [PubMed] [Google Scholar]
- 30.Larter CZ, Yeh MM. Animal models of NASH: getting both pathology and metabolic context right. J Gastroenterol Hepatol. 2008;23:1635–1648. doi: 10.1111/j.1440-1746.2008.05543.x. [DOI] [PubMed] [Google Scholar]
- 31.London RM, George J. Pathogenesis of NASH: animal models. Clin Liver Dis. 2007;11:55–74. viii. doi: 10.1016/j.cld.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 32.Choi YJ, Lee CH, Lee KY, Jung SH, Lee BH. Increased hepatic Fatty Acid uptake and esterification contribute to tetracycline-induced steatosis in mice. Toxicol Sci. 2015;145:273–282. doi: 10.1093/toxsci/kfv049. [DOI] [PubMed] [Google Scholar]
- 33.Larosche I, Letteron P, Fromenty B, Vadrot N, Abbey-Toby A, Feldmann G, Pessayre D, Mansouri A. Tamoxifen inhibits topoisomerases, depletes mitochondrial DNA, and triggers steatosis in mouse liver. J Pharmacol Exp Ther. 2007;321:526–535. doi: 10.1124/jpet.106.114546. [DOI] [PubMed] [Google Scholar]
- 34.Lelliott CJ, Lopez M, Curtis RK, Parker N, Laudes M, Yeo G, Jimenez-Linan M, Grosse J, Saha AK, Wiggins D, Hauton D, Brand MD, O’Rahilly S, Griffin JL, Gibbons GF, Vidal-Puig A. Transcript and metabolite analysis of the effects of tamoxifen in rat liver reveals inhibition of fatty acid synthesis in the presence of hepatic steatosis. FASEB J. 2005;19:1108–1119. doi: 10.1096/fj.04-3196com. [DOI] [PubMed] [Google Scholar]
- 35.Gudbrandsen OA, Rost TH, Berge RK. Causes and prevention of tamoxifen-induced accumulation of triacylglycerol in rat liver. J Lipid Res. 2006;47:2223–2232. doi: 10.1194/jlr.M600148-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Van Summeren A, Renes J, Lizarraga D, Bouwman FG, Noben JP, van Delft JH, Kleinjans JC, Mariman EC. Screening for drug-induced hepatotoxicity in primary mouse hepatocytes using acetaminophen, amiodarone, and cyclosporin a as model compounds: an omics-guided approach. OMICS. 2013;17:71–83. doi: 10.1089/omi.2012.0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le TT, Urasaki Y, Pizzorno G. Uridine prevents tamoxifen-induced liver lipid droplet accumulation. BMC Pharmacol Toxicol. 2014;15:27. doi: 10.1186/2050-6511-15-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deschamps D, DeBeco V, Fisch C, Fromenty B, Guillouzo A, Pessayre D. Inhibition by perhexiline of oxidative phosphorylation and the beta-oxidation of fatty acids: possible role in pseudoalcoholic liver lesions. Hepatology. 1994;19:948–961. [PubMed] [Google Scholar]
- 39.Berson A, De Beco V, Letteron P, Robin MA, Moreau C, Kahwaji JEl, Verthier N, Feldmann G, Fromenty B, Pessayre D. Steatohepatitis-inducing drugs cause mitochondrial dysfunction and lipid peroxidation in rat hepatocytes. Gastroenterology. 1998;114:764–774. doi: 10.1016/s0016-5085(98)70590-6. [DOI] [PubMed] [Google Scholar]
- 40.Tong V, Teng XW, Chang TK, Abbott FS. Valproic acid II: effects on oxidative stress, mitochondrial membrane potential, and cytotoxicity in glutathione-depleted rat hepatocytes. Toxicol Sci. 2005;86:436–443. doi: 10.1093/toxsci/kfi185. [DOI] [PubMed] [Google Scholar]
- 41.Amacher DE, Martin BA. Tetracycline-induced steatosis in primary canine hepatocyte cultures. Fundam Appl Toxicol. 1997;40:256–263. doi: 10.1006/faat.1997.2389. [DOI] [PubMed] [Google Scholar]
- 42.Sassa S, Sugita O, Galbraith RA, Kappas A. Drug metabolism by the human hepatoma cell, Hep G2. Biochem Biophys Res Commun. 1987;143:52–57. doi: 10.1016/0006-291x(87)90628-0. [DOI] [PubMed] [Google Scholar]
- 43.Choi JM, Oh SJ, Lee JY, Jeon JS, Ryu CS, Kim YM, Lee K, Kim SK. Prediction of Drug-Induced Liver Injury in HepG2 Cells Cultured with Human Liver Microsomes. Chem Res Toxicol. 2015;28:872–885. doi: 10.1021/tx500504n. [DOI] [PubMed] [Google Scholar]
- 44.Cui W, Chen SL, Hu KQ. Quantification and mechanisms of oleic acid-induced steatosis in HepG2 cells. Am J Transl Res. 2010;2:95–104. [PMC free article] [PubMed] [Google Scholar]
- 45.Donato MT, Martinez-Romero A, Jimenez N, Negro A, Herrera G, Castell JV, O’Connor JE, Gomez-Lechon MJ. Cytometric analysis for drug-induced steatosis in HepG2 cells. Chem Biol Interact. 2009;181:417–423. doi: 10.1016/j.cbi.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 46.Kamalian L, Chadwick AE, Bayliss M, French NS, Monshouwer M, Snoeys J, Park BK. The utility of HepG2 cells to identify direct mitochondrial dysfunction in the absence of cell death. Toxicol In Vitro. 2015;29:732–740. doi: 10.1016/j.tiv.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 47**.Gerets HH, Tilmant K, Gerin B, Chanteux H, Depelchin BO, Dhalluin S, Atienzar FA. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol Toxicol. 2012;28:69–87. doi: 10.1007/s10565-011-9208-4. This paper highlights the suboptimal relevance of cell lines when compared to primary human hepatocytes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, Lucas J, Trepo C, Guguen-Guillouzo C. Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci U S A. 2002;99:15655–15660. doi: 10.1073/pnas.232137699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lambert CB, Spire C, Renaud MP, Claude N, Guillouzo A. Reproducible chemical-induced changes in gene expression profiles in human hepatoma HepaRG cells under various experimental conditions. Toxicol In Vitro. 2009;23:466–475. doi: 10.1016/j.tiv.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 50.Antherieu S, Chesne C, Li R, Camus S, Lahoz A, Picazo L, Turpeinen M, Tolonen A, Uusitalo J, Guguen-Guillouzo C, Guillouzo A. Stable expression, activity, and inducibility of cytochromes P450 in differentiated HepaRG cells. Drug Metab Dispos. 2010;38:516–525. doi: 10.1124/dmd.109.030197. [DOI] [PubMed] [Google Scholar]
- 51.Pant A, Kocarek T. Farnesol suppresses lipid accumulation in a HepaRG-based model of hepatic steatosis (1142.8) The FASEB Journal. 2014;28:1142.1148. [Google Scholar]
- 52.Rogue A, Antherieu S, Vluggens A, Umbdenstock T, Claude N, de la Moureyre-Spire C, Weaver RJ, Guillouzo A. PPAR agonists reduce steatosis in oleic acid-overloaded HepaRG cells. Toxicol Appl Pharmacol. 2014;276:73–81. doi: 10.1016/j.taap.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 53.Hannan NR, Segeritz CP, Touboul T, Vallier L. Production of hepatocyte-like cells from human pluripotent stem cells. Nat Protoc. 2013;8:430–437. doi: 10.1038/nprot.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rashid ST, Corbineau S, Hannan N, Marciniak SJ, Miranda E, Alexander G, Huang-Doran I, Griffin J, Ahrlund-Richter L, Skepper J, Semple R, Weber A, Lomas DA, Vallier L. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J Clin Invest. 2010;120:3127–3136. doi: 10.1172/JCI43122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sirenko O, Hesley J, Rusyn I, Cromwell EF. High-content assays for hepatotoxicity using induced pluripotent stem cell-derived cells. Assay Drug Dev Technol. 2014;12:43–54. doi: 10.1089/adt.2013.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berger DR, Ware BR, Davidson MD, Allsup SR, Khetani SR. Enhancing the functional maturity of induced pluripotent stem cell-derived human hepatocytes by controlled presentation of cell-cell interactions in vitro. Hepatology. 2015;61:1370–1381. doi: 10.1002/hep.27621. [DOI] [PubMed] [Google Scholar]
- 57.Yu Y, Liu H, Ikeda Y, Amiot BP, Rinaldo P, Duncan SA, Nyberg SL. Hepatocyte-like cells differentiated from human induced pluripotent stem cells: relevance to cellular therapies. Stem Cell Res. 2012;9:196–207. doi: 10.1016/j.scr.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58**.Wilkening S, Stahl F, Bader A. Comparison of primary human hepatocytes and hepatoma cell line Hepg2 with regard to their biotransformation properties. Drug Metab Dispos. 2003;31:1035–1042. doi: 10.1124/dmd.31.8.1035. This paper describes the differences between primary hepatocytes and cell lines. [DOI] [PubMed] [Google Scholar]
- 59.Rodriguez-Antona C, Donato MT, Boobis A, Edwards RJ, Watts PS, Castell JV, Gomez-Lechon MJ. Cytochrome P450 expression in human hepatocytes and hepatoma cell lines: molecular mechanisms that determine lower expression in cultured cells. Xenobiotica. 2002;32:505–520. doi: 10.1080/00498250210128675. [DOI] [PubMed] [Google Scholar]
- 60**.Baker TK, Carfagna MA, Gao H, Dow ER, Li Q, Searfoss GH, Ryan TP. Temporal gene expression analysis of monolayer cultured rat hepatocytes. Chem Res Toxicol. 2001;14:1218–1231. doi: 10.1021/tx015518a. This paper demonstrates the rapid decline of liver specific phenotype of primary hepatocytes in conventional cultures. [DOI] [PubMed] [Google Scholar]
- 61.Boess F, Kamber M, Romer S, Gasser R, Muller D, Albertini S, Suter L. Gene expression in two hepatic cell lines, cultured primary hepatocytes, and liver slices compared to the in vivo liver gene expression in rats: possible implications for toxicogenomics use of in vitro systems. Toxicol Sci. 2003;73:386–402. doi: 10.1093/toxsci/kfg064. [DOI] [PubMed] [Google Scholar]
- 62.Suckale J, Solimena M. Pancreas islets in metabolic signaling–focus on the beta-cell. Front Biosci. 2008;13:7156–7171. doi: 10.2741/3218. [DOI] [PubMed] [Google Scholar]
- 63.Bertrand F, Philippe C, Antoine PJ, Baud L, Groyer A, Capeau J, Cherqui G. Insulin activates nuclear factor kappa B in mammalian cells through a Raf-1-mediated pathway. J Biol Chem. 1995;270:24435–24441. doi: 10.1074/jbc.270.41.24435. [DOI] [PubMed] [Google Scholar]
- 64.Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ke B, Zhao Z, Ye X, Gao Z, Manganiello V, Wu B, Ye J. Inactivation of NF-kappaB p65 (RelA) in Liver Improves Insulin Sensitivity and Inhibits cAMP/PKA Pathway. Diabetes. 2015;64:3355–3362. doi: 10.2337/db15-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66**.Dash A, Inman W, Hoffmaster K, Sevidal S, Kelly J, Obach RS, Griffith LG, Tannenbaum SR. Liver tissue engineering in the evaluation of drug safety. Expert Opin Drug Metab Toxicol. 2009;5:1159–1174. doi: 10.1517/17425250903160664. This review details the evolution of organotypic models designed to restore physiological relevance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kane BJ, Zinner MJ, Yarmush ML, Toner M. Liver-specific functional studies in a microfluidic array of primary mammalian hepatocytes. Anal Chem. 2006;78:4291–4298. doi: 10.1021/ac051856v. [DOI] [PubMed] [Google Scholar]
- 68.Xia L, Ng S, Han R, Tuo X, Xiao G, Leo HL, Cheng T, Yu H. Laminar-flow immediate-overlay hepatocyte sandwich perfusion system for drug hepatotoxicity testing. Biomaterials. 2009;30:5927–5936. doi: 10.1016/j.biomaterials.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 69.Domansky K, Inman W, Serdy J, Dash A, Lim MH, Griffith LG. Perfused multiwell plate for 3D liver tissue engineering. Lab Chip. 2010;10:51–58. doi: 10.1039/b913221j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hastings NE, Simmers MB, McDonald OG, Wamhoff BR, Blackman BR. Atherosclerosis-prone hemodynamics differentially regulates endothelial and smooth muscle cell phenotypes and promotes pro-inflammatory priming. Am J Physiol Cell Physiol. 2007;293:C1824–1833. doi: 10.1152/ajpcell.00385.2007. [DOI] [PubMed] [Google Scholar]
- 71**.Dash A, Simmers MB, Deering TG, Berry DJ, Feaver RE, Hastings NE, Pruett TL, LeCluyse EL, Blackman BR, Wamhoff BR. Hemodynamic flow improves rat hepatocyte morphology, function, and metabolic activity in vitro. Am J Physiol Cell Physiol. 2013;304:C1053–1063. doi: 10.1152/ajpcell.00331.2012. This paper was the first publication of the model described in this paper to demonstrate amiodarone induced steatohepatitis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dash A, Deering TG, Marukian S, Thomas J, Desbans C, Alexandre E, Richert L, Blackman BR, Wamhoff BR. 52nd Annual Meeting of the Society of ToxicologySan Antonio. Texas: 2013. Human Primary Hepatocytes Under Controlled Hemodynamics Elicit Induction Responses to Drugs at Clinical Cmax Concentrations; p. 10. [Google Scholar]
- 73.Dash A, Deering TG, Marukian S, Thomas J, Wamhoff BR, Blackman BR. Physiological Hemodynamics And Transport Restore Insulin and Glucagon Responses in a Normal Glucose Milieu in Hepatocytes In Vitro. 73rd Scientific Sessions of the American Diabetes AssociationChicago. 2013 [Google Scholar]
- 74.T JT, Deering TG, Blackman BR, Wamhoff BR, Dash A. Development of an In-vitro Model of Hepatic Steatosis Using Rat Hepatocytes Under Controlled Hemodynamics in a Diabetic Milieu. American Association for the Study of Liver Diseases (AASLD); Boston: 2012. [Google Scholar]
- 75.Terelius Y, Figler RA, Marukian S, Collado MS, Lawson MJ, Mackey AJ, Manka D, Qualls CW, Jr, Blackman BR, Wamhoff BR, Dash A. Transcriptional profiling suggests that Nevirapine and Ritonavir cause drug induced liver injury through distinct mechanisms in primary human hepatocytes. Chem Biol Interact. 2015 doi: 10.1016/j.cbi.2015.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reeder SB, Hu HH, Sirlin CB. Proton density fat-fraction: a standardized MR-based biomarker of tissue fat concentration. J Magn Reson Imaging. 2012;36:1011–1014. doi: 10.1002/jmri.23741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sasso M, Audiere S, Kemgang A, Gaouar F, Corpechot C, Chazouilleres O, Fournier C, Golsztejn O, Prince S, Menu Y, Sandrin L, Miette V. Liver Steatosis Assessed by Controlled Attenuation Parameter (CAP) Measured with the XL Probe of the FibroScan: A Pilot Study Assessing Diagnostic Accuracy. Ultrasound Med Biol. 2016;42:92–103. doi: 10.1016/j.ultrasmedbio.2015.08.008. [DOI] [PubMed] [Google Scholar]
- 78.Sasso M, Miette V, Sandrin L, Beaugrand M. The controlled attenuation parameter (CAP): a novel tool for the non-invasive evaluation of steatosis using Fibroscan. Clin Res Hepatol Gastroenterol. 2012;36:13–20. doi: 10.1016/j.clinre.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 79.Deffieux T, Gennisson JL, Bousquet L, Corouge M, Cosconea S, Amroun D, Tripon S, Terris B, Mallet V, Sogni P, Tanter M, Pol S. Investigating liver stiffness and viscosity for fibrosis, steatosis and activity staging using shear wave elastography. J Hepatol. 2015;62:317–324. doi: 10.1016/j.jhep.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 80.Eddowes P, Sasso M, Fournier C, Vuppalanchi R, Newsome PN. Steatosis and liver stiffness measurements using transient elastography. Hepatology. 2016 doi: 10.1002/hep.28515. [DOI] [PubMed] [Google Scholar]
- 81.Macaluso FS, Maida M, Camma C, Cabibbo G, Cabibi D, Alduino R, Di Marco V, Craxi A, Petta S. Steatosis affects the performance of liver stiffness measurement for fibrosis assessment in patients with genotype 1 chronic hepatitis C. J Hepatol. 2014;61:523–529. doi: 10.1016/j.jhep.2014.04.045. [DOI] [PubMed] [Google Scholar]
- 82.Ballestri S, Nascimbeni F, Romagnoli D, Lonardo A. The independent predictors of NASH and its individual histological features. Insulin resistance, serum uric acid, metabolic syndrome, ALT and serum total cholesterol are a clue to pathogenesis and candidate targets for treatment. Hepatol Res. 2016 doi: 10.1111/hepr.12656. [DOI] [PubMed] [Google Scholar]
- 83.Torres DM, Harrison SA. NAFLD: Predictive value of ALT levels for NASH and advanced fibrosis. Nat Rev Gastroenterol Hepatol. 2013;10:510–511. doi: 10.1038/nrgastro.2013.138. [DOI] [PubMed] [Google Scholar]
- 84.Harnois F, Msika S, Sabate JM, Mechler C, Jouet P, Barge J, Coffin B. Prevalence and predictive factors of non-alcoholic steatohepatitis (NASH) in morbidly obese patients undergoing bariatric surgery. Obes Surg. 2006;16:183–188. doi: 10.1381/096089206775565122. [DOI] [PubMed] [Google Scholar]
- 85.Losekann A, Weston AC, de Mattos AA, Tovo CV, de Carli LA, Espindola MB, Pioner SR, Coral GP. Non-Alcoholic Steatohepatitis (NASH): Risk Factors in Morbidly Obese Patients. Int J Mol Sci. 2015;16:25552–25559. doi: 10.3390/ijms161025552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fisher CD, Lickteig AJ, Augustine LM, Ranger-Moore J, Jackson JP, Ferguson SS, Cherrington NJ. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab Dispos. 2009;37:2087–2094. doi: 10.1124/dmd.109.027466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lake AD, Novak P, Fisher CD, Jackson JP, Hardwick RN, Billheimer DD, Klimecki WT, Cherrington NJ. Analysis of global and absorption, distribution, metabolism, and elimination gene expression in the progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos. 2011;39:1954–1960. doi: 10.1124/dmd.111.040592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chtioui H, Semela D, Ledermann M, Zimmermann A, Dufour JF. Expression and activity of the cytochrome P450 2E1 in patients with nonalcoholic steatosis and steatohepatitis. Liver Int. 2007;27:764–771. doi: 10.1111/j.1478-3231.2007.01524.x. [DOI] [PubMed] [Google Scholar]
- 89.Schattenberg JM, Czaja MJ. Regulation of the effects of CYP2E1-induced oxidative stress by JNK signaling. Redox Biol. 2014;3:7–15. doi: 10.1016/j.redox.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hardwick RN, Clarke JD, Lake AD, Canet MJ, Anumol T, Street SM, Merrell MD, Goedken MJ, Snyder SA, Cherrington NJ. Increased susceptibility to methotrexate-induced toxicity in nonalcoholic steatohepatitis. Toxicol Sci. 2014;142:45–55. doi: 10.1093/toxsci/kfu156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91**.Ijssennagger N, Janssen AW, Milona A, Ramos Pittol JM, Hollman DA, Mokry M, Betzel B, Berends FJ, Janssen IM, van Mil SW, Kersten S. Gene expression profiling in human precision cut liver slices in response to the FXR agonist obeticholic acid. J Hepatol. 2016 doi: 10.1016/j.jhep.2016.01.016. This recent paper highlights the challenges of rapid decline of metabolic activity in liver slices. [DOI] [PubMed] [Google Scholar]
- 92.Ding X, Thung SN, Grewal P. Steatohepatitis secondary to long-term glucocorticoid treatment for congenital adrenal hyperplasia: a potential diagnostic pitfall. Semin Liver Dis. 2013;33:389–392. doi: 10.1055/s-0033-1358526. [DOI] [PubMed] [Google Scholar]
- 93.He J, Li G, Chen J, Lin J, Zeng C, Chen J, Deng J, Xie P. Prolonged exposure to low-dose microcystin induces nonalcoholic steatohepatitis in mice: a systems toxicology study. Arch Toxicol. 2016 doi: 10.1007/s00204-016-1681-3. [DOI] [PubMed] [Google Scholar]